

Abstract
Chromatin remodelers use helicase-like ATPase domains to reorganize histone–DNA contacts within the nucleosome. Like other remodelers, the chromodomain helicase DNA-binding protein 1 (Chd1) remodeler repositions nucleosomes by altering DNA topology at its internal binding site on the nucleosome, coupling different degrees of DNA twist and DNA movement to distinct nucleotide-bound states of the ATPase motor. In this work, we used a competition assay to study how variations in the bound nucleotide, Chd1, and the nucleosome substrate affect stability of Chd1–nucleosome complexes. We found that Chd1–nucleosome complexes formed in nucleotide-free or ADP conditions were relatively unstable and dissociated within 30 s, whereas those with the nonhydrolyzable ATP analog AMP-PNP had a mean lifetime of 4.8 ± 0.7 min. Chd1–nucleosome complexes were remarkably stable with ADP·BeF3− and the transition state analogs ADP·AlFX and ADP·MgFX, being resistant to competitor nucleosome over a 24-h period. For the tight ADP·BeF3−–stabilized complex, Mg2+ was a critical component that did not freely exchange, and formation of these long-lived complexes had a slow, concentration-dependent step. The ADP·BeF3−–stabilized complex did not require the Chd1 DNA-binding domain nor the histone H4 tail and appeared relatively insensitive to sequence differences on either side of the Widom 601 sequence. Interestingly, the complex remained stable in ADP·BeF3− even when nucleosomes contained single-stranded gaps that disrupted most DNA contacts with the guide strand. This finding suggests that binding via the tracking strand alone is sufficient for stabilizing the complex in a hydrolysis-competent state.
Keywords: nucleosome, chromatin remodeling, DNA binding protein, nucleoside/nucleotide analog, ATPase, chromodomain helicase DNA-binding protein 1 (Chd1), motor protein, superfamily 2 (SF2) ATPase, transition state analog, twist defect
Introduction
Chromatin remodelers are members of a broad superfamily of helicase and helicase-like ATPases referred to collectively as superfamily 2 (SF2)3 (1). Like the related superfamily 1 ATPases, SF2 proteins have a bi-lobed architecture with a central ATP-binding cleft (2). Both halves of the ATPase motor engage with nucleic acid substrate (single or dsDNA or RNA), and conformational changes in the motor, dictated by the centrally bound nucleotide, in turn alter the structure of the bound substrate. For chromatin remodelers, the substrate is the nucleosome, which consists of an octameric histone core wrapped by ∼146 bp of duplex DNA. Nucleotide-dependent action of chromatin remodelers can stimulate histone octamer assembly of the nucleosome, full or partial histone disassembly/exchange, and repositioning the histone core along DNA (3). A central question in the field is how remodelers stabilize distinct intermediate structures of the nucleosome as they cycle through binding, hydrolysis, and release of ATP.
In addition to chromatin remodelers, SF2 ATPases include translocating helicases as well as the typically nontranslocating DEAD-box RNA helicases. Given their nontranslocating nature, many DEAD-box proteins are thought to act as clamps or switches, binding tightly to RNA in an ATP-bound state, with release coupled to ATP hydrolysis (4, 5). For several DEAD-box helicases, extremely tight RNA binding with stability >1 day has been observed when the protein is loaded with ADP·BeF3−, believed to mimic a ground state of ATP (6). Many DEAD-box helicases bind and then destabilize duplex RNA when bound to ATP or transition state analogs, favoring tight binding to single-stranded RNA (5).
SF2 ATPases that translocate along nucleic acids, in contrast, have more transient interactions with their substrates, because movement along the nucleic acid requires coordinated binding and release. To move processively, the two halves of the ATPase alternate between gripping and repositioning along the nucleic acid, resulting in an inchworm-like translocation (2). Interestingly, even translocating SF2 proteins like NHP-II and Rep helicases have been trapped in highly stable complexes with nucleic acids using ADP·BeF3− or transition state mimics such as ADP·AlFX (7, 8). Because by definition, the transition state during ATP hydrolysis is only fleetingly visited, the long-lived stability of these complexes simply reflects the high affinity at one step in the translocation process.
Chromatin remodelers share several features with translocating helicases but appear to remain in a fixed position on the nucleosome while shifting DNA past the histone core. For several families of chromatin remodelers, including SWI/SNF, ISWI, Chd1, and SWR1, the ATPase motor acts at an internal site on the nucleosome called SHL2 (superhelix location 2) (9–12). Recent work has pointed to the twist diffusion model as the likely mechanism for shifting DNA around the histone core (13–17). The twist diffusion model states that if a short segment of nucleosomal DNA were to absorb an additional bp, it would create a twist defect that could be passed to neighboring segments. Diffusion of a 1-bp twist defect all the way around the nucleosome would correspond to a 1-bp displacement of DNA past the histone core.
In support of the twist diffusion model, the Chd1 remodeler was shown to affect the DNA twist at its SHL2-binding site in a nucleotide-dependent manner (14). In apo (nucleotide-free) and ADP-bound states, the Chd1 remodeler can pull nucleosomal DNA toward itself upon binding, suggestive of a DNA bulge corresponding to a twist defect at the SHL2-binding site. In contrast, the ATPase motor bound to ADP·BeF3− and transition state mimics prefers a distinct twist of DNA that would be incompatible with the twist defect created in apo/ADP states (14). From recent cryo-EM structures of ISWI and SWI/SNF remodelers, the twist defect in apo/ADP states was discovered to only have a shift in one of the two DNA strands (15–17). To create a full twist defect that can reposition nucleosomes along DNA, both strands must be shifted, and therefore this one-stranded shift with apo/ADP states implied that the other strand likely shifts in a subsequent ATP-bound state (15). When bound to the nonhydrolyzable ATP analog AMP-PNP, Chd1 did not change DNA twist on canonical 601 nucleosomes; yet Chd1 bound to ATP analogs did induce a twist defect on a 601 variant nucleosome, although in that case it was unclear whether the twist defect involved one or both strands (14). A full twist defect has been observed on nucleosomes in complex with the SWR1 remodeler bound to ADP·BeF3− (18). The nucleosome bound to SWR1 showed a 1-bp DNA bulge at the SHL2-binding site, which differed from nucleosomes bound to Chd1, ISWI, and SWI/SNF remodelers, where no twist defects were observed with ADP·BeF3− (16, 17, 19, 20).
We previously reported how different nucleotide-bound states of the Chd1 ATPase motor altered both the interactions and stability with nucleosomes (12, 14, 21). Here, we developed a competition assay to determine the rate of remodeler–nucleosome dissociation as a means for studying Chd1–nucleosome stability in different nucleotide states. We found that in apo and ADP conditions, Chd1 rapidly dissociated from nucleosomes, whereas Chd1–nucleosome complexes dissociated much more slowly with AMP-PNP. In the presence of ADP·BeF3− and the transition state mimics ADP·AlFX and ADP·MgFX, Chd1–nucleosome complexes were remarkably stable, maintaining a significant fraction of bound species after more than 24 h. The long-lived ADP·BeF3− complex was resistant to EDTA, indicating that Mg2+ ions do not exchange in this stabilized complex. Interestingly, a fraction of the highly stable ADP·BeF3− complex formed slowly, with a concentration-dependent rate-limiting step. Formation of the stable ADP·BeF3− complex did not require the DNA-binding domain of Chd1, and the complex did not show a preference for a particular side of the 601 nucleosome, suggesting that formation of the complex was DNA sequence-independent. The ADP·BeF3− complex also did not require the histone H4 tail and was resistant to a ssDNA gap in the guide strand. In contrast, a guide strand gap at the binding site disrupted Chd1–nucleosome complexes in AMP-PNP, highlighting nucleotide-specific differences critical for stabilizing the complex. Taken together, these results describe how the stability of Chd1–nucleosome complexes are affected by nucleotide- and substrate-dependent changes, providing additional insight into how remodeler–nucleosome contacts evolve throughout the chromatin-remodeling cycle.
Results
Chd1–nucleosome complexes are extremely long-lived with transition state analogs
To examine the rate of Chd1 dissociation from nucleosomes, we developed a native gel competition assay. In native polyacrylamide gels, Chd1–nucleosome complexes are relatively stable, as evidenced by the appearance of discrete, supershifted nucleosome species correlated with increasing amounts of Chd1. Before entering the gel, these complexes can be disrupted by incubation with excess naked DNA. Such disruption is desirable and typically carried out for nucleosome sliding assays, where it is necessary to observe migration patterns of free nucleosomes (22). We reasoned that if the rate of dissociation were slow enough, it should be possible to observe Chd1–nucleosome complexes in the presence of competitor, because complexes would be both stabilized and separated from competitor upon entering the gel. Thus, the fraction of Chd1–nucleosome complexes visualized on a native gel should reflect the amount of nucleosomes stably bound to Chd1 at the time the sample is loaded on the gel.
Although naked DNA was an efficient competitor in some conditions, it was unable to efficiently outcompete nucleosomes in the presence of nucleotide analogs. Therefore, unlabeled nucleosomes were used as competitor. For these competition experiments, Chd1 was mixed with FAM-labeled nucleosome, in the presence or absence of nucleotide, and allowed to incubate for a defined time. Then, a 100-fold molar equivalent of competitor nucleosome was added to the reaction, and after a specific amount of time with competitor, each reaction was loaded onto a native acrylamide gel (Fig. 1A). Because the step of loading the sample on the gel was a critical aspect of preventing further competition, reactions were carried out in a time-reverse fashion, where the sample with the longest incubation with competitor was initiated first, and shortest competition was initiated last. This reversed order of incubation was necessary to consecutively load all samples on the gel in a short time window.
Figure 1.

A competition assay to determine stability of Chd1–nucleosome complexes. A, schematic workflow of a competition assay to determine off rates for Chd1. B, Chd1–nucleosome complexes are more stable in the presence of the nonhydrolyzable ATP analog AMP-PNP compared with ADP or no nucleotide (apo). As indicated, Chd1 (80 nm) was added to FAM-labeled 12N12 nucleosomes (20 nm), and after a 5-min incubation, reactions were competed with 12N9 unlabeled nucleosomes (2 μm). Lanes 2, 10, and 18 show reactions where unlabeled competitor nucleosome was first mixed with labeled nucleosome prior to addition of Chd1. Binding reactions were resolved on native acrylamide gels. Each gel is a representative of four or more experiments. C, quantification of Chd1–nucleosome dissociation over time. Disappearance of bound complexes in AMP-PNP was fit as a single exponential decay, giving an observed rate of 0.21 ± 0.03 min−1. Stably bound complexes were not detected under apo and ADP conditions. Each point represents the mean from four experiments, with error bars showing S.D. D, competition experiments reveal long-lived Chd1–nucleosome complexes in the presence of transition state analogs ADP·BeF3−, ADP·AlFX, and ADP·MgFX. Note that units of time are in hours. For these experiments, Chd1 (40 nm) was added to FAM-labeled 40N40 nucleosomes (10 nm), and after 2-h incubation, the reactions were competed with unlabeled 26N33 nucleosomes (1 μm). Lanes 1, 8, and 15 show reactions where unlabeled competitor nucleosome was first mixed with labeled nucleosome prior to addition of Chd1. Each gel is representative of five or more experiments. E, quantification of Chd1–nucleosome stability over time with transition state analogs. Each point represents an average of five or more experiments, with error bars showing S.D.
To determine how the nucleotide-bound state of the ATPase motor affects the rate of dissociation, we performed assays in the absence as well as the presence of different nucleotides: ADP, the nonhydrolyzable ATP analog AMP-PNP, and transition state mimics (Fig. 1, B–E). To ensure that the competitor nucleosome was capable of removing free Chd1, control reactions were performed where competitor nucleosome was first mixed with labeled nucleosome before addition of Chd1. With competitor and labeled nucleosome premixed, between 2 and 5% of binding sites on labeled nucleosome were bound by Chd1 (Fig. 1, B, lanes 2, 10, and 18, and D, lanes 1, 8, and 15). Performed with each experiment, these control reactions gave a baseline above which any observed signal was considered to be time-sensitive Chd1–nucleosome complex.
In nucleotide-free (apo) and ADP conditions, no appreciable amount of complex was observed after addition of competitor nucleosome (Fig. 1B, lanes 1–16). The evidence of supershifted nucleosome species in the absence of competitor (Fig. 1B, lanes 3 and 11) suggests that disappearance of the supershifted species was too fast to be measured under these conditions. In contrast, Chd1–nucleosome complexes were evident after addition of competitor in AMP-PNP conditions (lanes 17–24). The loss of Chd1–nucleosome complexes over time fit to a single exponential decay, with a rate of 0.21 ± 0.03 min−1 (Fig. 1C). Notably, the intercept for the single exponential decay (∼0.4) was far lower than the fraction observed in the absence of competitor (>0.95), indicating that a significant population of Chd1–nucleosome complexes dissociated too rapidly to be observed with this method. For these experiments, labeled nucleosomes had 12 bp of flanking DNA on each side (12N12) and competed with 12N9 unlabeled nucleosomes. Similar results for apo, ADP, and AMP-PNP were obtained for FAM-labeled nucleosomes with longer flanking DNA (40N40).
In ADP·BeF3−, the Chd1–nucleosome complex was much more stable. We previously reported that Chd1 bound more tightly to nucleosomes in ADP·BeF3− compared with AMP-PNP (12) but also that the rate of binding in ADP·BeF3− was slower (21). To account for the higher affinity and slower on-rate, we therefore expected the off-rate to be slower in ADP·BeF3−. Indeed, once formed, Chd1–nucleosome complexes were quite stable in ADP·BeF3−, even after incubation with competitor nucleosomes for 24 h (Fig. 1D, lanes 1–7).
The stability of Chd1–nucleosome complexes in ADP·BeF3− can be divided into three phases. In the first phase, a population of complexes dissociated rapidly, as evidenced by the reduced amount of complex with the first time point postcompetitor (lanes 2 and 3). This rapid dissociation occurred within 30 s and likely included Chd1 proteins bound nonspecifically. In a second phase, a portion of Chd1–nucleosome complexes dissociated slowly over the 24-h time course, often with an estimated mean lifetime of ∼2 h (compared with a mean lifetime in AMP-PNP of ∼0.08 h). The extent of dissociation during this phase was variable, depending on buffer and reagent conditions; in some cases the bound fraction was reduced up to 20%, whereas in other cases, there was no appreciable loss of complex between the first and the 24 h time points (Fig. 2B). Finally, in ADP·BeF3− conditions, a significant amount of complex remained intact at the 24-h time point, indicating a remarkably slow dissociation that resembled the highly stable DEAD Box-RNA clamped complexes specifically stabilized in ADP·BeF3− conditions (6).
Figure 2.
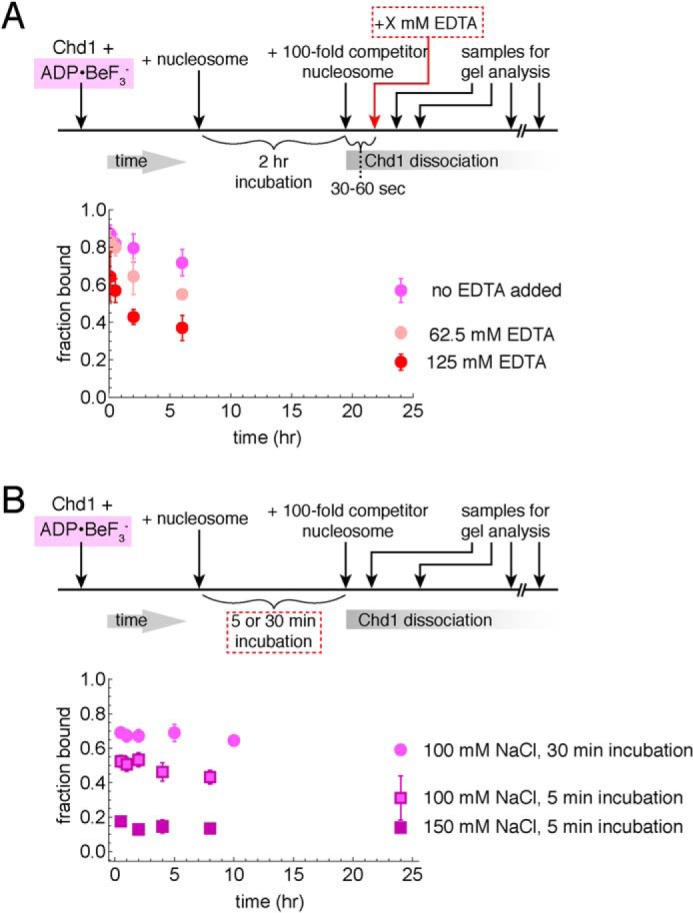
Effects of buffer components on stability of Chd1–nucleosome transition state complexes. A, excess EDTA does not disrupt long-lived complexes. Competition experiments were performed using 10 nm FAM-labeled 40N40 nucleosomes, 40 nm Chd1, and 1 μm unlabeled 26N33 nucleosomes in the presence of ADP·BeF3−. Data points are the mean values ± S.D. from three or more experiments. The “no EDTA” data are equivalent to those shown in Fig. 1D. B, salt and incubation time affect the initial amount of ADP·BeF3−–stabilized complex but not its lifetime. For these experiments, 60 nm Chd1 was bound to 20 nm FAM-labeled 12N12 nucleosomes and challenged with 2 μm of unlabeled 12N9 nucleosomes. Each point is the mean ± S.D. from three or more experiments.
ADP·BeF3− can represent different states of the hydrolysis cycle, from more ATP-like to a posthydrolysis ADP + Pi type state (23, 24). In contrast to the pyramidal shape of BeF3−, the planar geometry of AlFX and MgFX makes these γ-phosphate substitutes more specific for mimicking a pentacoordinate transition state achieved during hydrolysis of the γ-phosphate. We performed competition experiments with ADP·AlFX, and ADP·MgFX to see whether this long-lived complex could be recapitulated with the ATPase motor in a transition state geometry. Indeed, both ADP·AlFX and ADP·MgFX promoted >24-h stability of Chd1–nucleosome complexes (Fig. 1, D and E), indicating that a highly stable Chd1–nucleosome complex can be achieved by trapping the ATPase motor with transition state mimics.
Magnesium does not exchange in the long-lived complexes
Although, by definition, Chd1 does not exchange during the binding reaction in the presence of competitor, we wondered whether other components were also bound equally tightly in the complex. To determine whether critical magnesium ions exchanged, we performed experiments in the presence of excess EDTA, added after formation of Chd1–nucleosome complexes. Control experiments showed that addition of 62.5 mm EDTA prior to Chd1 blocked formation of stable Chd1–nucleosome complexes (Fig. S1). Thus, if Mg2+ were released during the binding reaction, EDTA added after formation of the complex should prevent exchanging Mg2+ from rebinding and thus destabilize the complex. We performed the competition experiment with increasing amounts of EDTA and found that even up to 125 mm EDTA, long-lived complexes were still apparent after 24 h (Fig. 2A). The starting amount of complex decreased with increasing EDTA concentrations, which we attribute to the intermediate forms of the complex being sensitive to Mg2+ exchange. However, the finding that even with high EDTA, complexes were still evident after 24 h suggests that bound Mg2+ does not exchange with the solution in the long-lived complex.
Ionic strength affects initial formation more strongly than longevity of the ADP·BeF3−–stabilized complex
We altered the NaCl concentration in the buffer, to see whether the complexes, once formed, were salt-sensitive. We found that the amount of complex forming initially was higher with lower salt; yet the relative amount of complex, once formed, remained constant (Fig. 2B). Therefore, these differences in ionic strength affected the fraction of bound Chd1 molecules capable of forming a long-lived complex but did not make an appreciable impact on the integrity of the complex over the limited range tested.
Formation of long-lived complexes has a slow, concentration-dependent phase
With increased incubation time, a higher fraction of nucleosomes was bound in the long-lived state (Fig. 2B). This suggests a time dependence in forming the highly stable, ADP·BeF3−–dependent complex. To investigate this further, we performed competition experiments where the incubation time was varied (Fig. 3). For these experiments, Chd1–nucleosome complexes were challenged with competitor nucleosome for a fixed time period prior to gel analysis. As shown in Fig. 3, the fraction of bound nucleosomes increased in a time-dependent, single exponential rise. These single exponential fits had significantly high nonzero intercepts, suggesting that up to half of the total fraction bound was established prior to the first time point. Binding therefore appeared biphasic, with extremely rapid binding followed by a much slower binding phase. Changing the time of the competitor challenge (1 min versus 2 h) only modestly affected the observed rates of time-dependent binding: 0.27 ± 0.08 min−1 for 1-min competitor and 0.18 ± 0.07 min−1 for 2-h competitor, both at 40 nm Chd1. In contrast, the slower phase increased roughly 4-fold with a 4-fold higher concentration of Chd1, with 320 nm yielding an observed rate of 1.0 ± 0.3 min−1 (Fig. 3). Formation of the ADP·BeF3−–dependent complexes therefore appeared to have a component that was both time-sensitive and concentration-dependent.
Figure 3.
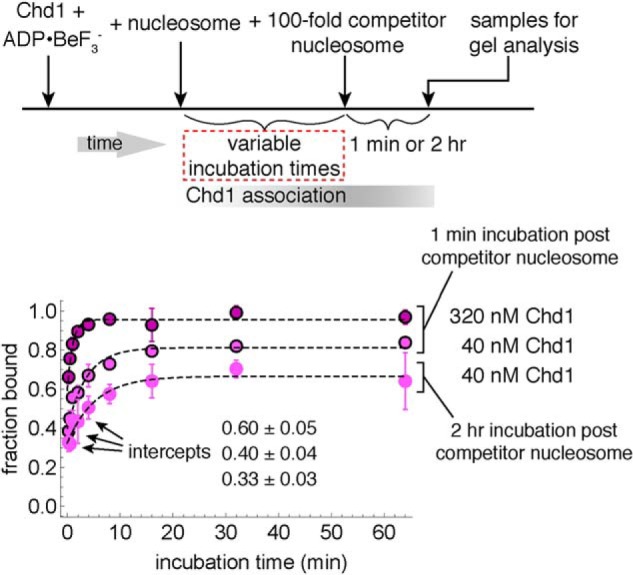
Time-dependent formation of long-lived Chd1–nucleosome complexes. For these experiments, 40 or 320 nm Chd1 was incubated with 10 nm FAM-labeled 40N40 nucleosomes and challenged with 1 μm unlabeled 26N33 nucleosomes. The apparent on-rates, fit with single exponential equations (dotted traces), were 1.0 ± 0.3 min−1 (320 nm Chd1, 1 min), 0.27 ± 0.08 min−1 (40 nm Chd1, 1 min), and 0.18 ± 0.07 min−1 (40 nm Chd1, 2 h). For the 1-min incubations with competitor, the values are averages of two separate experiments, with error bars indicating the ranges. For the 2-h incubation with competitor, the data are the mean values ± S.D. from four experiments.
The Chd1 DNA-binding domain is not required for forming stable, long-lived complexes with the nucleosome
We previously showed that Chd1 could interact with nucleosomes even when its DNA-binding domain (DBD) had been deleted (12). In cryoEM structures of Chd1–nucleosome complexes, however, the DNA-binding domain makes a large interface with both DNA flanking the nucleosome as well as the chromo-ATPase portion of the remodeler (19, 20). To investigate the extent that the DBD might contribute to stabilizing the long-lived Chd1–nucleosome complexes obtained in ADP·BeF3−, we performed competition experiments with Chd1(ΔDBD), which lacks the DBD. In the presence of competitor nucleosome, a significant fraction of Chd1(ΔDBD)–nucleosome complexes were evident even after 7 h (Fig. 4A). It was difficult to determine for certain whether Chd1(ΔDBD) could remain bound for >24 h because the starting fraction of bound nucleosomes was consistently low for Chd1(ΔDBD). In these types of competition experiments, we often observed up to a ∼20% loss of complex during a 24-h competition experiment (e.g. Fig. 1D), which could interfere with observing 24-h stable complexes when initial quantities of starting complex were low. Nonetheless, the clear presence of complex after 7 h demonstrates a long-lived species and indicates that the DBD is dispensable for forming highly stable Chd1–nucleosome complexes.
Figure 4.
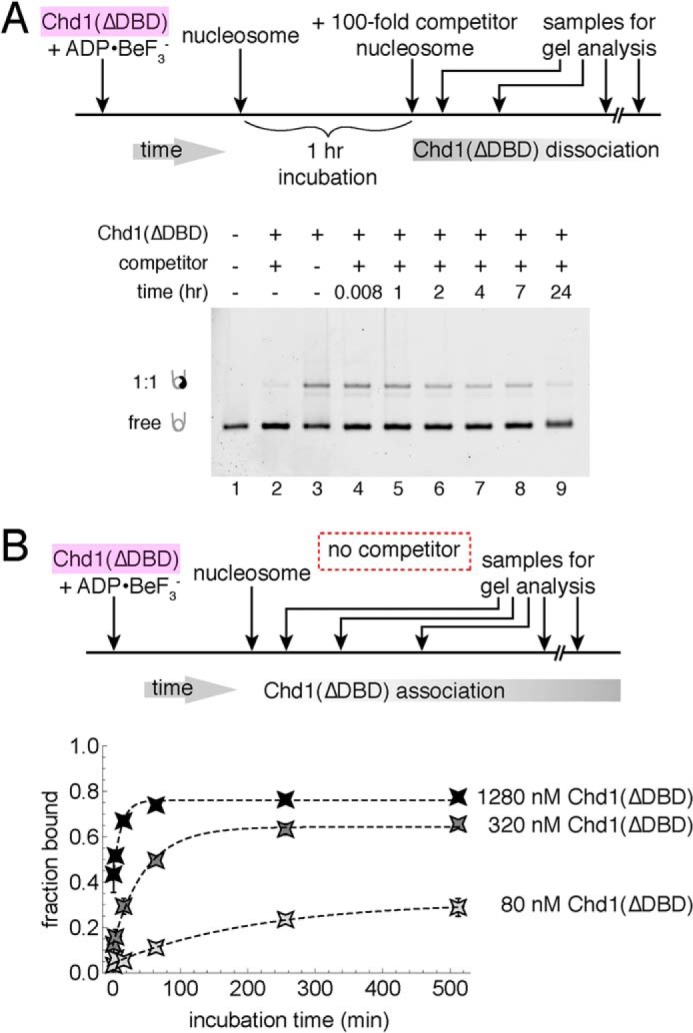
The Chd1 DNA-binding domain is not required for forming long-lived complexes. A, competition experiments showing stable binding of Chd1(ΔDBD) to nucleosomes in ADP·BeF3−. For these experiments, 80 nm Chd1 was incubated with 20 nm FAM-labeled 12N12 nucleosomes and challenged with 2 μm unlabeled 12N9 nucleosomes. Lane 2 shows a reaction where unlabeled competitor was mixed with labeled nucleosome prior to addition of Chd1(ΔDBD). The native acrylamide gel is representative of two experiments at this Chd1(ΔDBD) concentration. B, association of Chd1(ΔDBD) with nucleosomes is time- and concentration-dependent. At three different concentrations of Chd1(ΔDBD), the fraction bound increases over time, with each symbol indicating an average from four experiments, and error bars (often obscured by symbols) indicating S.D. Dotted lines show single exponential fits, with observed rates of 0.09 ± 0.02 min−1 (1280 nm Chd1), 0.021 ± 0.004 min−1 (320 nm Chd1), and 0.005 ± 0.002 min−1 (80 nm Chd1).
Interestingly, Chd1(ΔDBD) did not readily form higher order complexes with nucleosomes in the absence of competitor. In the competition experiment, the fraction of bound complex was similar at short time points in the absence and presence of competitor (Fig. 4A, compare lanes 3 and 4). Also, the addition of competitor to Chd1(ΔDBD) did not chase off slower migrating species in the gel, unlike the case for Chd1 (Fig. 1D, compare lanes 2 and 3). This result suggests that the DBD is largely responsible for the higher order complexes that rapidly disassemble in the presence of competitor nucleosomes. The Chd1(ΔDBD)–nucleosome complex observed without competitor therefore appears to represent the long-lived species.
The typical low amounts of stable Chd1(ΔDBD)–nucleosome complexes may have been due to weaker initial interactions of Chd1(ΔDBD) with the nucleosome. To better quantify how loss of the DBD impacts formation of long-lived complexes, we monitored time-dependent appearance of Chd1(ΔDBD)–nucleosome complexes in the absence of competitor (Fig. 4B). As observed for Chd1, longer incubation times allowed an increasing amount of Chd1(ΔDBD) to bind to nucleosomes. Like Chd1 with the DBD, the rate of Chd1(ΔDBD) binding was concentration-dependent but ∼50-fold slower, with 80, 320, and 1280 nm Chd1(ΔDBD) giving rates of 0.005 ± 0.002, 0.021 ± 0.004, and 0.09 ± 0.02 min−1, respectively. The DBD therefore increases the rate that Chd1 can form a stable ADP·BeF3− complex with the nucleosome, presumably because its association with DNA increases the effective concentration of the chromo-ATPase portion of Chd1 around the nucleosome.
Rapid formation of stable ADP·BeF3− complexes is largely DNA sequence-independent
With the 2-fold symmetry of the nucleosome, there are two SHL2 sites where the Chd1 ATPase motor can engage (12, 20). By native gel, we observed both 1:1 and 2:1 complexes; yet with short incubation times, the complex was typically undersaturated, at a 1:1 ratio. We wondered whether the 1:1 represented a random association of Chd1 to either SHL2, or whether one SHL2 site was preferentially bound. Nucleosomes made with the 601 sequence are well-known for asymmetric properties (25–29). One sequence motif that is important in defining the 601 asymmetry includes TpA dinucleotide (TA) steps that occur where the minor groove faces the histone core (26). The number of TA steps differs on either side of the central nucleosome dyad, and the two sides of the 601 sequence are therefore referred to as TA-poor and TA-rich.
We previously demonstrated that site-specific cross-linking of Chd1 to nucleosomal DNA could be achieved by introducing and labeling single cysteines with a photoreactive cross-linker (12). We used one of these cross-linking variants, N459C, to determine where Chd1 preferentially bound before and after exposure to competitor nucleosome. N459C, located on lobe 1 of the ATPase motor, cross-links to one strand of the DNA duplex, on the 5′ side and 15 nt from the nucleosome dyad (Fig. 5A). By labeling both DNA strands with two different fluorophores, each N459C cross-linking site can be independently monitored.
Figure 5.
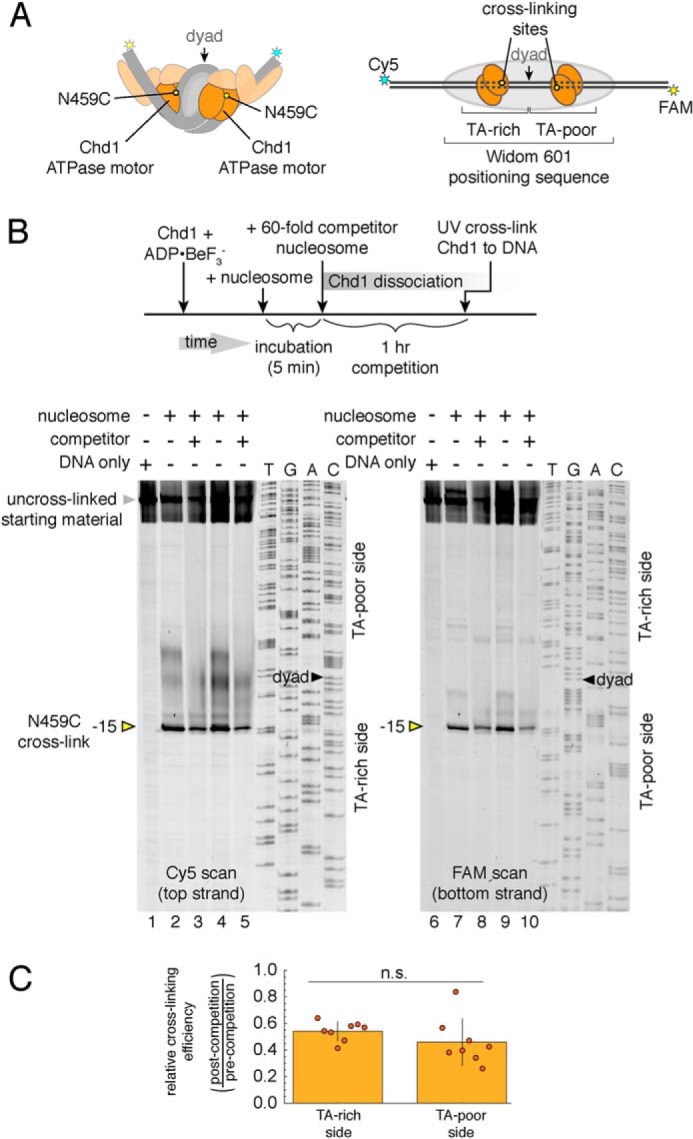
DNA sequence does not significantly influence formation of long-lived Chd1–nucleosome complexes. A, cartoon schematics of 2:1 Chd1–nucleosome complexes, highlighting the location of the N459C substitution (yellow ovals), which cross-links to nucleosomal DNA. B, cross-linking of Chd1(N459C) to 40N40 nucleosomes in the presence and absence of 12N9 competitor nucleosomes. For each experiment, 300 nm Chd1(N459C) was labeled with azidophencyl bromide, incubated with 150 nm nucleosome for 5 min, and then exposed to 0 or 9 μm 12N9 competitor nucleosome for 1 h before cross-linking. Shown are two experiments performed in 100 mm KCl (lanes 2, 3, 7, and 8) and 150 mm KCl (lanes 4, 5, 9, and 10). The two images show Cy5 (left panel) and FAM (right panel) scans of the same gel. C, quantification of N459C cross-linking observed after addition of competitor nucleosomes. The ratio of peak cross-linking intensities for plus-competitor relative to minus-competitor experiments are plotted as individual points, overlaid on the mean. TA-rich side gives the ratio for Cy5 scans, and TA-poor side gives the ratio for FAM scan. Error bars show S.D. values from eight experiments. n.s., not significant difference.
To see whether the Chd1 ATPase preferentially bound to one SHL2 site in the initial, the fast phase of binding, Chd1(N459C) was incubated for 5 min with FAM/Cy5–labeled 40N40 nucleosomes and then exposed to unlabeled competitor nucleosomes for 1 h prior to UV cross-linking (Fig. 5B). For comparison, parallel experiments were performed where no competitor was added. The presence of competitor nucleosome diminished band intensities from N459C cross-linking by ∼50% but roughly equal reduction was observed on the TA-rich (Cy5) and TA-poor (FAM) sides (Fig. 5, B and C). These results suggest that the rapid formation of tight, ADP·BeF3−–stabilized complexes occurred equally well on both sides of the nucleosome and thus did not appear to have a strong dependence on DNA sequence.
Chd1–nucleosome complexes can withstand loss of nucleosomal epitopes
Cross-linking and cryoEM structures have shown a close association of the Chd1 ATPase motor with both DNA strands at SHL2 (12, 19, 20). In addition, similar to both SWI/SNF and ISWI remodelers, the Chd1 ATPase motor directly binds to the H4 tail (19, 20, 30, 31). To probe how these interactions may impact the stability of the Chd1–nucleosome complex, we generated nucleosomes with specific disruptions in these epitopes (Fig. 6A).
Figure 6.

Chd1–nucleosome complexes are relatively stable in ADP·BeF3− despite loss of nucleosomal epitopes. A, molecular rendering of Chd1–nucleosome complex observed by cryoEM (Protein Data Bank code 6FTX) (20), highlighting the ATPase motor (brown and blue), histone H4 tail residues 11–19 (black), and the locations of 9-nt gaps in SHL2.5gapguide (green) and SHL2.5gaptracking (yellow) nucleosomes. In the lower right image, contacts from the ATPase motor to the H4 tail that would be lost are shown as gray surfaces, contacts to DNA that would be lost because of the gaps are shown as yellow or green surfaces, and ATPase contacts to DNA outside the gaps are shown as white surfaces. B, competition experiments in ADP·BeF3− reveal long-lived complexes with nucleosomes containing a ssDNA gap or H4 tail deletion. All FAM-labeled nucleosomes were 12N12 and used at 20 nm, and all competitor nucleosomes were 12N9 added to 2 μm final concentration. For H4Δtail nucleosomes, 80 nm Chd1 was incubated for 5 min prior to competition. For SHL2.5gaptracking nucleosomes, 80 nm Chd1 was incubated for 30 min prior to competition. For SHL2.5gapguide nucleosomes, 200 nm Chd1 was incubated for 30 min prior to competition. Data points are the mean values ± S.D. from three or more experiments. C, an example of a competition experiment with SHL2.5gapguide nucleosomes in ADP·BeF3−. Lane 2 shows the background signal when unlabeled competitor nucleosome was mixed with labeled nucleosomes before addition of Chd1. This gel is representative of three experiments, quantified in B. D, competition experiment with SHL2.5gapguide nucleosomes in AMP-PNP. Chd1 (200 nm) was preincubated with 20 nm SHL2.5gapguide nucleosomes for 30 min prior to addition of 2 μm competitor nucleosomes. This gel is representative of four experiments.
To test the importance of the H4 tail, FAM-labeled 12N12 nucleosomes lacking residues 1–19 of the H4 N terminus (H4Δtail) were tested in competition experiments with ADP·BeF3−. Competitor nucleosomes (12N9) were WT. In these experiments, Chd1–nucleosome(H4Δtail) complexes showed the same long-lived characteristics as WT nucleosomes, with a significant fraction of complex still present after 24 h (Fig. 6B). Thus, the contacts provided by the H4 tail are not required for stability of long-lived complexes.
Next, to test the importance of ATPase–DNA contacts, we introduced ssDNA gaps that partially overlapped the edge of the binding site. The two DNA strands bound by the ATPase motor are referred to as tracking and guide strands, with tracking referring to the nucleic acid strand bound by ssDNA and RNA helicases (19). Both strands are contacted by the ATPase motor along the phosphate backbone for ∼7 nt. We designed 9-nt gaps on each strand, located 22–30 bp from the nucleosome dyad, referred to as SHL2.5gaptracking and SHL2.5gapguide (Fig. 6A). These gaps encroach differently on the ATPase-binding site. The gap on the tracking strand is expected to disrupt backbone DNA contacts with two nucleotides, whereas the guide strand gap should eliminate backbone DNA contacts with five nucleotides (Fig. 6A, right panel).
Chd1 bound more poorly to both SHL2.5gaptracking and SHL2.5gapguide nucleosomes, and therefore longer incubation times and, for SHL2.5gapguide binding, higher Chd1 concentrations (10-fold above nucleosome) were used to obtain higher starting amounts of Chd1–nucleosome complex. In ADP·BeF3−, Chd1 formed complexes with both gapped nucleosomes that were resistant to competitor nucleosomes for greater than 8 h (Fig. 6B). Although the fraction of bound SHL2.5gapguide nucleosomes was extremely low after 24 h, a small amount of complex appeared to be above background after 24 h (Fig. 6C, compare lanes 2 and 10). We therefore conclude that the contacts lost because of these ssDNA gaps are not strictly required for maintaining a long-lived complex.
Despite the significant number of DNA contacts expected to be lost for SHL2.5gapguide nucleosomes (Fig. 6A), this region of the guide strand was not essential for long-lived complexes obtained in ADP·BeF3−. To see whether these contacts with the guide strand were important for stable interactions in AMP-PNP, Chd1 and SHL2.5gapguide nucleosomes were incubated in AMP-PNP conditions and exposed to competitor nucleosomes. Under these conditions, no complex was observed (Fig. 6D). These results suggest that as the ATPase motor transitions from ATP-bound state to one where hydrolysis takes place, energetically important contacts with DNA also shift, with the guide strand playing a more critical role in ATP-bound state prior to hydrolysis.
Discussion
Here we show how stabilities of Chd1–nucleosome complexes depend on the nucleotide-bound state. Our competition experiments revealed vastly different time scales for dissociation of Chd1 from nucleosomes, with rapid dissociation occurring in apo and ADP states and longer-lived complexes stabilized by transition state and ATP mimics (Fig. 1). Like other chromatin remodelers, Chd1 appears to reposition nucleosomes by creating and then eliminating twist defects in nucleosomal DNA where the ATPase motor binds (14–17). Combined with recent structural and biochemical findings, the nucleotide-dependent stabilities described here help explain how the cycle of ATP binding and hydrolysis promote key structural states that ratchet DNA past the histone core.
Our experiments show a remarkably tight association between nucleosomes and Chd1 in the presence of ADP·BeF3− and transition state mimics. Although it is unclear why ADP·BeF3− and transition state mimics stabilize Chd1–nucleosome complexes much more than AMP-PNP, we speculate that the differences in affinity stem from the range of conformational states available to each nucleotide-bound state of the ATPase. For all SF2 ATPases, different conformations of the protein are expected to be driven by the nature of the bound nucleotide. However, each conformation of the ATPase is not necessarily restricted to only one nucleotide-bound state. For the DEXH-type flavivirus helicase NS3, nearly identical closed conformations of the ATPase motor were crystallized with AMP-PNP and transition-state analogs (32). Similarly, crystal structures of the DEAD-box RNA helicase Mss116 bound to RNA and AMP-PNP showed essentially the same ATPase conformation when bound to RNA and ADP·AlF4− (33). Therefore, the closed conformation of the ATPase that makes intimate interactions with the nucleic acid is not unique to analogs that mimic the transition state but is accessible in other nucleotide-bound states as well. For a remodeler bound to the nucleosome, an ability to sample different conformational states is in agreement with dynamic observations of the SF2-type helicase NPH-II bound to RNA, which was found to visit different conformations with a single, nucleotide-bound state (7).
We propose that when bound to AMP-PNP, a range of different conformational states is likely available to the Chd1 ATPase motor and that excursions to states incompatible with binding canonical nucleosomes stimulate faster dissociation. With the same logic, much tighter binding caused by ADP·BeF3− and transition state mimics should result from a more limited range of conformational states, where the ATPase maintains structures that are compatible with binding to canonical nucleosomal DNA at SHL2.
Likewise, the relatively fast dissociation of Chd1 in apo and ADP-bound states may also arise from conformational mismatches of the remodeler with its nucleosome substrate. The apo and ADP states of the remodeler ATPase initiate the nucleosome sliding reaction by shifting entry-side DNA upon binding to the nucleosome (14). Although these states appear to create strain in the DNA by only shifting the tracking strand of the DNA duplex, a key anticipated intermediate should also have a shift of the guide strand (15–17). A shift of both guide and tracking strands has been visualized for nucleosomes bound by the SWR1 remodeler loaded with ADP·BeF3−, with the closed form of the ATPase motor apparently essential for stabilizing this bulged DNA structure (18). We expect that the apo and ADP states of remodeler–nucleosome complexes transiently sample a fully bulged state, although such a conformation would be unstable. Without ATP, the remodeler–nucleosome complex would either collapse back to a structure in which only the tracking strand is shifted or would dissociate. However, in the presence of ATP, a closed form of the ATPase bound to DNA fully bulged by 1 bp would be stabilized by ATP binding. Assuming that a shift in only the tracking strand is not favored by the ATP-bound state, such a conformational hand-off could allow ATP binding to enforce directionality in the formation of twist defects.
Directionality in the twist defect cycle is also supported by the extremely stable binding with ADP·BeF3− and transition state mimics. Following formation of a twist defect, spontaneous recovery of canonical DNA twist can be achieved by a corkscrew-like motion of DNA that transfers twist to a neighboring segment of the nucleosome. Because the closed, hydrolysis-competent state of the ATPase motor matches nucleosomal DNA in a canonical conformation, a return of the remodeler-bound DNA to a canonical twist would be expected to stimulate ATP hydrolysis. By disrupting the ATP-bound state, hydrolysis will both prevent the ATPase motor from potentially reestablishing the twist defect that was previously transferred toward the dyad and also initiate the next round of DNA movement (in the ADP-bound state) through a single nucleotide shift of the tracking strand. Hydrolysis therefore would be expected to enforce directionality after elimination of twist defects, consistent with the role of remodeler ATPases as Brownian ratchets.
An unexpected finding was the slow establishment of the stable ADP·BeF3− complex. The rate of complex formation was concentration-dependent (Fig. 3), and although the DNA-binding domain was not essential, formation of a stable complex was much slower in its absence (Fig. 4). The concentration dependence could reflect rate-limiting formation of a Chd1–nucleosome complex prior to BeF3− binding. Because BeF3− coordination relies on the ATPase domain organization, the critical BeF3− component may not stably bind unless ADP-bound Chd1 achieves a closed conformation, which would presumably be coupled to a further distortion of nucleosomal DNA. Cryo-EM structures show ADP-bound remodelers in open conformations (16, 17), suggesting that such a closed conformation of the ATPase motor (competent for BeF3− binding) would likely be short-lived.
Finally, our experiments with the guide strand gap revealed that the guide strand is required for stable binding of the remodeler with AMP-PNP, yet not with ADP·BeF3− (Fig. 6). In addition to disrupting stable binding with AMP-PNP, the guide strand gap also appeared to antagonize remodeler binding with ADP, as judged from the significantly reduced amount of initial complex obtained with ADP·BeF3−, even with higher Chd1 concentrations and longer incubation times. On nucleosomes containing gaps that removed most of the guide strand contacts, the tight ADP·BeF3−–dependent binding suggests, somewhat unexpectedly, that the remodeler is relatively insensitive to the conformation of the guide strand when the ATPase is poised for hydrolysis. Such a dependence of the remodeler on only the tracking strand for hydrolysis is reminiscent of the remodeler's roots as an SF2-type ATPase. For most SF2 proteins, the ATPase core strongly or preferentially interacts with ssDNA or RNA, corresponding to the tracking strand. All SF2 proteins maintain strong sequence and structural conservation of the ATP-binding pocket, and our results with Chd1 suggest that coordination via the tracking strand is likely sufficient for all SF2 proteins to organize their catalytic site for hydrolysis, despite some having double-stranded nucleic acid substrates.
In contrast, SF2 proteins would be expected to naturally diverge in how they interact with nucleic acids when in an ATP-bound state not configured for hydrolysis. The ATP-bound state typically enables ATPases to bind tightly to their substrates and can be accompanied by otherwise energetically unfavorable changes in substrate conformation. For many DEAD-box helicases, the ATP-bound state appears to typically bind to single-stranded RNA to the exclusion of the complementary strand, disrupting duplexes without ATP hydrolysis (4, 34). For chromatin remodelers, the ATP-bound state likely favors a DNA bulge in the form of a twist defect on the nucleosome. Our data show that the ATP-bound state of Chd1 relies on the guide strand for stable interactions, and we postulate that stabilizing a full twist defect on the nucleosome will likewise require intimate interactions between the ATP-bound state of the motor and the guide strand.
Experimental procedures
Protein reagents
The Saccharomyces cerevisiae Chd1 construct referred to here as Chd1 consisted of residues 118–1274. This construct results from N- and C-terminal truncations and was previously characterized (12, 35). The Chd1 construct lacking the DNA-binding domain (ΔDBD) removed an additional 260 residues from the C terminus, resulting in a protein spanning residues 118–1014. The N459C variant used for site-specific cross-linking, as originally reported in Ref. 12, was in a Chd1118–1274 background where all native cysteines had been mutated to alanine. All Chd1 constructs were purified as previously described (12, 36). All nucleosomes in this study contained Xenopus laevis histones. Histone proteins were purified individually and reconstituted into octamer as described previously (12, 37).
DNA reagents
The Widom 601 sequence (38) was used for all nucleosomes in this study. Most FAM-labeled DNA was generated by large-scale PCR, as previously described (12). For the SHL2.5gaptracking DNA, long primers containing uracils corresponding to positions 22–28 from the dyad were used for PCR and subsequently treated with USER enzyme (NEB) to generate the single-stranded gaps. The SHL2.5gapguide DNA was created by annealing four oligonucleotides that left gaps on the guide strand (3′ from dyad), 22–28 nt on each side of the dyad. DNA for competitor nucleosomes (12N9 or 26N33) was generated by digesting 601 array plasmids with EcoRV, purified by a Bio-Rad Prep Cell (12). Primers and 601 constructs are given in Table S1. Nucleosomes were reconstituted by mixing the histone octamer with nucleosomal DNA at 1:1 ratio and using the salt dialysis method and then purified with a Bio-Rad Prep or mini-Prep Cell as previously described (37).
Native gel competition experiments
The competition experiments were carried out using either 20 nm FAM-labeled 12N12 nucleosomes with 2 μm 12N9 unlabeled competitor, or 10 nm FAM-labeled 40N40 nucleosomes with 1 μm unlabeled 26N33 competitor. Chd1 was incubated for a defined time with labeled nucleosomes (preincubation), followed by the addition of unlabeled competitor nucleosome, also for a defined time. The samples were separated on 3.5% native acrylamide gels (60:1 acrylamide to bis-acrylamide, 1× TBE) and electrophoresed at 4 °C, and the time of loading was considered to terminate the competition reaction and reveal the different bound and unbound species. Each time point was a separate reaction, and the components were mixed in a reverse chronological order (longest incubations or competition conditions carried out first) so that all samples could be loaded on the gel in the same ∼2-min window. For each condition, control reactions were carried out where labeled, and competitor nucleosome were premixed before addition of Chd1. These controls indicated the expected fraction of Chd1 bound to labeled nucleosomes after equilibration. Unless otherwise noted, all reactions were carried out in 1× binding buffer (10 mm HEPES, pH 7.5, 1 mm DTT, 0.04 mg/ml BSA, 5% sucrose, 100 mm NaCl). Nucleotides and mimics were used at following concentrations: 1 mm ADP, 1 mm AMP-PNP, 1 mm ADP·BeF3− (1 mm ADP, 1.2 mm BeCl2, 6 mm NaF, 2.5 mm MgCl2), 1 mm ADP·AlFX (1 mm ADP, 1.2 mm AlCl3, 6 mm NaF, 2.5 mm MgCl2), and 2 mm ADP·MgClX (2 mm ADP, 15 mm NaF, 5 mm MgCl2). The gels were visualized using a GE Typhoon 9410 variable mode imager and quantified with ImageJ (National Institutes of Health). Because each nucleosome has two binding sites, the 1:1 species was considered to be half bound and half unbound. Single exponential fits were performed in Mathematica using the equation a1*(Exp[−k1*x] + c, where k1 is the rate, a1 is the amplitude, and c is a constant.
Chd1 site-specific cross-linking
Cross-linking experiments were carried out as previously described (12). Labeling of Chd1(N459C) was carried out by incubating a stock of 7.5 μm Chd1 with 400 μm azidophencyl bromide for 2–3 h at room temperature in the dark. For the competition cross-linking experiments, 300 nm labeled Chd1(N459C) was incubated with 150 nm 40N40 601 nucleosome (FAM/Cy5-labeled) for 5 min in ADP·BeF3− conditions (1 mm ADP, 1.2 mm BeCl2, 6 mm NaF, 2.5 mm MgCl2) and 1× slide buffer (20 mm Tris-HCl (pH 7.5), 100 mm KCl or 150 mm KCl, 0.1 mg/ml BSA, 1 mm DTT, 5% sucrose). After the 5-min incubation at room temperature, a 60-fold equivalent (9 μm) of unlabeled 12N9 competitor nucleosome was added to compete for 1 h. As controls, Chd1(N459) was separately incubated with 40N40 without competitor nucleosome or with 150 nm naked DNA. Samples (50 μl) were transferred onto a silanized coverslip; UV-irradiated for 15 s; subsequently quenched with 100 μl of 20 mm Tris-HCl (pH 7.5), 50 mm KCl, 0.1 mg/ml BSA, 5 mm DTT, and 5 mm EDTA; and further processed as described (39). Cleaved DNA fragments, indicating the locations and efficiencies of cross-linking, were resolved on urea denaturing gels.
Author contributions
R. R. and G. D. B. conceptualization; R. R., S. G. K., and G. D. B. formal analysis; R. R. and S. G. K. investigation; R. R., S. G. K., and G. D. B. writing-review and editing; G. D. B. supervision; G. D. B. funding acquisition; G. D. B. visualization; G. D. B. writing-original draft.
Supplementary Material
This work was supported by National Institutes of Health Grant R01-GM084192 (to G. D. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Fig. S1.
- SF
- superfamily
- DBD
- DNA-binding domain
- nt
- nucleotide(s)
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- SHL2
- superhelix location 2
- FAM
- Fluorescein
- TA
- TpA dinucleotide
- ssDNA
- single-stranded DNA.
References
- 1. Flaus A., Martin D. M., Barton G. J., and Owen-Hughes T. (2006) Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 34, 2887–2905 10.1093/nar/gkl295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singleton M. R., Dillingham M. S., and Wigley D. B. (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 10.1146/annurev.biochem.76.052305.115300 [DOI] [PubMed] [Google Scholar]
- 3. Narlikar G. J., Sundaramoorthy R., and Owen-Hughes T. (2013) Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490–503 10.1016/j.cell.2013.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu F., Putnam A., and Jankowsky E. (2008) ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc. Natl. Acad. Sci. U.S.A. 105, 20209–20214 10.1073/pnas.0811115106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Putnam A. A., and Jankowsky E. (2013) DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta 1829, 884–893 10.1016/j.bbagrm.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu F., Putnam A. A., and Jankowsky E. (2014) DEAD-box helicases form nucleotide-dependent, long-lived complexes with RNA. Biochemistry 53, 423–433 10.1021/bi401540q [DOI] [PubMed] [Google Scholar]
- 7. Fairman-Williams M. E., and Jankowsky E. (2012) Unwinding initiation by the viral RNA helicase NPH-II. J. Mol. Biol. 415, 819–832 10.1016/j.jmb.2011.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wong I., and Lohman T. M. (1997) A two-site mechanism for ATP hydrolysis by the asymmetric rep dimer P2S as revealed by site-specific inhibition with ADP-AlF4. Biochemistry 36, 3115–3125 10.1021/bi9621977 [DOI] [PubMed] [Google Scholar]
- 9. Saha A., Wittmeyer J., and Cairns B. R. (2005) Chromatin remodeling through directional DNA translocation from an internal nucleosomal site. Nat. Struct. Mol. Biol. 12, 747–755 10.1038/nsmb973 [DOI] [PubMed] [Google Scholar]
- 10. Dang W., and Bartholomew B. (2007) Domain architecture of the catalytic subunit in the ISW2-nucleosome complex. Mol. Cell. Biol. 27, 8306–8317 10.1128/MCB.01351-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ranjan A., Wang F., Mizuguchi G., Wei D., Huang Y., and Wu C. (2015) H2A histone-fold and DNA elements in nucleosome activate SWR1-mediated H2A.Z replacement in budding yeast. Elife 4, e06845 10.7554/eLife.06845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nodelman I. M., Bleichert F., Patel A., Ren R., Horvath K. C., Berger J. M., and Bowman G. D. (2017) Interdomain communication of the Chd1 chromatin remodeler across the DNA gyres of the nucleosome. Mol. Cell 65, 447–459.e6 10.1016/j.molcel.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Holde K. E., and Yager T. D. (1985) in Structure and Function of the Genetic Apparatus (Nicolini C., ed) pp. 35–53, Springer-Verlag, New York Inc., New York [Google Scholar]
- 14. Winger J., Nodelman I. M., Levendosky R. F., and Bowman G. D. (2018) A twist defect mechanism for ATP-dependent translocation of nucleosomal DNA. Elife 7, e34100 10.7554/eLife.34100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bowman G. D. (2019) Uncovering a new step in sliding nucleosomes. Trends Biochem. Sci. 44, 643–645 10.1016/j.tibs.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li M., Xia X., Tian Y., Jia Q., Liu X., Lu Y., Li M., Li X., and Chen Z. (2019) Mechanism of DNA translocation underlying chromatin remodelling by Snf2. Nature 567, 409–413 10.1038/s41586-019-1029-2 [DOI] [PubMed] [Google Scholar]
- 17. Yan L., Wu H., Li X., Gao N., and Chen Z. (2019) Structures of the ISWI-nucleosome complex reveal a conserved mechanism of chromatin remodeling. Nat. Struct. Mol. Biol. 26, 258–266 10.1038/s41594-019-0199-9 [DOI] [PubMed] [Google Scholar]
- 18. Willhoft O., Ghoneim M., Lin C. L., Chua E. Y. D., Wilkinson M., Chaban Y., Ayala R., McCormack E. A., Ocloo L., Rueda D. S., and Wigley D. B. (2018) Structure and dynamics of the yeast SWR1-nucleosome complex. Science 362, aat7716 10.1126/science.aat7716 [DOI] [PubMed] [Google Scholar]
- 19. Farnung L., Vos S. M., Wigge C., and Cramer P. (2017) Nucleosome–Chd1 structure and implications for chromatin remodelling. Nature 550, 539–542 10.1038/nature24046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sundaramoorthy R., Hughes A. L., El-Mkami H., Norman D. G., Ferreira H., and Owen-Hughes T. (2018) Structure of the chromatin remodelling enzyme Chd1 bound to a ubiquitinylated nucleosome. Elife 7, e35720 10.7554/eLife.35720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tokuda J. M., Ren R., Levendosky R. F., Tay R. J., Yan M., Pollack L., and Bowman G. D. (2018) The ATPase motor of the Chd1 chromatin remodeler stimulates DNA unwrapping from the nucleosome. Nucleic Acids Res. 46, 4978–4990 10.1093/nar/gky206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eberharter A., Längst G., and Becker P. B. (2004) A nucleosome sliding assay for chromatin remodeling factors. Methods Enzymol. 377, 344–353 [DOI] [PubMed] [Google Scholar]
- 23. Kagawa R., Montgomery M. G., Braig K., Leslie A. G., and Walker J. E. (2004) The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J. 23, 2734–2744 10.1038/sj.emboj.7600293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomsen N. D., and Berger J. M. (2009) Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534 10.1016/j.cell.2009.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hall M. A., Shundrovsky A., Bai L., Fulbright R. M., Lis J. T., and Wang M. D. (2009) High-resolution dynamic mapping of histone–DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 16, 124–129 10.1038/nsmb.1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chua E. Y., Vasudevan D., Davey G. E., Wu B., and Davey C. A. (2012) The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 40, 6338–6352 10.1093/nar/gks261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ngo T. T., Zhang Q., Zhou R., Yodh J. G., and Ha T. (2015) Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell 160, 1135–1144 10.1016/j.cell.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levendosky R. F., Sabantsev A., Deindl S., and Bowman G. D. (2016) The Chd1 chromatin remodeler shifts hexasomes unidirectionally. Elife 5, e21356 10.7554/eLife.21356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Winger J., and Bowman G. D. (2017) The direction that the Chd1 chromatin remodeler slides nucleosomes can be influenced by DNA sequence. J. Mol. Biol. 429, 808–822 10.1016/j.jmb.2017.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan L., Wang L., Tian Y., Xia X., and Chen Z. (2016) Structure and regulation of the chromatin remodeller ISWI. Nature 540, 466–469 10.1038/nature20590 [DOI] [PubMed] [Google Scholar]
- 31. Liu X., Li M., Xia X., Li X., and Chen Z. (2017) Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 544, 440–445 10.1038/nature22036 [DOI] [PubMed] [Google Scholar]
- 32. Luo D., Xu T., Watson R. P., Scherer-Becker D., Sampath A., Jahnke W., Yeong S. S., Wang C. H., Lim S. P., Strongin A., Vasudevan S. G., and Lescar J. (2008) Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 27, 3209–3219 10.1038/emboj.2008.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Del Campo M., and Lambowitz A. M. (2009) Structure of the yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol. Cell 35, 598–609 10.1016/j.molcel.2009.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jarmoskaite I., and Russell R. (2011) DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip. Rev. RNA 2, 135–152 10.1002/wrna.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patel A., Chakravarthy S., Morrone S., Nodelman I. M., McKnight J. N., and Bowman G. D. (2013) Decoupling nucleosome recognition from DNA binding dramatically alters the properties of the Chd1 chromatin remodeler. Nucleic Acids Res. 41, 1637–1648 10.1093/nar/gks1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nodelman I. M., Horvath K. C., Levendosky R. F., Winger J., Ren R., Patel A., Li M., Wang M. D., Roberts E., and Bowman G. D. (2016) The Chd1 chromatin remodeler can sense both entry and exit sides of the nucleosome. Nucleic Acids Res. 44, 7580–7591 10.1093/nar/gkw406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 10.1016/S0076-6879(99)04003-3 [DOI] [PubMed] [Google Scholar]
- 38. Lowary P. T., and Widom J. (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 10.1006/jmbi.1997.1494 [DOI] [PubMed] [Google Scholar]
- 39. Kassabov S. R., and Bartholomew B. (2004) Site-directed histone–DNA contact mapping for analysis of nucleosome dynamics. Methods Enzymol. 375, 193–210 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.