

Abstract
A myriad of protein partners modulate the activity of the human DNA methyltransferase 3A (DNMT3A), whose interactions with these other proteins are frequently altered during oncogenesis. We show here that the tumor suppressor p53 decreases DNMT3A activity by forming a heterotetramer complex with DNMT3A. Mutational and modeling experiments suggested that p53 interacts with the same region in DNMT3A as does the structurally characterized DNMT3L. We observed that the p53-mediated repression of DNMT3A activity is blocked by amino acid substitutions within this interface, but surprisingly, also by a distal DNMT3A residue, R882H. DNMT3A R882H occurs frequently in various cancers, including acute myeloid leukemia, and our results suggest that the effects of R882H and other DNMT3A mutations may go beyond changes in DNMT3A methylation activity. To further understand the dynamics of how protein-protein interactions modulate DNMT3A activity, we determined that p53 has a greater affinity for DNMT3A than for DNMT3L and that p53 readily displaces DNMT3L from the DNMT3A:DNMT3L heterotetramer. Interestingly, this occurred even when the preformed DNMT3A:DNMT3L complex was actively methylating DNA. The frequently identified p53 substitutions (R248W and R273H), whereas able to regulate DNMT3A function when forming the DNMT3A:p53 heterotetramer, no longer displaced DNMT3L from the DNMT3A:DNMT3L heterotetramer. The results of our work highlight the complex interplay between DNMT3A, p53, and DNMT3L and how these interactions are further modulated by clinically derived mutations in each of the interacting partners.
Keywords: epigenetics, nucleic acid enzymology, protein-protein interaction, DNA demethylation, allosteric regulation, DNMT3A, p53
Introduction
Transcriptional regulation, genomic imprinting, and cellular differentiation, including 5-methylcytosine patterning on DNA (1–7), relies on diverse protein-protein and protein-nucleic acid interactions. In the crowded intracellular environment, formation of biologically significant complexes relies on the kinetic accessibility to specific protein surfaces and the thermodynamic stability of the resultant complexes (8–11). For example, de novo DNA methylation by the DNA methyltransferase 3A (DNMT3A) involves the formation of complexes that include a wide-range of regulatory partners, such as histones, histone-modifying enzymes, transcription factors, and RNA (2, 4, 12, 13). Such interactions are frequently altered during oncogenesis, resulting in the disruption to DNMT3A genomic localization and/or regulation of enzyme activity (14). The tumor suppressor p53 is well-known to interact with components of the epigenetic machinery, including DNMT3A; however, a functional understanding of p53-DNMT3A interactions remains largely unknown (15, 16).
In addition to directly activating transcription of genes essential for cell cycle arrest and apoptosis in response to genotoxic stress, the interactions between p53 and histone modifying enzymes are a key driver of gene activation (17, 18). The progressive accumulation of p53 mutations leads to the recruitment of histone modifying enzymes (19–21). Several studies suggest a link between p53 and DNA methylation. For example, whereas DNMT12 (the maintenance DNA methyltransferase) represses expression of the p53 gene (22), p53 binding to DNMT1 stimulates DNMT1-mediated methylation (23). In addition, p53 represses the expression of DNMT3A and DNMT3B, whereas DNMT3A has been shown to repress the transcriptional activity of p53 (24, 25). However, definitive evidence of the functional consequences of a DNMT3A:p53 complex on DNMT3A activity remains elusive. Our interest lies in exploring whether p53 binding to DNMT3A alters DNMT3A activity, and if well-studied DNMT3A and p53 mutants, such as R882H (DNMT3A) and p53 mutants R248W and R273H, alter this regulation.
Located at the dimer interface, the major DNA-binding site, the R882H substitution in DNMT3A disrupts tetramerization and processive catalysis, both of which can be restored by DNA-methyltransferase 3-like (DNMT3L) (26–28). Additionally, R882H displays altered interactions with components of the Polycomb repressive complex 1 compared with WT DNMT3A, thereby leading to transcriptional silencing in a DNA methylation-independent manner (29). These observations suggest that compared with WT DNMT3A, the R882H substitution may lead to altered binding and/or regulation by partner proteins. Given that the p53 gene is a recurring target for mutations in a wide-range of human cancers, there is growing interest in understanding how mutations in p53 contribute to disease onset and progression (30, 31). In addition to a high mutation frequency, p53 R273H and R248W form aberrant protein complexes that affect the activity of interacting partner proteins (32–34). Our goal is to understand the dynamics and functional consequences of complex assembly involving the WT catalytic domain of DNMT3A (DNMT3AWT) and the R882H substitution (DNMT3AR882H) under a variety of conditions. Furthermore, we seek to better understand the functional consequences of protein complexes involving two or more proteins to better understand the cellular basis of enzyme function.
Results
WT (p53WT) and mutant p53 inhibit the DNA methylation activity of full-length and catalytic domain DNMT3AWT
Previous cell-based evidence implicates direct and indirect DNMT3A and p53 interactions (24, 25). In mouse embryonic stem cells, p53 indirectly regulates DNMT3A-mediated methylation by restricting the expression of DNMT3A (24). Alternatively, direct binding of DNMT3A to p53 suppresses p53-mediated transcription of p21 in a DNA methylation-independent manner, implying that DNMT3A may allosterically regulate p53 activity (25). Based on this evidence, we sought to determine whether p53 has any effect on the DNA methylation activity of DNMT3A. Given that the DNMT3A catalytic domain and full-length enzyme have comparable kinetic parameters (kcat, KmDNA, KmAdoMet, processivity, and DNMT3L stimulation), the catalytic domain of DNMT3A has proven to be a suitable model for in vitro studies and is commonly employed (35, 36). However, the N-terminal domains in full-length DNMT3A, including the ATX-DNMT3A-DNMT3L (ADD) and PWWP domains, are known to interact with numerous partner proteins that may modulate the enzymatic activities of DNMT3A (37, 38). Therefore, we compared the effect of p53WT on the methylation activity of the DNMT3A catalytic domain and full-length enzymes by preincubating p53 with equimolar concentrations of DNMT3A for 1 h prior to initiating the reaction by the addition of poly(dI-dC). We observed comparable levels of p53WT-mediated DNMT3A inhibition with the catalytic domain (Fig. 1, A and B) or the full-length DNMT3A (Fig. 1, A and C). The comparable inhibition indicates that the N-terminal portion of DNMT3A is not essential for p53WT-DNMT3A interactions and does not perturb p53WT acting on the catalytic domain of DNMT3A. The consensus DNA binding sequence of p53 (5′-RRRC(A/T)(A/T)GYYY(0–14 bases)RRRC(A/T)(A/T)GYYY-3′; A= adenine, G= guanine, C= cytosine and T= thymine) differs from that of DNMT3A, which displays a strong preference toward CpG sites (39). However, it is possible that p53WT inhibition of DNMT3A (full-length and catalytic domain) enzymatic activity is attributable to the DNA-binding ability of p53. To assess whether the inhibitory effect of p53WT on DNA methylation is specific to DNMT3A, the activity of the bacterial methyltransferase M.HhaI, which recognizes 5′-GCGC-3′ sites in dsDNA, was challenged with increasing concentrations of p53WT. Although p53WT led to roughly a 50% decrease in DNMT3A (full-length and catalytic domain) activity (Fig. 1), DNA methylation by M.HhaI was unaltered in similar reactions involving p53 at 1:1 and 3:1 relative to M.HhaI (Fig. 1, A and D). Like DNMT3A, M.HhaI is a C-5 cytosine-specific methyltransferase that possesses a remarkably similar structure to that of the DNMT3A catalytic domain (40). Despite the shared similarities of DNMT3A and M.HhaI, here we show that p53 inhibition is specific to DNMT3A.
Figure 1.

p53WT-dependent inhibition of DNA methylation is specific to DNMT3A. A, fold-inhibition calculated by product formed by WT DNMT3A (full-length or catalytic domain enzymes) or M.HhaI divided by product formed by DNMT3A (full-length or catalytic domain enzymes) or M.HhaI without p53WT from reactions in B–D. Co-incubation of DNMT3A full-length (A, blue square calculated from B) and catalytic domain (A, orange square calculated from C) enzymes with p53WT (1:1 at 150 nm) leads to comparable levels of inhibition. Similar reactions involving the bacterial methyltransferase M.HhaI (A, green square calculated from D) with excess p53WT (1:3) failed to inhibit DNA methylation. In all co-incubations, proteins were held at 37 °C for 1 h prior to the start of the reaction by the addition of DNA (5 μm bp poly(dI-dC)). Data reflect the mean ± S.D. of 3 experiments; one-way analysis of variance was used to compare values of all three reactions; ***, p < 0.001; ns, p > 0.05.
To further characterize the interactions between DNMT3AWT and p53, the apparent binding affinities (KD,app) of p53WT and p53R248W for DNMT3AWT were determined. For comparison, we also determined the KD,app of DNMT3L for DNMT3AWT, which has a well-characterized interaction. KD,app was assessed by measuring the activity of DNMT3AWT with increasing levels of DNMT3L (Fig. S1A) or p53 (Fig. S1B), and subsequently determining the fold-stimulation or inhibition by DNMT3L or p53WT and p53R248W, respectively. Although DNMT3L resulted in a KD,app of 80 ± 12 nm, p53WT displayed a nearly 5-fold stronger binding affinity for DNMT3AWT with a KD,app of 17 ± 3 nm, followed by p53R248W, which resulted in a KD,app of 41 ± 6 nm (calculated from supporting Fig. 1, A and B). Thus, compared with the DNMT3AWT-DNMT3L complex, DNMT3AWT-p53WT and -p53R248W complexes are more energetically favorable.
Mutational mapping suggests p53WT interacts with the tetramer interface DNMT3A
Previous work by Wang et al. (25) suggests that DNMT3A interacts with the C-terminal tetramerization domain of p53 (amino acids 319–393, Fig. S3). No such information is available for the region on DNMT3A that is associated. We previously used alanine scanning to identify residues on the DNMT3A tetramer interface that largely contribute to tetramer stability and alter the ability of DNMT3A to form higher order complexes (41). In a similar manner, we employed docking-based modeling of protein-protein interfaces using monomeric forms of DNMT3A (PDB code 5YX2; residues 628–914) and p53 (PDB code 3TS8; 94–356) to predict a surface on DNMT3A for interactions with p53. Computational models generated in ZDOCK and RosettaDock servers were used to predict the DNMT3A tetramer interface as a likely surface for DNMT3A-p53 interactions (Fig. S2) (42, 43). Based on this rationale, p53WT inhibition of the DNMT3A catalytic activity was assessed in a subset of alanine mutations on the DNMT3A tetramer interface (R729A, E733A, R736A, R771A), which are also commonly observed in acute myeloid leukemia (AML) (26) (Fig. 2A). The extent of p53WT inhibition varied across the mutations examined in this study: the fold-inhibition of DNMT3AR771A (Fig. 2, B and C, green square) and DNMT3AE733A (Fig. 2, B and C, blue square) was greater than WT, whereas the fold-inhibition of DNMT3AR729A (Fig. 2, B and C, orange square) was less than WT and DNMT3AR736A (Fig. 2, B and C, red square) displayed no inhibition. The results obtained implicate the DNMT3A tetramer interface as a potential surface on DNMT3A for interactions with p53 and suggest that residue Arg-736 on the DNMT3A tetramer interface contributes to the necessary contacts for p53 inhibition of DNMT3A.
Figure 2.

DNMT3AWT tetramer interface mutants show highly variable response to p53WT inhibition. Crystal structure of a DNMT3AWT-DNMT3L complex (adapted from PDB code 5YX2) denoting critical residues for DNMT3AWT oligomerization (A) (36, 50). Although the extent of p53WT inhibition varies across DNMT3AWT mutants harboring a single alanine substitution within the tetramer interface, the DNMT3AR736A was unresponsive to p53WT inhibition (B and C). All reactions consisted of 150 nm DNMT3AWT and were initiated by the addition of 5 μm bp poly(dI-dC). For co-incubations, DNMT3AWT and p53WT (1:1) were preincubated for 1 h at 37 °C prior to starting the reaction by the addition of substrate DNA. Fold-inhibition was calculated by product formed by DNMT3A (WT and mutants) divided by product formed by DNMT3A (WT and mutants) without p53WT. All reactions were performed in duplicates. In B, a Student's unpaired t test was used to compare values within each set of reactions; **, p < 0.01; ns, p > 0.05. For C, a one-way analysis of variance was used to compare the values of each mutant to WT (****, p < 0.001) and across all samples (orange ****, p < 0.001). Data reflect the mean ± S.D. of 3 experiments.
The regulation of DNMT3AWT by p53WT is dominant to that of DNMT3L
Given that DNMT3A exists in several multiprotein complexes associated with transcriptional regulation, we sought to assess the extent of DNMT3A modulation in the presence of multiple regulatory partner proteins (5, 44, 45). Our mutational mapping and modeling results suggest that the DNMT3A tetramer interface (Fig. 2 and Fig. S2) interacts with both DNMT3L and p53. Given that the DNMT3A-DNMT3L co-crystal structure (46) presents the DNMT3A tetramer interface as an established surface for regulation of DNMT3A activity by an additional partner protein, we assessed whether DNMT3L and p53WT regulation of DNMT3AWT is mutually exclusive using poly(dI-dC) as a substrate. After demonstrating DNMT3AWT is responsive to DNMT3L (Fig. 3A, green square) and p53WT (Fig. 3A, blue square), we observed that p53WT modulation of DNMT3AWT is dominant over that of DNMT3L in DNMT3AWT-DNMT3L-p53WT co-incubations (Fig. 3A, red square). We previously showed that p53WT inhibition of DNMT3AWT does not rely on DNA binding by p53 (Fig. 1). Therefore, the use of poly(dI-dC) as a substrate allowed us to investigate the isolated effects of p53-mediated inhibition of DNMT3A activity. Inspired by our previous studies using human promoters, we also studied the Cyclin-dependent Kinase Inhibitor 1A/P21 promoter, which is a common target for DNMT3A and p53 (25, 102). As we observed for poly(dI-dC), p53WT-dependent inhibition of DNMT3AWT activity is dominant over DNMT3L stimulation as DNMT3AWT-p53WT-DNMT3L co-incubations (Fig. 3B, yellow square) displayed comparable levels of activity as reactions consisting of DNMT3AWT-p53WT (Fig. 3B, red square) with the Cyclin-dependent Kinase Inhibitor 1A/P21 promoter as a DNA substrate. Furthermore, equimolar concentrations of all proteins were used in DNMT3AWT-DNMT3L-p53WT co-incubations (Fig. 3, A and B), suggesting that the dominant modulation of DNMT3AWT activity by p53WT over DNMT3L was not due to stoichiometric differences. The stability of the DNMT3AWT:p53WT complex is greater than the DNMT3AWT:DNMT3L complex (Fig. S1) and DNMT3L and p53 likely share a binding interface on DNMT3A (Fig. 2). To further investigate the dynamics of DNMT3AWT-DNMT3L-p53WT interactions, we next assessed the effect of adding regulatory proteins to actively catalyzing heterotetrameric complexes, which is arguably a better representation of what occurs within the cell. We observed that addition p53WT disrupts the DNMT3AWT:DNMT3L complex during catalysis (Fig. 3C, red square). This not only supports our observation that the DNMT3AWT:p53WT complex is more stable, but that it can access the DNMT3AWT:DNMT3L complex and displace DNMT3L during catalysis. In contrast, adding DNMT3L to an actively methylating DNMT3AWT:p53WT complex failed to disrupt the modulatory effect of p53WT (Fig. 3E, red square). In fact, comparable levels of DNMT3AWT-dependent methylation activity were observed in reactions consisting of DNMT3AWT-p53WT (Fig. 3E, blue square) and functional DNMT3AWT:p53WT complexes to which DNMT3L was added (Fig. 3E, red square). In sum, our results consistently suggest the regulatory effect of p53WT on DNMT3AWT is dominant compared with DNTM3L regulation and provides insights into the dynamics of p53WT and DNMT3L binding on DNMT3AWT.
Figure 3.

DNMT3AWT and DNMT3AR882H are differentially responsive to modulation by p53WT. p53WT-dependent inhibition of DNMT3AWT activity is dominant in DNMT3AWT-p53WT-DNMT3L co-incubations (A, red square), whereas p53WT fails to inhibit DNMT3AR882H in DNMT3AR882H-p53WT (A, blue square) and DNMT3AR882H-p53WT-DNMT3L co-incubations (A, red square) using poly(dI-dC) (5 μm bp) as a substrate. B, p53WT-dependent inhibition of DNMT3AWT activity is dominant in DNMT3AWT-p53WT-DNMT3L co-incubations (yellow square) with p21-pCpGL (10 μm) as a substrate. p53WT (C, red square) disrupts DNMT3L stimulation of DNMT3AWT in catalytically active DNMT3AWT:DNMT3L complexes (C, green square), whereas catalytically active DNMT3AR882H-DNMT3L (D, green square) are unaltered by the addition of p53WT (D, red square). Reactions consisting of catalyzing p53WT-DNMT3AWT (E, blue square) or DNMT3AR882H-p53WT (F, blue square) were unaltered by the addition of DNMT3L (E and F, red square). The following reactions were also performed as controls: DNMT3AWT (A, C, and E, ■), DNMT3AR882H (A. D, and F, ■), DNMT3AWT-DNMT3L co-incubations (A and C, green square), DNMT3AR882H-DNMT3L co-incubations (A and D, green square), DNMT3AWT-p53WT co-incubations (A and E, blue square) and DNMT3AR882H-p53WT co-incubations (A and F, blue square). In all reactions performed, protein concentrations were 150 nm and were initiated by the addition of substrate DNA. For co-incubations, proteins were placed at 37 °C for 1 h prior to the addition of substrate DNA. All reactions were performed in triplicates. Values (A, green, blue, and red) were compared with either DNMT3AWT (A, ■) or DNMT3AR882H (A, ■) using a one-way analysis of variance; ****, p < 0.001. Data reflect the mean ± S.D. of 3 experiments.
p53WT fails to inhibit the methylation activity of DNMT3AR882H
Identified as the most common mutation in DNMT3A in AML patients, in vitro evidence suggests the R882H substitution leads to altered binding and/or regulation by partner proteins (26). Although R882H is unable to form homotetramers, the addition of DNMT3L leads to the formation of heterotetramers and restores processive catalysis (28). In addition, immunoprecipitation experiments using HEK293T cells reveal R882H displays increased binding to components of the Polycomb repressive complex 1 compared with WT DNMT3A (29). These observations suggest that whereas R882H is located at the dimer interface (Fig. 2A), which is distal from the tetramer interface and proximal to the DNA interface (Fig. 2A), R882H appears to allosterically affect the ability of DNMT3A to interact with partner proteins. Although DNMT3AR882H is responsive to DNMT3L activation (Fig. 3A, green square), it is unresponsive to the modulatory effect of p53WT in DNMT3AR882H-p53WT (Fig. 3A, blue square) and mixed DNMT3AR882H-p53WT-DNMT3L co-incubations (Fig. 3A, red square). In fact, DNMT3AR882H-p53WT (Fig. 3A, blue square) co-incubations led to comparable levels of product formed as DNMT3AR882H only (Fig. 4A, ■) and mixed DNMT3AR882H-p53WT-DNMT3L co-incubations (Fig. 3A, red square) reflected similar levels of activity as DNMT3AR882H-DNMT3L co-incubations (Fig. 3A, green square). Thus, the R882H substitution appears to disrupt the modulatory effect of p53WT on DNA methylation. To additionally challenge this notion, we evaluated the effect of adding DNMT3L or p53WT to actively methylating DNMT3AR882H-p53WT or DNMT3AR882H-DNMT3L co-incubations. Consistent with the hypothesis that p53WT fails to modulate DNMT3AR882H activity, the addition of p53WT failed to disrupt functional DNMT3AR882H:DNMT3L heterotetramers (Fig. 3D, red square) unlike that observed in similar reactions involving DNMT3AWT (Fig. 3C, red square). However, the addition of DNMT3L failed to stimulate DNMT3AR882H in actively catalyzing DNMT3AR882H-p53WT co-incubations (Fig. 4F, red square).
Figure 4.

p53R248W and p53R273H fail to disrupt stimulation of DNMT3AWT by DNMT3L. The stimulatory effect of DNMT3AWT activity by DNMT3L is dominant in DNMT3AWT-p53R248-DNMT3L (A, red square) or DNMT3AWT-p53R273H-DNMT3L co-incubations (A, blue square). Catalytically active DNMT3AWT-DNMT3L heterotetramers are unaffected by the addition of p53R248W (B, red square) or p53R273H (D, blue square), whereas the addition of DNMT3L leads to an increase in DNMT3AWT-p53R248W (C, yellow square) or DNMT3AWT-p53R273H (C, teal square) co-incubations. The following reactions were also performed as controls: DNMT3AWT (A–E, ■), DNMT3AWT-DNMT3L co-incubations (A, B, and D, green square), DNMT3AWT-p53R248W co-incubations (A and C, red square), and DNMT3AWT-p53R273H co-incubations (A and C, blue square). Protein concentrations were 150 nm for all reactions and were initiated by the addition of 5 μm poly(dI-dC) as a substrate. For co-incubations, proteins were preincubated at 37 °C for 1 h prior to the addition of DNA. All reactions were performed in triplicates and all values in A were compared with WT (A, ■) using a one-way analysis of variance; ****, p < 0.001. Data reflect the mean ± S.D. of 3 experiments.
p53R248W and p53R273H display altered regulation of DNMT3AWT in the presence of DNMT3L
Mutated in over half of all human cancers, the majority of p53 mutations are missense mutations throughout the p53 DNA-binding domain (47–50). Two of these “hot spot” substitutions, p53 R248W and R273H, display altered interactions and regulation of several partner proteins compared with WT p53 (32–34). Although p53 R248W and R273H are outside of the region on p53 known to interact with DNMT3A (Fig. S3), we compared the effect of p53WT with p53R248W and p53R273H substitutions on the enzymatic activity of DNMT3AWT based on their recurrence in a wide range of cancers. Although p53WT (Fig. 3A, blue square), p53R248W (Fig. 4A, red square), and p53R273H (Fig. 4A, blue square) displayed comparable levels of DNMT3AWT inhibition, p53R248W and p53R273H failed to reverse the stimulatory effect of DNMT3L in DNMT3AWT-p53R248-DNMT3L co-incubations (Fig. 4A, yellow square) or in DNMT3AWT-p53R273H-DNMT3L co-incubations (Fig. 4A, teal square) as previously observed in similar reactions involving DNMT3AWT and p53WT (Fig. 3A, red square). In fact, DNMT3AWT-DNMT3L co-incubations with p53R248W (Fig. 4A, red square) or p53R273H (Fig. 4A, teal square) displayed comparable levels of product formation as DNMT3AWT-DNMT3L co-incubations (Fig. 4A, green square). To further challenge the dominant regulatory effect of DNMT3L over p53R248W and p53R273H on DNMT3AWT observed, we then assessed the outcome of adding DNMT3L to DNMT3AWT-p53R248W (or-p53R273H) co-incubations as well as the addition of p53R248W (or p53R273H) to catalyzing DNMT3AWT:DNMT3L complexes. In contrast to similar experiments involving p53WT (Fig. 3, C and E, red square), the addition of p53R248W (Fig. 4B, red square), or p53R273H (Fig. 4C, blue square) to catalyzing DNMT3AWT:DNMT3L complexes did not disrupt DNMT3L-mediated stimulation, whereas the addition of DNMT3L to functional DNMT3AWT-p53R248W (Fig. 4D, yellow square) or DNMT3AWT-p53R273H (Fig. 4E, teal square) assembles led to an increase in DNMT3AWT activity. These findings support the notion that the regulatory effect of DNMT3L on DNMT3AWT activity is dominant over that of p53R248W or p53R273H. Here we show that whereas p53WT, p53R248W, and p53R273H display comparable levels of DNMT3AWT inhibition, p53R248W and p53R273H displayed altered regulation of DNMT3AWT in the presence of DNMT3L compared with p53WT. This presents an example in which p53R248W and p53R273H display altered protein-protein interactions compared with p53WT in the context of DNA methylation and in addition to those previously reported (32–34).
p53 binds DNMT3AWT and DNMT3AR882H to form of heterotetramers
The ability of partner proteins to impact DNMT3A function depends on the formation of a complex, although this may not be sufficient. Anisotropy measurements are widely employed to assess protein-DNA, protein-protein interactions, and estimate the oligomeric state of proteins (51–58). We previously used a 30-bp 5′ 6-FAM-labeled duplex DNA (GCbox30), which contains a single recognition site for DNMT3A, to resolve the oligomeric state of DNMT3A (28, 41). We relied on this approach and fluorescence anisotropy to assess the dynamics of DNMT3AWT-p53WT interactions on DNA (GCbox30). Increasing concentrations of p53WT or DNMT3L (Fig. 5A, purple square) to a fixed concentration of DNA-bound DNMT3AR882H (Fig. 5A, ▾) resulted in a corresponding increase to the initial anisotropy value, thereby suggesting the formation of higher order complexes on DNA. Under identical conditions, we observed that increasing concentrations of DNMT3L (Fig. 5A, green circle), p53WT (Fig. 5A, red triangle), or p53R248W (Fig. 5A, blue diamond) did not result in a detectable change to the initial anisotropy value of DNMT3AWT. DNMT3AR882H binds DNA as a homodimer and forms heterotetramers with DNMT3L on DNA (28, 36). The titration of p53WT to DNMT3AR882H led to similar final anisotropy values as those observed for DNMT3AWT in the absence and presence of DNMT3L, p53WT, and p53R248W (∼0.19; Fig. 5A). To confirm whether the increased anisotropy observed with DNMT3AR882H (Fig. 5A, ▾) is due to formation of a higher order structure (likely a tetramer), we performed gel shift assays of DNMT3AR882H with varying concentrations of p53WT using GCbox30 as a substrate. Consistent with the results of the fluorescence anisotropy assays, an increase in the concentration of p53WT to a fixed concentration of DNMT3AR882H (Fig. 5, B and C, lanes 3-5) led to a supershift and disappearance of the band corresponding to DNA-bound DNMT3AR882H (Fig. 5B, lane 1). In sum, our results indicate that p53 interacts with DNMT3A to form heterotetramers and that inhibition does not arise from disrupting the ability of DNMT3A to bind DNA.
Figure 5.

p53 heterodimerizes with WT and R882H DNMT3A. A, increasing concentrations of DNMT3L (purple square) or p53WT (▾) to a fixed concentration of DNMT3AR882H led to a robust increase in anisotropy, whereas DNMT3AWT did not display a significant change in anisotropy by the titration of DNMT3L (green circle), p53WT (red triangle), or p53R248W (blue diamond). B, EMSA and C, EMSA band densitometry (lanes 1 and 5) show increasing concentrations of p53WT (lanes 3-5) to a constant concentration of DNMT3AR882H leads to disappearance of the DNMT3AR882H band and formation of a higher order structure (see arrows, lanes 3-5). In A, DNMT3AWT and DNMT3AR882H concentrations were 2.5 μm. Single point anisotropy measurements were taken after increasing concentrations of DNMT3L (green circle), p53WT (red triangle), and p53R248W (blue diamond) were added to DNA bound (250 nm 5′ 6-FAM-labeled GCbox30) DNMT3AWT and DNMT3AR882H and allowed to incubate at room temperature for 5 min. Measurements were taken using a fluorometer equipped with polarizing filters (excitation, 485 nm; emission, 520 nm). In B, gel shift assays were carried out as described in Holz-Schietinger et al. (28) and other than samples were run on native 4.5% polyacrylamide gels and binding reactions were performed at 37 °C (lane 1). For p53WT supershifting, varying concentrations of p53WT were preincubated for 30 min at 37 °C with DNMT3AR882H before the addition of DNA. A and C reflect the results (mean ± S.D.) of 2 independent experiments.
Discussion
Although p53 has been extensively investigated, much less is known about whether or how this protein influences epigenetic pathways, particularly DNA methylation (17, 18, 30, 47–50). Reports on the cross-talk between p53 and members of the DNMT family include the loss of global methylation by 5-aza-2′-deoxycytidine treatment induces a p53 DNA damage response pathway (59), deletion of the DNMT1 gene activates p53-mediated apoptosis (60) and p53 directly stimulates DNMT1-mediated methylation (23). In the context of the de novo DNA methyltransferases, p53 transcriptionally represses DNMT3A and DNMT3B, whereas DNMT3A inhibits p53-mediated transcription (24). Based on this evidence and the association of DNMT3A and p53 in various human cancers (26, 47–50), we sought to characterize the dynamics and regulation of DNMT3A activity by p53 under various conditions. We show that p53 and the well-characterized DNMT3L bind to the same region on DNMT3A, resulting in roughly a 2-fold inhibition of DNMT3A activity. The DNMT3A-p53 interaction is modulated by well-known mutations in DNMT3A and p53. Our results provide insights into the complexity of mutation-specific variation in the regulation of protein function and elucidates a molecular basis for the distinguishing DNA methylation phenotypes associated with the R882H substitution in DNMT3A (61, 62).
The interaction interface between proteins relies on well-defined single residue interactions within flat surfaces (63, 64). Factors like kinetic accessibility to specific protein surfaces, thermodynamic stability of the resultant complexes, along with structural properties of protein complexes, contribute to the formation of biologically significant assembles (8–11, 65). Complexes with partner proteins regulate DNMT3A activity (12, 13, 37, 38, 46), thereby contributing to normal and aberrant tissue-specific methylation patterns (2–5). Given that DNMT3A binds the C-terminal tetramerization domain of p53 (residues 319–393, Fig. S3) (25), we sought to identify the surface on DNMT3A that binds p53 and whether p53 directly impacts DNMT3A activity. Like DNMT3L (36), our results suggest that surfaces on the catalytic domain of DNMT3A are sufficient for p53-mediated inhibition of DNMT3A (Fig. 1). Using docking-based computational models of DNMT3A (PDB 5YX2; residues 628–914) and p53 (PDB code 3TS8; 94–356) monomers, we identified the tetramer interface on DNMT3A as a likely surface for DNMT3A-p53 interactions (Fig. S2). We challenged this finding by determining if mutations at this interface (Fig. 2A) (41) interfered with p53 interactions (Fig. 2, B and C). Our results suggest an overlap between DNMT3L and p53 for binding and allosteric regulation of DNMT3A activity (Tetramer interface) (Fig. 2 and Fig. S2) through the formation of heterotetramers with DNMT3AWT (Fig. 5). Our proposed complex for DNMT3AWT:p53WT is consistent with previously resolved p53 co-crystal structures (PDB code 1KZY and 5ECG), which consist of p53 dimers bound to interacting partner proteins (66, 67). The structure and functional studies on DNMT3A interactions with another protein, DNMT3L, provides a reliable “metric” to investigate a common surface on DNMT3A that facilitates allosteric regulation of enzymatic activity. We show that DNMT3AWT:p53WT complexes (KD,app of 17 ± 3 nm) are more thermodynamically stable than DNMT3AWT:DNMT3L complexes (KD,app of 80 ± 12 nm). Consistent with these relative stabilities, DNMT3AWT:p53WT complexes appear to be more kinetically stable when all three proteins are combined (DNMT3A, DNMT3L, and p53). Moreover, p53WT can displace DNMT3L from the DNMT3A:DNMT3L heterotetrameric complex under catalytic conditions, which is arguably more relevant (Fig. 3). We propose that p53WT replaces the outer pair of DNMT3AWT monomers (Fig. 6A, III) or DNMT3L (Fig. 6A, V) monomers to allosterically inhibit the enzymatic activity of DNMT3AWT.
Figure 6.
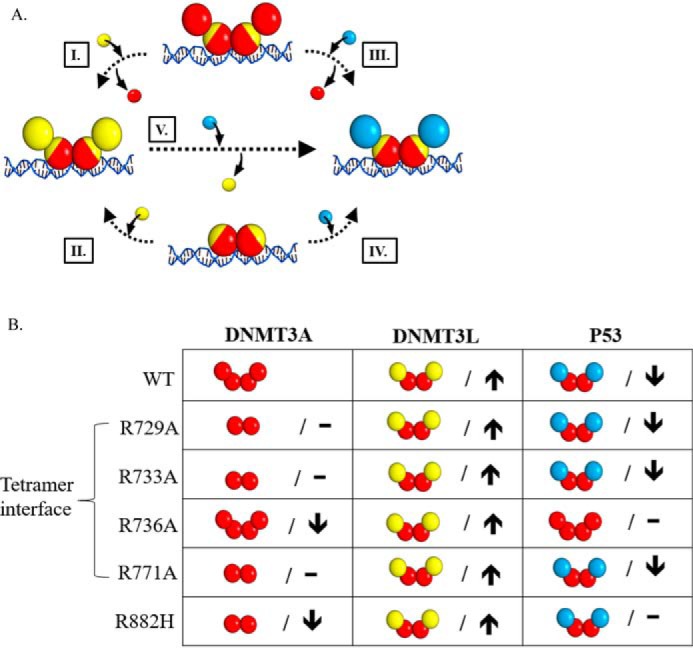
Mutations in DNMT3A lead to diverse interactions with p53. A, the addition of DNMT3L (yellow square) to DNMT3A homotetramers or homodimers (red square tetramer interface depicted in yellow) leads to the formation of DNMT3A (red square)-DNMT3L (yellow square) heterotetramers (I and II). Similarly, p53 (blue square) interacts with DNMT3A homotetramers or homodimers (red square) to form DNMT3A (red square)-p53 (blue square) heterotetramers (III and IV). Furthermore, the addition of p53 (blue square) to DNMT3A (red square)-DNMT3L (yellow square) heterotetramers displaces monomers at the tetramer interface and leads to the formation of DNMT3A-P53 (blue square) heterotetramers (V). B, summary of the oligomeric states of DNMT3A mutants in complex with DNMT3L (28, 36) and p53 as well as the effects on the catalytic function of DNMT3A. DNMT3A mutants display no change (–) or decreased (↓) activity (kcat) relative to WT. Although all the DNMT3A mutants are responsive to DNMT3L stimulation (↑), DNMT3A mutants display varying p53 inhibition (↓). Although DNMT3A R736A and R882H are unresponsive to p53 inhibition (–), p53 binds R882H to form heterotetramer.
Due to the energetic contributions of specific residues to protein-protein interactions (63, 64, 68, 69), it is not surprising that mutations that alter protein complex formation have been linked to various human disorders and are over-represented among disease-causing mutations (70, 71). The R882H substitution in DNMT3A is the most common recurrent mutation in AML patients (26) and DNMT3AR882H may be disruptive to protein-protein interactions (29, 72, 73). Although DNMT3AR882H is mildly impacted in function (28), it seems reasonable that altered interactions with partner proteins contribute to the aberrant methylation patterns observed in AML (61, 62). Previous work from our lab has shown that although R882H is a functional dimer on DNA, the addition of DNMT3L restores the formation of heterotetramers, and near-normal levels of catalysis (Fig. 6A, II) (28). The comparable increase in anisotropy observed by the addition of DNMT3L or p53 to DNA-bound DNMT3AR882H (Fig. 5) supports the notion that p53WT binds DNMT3AR882H (Fig. 5) to form heterotetramers but is unable to allosterically inhibit DNMT3AR882H activity (Figs. 3 and 6A, IV). Furthermore, p53WT fails to displace DNMT3L monomers in DNMT3AR882H:DNMT3L heterotetramers (Fig. 3) (Fig. 6A, V). Like DNMT3AR882H, certain mutations in p53 are disruptive to protein-protein interactions and alter regulation of partner proteins relative to WT p53 (32–34, 74, 75). In addition, several observations suggest an interplay between components of the epigenetic machinery and mutations in p53 (76, 77). Although located outside of the DNMT3AWT-p53WT interface (residues 319–393, Fig. S3) (25), we investigated how p53R248W and p53R273H mutations alter interactions with DNMT3AWT based on our previous findings on DNMT3AWT-p53WT interactions and high incidence in human cancers (47–50). Despite the greater stability of the DNMT3AWT:p53R248W heterotetramers (Fig. 5) (KD,app of 41 ± 6 nm) compared with the DNMT3AWT-DNMT3L complexes (KD,app of 80 ± 12 nm), DNMT3L modulation of DNMT3AWT activity is dominant over that of p53R248W and p53R273H (Fig. 4). We propose that p53 mutations do not compromise modulation of DNMT3AWT (Fig. 6A, III). These mutations may allosterically affect the DNMT3A-interacting interface (p53 residues 319–393, Fig. S3) (25) such that the affinity of mutant p53 in DNMT3AWT:mutant p53 complexes is compromised when presented with DNMT3L (Fig. 6A, V).
The ongoing discovery of hot spots in protein-protein interfaces, discrete regions that confer most of the binding energy, has sparked an interest in the pharmacological intervention of protein-protein interactions (78–82). In fact, successful modulation of protein-protein interactions by small molecules has been reported in p53 (83–89). We describe how, in the context of DNMT3A interactions with partner proteins, mutations at protein-protein interfaces may lead to diverse changes in protein interactions and modulation of protein activity. In addition, we provide examples in two important cancer-related proteins of how mutations located distally from protein-protein interfaces may affect modulation of enzymatic activity, thereby contributing to the diverse phenotypic consequences of mutations in epigenetic enzymes like DNMT3A (61, 62). The findings in this study broaden our understanding of regulation of DNMT3A activity and emphasize the potential use of small molecules to target protein-protein interactions in diseases, like AML, where DNMT3A and p53 are implicated (26, 90).
Experimental procedures
Expression constructs
The plasmids used for expression of recombinant DNMT3A full-length and catalytic domain (WT and R882H) proteins include pET28a-hDNMT3ACopt and pET28a-hDNMT3A_catalytic_domain (Δ1–611) (37). pTYB1–3L was used to express full-length human DNMT3L (38). pET15b-human p53(1–393) (Addgene) was used for expression of recombinant full-length human p53 as a template to generate R248W and R273H substitutions by site-directed mutagenesis (91).
Protein expression
DNMT3A full-length and catalytic domain (WT and R882H), DNMT3L and P53 (WT, R248W, and R273H) were expressed in NiCo21(DE3) Competent Escherichia coli cells (New England Biolabs). Cells were grown in LB media at 37 °C to an A600 nm of 0.9 (DNMT3A full-length), 0.7 (DNMT3A catalytic domain (WT and R882H)), 0.4 (DNMT3L), and 0.6 (P53 (WT, R248W, and R273H)). Protein expression was induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside (GoldBio) after lowering the temperature to 28 °C. Induction was 5 h for DNMT3A full-length and catalytic domains (WT and R882H) and 16 h for DNMT3L and P53 (WT, R248W, and R273H). Cell pellets were harvested by centrifugation at 5,000 × g for 15 min and stored at −80 °C.
Protein purification
Cell pellets from 1 liter of bacterial culture were resuspended in 30 ml of lysis buffer (50 mm HEPES, pH 7.8, 500 mm NaCl, 50 mm imidazole, 10% glycerol and phenylmethylsulfonyl fluoride) and lysed by sonication. Following sonication, lysates were centrifuged at 11,000 × g for 1 h and the supernatant was retained for affinity chromatography. Recombinant proteins were purified using ÄKTA Fast Protein Liquid Chromatography (FPLC) system (GE Healthcare) containing a 5-ml HisTrap HP nickel-charged IMAC column (GE Healthcare). Columns were preequilibrated with 50 ml of loading buffer (50 mm HEPES, pH 7.8, 500 mm NaCl, 50 mm imidazole, 10% glycerol). After flowing the supernatant through the column, resins were washed using 47.5 ml of wash buffer (50 mm HEPES, pH 7.8, 500 mm NaCl, 75 mm imidazole, 10% glycerol). Fractions of 0.5-ml were eluted with elution buffer (50 mm HEPES, pH 7.8, 500 mm NaCl, 500 mm imidazole, 10% glycerol) using a linear imidazole gradient (0–100%) over 15 ml. The eluate containing the proteins of interest was desalted and concentrated into storage buffer (50 mm Tris-Cl, 200 mm NaCl, 1 mm EDTA, 20% (v/v) glycerol, pH 7.8, with 0.5 mm DTT) using a 0.5-ml centrifugal filter (10K device) supplied by Millipore. Proteins were stored at −80 °C for later use.
Computational modeling
Using a DNMT3A (PDB code 5YX2, chain A) (92) and a P53 monomer (PDB code 3TS8, chain B) (93), the protein docking server ZDOCK was initially used to predict the interface on DNMT3A involved with DNMT3A-P53 interactions (42). In addition, identical modeling was performed using a DNMT3A monomer (PDB code 5YX2, chain A) (92) and a DNMT3L monomer (PDB code 2QRV, chain B) (47) for comparison as the interface on DNMT3A for DNMT3A-DNMT3L interactions has been previously resolved (47, 92). ZDOCK performs a rigid-body search of possible docking orientations between the proteins of interest (42). This docking server relies on the fast Fourier transform algorithm to perform a global docking search and explores all possible binding modalities by combining translation and rotation of the ligand. To rank each possible docking pose, ZDOCK applies a combination of shape complementarity, electrostatics, and statistical potential terms (94, 95). To predict the interface on DNMT3A involved with DNMT3A-P53 interactions, known contacting residues on P53 were considered (residues 319–393) (25). The local refinement on the RosettaDock server was then employed to perform rigid-body minimization and side chain conformation optimization (43). The local refinement function involves side chain repacking to improve rotameric side chain conformations and a Monte Carlo-based recovery of near-native protein structures (43). RosettaDock employs an energy-based scoring function that calculates the energy of interactions by amino acids (43).
Methylation assays
Assays were carried out to measure the ability of DNMT3A to incorporate tritiated methyl groups transferred from cofactor AdoMet onto DNA substrate under various experimental conditions. Reactions were carried out at 37 °C in a buffer consisting of 50 mm KH2PO4/K2HPO4, 1 mm EDTA, 1 mm DTT, 0.2 mg/ml of BSA, 20 mm NaCl with saturating AdoMet (15 μm) at pH 7.8. 50 μm ([3H]methyl-labeled:unlabeled) AdoMet stocks were made using 32 mm unlabeled AdoMet (New England Biolabs) and [3H]methyl-labeled AdoMet (X Ci/mmol) supplied by PerkinElmer in 10 mm H2SO4. In all assays, 15-μl aliquots were taken from a larger reaction and quenched by mixing with 0.1% SDS (1:1). Samples were spotted onto Hybond-XL membranes (GE Healthcare), washed, dried, and methylation was using a Beckman LS 6000 liquid scintillation Counter as previously established (96). Due to the large number of potential methylation sites, poly(dI-dC) is commonly used as a DNA substrate to study the enzymatic activity of DNA modifying enzymes (97–101). In addition, the use of poly(dI-dC) as a DNA substrate allowed us to investigate the isolated modulatory effect of p53 on DNMT3A activity. On this basis, 5 μm poly(dI-dC) (Sigma-Aldrich) was used as a DNA substrate. Previous work from our lab has provided insights into the mechanism of DNMT3A activity on human promoters (102). Given that Cyclin-dependent Kinase Inhibitor 1A/P21 is a common target for DNMT3A and p53 (25, 102), we additionally employed Cyclin-dependent Kinase Inhibitor 1A/P21-pCpGL as a substrate. All radiochemical assays were performed in triplicates from a single purification and statistical analysis was performed using Prism version 7 (GraphPad).
P53 assays
To test modulation of DNMT3A (full-length, catalytic domain, and mutant enzymes) or M.HhaI methylation activity by P53WT, proteins were preincubated in reaction buffer with AdoMet for 1 h at 37 °C before initiating the reaction by the addition of substrate DNA. Reactions were then run for 1 h, stopped as stated above, and methylation was counted. All proteins (DNMT3A, M.HhaI, or P53WT) were at a ratio of 1:1 (150 nm) in the 1-h preincubation. Fold-inhibition was calculated by product formed by DNMT3A variants (or M.HhaI) divided by product formed by DNMT3A variants (or M.HhaI) without P53WT. Statistical analysis was performed using GraphPad software (version 6.0).
DNMT3L and P53 assays
To evaluate regulation of DNMT3A activity by DNMT3L or P53 under a binary (protein pairs) approach, proteins (1:1:1 at 150 nm) were preincubated in reaction buffer with AdoMet for 1 h at 37 °C before starting the reaction by the addition of substrate DNA. To assess modulation of DNMT3A activity under a co-complex (groups of proteins) approach, DNMT3A was preincubated with DNMT3L (or P53) (1:1 at 150 nm) for 1 h at 37 °C prior to initiating the reaction by the addition of substrate DNA and enzyme were allowed to carry out catalysis for 30 min before the addition of P53 (or DNMT3L) (150 nm). Reactions were then monitored for an additional hour, stopped, and methylation was counted as stated above.
Apparent binding affinities (KD,app)
Modulation of DNMT3A activity by varying DNMT3L, P53WT, or P53R248W concentrations from 10 to 300 nm with 10 nm DNMT3A was tested to determine apparent affinities (KD,app). DNMT3AWT was preincubated with varying concentrations of DNMT3L, P53WT, or P53R248W in reaction buffer with AdoMet for 1 h at 37 °C prior initiating the reaction by the addition of substrate DNA. Following the addition of DNA, reactions were run for 1 h at 37 °C, stopped, and methylation was counted as stated above. Fold-stimulation was calculated by product formed by DNMT3AWT with DNMT3L divided by product formed by DNMT3AWT without DNMT3L. Fold-inhibition by P53WT or P53R248W was calculated as stated above. The data were fit to a one site-specific binding equation using GraphPad software (version 6.0).
Fluorescence anisotropy
Fluorescence anisotropy measurements were performed using a Horiba Fluoromax fluorescence spectrophotometer equipped with excitation and emission polarizers (excitation, 485 nm; emission, 520 nm). Fluorescence anisotropy measurements of DNA-bound DNMT3AWT or DNMT3AR882H (both at 2.5 μm) were taken following the titration of DNMT3L (DNMT3AWT and DNMT3AR882H reactions), P53WT (DNMT3AWT and DNMT3AR882H reactions), or P53R248W (DNMT3AWT reactions). Anisotropy values were obtained following a 5-min preincubation at room temperature. The substrate DNA (Gcbox30) consisted of a fluorescein (6-FAM) label on the 5′ end of the top strand of the duplex (5′/6-FAM/TGGATATCTAGGGGCGCTATGATATCT-3′) and was supplied by Integrated DNA Technologies; the recognition site for DNMT3A is underlined.
Electrophoretic mobility shift assay (EMSA)
Experiments were carried out as described in Holz-Schietinger et al. (28). In brief, DNMT3AR882H (150 nm) was incubated at 37 °C for 15 min with 200 nm duplex 5′ 6-FAM GCbox30 in reaction buffer with 50 μm Sinefungin (Sigma-Aldrich) and 10% glycerol. For P53WT super shifting, varying concentrations of P53WT were preincubated with DNMT3AR882H under identical conditions for 30 min at 37 °C before the addition of GCbox30. Samples were run on a native 4.5% (75:1) polyacrylamide gel in 0.25× Tris boric acid/EDTA, pH 7.8, at 250 V for 50 min. Gels were visualized for fluorescein using a Typhoon scanner and data were analyzed using ImageJ.
Author contributions
J. E. S. data curation; J. E. S. and N. O. R. formal analysis; J. E. S. investigation; J. E. S. and N. O. R. methodology; J. E. S. and N. O. R. writing-original draft; J. E. S. and N. O. R. project administration; J. E. S. and N. O. R. writing-review and editing; N. O. R. conceptualization; N. O. R. resources; N. O. R. supervision; N. O. R. funding acquisition.
Supplementary Material
This work was supported by Center for Hierarchical Manufacturing National Science Foundation award 1808775. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
- DNMT
- DNA methyltransferase
- DNMT3L
- DNA-methyltransferase 3-like
- PDB
- Protein Data Bank
- AML
- acute myeloid leukemia
- AdoMet
- S-adenosylmethionine.
References
- 1. Vertino P. M., Sekowski J. A., Coll J. M., Applegreen N., Han S., Hickey R. J., and Malkas L. H. (2002) DNMT1 is a component of a multiprotein DNA replication complex. Cell Cycle 1, 416–423 [DOI] [PubMed] [Google Scholar]
- 2. Viré E., Brenner C., Deplus R., Blanchon L., Fraga M., Didelot C., Morey L., Van Eynde A., Bernard D., Vanderwinden J. M., Bollen M., et al. (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874 10.1038/nature04431 [DOI] [PubMed] [Google Scholar]
- 3. Datta J., Majumder S., Bai S., Ghoshal K., Kutay H., Smith D. S., Crabb J. W., and Jacob S. T. (2005) Physical and functional interaction of DNA methyltransferase 3A with Mbd3 and Brg1 in mouse lymphosarcoma cells. Cancer Res. 65, 10891–10900 10.1158/0008-5472.CAN-05-1455 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4. Brenner C., Deplus R., Didelot C., Loriot A., Viré E., De Smet C., Gutierrez A., Danovi D., Bernard D., Boon T., Pelicci P. G., et al. (2005) Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 24, 336–346 10.1038/sj.emboj.7600509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thillainadesan G., Chitilian J. M., Isovic M., Ablack J. N., Mymryk J. S., Tini M., and Torchia J. (2012) TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol. Cell 46, 636–649 10.1016/j.molcel.2012.03.027 [DOI] [PubMed] [Google Scholar]
- 6. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- 7. Reik W., Dean W., and Walter J. (2001) Epigenetic reprogramming in mammalian development. Science 293, 1089–1093 10.1126/science.1063443 [DOI] [PubMed] [Google Scholar]
- 8. Ellis R. J. (2001) Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11, 114–119 10.1016/S0959-440X(00)00172-X [DOI] [PubMed] [Google Scholar]
- 9. Levy E. D., De S., and Teichmann S. A. (2012) Cellular crowding imposes global constraints on the chemistry and evolution of proteomes. Proc. Natl. Acad. Sci. U.S.A. 109, 20461–20466 10.1073/pnas.1209312109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schreiber G., Haran G., and Zhou H. X. (2009) Fundamental aspects of protein-protein association kinetics. Chem. Rev. 109, 839–860 10.1021/cr800373w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chakravarty D., Janin J., Robert C. H., and Chakrabarti P. (2015) Changes in protein structure at the interface accompanying complex formation. IUCr J. 2, 643–652 10.1107/S2052252515015250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo X., Wang L., Li J., Ding Z., Xiao J., Yin X., He S., Shi P., Dong L., Li G., Tian C., et al. (2015) Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517, 640–644 10.1038/nature13899 [DOI] [PubMed] [Google Scholar]
- 13. Holz-Schietinger C., and Reich N. O. (2012) RNA modulation of the human DNA methyltransferase 3A. Nucleic Acids Res. 40, 8550–8557 10.1093/nar/gks537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang L., Rau R., and Goodell M. A. (2015) DNMT3A in haematological malignancies. Nat. Rev. Cancer 15, 152–165 10.1038/nrc3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bieging K. T., Mello S. S., and Attardi L. D. (2014) Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370 10.1038/nrc3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kastenhuber E. R., and Lowe S. W. (2017) Putting p53 in context. Cell 170, 1062–1078 10.1016/j.cell.2017.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Espinosa J. M., Verdun R. E., and Emerson B. M. (2003) p53 functions through stress-and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol. Cell 12, 1015–1027 10.1016/S1097-2765(03)00359-9 [DOI] [PubMed] [Google Scholar]
- 18. Williams A. B., and Schumacher B. (2016) p53 in the DNA-damage-repair process. Cold Spring Harbor Perspect. Med. 6, a026070 10.1101/cshperspect.a026070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xie P., Tian C., An L., Nie J., Lu K., Xing G., Zhang L., and He F. (2008) Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell. Signal. 20, 1671–1678 10.1016/j.cellsig.2008.05.012 [DOI] [PubMed] [Google Scholar]
- 20. Agalioti T., Chen G., and Thanos D. (2002) Deciphering the transcriptional histone acetylation code for a human gene. Cell 111, 381–392 10.1016/S0092-8674(02)01077-2 [DOI] [PubMed] [Google Scholar]
- 21. Kaeser M. D., and Iggo R. D. (2004) Promoter-specific p53-dependent histone acetylation following DNA damage. Oncogene 23, 4007–4013 10.1038/sj.onc.1207536 [DOI] [PubMed] [Google Scholar]
- 22. Georgia S., Kanji M., and Bhushan A. (2013) DNMT1 represses p53 to maintain progenitor cell survival during pancreatic organogenesis. Genes Dev. 27, 372–377 10.1101/gad.207001.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Estève P. O., Chin H. G., and Pradhan S. (2005) Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc. Natl. Acad. Sci. U.S.A. 102, 1000–1005 10.1073/pnas.0407729102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tovy A., Spiro A., McCarthy R., Shipony Z., Aylon Y., Alton K., Ainbinder E., Furth N., Tanay A., Barton M., and Oren M. (2017) p53 is essential for DNA methylation homeostasis in naïve embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 31, 959–972 10.1101/gad.299198.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y. A., Kamarova Y., Shen K. C., Jiang Z., Hahn M. J., Wang Y., and Brooks S. C. (2005) DNA methyltransferase-3a interacts with p53 and represses p53-mediated gene expression. Cancer Biol. Ther. 4, 1138–1143 10.4161/cbt.4.10.2073 [DOI] [PubMed] [Google Scholar]
- 26. Ley T. J., Ding L., Walter M. J., McLellan M. D., Lamprecht T., Larson D. E., Kandoth C., Payton J. E., Baty J., Welch J., Harris C. C., et al. (2010) DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 10.1056/NEJMoa1005143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Russler-Germain D. A., Spencer D. H., Young M. A., Lamprecht T. L., Miller C. A., Fulton R., Meyer M. R., Erdmann-Gilmore P., Townsend R. R., Wilson R. K., and Ley T. J. (2014) The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 25, 442–454 10.1016/j.ccr.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holz-Schietinger C., Matje D. M., and Reich N. O. (2012) Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 287, 30941–30951 10.1074/jbc.M112.366625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koya J., Kataoka K., Sato T., Bando M., Kato Y., Tsuruta-Kishino T., Kobayashi H., Narukawa K., Miyoshi H., Shirahige K., and Kurokawa M. (2016) DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat. Commun. 7, 10924 10.1038/ncomms10924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vogelstein B., Lane D., and Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- 31. Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S. V., Hainaut P., and Olivier M. (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629 10.1002/humu.20495 [DOI] [PubMed] [Google Scholar]
- 32. Song H., Hollstein M., and Xu Y. (2007) p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 9, 573–580 10.1038/ncb1571 [DOI] [PubMed] [Google Scholar]
- 33. Strano S., Fontemaggi G., Costanzo A., Rizzo M. G., Monti O., Baccarini A., Del Sal G., Levrero M., Sacchi A., Oren M., and Blandino G. (2002) Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 277, 18817–18826 10.1074/jbc.M201405200 [DOI] [PubMed] [Google Scholar]
- 34. Gaiddon C., Lokshin M., Ahn J., Zhang T., and Prives C. (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 21, 1874–1887 10.1128/MCB.21.5.1874-1887.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Purdy M. M., Holz-Schietinger C., and Reich N. O. (2010) Identification of a second DNA binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion. Arch. Biochem. Biophys. 498, 13–22 10.1016/j.abb.2010.03.007 [DOI] [PubMed] [Google Scholar]
- 36. Kareta M. S., Botello Z. M., Ennis J. J., Chou C., and Chédin F. (2006) Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 281, 25893–25902 10.1074/jbc.M603140200 [DOI] [PubMed] [Google Scholar]
- 37. Galonska C., Charlton J., Mattei A. L., Donaghey J., Clement K., Gu H., Mohammad A. W., Stamenova E. K., Cacchiarelli D., Klages S., Timmermann B., Cantz T., Schöler H. R., Gnirke A., Ziller M. J., and Meissner A. (2018) Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun. 9, 597 10.1038/s41467-017-02708-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lei Y., Huang Y. H., and Goodell M. A. (2018) DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. 19, 187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. el-Deiry W. S., Kern S. E., Pietenpol J. A., Kinzler K. W., and Vogelstein B. (1992) Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49 10.1038/ng0492-45 [DOI] [PubMed] [Google Scholar]
- 40. Cheng X., and Blumenthal R. M. (2008) Mammalian DNA methyltransferases: a structural perspective. Structure 16, 341–350 10.1016/j.str.2008.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Holz-Schietinger C., Matje D. M., Harrison M. F., and Reich N. O. (2011) Oligomerization of DNMT3A controls the mechanism of de novo DNA methylation. J. Biol. Chem. 286, 41479–41488 10.1074/jbc.M111.284687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lyskov S., and Gray J. J. (2008) The RosettaDock server for local protein–protein docking. Nucleic Acids Res. 36, W233–W238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Datta J., Ghoshal K., Sharma S. M., Tajima S., and Jacob S. T. (2003) Biochemical fractionation reveals association of DNA methyltransferase (Dnmt) 3b with Dnmt1 and that of Dnmt 3a with a histone H3 methyltransferase and Hdac1. J. Cell. Biochem. 88, 855–864 10.1002/jcb.10457 [DOI] [PubMed] [Google Scholar]
- 45. Neri F., Krepelova A., Incarnato D., Maldotti M., Parlato C., Galvagni F., Matarese F., Stunnenberg H. G., and Oliviero S. (2013) Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 155, 121–134 10.1016/j.cell.2013.08.056 [DOI] [PubMed] [Google Scholar]
- 46. Jia D., Jurkowska R. Z., Zhang X., Jeltsch A., and Cheng X. (2007) Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251 10.1038/nature06146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hollstein M., Sidransky D., Vogelstein B., and Harris C. C. (1991) p53 mutations in human cancers. Science 253, 49–53 10.1126/science.1905840 [DOI] [PubMed] [Google Scholar]
- 48. Levine A. J., and Oren M. (2009) The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758 10.1038/nrc2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harris C. C., and Hollstein M. (1993) Clinical implications of the p53 tumor-suppressor gene. N. Engl. J. Med. 329, 1318–1327 10.1056/NEJM199310283291807 [DOI] [PubMed] [Google Scholar]
- 50. Cho Y., Gorina S., Jeffrey P. D., and Pavletich N. P. (1994) Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346–355 10.1126/science.8023157 [DOI] [PubMed] [Google Scholar]
- 51. Anderson B. J., Larkin C., Guja K., and Schildbach J. F. (2008) Using fluorophore-labeled oligonucleotides to measure affinities of protein–DNA interactions. Methods Enzymol. 450, 253–272 10.1016/S0076-6879(08)03412-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gradinaru C. C., Marushchak D. O., Samim M., and Krull U. J. (2010) Fluorescence anisotropy: from single molecules to live cells. Analyst 135, 452–459 10.1039/b920242k [DOI] [PubMed] [Google Scholar]
- 53. Heyduk T., Ma Y., Tang H., and Ebright R. H. (1996) Fluorescence anisotropy: rapid, quantitative assay for protein-DNA and protein-protein interaction. Methods Enzymol. 274, 492–503 10.1016/S0076-6879(96)74039-9 [DOI] [PubMed] [Google Scholar]
- 54. Emperle M., Rajavelu A., Reinhardt R., Jurkowska R. Z., and Jeltsch A. (2014) Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J. Biol. Chem. 289, 29602–29613 10.1074/jbc.M114.572032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yan Y., and Marriott G. (2003) Analysis of protein interactions using fluorescence technologies. Curr. Opin. Chem. Biol. 7, 635–640 10.1016/j.cbpa.2003.08.017 [DOI] [PubMed] [Google Scholar]
- 56. Kreida S., Roche J. V., Olsson C., Linse S., and Törnroth-Horsefield S. (2018) Protein–protein interactions in AQP regulation: biophysical characterization of AQP0–CaM and AQP2–LIP5 complex formation. Faraday Disc. 209, 35–54 10.1039/C8FD00065D [DOI] [PubMed] [Google Scholar]
- 57. Oi C., Treado J. D., Levine Z. A., Lim C. S., Knecht K. M., Xiong Y., O'Hern C. S., and Regan L. (2018) A threonine zipper that mediates protein–protein interactions: structure and prediction. Protein Sci. 27, 1969–1977 10.1002/pro.3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Singh S. P., Kukshal V., and Galletto R. (2019) A stable tetramer is not the only oligomeric state that mitochondrial single-stranded DNA-binding proteins can adopt. J. Biol. Chem. 294, 4137–4144 10.1074/jbc.RA118.007048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Karpf A. R., Moore B. C., Ririe T. O., and Jones D. A. (2001) Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2′-deoxycytidine. Mol. Pharmacol. 59, 751–757 10.1124/mol.59.4.751 [DOI] [PubMed] [Google Scholar]
- 60. Jackson-Grusby L., Beard C., Possemato R., Tudor M., Fambrough D., Csankovszki G., Dausman J., Lee P., Wilson C., Lander E., and Jaenisch R. (2001) Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 27, 31–39 10.1038/83730 [DOI] [PubMed] [Google Scholar]
- 61. Spencer D. H., Russler-Germain D. A., Ketkar S., Helton N. M., Lamprecht T. L., Fulton R. S., Fronick C. C., O'Laughlin M., Heath S. E., Shinawi M., Westervelt P., et al. (2017) CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 168, 801–816.e13 10.1016/j.cell.2017.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen D., Christopher M., Helton N. M., Ferguson I., Ley T. J., and Spencer D. H. (2018) DNMT3A R882-associated hypomethylation patterns are maintained in primary AML xenografts, but not in the DNMT3A R882C OCI-AML3 leukemia cell line. Blood Cancer J. 8, 38 10.1038/s41408-018-0072-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rajamani D., Thiel S., Vajda S., and Camacho C. J. (2004) Anchor residues in protein–protein interactions. Proc. Natl. Acad. Sci. U.S.A. 101, 11287–11292 10.1073/pnas.0401942101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li X., Keskin O., Ma B., Nussinov R., and Liang J. (2004) Protein–protein interactions: hot spots and structurally conserved residues often locate in complemented pockets that pre-organized in the unbound states: implications for docking. J. Mol. Biol. 344, 781–795 10.1016/j.jmb.2004.09.051 [DOI] [PubMed] [Google Scholar]
- 65. Sowmya G., Breen E. J., and Ranganathan S. (2015) Linking structural features of protein complexes and biological function. Protein Sci. 24, 1486–1494 10.1002/pro.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Joo W. S., Jeffrey P. D., Cantor S. B., Finnin M. S., Livingston D. M., and Pavletich N. P. (2002) Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes Dev. 16, 583–593 10.1101/gad.959202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baldock R. A., Day M., Wilkinson O. J., Cloney R., Jeggo P. A., Oliver A. W., Watts F. Z., and Pearl L. H. (2015) ATM localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with γH2AX. Cell Rep. 13, 2081–2089 10.1016/j.celrep.2015.10.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jubb H. C., Pandurangan A. P., Turner M. A., Ochoa-Montaño B., Blundell T. L., and Ascher D. B. (2017) Mutations at protein-protein interfaces: small changes over big surfaces have large impacts on human health. Prog. Biophys. Mol. Biol. 128, 3–13 10.1016/j.pbiomolbio.2016.10.002 [DOI] [PubMed] [Google Scholar]
- 69. David A., Razali R., Wass M. N., and Sternberg M. J. (2012) Protein–protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat. 33, 359–363 10.1002/humu.21656 [DOI] [PubMed] [Google Scholar]
- 70. Engin H. B., Kreisberg J. F., and Carter H. (2016) Structure-based analysis reveals cancer missense mutations target protein interaction interfaces. PLoS ONE 11, e0152929 10.1371/journal.pone.0152929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yates C. M., and Sternberg M. J. (2013) The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein–protein interactions. J. Mol. Biol. 425, 3949–3963 10.1016/j.jmb.2013.07.012 [DOI] [PubMed] [Google Scholar]
- 72. Trowbridge J. J., and Orkin S. H. (2011) Dnmt3a silences hematopoietic stem cell self-renewal. Nat. Genet. 44, 13–14 10.1038/ng.1043 [DOI] [PubMed] [Google Scholar]
- 73. Xu J., Wang Y. Y., Dai Y. J., Zhang W., Zhang W. N., Xiong S. M., Gu Z. H., Wang K. K., Zeng R., Chen Z., and Chen S. J. (2014) DNMT3A Arg882 mutation drives chronic myelomonocytic leukemia through disturbing gene expression/DNA methylation in hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 111, 2620–2625 10.1073/pnas.1400150111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Freed-Pastor W. A., and Prives C. (2012) Mutant p53: one name, many proteins. Genes Dev. 26, 1268–1286 10.1101/gad.190678.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim M. P., and Lozano G. (2018) Mutant p53 partners in crime. Cell Death Differ. 25, 161–168 10.1038/cdd.2017.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhu J., Sammons M. A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Al-awar R., Katona B. W., Shilatifard A., Huang J., Hua X., Arrowsmith C. H., and Berger S. L. (2015) Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 10.1038/nature15251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nieto M., Samper E., Fraga M. F., González de Buitrago G. G., Esteller M., and Serrano M. (2004) The absence of p53 is critical for the induction of apoptosis by 5-aza-2′-deoxycytidine. Oncogene 23, 735–743 10.1038/sj.onc.1207175 [DOI] [PubMed] [Google Scholar]
- 78. Jin L., Wang W., and Fang G. (2014) Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 54, 435–456 10.1146/annurev-pharmtox-011613-140028 [DOI] [PubMed] [Google Scholar]
- 79. Ivanov A. A., Khuri F. R., and Fu H. (2013) Targeting protein–protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 34, 393–400 10.1016/j.tips.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Petta I., Lievens S., Libert C., Tavernier J., and De Bosscher K. (2016) Modulation of protein–protein interactions for the development of novel therapeutics. Mol. Ther. 24, 707–718 10.1038/mt.2015.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Arkin M. R., and Wells J. A. (2004) Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 3, 301–317 10.1038/nrd1343 [DOI] [PubMed] [Google Scholar]
- 82. Clackson T., and Wells J. A. (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267, 383–386 [DOI] [PubMed] [Google Scholar]
- 83. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., and Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 10.1126/science.1092472 [DOI] [PubMed] [Google Scholar]
- 84. Vu B., Wovkulich P., Pizzolato G., Lovey A., Ding Q., Jiang N., Liu J. J., Zhao C., Glenn K., Wen Y., Tovar C., Packman K., Vassilev L., and Graves B. (2013) Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469 10.1021/ml4000657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ray-Coquard I., Blay J. Y., Italiano A., Le Cesne A., Penel N., Zhi J., Heil F., Rueger R., Graves B., Ding M., Geho D., Middleton S. A., Vassilev L. T., Nichols G. L., and Bui B. N. (2012) Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 13, 1133–1140 10.1016/S1470-2045(12)70474-6 [DOI] [PubMed] [Google Scholar]
- 86. Ding Q., Zhang Z., Liu J. J., Jiang N., Zhang J., Ross T. M., Chu X. J., Bartkovitz D., Podlaski F., Janson C., Tovar C., Filipovic Z. M., Higgins B., Glenn K., Packman K., et al. (2013) Discovery of RG7388, a potent and selective p53–MDM2 inhibitor in clinical development. J. Med. Chem. 56, 5979–5983 10.1021/jm400487c [DOI] [PubMed] [Google Scholar]
- 87. Zhao Y., Aguilar A., Bernard D., and Wang S. (2015) Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 inhibitors) in clinical trials for cancer treatment: miniperspective. J. Med. Chem. 58, 1038–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang S., Sun W., Zhao Y., McEachern D., Meaux I., Barrière C., Stuckey J. A., Meagher J. L., Bai L., Liu L., Hoffman-Luca C. G., Lu J., Shangary S., Yu S., Bernard D., et al. (2014) SAR405838: an optimized inhibitor of MDM2–p53 interaction that induces complete and durable tumor regression. Cancer Res. 74, 5855–5865 10.1158/0008-5472.CAN-14-0799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sun D., Li Z., Rew Y., Gribble M., Bartberger M. D., Beck H. P., Canon J., Chen A., Chen X., Chow D., Deignan J., Duquette J., Eksterowicz J., Fisher B., Fox B. M., et al. (2014) Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2–p53 inhibitor in clinical development. J. Med. Chem. 57, 1454–1472 10.1021/jm401753e [DOI] [PubMed] [Google Scholar]
- 90. Cancer Genome Atlas Research Network, Ley T. J., Miller C., Ding L., Raphael B. J., Mungall A. J., Robertson A., Hoadley K., Triche T. J. Jr., Laird P. W., Baty J. D., Fulton L. L., et al. (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 10.1056/NEJMoa1301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ayed A., Mulder F. A., Yi G. S., Lu Y., Kay L. E., and Arrowsmith C. H. (2001) Latent and active p53 are identical in conformation. Nat. Struct. Mol. Biol. 8, 756–760 10.1038/nsb0901-756 [DOI] [PubMed] [Google Scholar]
- 92. Zhang Z. M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S. B., Wang Y., Wang G. G., and Song J. (2018) Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554, 387–391. 10.1038/nature25477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Emamzadah S., Tropia L., and Halazonetis T. D. (2011) Crystal structure of a multidomain human p53 tetramer bound to the natural CDKN1A (p21) p53-response element. Mol. Cancer Res. 9, 1493–1499 10.1158/1541-7786.MCR-11-0351 [DOI] [PubMed] [Google Scholar]
- 94. Mintseris J., Pierce B., Wiehe K., Anderson R., Chen R., and Weng Z. (2007) Integrating statistical pair potentials into protein complex prediction. Proteins 69, 511–520 10.1002/prot.21502 [DOI] [PubMed] [Google Scholar]
- 95. Chen R., Li L., and Weng Z. (2003) ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87 10.1002/prot.10389 [DOI] [PubMed] [Google Scholar]
- 96. Peterson S. N., and Reich N. O. (2006) GATC flanking sequences regulate Dam activity: evidence for how Dam specificity may influence pap expression. J. Mol. Biol. 355, 459–472 10.1016/j.jmb.2005.11.003 [DOI] [PubMed] [Google Scholar]
- 97. Zhang G., Estève P. O., Chin H. G., Terragni J., Dai N., Corrêa I. R. Jr., and Pradhan S. (2015) Small RNA-mediated DNA (cytosine-5) methyltransferase 1 inhibition leads to aberrant DNA methylation. Nucleic Acids Res. 43, 6112–6124 10.1093/nar/gkv518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ponnaluri V. C., Estève P. O., Ruse C. I., and Pradhan S. (2018) S-Adenosylhomocysteine hydrolase participates in DNA methylation inheritance. J. Mol. Biol. 430, 2051–2065 10.1016/j.jmb.2018.05.014 [DOI] [PubMed] [Google Scholar]
- 99. Mellini P., Marrocco B., Borovika D., Polletta L., Carnevale I., Saladini S., Stazi G., Zwergel C., Trapencieris P., Ferretti E., and Tafani M. (2018). Pyrazole-based inhibitors of enhancer of zeste homologue 2 induce apoptosis and autophagy in cancer cells. Philos. Trans. R. Soc. B Biol. Sci. 373, 20170150 10.1098/rstb.2017.0150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mao S. Q., Ghanbarian A. T., Spiegel J., Cuesta S. M., Beraldi D., Di Antonio M., Marsico G., Hänsel-Hertsch R., Tannahill D., and Balasubramanian S. (2018) DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol. 25, 951–957 10.1038/s41594-018-0131-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Balakrishnan A., Guruprasad K. P., Satyamoorthy K., and Joshi M. B. (2018) Interleukin-6 determines protein stabilization of DNA methyltransferases and alters DNA promoter methylation of genes associated with insulin signaling and angiogenesis. Lab. Invest. 98, 1143–1158 10.1038/s41374-018-0079-7 [DOI] [PubMed] [Google Scholar]
- 102. Holz-Schietinger C., and Reich N. O. (2010) The inherent processivity of the human de novo methyltransferase 3A (DNMT3A) is enhanced by DNMT3L. J. Biol. Chem. 285, 29091–29100 10.1074/jbc.M110.142513 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.