

Abstract
Previous work with the classic T4 endonuclease V digestion of DNA from irradiated Drosophila cells followed by Southern hybridization led to the conclusion that Drosophila lacks transcription-coupled repair (TCR). This conclusion was reinforced by the Drosophila Genome Project, which revealed that Drosophila lacks Cockayne syndrome WD repeat protein (CSA), CSB, or UV-stimulated scaffold protein A (UVSSA) homologs, whose orthologs are present in eukaryotes ranging from Arabidopsis to humans that carry out TCR. A recently developed in vivo excision assay and the excision repair-sequencing (XR-Seq) method have enabled genome-wide analysis of nucleotide excision repair in various organisms at single-nucleotide resolution and in a strand-specific manner. Using these methods, we have discovered that Drosophila S2 cells carry out robust TCR comparable with that observed in mammalian cells. Our findings provide critical new insights into the mechanisms of TCR among various different species.
Keywords: nucleotide excision repair, UV DNA damage, DNA transcription, Drosophila, RNA polymerase II, cyclobutane pyrimidine dimers, Cockayne syndrome WD repeat protein (CSA), CSB, excision repair sequencing (XR-Seq), transcription-coupled repair (TCR), Rad28, Rad26
Introduction
Nucleotide excision repair (excision repair) is the main mechanism used by many organisms to remove cyclobutane pyrimidine dimers (CPDs)5 that are induced in DNA by UV light (1, 2). The mechanism of excision repair has been investigated in considerable detail in several model organisms including Escherichia coli, Saccharomyces cerevisiae, mice, humans, and more recently Arabidopsis thaliana (3, 4). In contrast, although Drosophila melanogaster is an extensively used model organism that has contributed significantly to the development of modern views on ionizing radiation and chemical mutagenesis and to the development of double-strand break repair and recombination models (5), there is rather limited information on nucleotide excision repair in this organism.
Work on excision repair in Drosophila is of special interest because, of all of the model organisms studied, ranging from E. coli to humans, it is the only one that has been reported to lack transcription-coupled repair (TCR) (6–10). This conclusion was further supported by genomic analyses that revealed that the CSA and CSB proteins, which are necessary for TCR (11) and are present in organisms ranging from yeast (Rad28 and Rad26, respectively) to humans, are missing in Drosophila and other insects in the order Diptera (5, 12, 13). Considering the importance of TCR in mutation avoidance (14), the apparent absence of TCR in Drosophila is rather surprising. Therefore, we decided to investigate this phenomenon with new biochemical tools that have become available since the previous experimental studies on the subject nearly 3 decades ago.
In transcription-coupled repair, damage in the template (transcribed strand (TS)), but not the nontranscribed strand (NTS), causes arrest of RNA polymerase (RNAP). The arrested RNAP is recognized by Mfd in E. coli (15, 16) and by CSB in eukaryotes (17). The bacterial TCR process is well-characterized and involves concerted removal of RNAP by the Mfd translocase and Mfd-mediated delivery of repair proteins, resulting in repair at an accelerated rate compared with repair of the NTS (15). In contrast, TCR is not well-understood in higher organisms. The mechanism is unlike that in E. coli, because the CSB translocase does not remove the stalled RNAP (18). Nevertheless, CSB, along with CSA, a WD repeat–containing protein with ubiquitin ligase activity (19), somehow enables the eukaryotic basal repair proteins (which are not homologous to proteins in E. coli) to produce a commonly 3–10-fold accelerated rate of TS repair, which varies depending upon the level of transcription (14, 16, 20).
In the classic TCR assay (14), UV-irradiated cells are incubated for various times to allow repair and then harvested. Genomic DNA is isolated and digested with appropriate restriction enzymes, and then duplicate aliquots from each time point are incubated with or without T4 endonuclease V, which incises sites of CPDs. Following separation of the DNA on a denaturing agarose gel, the gel is probed with gene-specific and strand-specific probes. The level of damage at each time point is calculated from the decrease in full-length fragment caused by T4 endonuclease. The loss of damage with time represents repair. Although this method has served to characterize essential properties of strand-specific repair, by its very nature, it is low-resolution and has the potential of missing TCR because it is an indirect method that is based on subtracting two large numbers from one another and because it is limited to analyzing one gene at a time. In contrast, the recently developed XR-Seq (excision repair-sequencing) assay directly captures and identifies the excised oligomers, and thus it directly and simultaneously measures repair throughout the genome (4, 20). Using XR-Seq, we find that Drosophila is proficient at TCR despite the previous reports and the absence of CSA and CSB in this organism.
Results
Excision by dual incision in Drosophila
Drosophila S2 cells were irradiated with 20 J m−2 of 254-nm UV light and incubated for various times at 27 °C. Then cells were lysed, and the low-molecular weight DNA was separated from genomic DNA by the Hirt procedure (4). After immunoprecipitation with anti-CPD antibodies, the samples were mixed with an internal control 50-mer DNA oligonucleotide, 3′-end–labeled, and separated on a sequencing gel. As seen in Fig. 1A, excision products in the range of 24–30 nt were detected with a median of 27–28 nt.
Figure 1.

In vivo excision assay and XR-Seq analysis in Drosophila. A, in vivo excision assay. S2 cells were irradiated with 20 J/m2 UV and incubated at 27 °C for 0–5 h. Then cells were lysed by the Hirt procedure, and low-molecular weight DNA in the supernatant was immunoprecipitated with anti-CPD antibodies. The oligonucleotides recovered were mixed with a 50-mer internal control oligonucleotide, 3′-end–labeled, and separated on a DNA-sequencing gel along with size markers. The excised oligomers are 24–32 nt in length, as is the case in human excision repair. B, size distribution of excised oligonucleotides as determined by XR-Seq analysis. A range of 24–32 and median of 27–28 nt was found. C, dual incision site determination by the distribution of thymine nucleotide in the reads. Frequency distribution of the 4 nucleotide bases in the 26-, 27-, 28-, and 29-nt-long reads as determined by XR-Seq is plotted. Potential T-T dinucleotides peak ∼6–8 nucleotides from the 3′-end and 21–23 nucleotides from the 5′-terminus.
Irradiated S2 cells were also processed for XR-Seq, in which the excision products are isolated, repaired by CPD photolyase, sequenced by next-generation sequencing (NGS), and aligned to the genome to create a map of the entire genome showing sites of repair in both strands at single-nucleotide resolution. The lengths of excision products sequenced (reads) after 10 min of repair were recovered with the frequency distribution shown in Fig. 1B, which shows a peak at 27 and 28 nt, in agreement with Fig. 1A results. Reads for products 26, 27, 28, and 29 nt in length were individually analyzed for nucleotide distribution as shown in Fig. 1C. The results show enrichment of pyrimidines 7–9 nt from the 3′-end. Thymines are more common than cytosines, consistent with the prevalence of cyclobutane thymine dimer photoproducts produced by UV. Thus, the location of the dual incisions made by the excision nuclease with respect to the CPD in S2 cells is essentially the same as in humans (3, 20). This finding is not surprising, considering that Drosophila possesses the complete set of orthologs to the basal excision repair factors present in mammals (5). It is worth noting that in human cells, it has been shown that the same excision pattern is produced by the transcription-coupled and global repair pathways (20).
Processing of the excised oligomers
In humans (20, 21), yeast (22), and Arabidopsis (23, 24), the CPD-containing oligomers are rapidly processed by exonuclease(s) such that the primary excision product has a half-life of about 1 h. As repair progresses, two populations of excision products, one ranging from 24 to 32 nt and centering at 26–27 nt and a second in the range of 15–20 nt and centering at 17–18 nt, are observed. These products as produced in NHF1 cells are illustrated in Fig. 2A. The 15–20-nt degradation products can be seen even at the earliest time point of 30 min and become prominent at 2 h. Thereafter, both the primary and secondary excision products detectable by radiolabeling decline, presumably due to low recovery and labeling efficiency of smaller degraded CPD-containing oligonucleotides (not shown). A comparison of the fate of excised oligomers in human and Drosophila cells (Fig. 2B) reveals an interesting contrast; in S2 cells, no degradation products are detectable in the first 2 h and, in fact, up to at least 12 h (data not shown). This indicates that Drosophila either lacks a nuclease that is present in organisms ranging from yeast to Arabidopsis to humans and that generates a similar degradation pattern in all of these other species, or this degradation intermediate is not stabilized in Drosophila as it is in other organisms.
Figure 2.
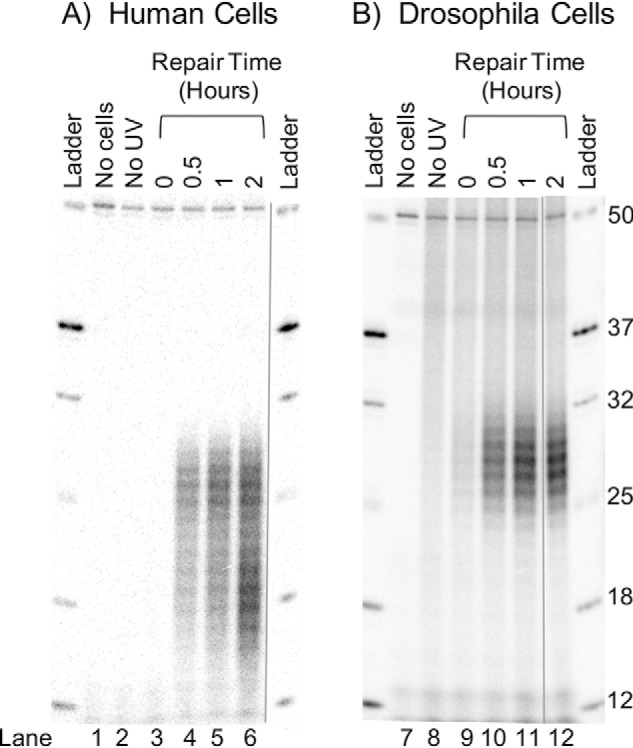
Nucleolytic processing of the excised oligonucleotides in human and Drosophila cells. Human NHF1 and Drosophila S2 cells were irradiated with 10 Jm−2 of 254-nm UV and incubated at 37 °C (human) or 27 °C (Drosophila) for the indicated times. Then the excised oligonucleotides were isolated, mixed with a 50-mer internal control, 3′-end labeled, and separated on DNA-sequencing gels. Shown are NHF1 (A) and S2 (B) cell excision products. Note that in NHF1 cells, the excised oligomers are degraded significantly by 2 h so that at this time point two classes of oligomers are seen, those centering at 26–27 nt (primary excision product) and those centering at 18–19 nt (degradation products). In contrast, in S2 cells, the size distribution of the excision products does not change with time.
Transcription-coupled repair in Drosophila
Previous work using strand-specific Southern hybridization did not detect a difference in the rates of repair of TS and NTS in individual Drosophila genes (6–8), and it was concluded that Drosophila does not perform TCR. This notion was further strengthened by genome analyses, which revealed that Drosophila and other Dipteran insects lacked CSA and CSB (5, 12, 13) and UVSSA (25) orthologs. However, it was also pointed out that in the absence of TCR, RNAPII stalled at damage sites in the TS would be expected to interfere with excision repair, as has been shown in E. coli (26), resulting in preferential repair of the NTS (5), which was not observed (6–10). To clarify these conflicting observations and theoretical expectations, we analyzed our XR-Seq data from S2 cells to obtain a global view of strand-specific repair of the Drosophila genome.
From our genomic repair maps of Drosophila, we initially focused on the betaTub60D and ade3 genes for TCR analysis because the lack of strand-specific repair in these genes, as determined by Southern blotting, was considered definitive evidence for lack of TCR in Drosophila (6–8). Fig. 3A shows screenshots of repair of the betaTub60D and ade3 genes in Drosophila as well as the human orthologues of these genes (TUBB3 and GART, respectively) in NHF1 and CSB cells, and Fig. 3B shows quantitative analysis of the TS and NTS repair of these genes. As is visually apparent from the screenshots, the TS of betaTub60D is repaired more efficiently than the NTS. A similar result is seen with repair of ade3. The quantitative data for TS and NTS reads in Fig. 3B clearly show that Drosophila performs TCR similar in magnitude to that seen in NHF1 human cells. A gene-by-gene survey of the genomic repair map (not shown) reveals varied levels of TCR in many but not all Drosophila genes, presumably reflecting varied levels or absence of transcription. For comparative purposes, Fig. 3 shows a screenshot and quantitation of repair for a Drosophila gene, CG10348, in which TCR is essentially absent.
Figure 3.

Transcription-coupled repair in human NHF1 and Drosophila S2 cells at the single gene level. Screenshots (A) of XR-Seq data of select genes are shown along with quantitative analysis (B) of the read numbers. Genes analyzed include the betaTub60D gene from Drosophila and the human ortholog TUBB3, as well as the Drosophila ade3 gene and the human ortholog GART. Preferential repair in the TS as compared with the NTS of these genes is seen in the S2 and NHF1 cells but is absent in CSB cells, which in fact show the opposite preference, perhaps due to inhibition of TS repair by RNAP stalled at the damage. The Drosophila CG10348 gene is also analyzed in the bottom panels of A and B to illustrate an example of the absence of TCR. The y axis in each case is RPKM. The repair signal for Drosophila appears stronger because it has a much smaller genome and thus more reads per kbp per million reads. Time points are 30 min (S2 cells), 36 min (NHF1), and 1 h (CSB).
Interestingly, in CSB cells (Fig. 3), there is not only a lack of TCR, but a preferential repair of the NTS in both the TUBB3 and GART genes. This result parallels the preferred NTS repair seen in E. coli cells lacking the TCR factor Mfd (27). This result in CSB cells is consistent with inhibition of repair by the stalled polymerase and merits more systematic investigation.
Genome-wide TCR in Drosophila
To examine TCR in a large sample of Drosophila genes, we analyzed the CPD repair of all nonoverlapping genes over 1 kbp (5,706 genes). The results are shown in Fig. 4A, with results for different repair time points for S2 cells in the top row, and corresponding time points for NHF1 cells (10,100 genes over 5 kbp) in the row below. Each panel illustrates the averaged repair from the transcription start site (TSS) to the transcription end site (TES) for all of the genes considered, plus the average repair 2 kbp upstream and downstream. As is clear from this figure, in both species, TCR across the gene bodies appears to have begun by 10 min, and TS repair is dominant at 30–36 min. The magnitudes of TCR in human and Drosophila cell lines are at comparable levels. At later time points, after the TS is cleared of damage, the preference shifts to the NTS strand. It takes longer to clear TS damage (over 8 h) in human cells, probably due to longer genes in humans. The robust TCR signal exhibited by S2 cells, which lack CS proteins, contrasts starkly with the complete absence of TCR in mutant human CSB cells, which are compared with NHF1 cells in Fig. 4B. In fact, the CSB cells exhibit a trend toward preferential NTS repair, as seen in the individual TUBB3 and GART genes (Fig. 3).
Figure 4.

Genome-wide analysis of TCR of transcribed genes in Drosophila and human cells. A, comparison of Drosophila S2 and human NHF1 cells. XR-Seq data from the indicated time points are plotted as average repair reads RPKM (y axis) along the length of a “unit gene.” For constructing a unit gene, a Drosophila data set of 5,706 genes was selected to include all genes >1 kbp with no genes that overlap or have a distance of less than 100 bp between adjacent genes. The human gene set includes 10,100 genes of >5 kbp in length with no overlaps and with a distance of at least 5 kbp between genes. For each species, the “unit gene” is 100 bins in length, and values for average repair were obtained by dividing each gene into 100 bins and averaging the repair values for each successive bin from 1 to 100. B, comparison of WT human NHF1 cells with cells from a patient with CSB. Data were analyzed as in A.
An interesting difference between the human and Drosophila repair profiles is seen in Fig. 4. As reported previously for NHF1 cells (20), and seen here at the 36-min and 1-h time points, there is a switch in strand preference for repair, from TS within the gene body to the NTS immediately upstream of the TSS. This is because of the well-known fact that upstream of the mammalian TSS, there is promoter- and enhancer-specific transcription “antisense” relative to the gene body (28). These “antisense” transcripts are not found in Arabidopsis (24), and our data suggest that they are absent in Drosophila as well. Instead, a dip in repair of both strands is observed immediately upstream of the TSS in Drosophila, presumably because of interference by transcription factors bound immediately upstream of coding sequences.
Discussion
Contrary to the view held for nearly 3 decades, we show that Drosophila performs TCR. In fact, the magnitude of the TS/NTS repair ratio in transcribed genes is comparable to the ratios of corresponding genes in human cells. We ascribe the previous failure to observe TCR in Drosophila partly to the limitation of the T4 endonuclease V digestion/Southern assay used in previous studies. An important additional consideration is the particular time points at which repair was analyzed. The earliest repair time point commonly used in the previous studies was 4 h post-UV treatment (6–10). TCR may be detected in mammalian cells following 4 h of repair (Fig. 4); however, in yeast, which have much smaller genes, TCR is complete within 1 h (22). It appears that a similar rapid kinetic profile in Drosophila, which also has relatively small genes, likely contributes to the discrepancy between our findings and previous studies.
Our finding of TCR in Drosophila raises a new question: analysis of the Drosophila genome and the genomes of other Dipteran insects has failed to reveal CSA, CSB, or UVSSA orthologs, which are known to be essential for TCR in organisms ranging from plants to humans. The E. coli TCR factor Mfd, which has a different evolutionary history than CSB, exhibits considerable sequence homology in the ATPase/translocase domain of the protein. It is possible that another translocase with great sequence divergence from all known TCR translocases couples transcription to repair upon the encounter of RNAP with a transcription-blocking DNA lesion in Drosophila. In fact, in Caenorhabditis elegans, other chromatin-remodeling factors have been suggested to function in place of CSB for TCR during certain developmental stages (29). There is a need for further work to identify such potential transcription-coupled repair factors in Drosophila.
A passive mechanism for TCR has been suggested in which the stalled RNAPII reveals to the basal repair factors lesions that would otherwise be concealed by nucleosomes. This mechanism is based partly upon observations of inhibition of repair by nucleosomes, seen in vitro (30), and repair of transcription-blocking lesions unimpeded by the presence of the stalled RNAPII, also seen in vitro (11, 31–33). This particular model as related to Drosophila is attractive in its independence of CS proteins. Further work is needed to clarify the involvement of CS proteins in TCR as well as in the developmental deficiencies associated with Cockayne syndrome (34).
Experimental procedures
Excision and XR-Seq assays
S2-DGRC cells were obtained from the Drosophila Genetic Resources Center. Cells were cultured at 27 °C in Schneider's medium with heat-inactivated fetal bovine serum at 10% (v/v). For UV treatments, cells were inoculated into R-150 plates and grown to about 25–80% confluence. Medium was gently removed from the semi-adherent monolayer, cells were irradiated with UV-C, and then fresh medium containing sterilized, conditioned medium was added to cells, and cells were incubated at 27 ºC. Excision assays employed 20 J m−2 (Fig. 1) or 10 J m−2 (Fig. 2), and XR-Seq assays employed 20 J m−2. Following predetermined repair times, plates were placed on ice, and cooled cells were harvested by scraping and rinsing with ice-cold PBS. Cells were pelleted, transferred to Eppendorf tubes, washed with cold PBS, and resuspended in 320–340 μl of cold TE. Samples were processed for excision and XR-Seq assays as described (27), using 5 μl of RNase A and proteinase K and using 9.2% sequencing gels. For both assays, samples were immunoprecipitated with anti-CPD antibody and then either radiolabeled (excision assay) or ligated to adapters and processed for sequencing (XR-Seq). Approximately 13% of the input CPD-containing excision products are recovered using this standard excision assay/XR-Seq immunoprecipitation procedure. One plate of S2 cells per repair time point was sufficient for excision or XR-Seq assay. Drosophila possesses photolyases, so cells were kept in the dark or under dim yellow illumination from the time of irradiation to the time of addition of NaCl to cell lysates. XR-Seq of NHF1 (normal human fibroblast) cells was performed as described previously (4). XR-Seq analysis of CSB cell repair utilized data deposited in a prior study (20).
Data analysis
At least 6 million unique mapped reads were obtained for each sample. Analysis of sequencing reads and data visualization were as described previously (35). The alignment genome version was dm6_UCSC for S2 cells and hg38_UCSC for human cell lines. For plotting average repair profiles as a unit gene, we chose the genes with length >1 kbp for Drosophila and >5 kbp for Homo sapiens, and the distance between genes was at least 100 bp for Drosophila and 5 kbp for Homo sapiens. With these criteria, the total number of genes selected was 5,706 for Drosophila and 10,100 for Homo sapiens. Each gene was evenly divided into 100 bins from the TSS to the TES, and 2 kbp (25 bins) upstream of TSS, 2 kbp (25 bins) downstream of TES, and for each bin, from first to last, an average value for each of the selected genes was obtained and plotted. The y axis average reads per kbp per million total reads (RPKM) for each bin was plotted with R. The raw data and alignment data have been deposited in the Gene Expression Omnibus under accession numbers GSE76391, GSE67941, and GSE138846.
Author contributions
N. D., Y. Y., A. S., and C. P. S. conceptualization; N. D., Y. Y., A. S., and C. P. S. formal analysis; N. D., Y. Y., A. S., and C. P. S. investigation; N. D., Y. Y., and C. P. S. methodology; N. D., Y. Y., L. A. L.-B., A. S., and C. P. S. writing-original draft; N. D., Y. Y., L. A. L.-B., A. S., and C. P. S. writing-review and editing; Y. Y. data curation; Y. Y., L. A. L.-B., and C. P. S. supervision; Y. Y. and C. P. S. visualization; L. A. L.-B., A. S., and C. P. S. project administration; A. S. funding acquisition.
Acknowledgment
Cells were provided by the Drosophila Genomics Resource Center, supported by National Institutes of Health Grant 2P40OD010949.
This work was supported by National Institutes of Health Grants GM118102 and ES027255 (to A. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CPD
- cyclobutane pyrimidine dimer
- TCR
- transcription-coupled repair
- XR-Seq
- excision repair sequencing
- TS
- transcribed strand or template strand
- NTS
- nontranscribed strand or coding strand
- NGS
- next-generation sequencing
- TSS
- transcription start site
- TES
- transcription end site
- CSA and CSB
- Cockayne Syndrome group A and B, respectively
- RPKM
- reads per kbp per million total reads
- RNAP
- RNA polymerase
- nt
- nucleotide(s)
- UVSSA
- UV-stimulated scaffold protein A.
References
- 1. Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 10.1146/annurev.bi.65.070196.000355 [DOI] [PubMed] [Google Scholar]
- 2. Wood R. D. (1997) Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468 10.1074/jbc.272.38.23465 [DOI] [PubMed] [Google Scholar]
- 3. Sancar A. (2016) Mechanisms of DNA repair by photolyase and excision nuclease (Nobel Lecture). Angew Chem. Int. Ed. Engl. 55, 8502–8527 10.1002/anie.201601524 [DOI] [PubMed] [Google Scholar]
- 4. Hu J., Li W., Adebali O., Yang Y., Oztas O., Selby C. P., and Sancar A. (2019) Genome-wide mapping of nucleotide excision repair with XR-seq. Nat. Protoc. 14, 248–282 10.1038/s41596-018-0093-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sekelsky J. (2017) DNA Repair in Drosophila: mutagens, models, and missing genes. Genetics 205, 471–490 10.1534/genetics.116.186759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Cock J. G., Klink E. C., Ferro W., Lohman P. H., and Eeken J. C. (1991) Repair of UV-induced pyrimidine dimers in the individual genes Gart, Notch and white from Drosophila melanogaster cell lines. Nucleic Acids Res. 19, 3289–3294 10.1093/nar/19.12.3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Cock J. G., Klink E. C., Ferro W., Lohman P. H., and Eeken J. C. (1992) Neither enhanced removal of cyclobutane pyrimidine dimers nor strand-specific repair is found after transcription induction of the β 3-tubulin gene in a Drosophila embryonic cell line Kc. Mutat. Res. 293, 11–20 10.1016/0921-8777(92)90003-L [DOI] [PubMed] [Google Scholar]
- 8. de Cock J. G., Klink E. C., Lohman P. H., and Eeken J. C. (1992) Absence of strand-specific repair of cyclobutane pyrimidine dimers in active genes in Drosophila melanogaster Kc cells. Mutat. Res. 274, 85–92 10.1016/0921-8777(92)90055-8 [DOI] [PubMed] [Google Scholar]
- 9. de Cock J. G., van Hoffen A., Wijnands J., Molenaar G., Lohman P. H., and Eeken J. C. (1992) Repair of UV-induced (6–4)photoproducts measured in individual genes in the Drosophila embryonic Kc cell line. Nucleic Acids Res. 20, 4789–4793 10.1093/nar/20.18.4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Helm P. J., Klink E. C., Lohman P. H., and Eeken J. C. (1997) The repair of UV-induced cyclobutane pyrimidine dimers in the individual genes Gart, Notch and white from isolated brain tissue of Drosophila melanogaster. Mutat. Res. 383, 113–124 10.1016/S0921-8777(96)00050-X [DOI] [PubMed] [Google Scholar]
- 11. Venema J., van Hoffen A., Karcagi V., Natarajan A. T., van Zeeland A. A., and Mullenders L. H. (1991) Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes. Mol. Cell. Biol. 11, 4128–4134 10.1128/MCB.11.8.4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sekelsky J. J., Hollis K. J., Eimerl A. I., Burtis K. C., and Hawley R. S. (2000) Nucleotide excision repair endonuclease genes in Drosophila melanogaster. Mutat. Res. 459, 219–228 10.1016/S0921-8777(99)00075-0 [DOI] [PubMed] [Google Scholar]
- 13. Sekelsky J. J., Brodsky M. H., and Burtis K. C. (2000) DNA repair in Drosophila: insights from the Drosophila genome sequence. J. Cell Biol. 150, F31–F36 10.1083/jcb.150.2.F31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanawalt P. C., and Spivak G. (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 10.1038/nrm2549 [DOI] [PubMed] [Google Scholar]
- 15. Selby C. P., and Sancar A. (1993) Molecular mechanism of transcription-repair coupling. Science 260, 53–58 10.1126/science.8465200 [DOI] [PubMed] [Google Scholar]
- 16. Selby C. P. (2017) Mfd protein and transcription-repair coupling in Escherichia coli. Photochem. Photobiol. 93, 280–295 10.1111/php.12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu J., Lahiri I., Wang W., Wier A., Cianfrocco M. A., Chong J., Hare A. A., Dervan P. B., DiMaio F., Leschziner A. E., and Wang D. (2017) Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature 551, 653–657 10.1038/nature24658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Selby C. P., and Sancar A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 272, 1885–1890 10.1074/jbc.272.3.1885 [DOI] [PubMed] [Google Scholar]
- 19. Epanchintsev A., Costanzo F., Rauschendorf M. A., Caputo M., Ye T., Donnio L. M., Proietti-de-Santis L., Coin F., Laugel V., and Egly J. M. (2017) Cockayne's syndrome A and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Mol. Cell 68, 1054–1066.e6 10.1016/j.molcel.2017.11.009 [DOI] [PubMed] [Google Scholar]
- 20. Hu J., Adar S., Selby C. P., Lieb J. D., and Sancar A. (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 29, 948–960 10.1101/gad.261271.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu J., Choi J. H., Gaddameedhi S., Kemp M. G., Reardon J. T., and Sancar A. (2013) Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 10.1074/jbc.M113.482257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li W., Adebali O., Yang Y., Selby C. P., and Sancar A. (2018) Single-nucleotide resolution dynamic repair maps of UV damage in Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. U.S.A. 115, E3408–E3415 10.1073/pnas.1801687115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Canturk F., Karaman M., Selby C. P., Kemp M. G., Kulaksiz-Erkmen G., Hu J., Li W., Lindsey-Boltz L. A., and Sancar A. (2016) Nucleotide excision repair by dual incisions in plants. Proc. Natl. Acad. Sci. U.S.A. 113, 4706–4710 10.1073/pnas.1604097113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oztas O., Selby C. P., Sancar A., and Adebali O. (2018) Genome-wide excision repair in Arabidopsis is coupled to transcription and reflects circadian gene expression patterns. Nat. Commun 9, 1503 10.1038/s41467-018-03922-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Al Khateeb W. M., Sher A. A., Marcus J. M., and Schroeder D. F. (2019) UVSSA, UBP12, and RDO2/TFIIS contribute to Arabidopsis UV tolerance. Front. Plant Sci. 10, 516 10.3389/fpls.2019.00516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Selby C. P., and Sancar A. (1990) Transcription preferentially inhibits nucleotide excision repair of the template DNA strand in vitro. J. Biol. Chem. 265, 21330–21336 [PubMed] [Google Scholar]
- 27. Adebali O., Chiou Y. Y., Hu J., Sancar A., and Selby C. P. (2017) Genome-wide transcription-coupled repair in Escherichia coli is mediated by the Mfd translocase. Proc. Natl. Acad. Sci. U.S.A. 114, E2116–E2125 10.1073/pnas.1700230114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Core L. J., Martins A. L., Danko C. G., Waters C. T., Siepel A., and Lis J. T. (2014) Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 46, 1311–1320 10.1038/ng.3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lans H., Marteijn J. A., Schumacher B., Hoeijmakers J. H., Jansen G., and Vermeulen W. (2010) Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet. 6, e1000941 10.1371/journal.pgen.1000941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hara R., Mo J., and Sancar A. (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol. 20, 9173–9181 10.1128/MCB.20.24.9173-9181.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selby C. P., Drapkin R., Reinberg D., and Sancar A. (1997) RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res. 25, 787–793 10.1093/nar/25.4.787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tremeau-Bravard A., Riedl T., Egly J. M., and Dahmus M. E. (2004) Fate of RNA polymerase II stalled at a cisplatin lesion. J. Biol. Chem. 279, 7751–7759 10.1074/jbc.M309853200 [DOI] [PubMed] [Google Scholar]
- 33. Lainé J. P., and Egly J. M. (2006) Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J. 25, 387–397 10.1038/sj.emboj.7600933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brooks P. J. (2013) Blinded by the UV light: how the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair 12, 656–671 10.1016/j.dnarep.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang Y., Adebali O., Wu G., Selby C. P., Chiou Y. Y., Rashid N., Hu J., Hogenesch J. B., and Sancar A. (2018) Cisplatin-DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues. Proc. Natl. Acad. Sci. U.S.A. 115, E4777–E4785 10.1073/pnas.1804493115 [DOI] [PMC free article] [PubMed] [Google Scholar]