

Abstract
Alginate is a polymer containing two uronic acid epimers, β-d-mannuronate (M) and α-l-guluronate (G), and is a major component of brown seaweed that is depolymerized by alginate lyases. These enzymes have diverse specificity, cleaving the chain with endo- or exotype activity and with differential selectivity for the sequence of M or G at the cleavage site. Dp0100 is a 201-kDa multimodular, broad-specificity endotype alginate lyase from the marine thermophile Defluviitalea phaphyphila, which uses brown algae as a carbon source, converting it to ethanol, and bioinformatics analysis suggested that its catalytic domain represents a new polysaccharide lyase family, PL39. The structure of the Dp0100 catalytic domain, determined at 2.07 Å resolution, revealed that it comprises three regions strongly resembling those of the exotype lyase families PL15 and PL17. The conservation of key catalytic histidine and tyrosine residues belonging to the latter suggests these enzymes share mechanistic similarities. A complex of Dp0100 with a pentasaccharide, M5, showed that the oligosaccharide is located in subsites −2, −1, +1, +2, and +3 in a long, deep canyon open at both ends, explaining the endotype activity of this lyase. This contrasted with the hindered binding sites of the exotype enzymes, which are blocked such that only one sugar moiety can be accommodated at the −1 position in the catalytic site. The biochemical and structural analyses of Dp0100, the first for this new class of endotype alginate lyases, have furthered our understanding of the structure–function and evolutionary relationships within this important class of enzymes.
Keywords: alginate lyase, carbohydrate-binding protein, oligosaccharide, crystal structure, metalloenzyme, substrate specificity, structure–function
Introduction
The growth of brown algae represents an important carbon sink in the oceans of the world, and as a result, the polysaccharide alginate, a major component of the algal cell wall, is one of the most abundant carbohydrates in the ocean (1, 2). The abundance of these seaweeds has made them an attractive and important source of renewable biomass for biofuel production (3, 4). Alginate is a linear polysaccharide consisting of ∼1000 residues of a mixture of the two uronic acid epimers, α-l-guluronate (G)4 and β-d-mannuronate (M), that are connected by α-(1,4) O-linked glycosidic bonds (5, 6). The sequence of the sugars in the polymer is not random but instead consists of regions where the sugars can be found in M- or G-rich regions or in stretches of alternating M and G residues. An unusual property of this polymer is its ability to self-assemble into crystalline or less structured regions depending on the nature of the sequence in a process that requires the addition of divalent cations, particularly calcium. The organization and spacing of the structured and unstructured regions provide an extracellular matrix in which the algal cells are embedded in macrocellular arrays in pockets that have been likened to the chambers in an egg-box, the so-called egg-box model (7). As a natural polysaccharide, alginate and oligosaccharides derived from it by depolymerization have been widely used in the food, pharmaceutical, and biomaterial industries (8). Thus, the enzymatic degradation of alginate is of significant biotechnological importance.
Alginate lyases catalyze the depolymerization of alginate cleaving the polysaccharide chain in an endo- or exo-specific manner. These enzymes have also been shown to exhibit differential substrate specificity with three types of activity being recognized, mannuronate-specific alginate lyase (EC 4.2.2.3), guluronate-specific alginate lyase (EC 4.2.2.11), and a bifunctional enzyme that cleaves regions of alginate containing both M and G. The reaction results in the cleavage of the glycosidic bond leaving an unsaturated monosaccharide, 4-deoxy-l-erythro-hex-4-enepyranosyl uronic acid, at the nonreducing end of the oligosaccharide chain (9, 10). Of the 37 polysaccharide lyase (PL) families identified to date in the Carbohydrate-Active enZYmes (CAZy) database (11), nine are alginate lyases, and the structures of representative members of seven of these have been determined. The structural studies have resulted in the recognition of 4-fold classes, including simple single domain alginate lyases with an (α/α)n toroid (PL5), a β-helix (PL6), or a β-jelly-roll (PL7, PL14, and PL18) fold, and the more complex multidomain alginate lyases, which combine domains with an (α/α)n toroid fold with antiparallel β-sandwich domains (PL15 and PL17) (12). Metal ions, including calcium and zinc, have been identified in the structures of some of these enzymes; however, their roles in catalysis, specificity, and stability are not yet fully understood.
Structural and biochemical studies of representative exotype alginate lyases belonging to families PL7, PL15, and PL17 have identified that the exotype cleavage arises as a result of the relative position of the catalytic site compared with that of the binding site for oligosaccharides where the nonreducing end of the sugar occupies a sterically-hindered pocket that can only accommodate a single sugar moiety (13–15). In contrast, in the single domain endotype PL7 family lyase from Zobellia galactanivorans, the oligosaccharide-binding site is more open, providing a molecular explanation for the endolytic activity.
Proposals for the mechanism of alginate lyases suggest that the reaction involves the removal of the acidic proton at the C5 position of the +1 subsite uronic acid (sugar-binding subsites are numbered according to the nomenclature proposed by Davies et al. (16)) leading to the formation of a stabilized aci-carboxylate intermediate (17). Collapse of the intermediate leads to the formation of a double bond between C4 and C5 and the cleavage of the glycosidic linkage between −1 and +1 subsites (12). Residues identified as important roles for catalysis include tyrosine as a Brønsted acid, tyrosine or histidine as a Brønsted base, and Gln/Asn and His/Arg as playing important roles in the stabilization of the aci-carboxylate (12, 18).
To contribute to a better understanding of the structural specificity, catalysis and evolution of multidomain alginate lyases, this paper reports studies on a novel endolytic alginate lyase Dp0100 from the thermophilic bacterium Defluviitalea phaphyphila. This microbe is a moderate marine thermophile capable of direct utilization of brown algae that can ferment mannitol, laminarin, and alginate to ethanol with high yield, making this bacterium well-suited for bioconversion (19, 20). D. phaphyphila contains at least four alginate lyases, including examples belonging to PL6 (Dp0084) and PL7 (Dp2072) together with a number of novel lyases that represent totally new PL families with both endo- and exo-type activities (Dp0100 and Dp1761, respectively) (19, 21). Dp0100 is a 201-kDa polypeptide with bifunctional alginate lyase activity cleaving either polyM or polyG substrates in an endotype manner, but showing no activity against related polysaccharides such as heparin, heparan sulfate, dermatan sulfate, and chondroitin sulfate. The catalytic domain shows limited sequence similarity to the members of the multidomain PL15 and PL17 families but with many additional poorly-characterized domains.
In this paper, we report an analysis of the functions of the different domains of Dp0100 together with the crystal structure of the catalytic domain and its complexes with substrates. Together, these data have allowed us to unravel the molecular basis of its endolytic activity and establish the relationship of this enzyme to the broader superfamily, thereby deepening our understanding of these important enzymes that depolymerize alginate.
Results and discussion
Bioinformatic analysis of Dp0100
Based on the BLAST sequence analysis, Dp0100 has a full length of 1825 amino acids, including a 26-amino acid signal peptide at the N terminus. The enzyme appears to contain eight conserved domains which include a DUF4962 domain (pfam16332), a Hepar_II_III domain (pfam07940), a CBM35 carbohydrate-binding module (cd04086), a CBM32 discoidin domain (also known as an F5/8-type C domain) (pfam00754), and four fibronectin type III domains (cd00063/COG3401) (Fig. 1A). Further consideration of the limited sequence similarity between Dp0100 and the PL15 alginate lyase Atu3025 from Agrobacterium tumefaciens, the PL17 exotype alginate lyase Alg17c from Saccharophagus degradans, and the PL21 heparinase HepII from Pedobacter heparinus suggests that the catalytic site of Dp0100 includes both the DUF4962 domain and a block of sequence similar to the Hepar_II_III region of the heparinase II/III enzymes that together form the activity site of these related enzymes (13, 14, 22). The closest sequence similarities of the catalytic domain of Dp0100 are to a predicted heparinase II/III family protein from Rhodopirellula sp. SWK7 isolated from the surface of a macroalgae (GenBankTM accession number EMI43857) and a hypothetical protein from Verrucomicrobiae bacterium DG1235 isolated from a dinoflagellate (GenBankTM accession number EDY81874). Phylogenetic analysis shows that the catalytic domain of Dp0100 and its relatives constitute a new PL family, PL39 (Fig. 1B). An unusual feature of the Dp0100 sequence is the combination of the catalytic domain with so many accessory noncatalytic modules, including the CBM35 domain and the CBM32 discoidin domain raising questions as to the precise roles of these domains in alginate recognition.
Figure 1.

A, schematic diagram of the modular structure of Dp0100 and its truncation mutants. B, phylogenetic analysis of the catalytic domain of Dp0100 (TM5) and its close relatives from PL8, PL15, PL17, and PL21. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches (57). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree.
Enzymatic properties of Dp0100 and its truncated derivatives
To analyze the function of the different domains of Dp0100, constructs covering the full-length WT protein without the N-terminal signal peptide and a series of truncation mutants (TM1 to TM7) were produced (Fig. 1A). The bioinformatics analysis was used to design truncated constructs that sought to isolate fragments to identify the location of the active-site region. These constructs were expressed as soluble proteins and purified to homogeneity. Activity assays showed that the combination of the DUF4962 domain and Hepar_II_III region were necessary for the alginate lyase activity of Dp0100. The full-length enzyme has a pH optimum of 5.8 and shows considerable thermostability with a temperature optimum of 65 °C with a half-life of 45 min (Fig. S1). These properties were similarly reflected in the TM1–TM5 constructs. Constructs TM6 and TM7, which lack the DUF4962 domain and the Hepar_II_III region, showed no catalytic activity. These results suggest that the TM5 construct contains the minimal unit required for activity and that the other domains must be involved in aspects of substrate recognition in the context of the alginate matrix. Comparison of the kinetic parameters of Dp0100 and its truncation mutants showed that the Km for Dp0100 is smaller than that for other truncated derivatives, and the catalytic efficiency (kcat/Km) is higher again possibly indicating that the noncatalytic domains function to improve substrate processing (Table S2). Analysis of the incubation of the enzyme with alginate showed that the major products of cleavage were di-, tri-, and tetra-saccharides (Fig. 2).
Figure 2.

TLC analysis of the products released from alginate and affinity of Dp0100 and its TM1–TM7 constructs for soluble alginate by native affinity gel electrophoresis. A and B, marker sugars (1st lane, sodium d-mannuronate (S1); 2nd lane, sodium d-dimannuronate (S2); 3rd lane, sodium d-trimannuronate (S3); and 4th lane, sodium d-tetramannuronate (S4)) and the products of TM5 (unsaturated U2 and U3, 5th lane) after 24 h of incubation with alginate stained by the sulfuric acid/ethanol and TBA methods, respectively. Note that unsaturated sugars on the plate run faster than those of the equivalent saturated oligosaccharides (S1–S4) as reported previously (58, 59), and the saturated sugars are not stained by the TBA method (35, 59). C, unsaturated oligosaccharide products (U2, U3, and U4) of Dp0100 and TM1–TM5. 1st to 7th lanes represent incubation for 0, 5, and 30 min and 2, 4, 6, and 24 h, respectively. Plates in C were visualized by spraying with sulfuric acid in ethanol. D, 10% native-PAGE without alginate. E, 10% native-PAGE supplemented with 0.1% (w/v) alginate. 1st to 8th lanes represent Dp0100 and TM1–TM7 respectively; 9th lane contains BSA (0.2 μg) as a control.
Analysis of the alginate-binding properties of the noncatalytic domains
Extensive biochemical data and sequence comparisons have indicated that enzymes involved in carbohydrate chemistry possess a modular structure, as a part of which a catalytic module is associated with one or more noncatalytic modules in a multifunctional polypeptide chain (23). This type of architecture is common for many PL family enzymes, but it is less common for alginate lyases, although in some but not all enzymes belonging to the PL7 family, accessory modules belonging to CBM13, CBM16, and CBM32 have been identified (24–26). Currently, the precise roles of these domains in alginate lyases are poorly understood with only weak binding of the CBM32 domains to the oligosaccharide being reported (25, 27). Moreover, nothing is known about the function of the far more complex arrangement of accessory domains in Dp0100.
To investigate the substrate-binding properties of different domains of Dp0100, negative stain EM was used to show that, when mixed with alginate, Dp0100 forms dense aggregates along the alginate chain (Fig. S2). The affinity of Dp0100 and TM1–TM7 for soluble alginate was qualitatively evaluated by native-affinity PAGE (Fig. 2) and showed that the electrophoretic mobility of the constructs containing the CBM35 domain (Dp0100, TM1, TM2, TM6, and TM7) were dramatically retarded by inclusion of 0.1% (w/v) alginate, although some retardation was also noted with TM4 and TM5 (Fig. 2E). These results suggest that within the boundaries of the TM6 construct (residues 1020–1466), which include the CBM35 domain, the Dp0100 contains one or more domains with considerable affinity for alginate. As an extracellular carbohydrate-active enzyme, the efficient recognition by Dp0100 of the substrate would facilitate polysaccharide degradation of the cell walls of brown algae by D. phaphyphila to permit the subsequent utilization of the degradation products as a carbon source for the bacterium (20). The higher catalytic efficiency (kcat/Km) of the full-length Dp0100 enzyme, compared with all other constructs that involve deletion of the various accessory domains, indicates that these noncatalytic modules play a role in assisting the enzyme in recognizing the substrate under natural conditions (Table S2).
Structure of the catalytic domain
Consistent with gel-filtration studies, structure determination of TM5 at 2.07 Å showed that the four subunits in the asymmetric unit of the crystals of the apo-protein form independent monomers with an overall subunit architecture constructed of three domains (Fig. 3). The N-terminal domain (Ala-1–Pro-364) is largely helical being formed from an incomplete (α/α)6 toroid (Fig. 3B). The central domain (Asp-365–Tyr-616) is formed from 16 antiparallel β-strands arranged in two β-sheets together with a distorted α-helix (Fig. 3C and Fig. S3). The C-terminal domain (Ala-617–Gly-771) is composed of two antiparallel β-sheets that form a typical β-sandwich comprising 16 β-strands (Fig. 3D and Fig. S3). One face of the central domain packs against the C-terminal domain to form a four-layered β-sheet stack with the other face packing against the helical N-terminal domain (Fig. 3A).
Figure 3.

Structure of the catalytic domain of Dp0100 (TM5). A, schematic diagram of the fold of TM5 showing its N-terminal (orange), central (green), and C-terminal domains (sky blue). The positions of the Mn2+, Ca2+, and Mg2+ ions are shown as purple, green, and yellow spheres, respectively. B–D, schematic diagrams of the N-terminal, central, and C-terminal domains drawn in a rainbow format (blue to red from the N to C termini) to show their secondary structure elements, which are labeled. Figure was prepared using PyMOL (56).
Structural comparison using the Dali structural alignment server (28) identified matches between TM5 and domains found in enzymes belonging to a number of PL families, including PL5, PL8, PL12, PL15, PL17, and PL21 (Table 1). The N-terminal domain most closely resembles the structure of the PL5 endotype alginate lyase from Sphingomonas sp. (PDB code 4E1Y) with an RMSD of 3.2 Å. However, the latter is a much simpler structure lacking the other two domains of the catalytic region, and all of the accessory binding domains found in Dp0100 (Fig. S5). The more complex architecture of the catalytic domain of TM5 is mirrored in representative exotype alginate lyases belonging to family PL17 (PDB code 4OJZ) and the endotype PL21 family heparinase (PDB code 2FUQ), which contain all three domains (Fig. S5) (14, 22). Extensive similarities were also observed in the exotype alginate lyase belonging to family PL15 (PDB code 3AFL), which contains equivalents of the N-terminal and central domains but with a much smaller C-terminal domain and also has an additional N-terminal β-sheet extension that is not found in the TM5 structure (Fig. S5) (13). Although the sequence similarities between all these enzymes and Dp0100 only fall in the region of 13–16% identity (Table 1), the superpositions show that many of the conserved residues occupy equivalent positions in the structures (Fig. 7).
Table 1.
Structural comparison using the Dali server
| PDB code | PL family | Z score | RMSD | LALIa | Identity | Function |
|---|---|---|---|---|---|---|
| Å | % | |||||
| 3AFL | PL15 | 27 | 3.6 | 587 | 14 | Alginate lyase |
| 4OJZ | PL17 | 25.9 | 3.3 | 572 | 16 | Alginate lyase |
| 4FNV | PL12 | 24.9 | 4.4 | 494 | 15 | Heparinase III |
| 2FUQ | PL21 | 24.2 | 4.6 | 588 | 16 | Heparinase II |
| 4E1Y | PL5 | 21 | 3.2 | 288 | 13 | Alginate lyase |
| 6F2P | PL8 | 14.9 | 6.3 | 570 | 6 | Xanthan lyase |
a Total number of the equivalenced residues is shown.
Figure 7.
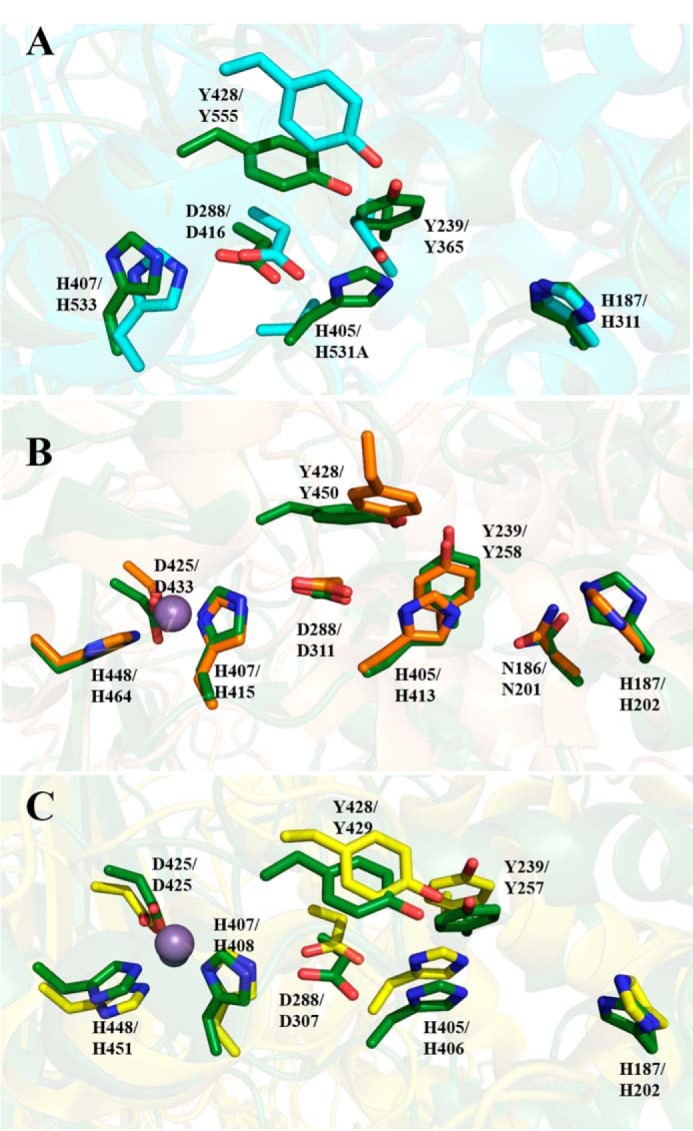
Superposition of Dp0100 and related PL family enzymes. A–C, conserved residues around the catalytic center between TM5 (6JP4), PL15 alginate lyase (3AFL), PL17 (4NEI), and PL21 (2FUT), respectively. The conserved residues of these structures are drawn in oxygen, red; nitrogen, blue; and carbon, as green, teal, orange, and yellow, respectively. Residue numbers of Dp0100 are shown first alongside those of their counterparts in the other PL family enzymes. Figure was prepared using PyMOL (56).
Metal-binding sites
ICP-MS and MCA analysis indicated the presence of Ca2+, Mn2+, and Fe2+ in purified TM5 (Table S3 and Fig. S4). Consistent with this, the initial SAD electron density map showed three very strong peaks in identical positions in each of the three subunits in the asymmetric unit that are associated with good electron density indicating that these were metal ions. Examination of the bond lengths, coordination chemistry, and the nature of the ligands led to the assignment of two of these peaks as Ca2+ consistent with the number of Ca2+ ions per subunit from ICP-MS. Both Ca2+ ions are associated with the central domain with one of them (Ca-1) stabilizing the interaction between its β-sheet and that in the C-terminal domain, and the other (Ca-2) lying at the interface with the N-terminal domain. The carboxyl oxygens of Glu-287, Glu-401, and Asp-409, the carbonyl oxygen of His-407, and two water molecules (w10 and w11) act as the ligands of Ca-1 (Fig. 4A), whereas the carboxyl oxygens of Asp-479 and Asp-474, the side-chain carbonyl oxygen of Gln-513, the main-chain carbonyl oxygen of Thr-475, and two water molecules (w96 and w477) coordinate Ca-2 (Fig. 4B and Table 2). The third metal peak lies close to Ca-1 and is octahedrally coordinated by His-407, Asp-425, His-488, and three water molecules (w1, w5, and w6) with distances of 2.1–2.3 Å (Table 2). Given the metal-ligand geometry and calculation of the bond-valence sum (29), the observation from ICP-MS that preparations of TM5 contains manganese and iron (Table S3), and the identification of both these elements in the MCA spectrum of a TM5 crystal (Fig. S4), it is likely that in the crystal this site may be occupied by a mixture of both Mn2+ and Fe2+. This assignment is consistent with the behavior of the metal ion in refinement where, for example, its refined temperature factor in subunit A of the apo structure (24 Å2) is similar to the average of the six ligands (22 Å2). We suggest that under the growth conditions of D. phaphyphila, this metal is probably a Mn2+ ion. We note this metal site is essentially identical in position and in the nature of the ligand to that reported for a Zn2+ ion in the structures of a PL17 family alginate lyase and a PL21 family heparinase (Fig. 7, B and C) (14, 22). However, our enzyme does not contain zinc, and the observed octahedral geometry of the metal in these other enzymes, rather than tetrahedral coordination that would be expected for Zn2+, suggests that the previous assignment of a Zn2+ ion to this site is incorrect. A fourth metal ion, Mg2+, has been identified in the electron density map of TM5 ligated by Asp-132, the carbonyl oxygen of Trp-180, and the four water molecules (w37, w160, w440, and w443) (Fig. 4C). This assignment is consistent with the ligand geometry, the metal-ligand distances, and the fact that the refined B-factors are comparable with those of neighboring atoms. This metal is thought to arise from the crystallization solution that contains magnesium chloride and is not thought to be biologically significant.
Figure 4.

Metal-binding sites in the catalytic domain of Dp0100. A, binding site of the Mn2+ ion (Mn-1, purple sphere) and its neighboring Ca2+ ion (Ca-1, green sphere). Amino acid residues (drawn in atom colors (oxygen, red; carbon, gray; and nitrogen, blue)) and water molecules (red spheres) in the coordination spheres of the metal ions are shown. B, second Ca2+ ion–binding site (Ca-2) colored as in A. C, Mg2+ ion–binding site (Mg-1, yellow sphere) drawn as in A. Dotted lines indicate interactions between metal ions and ligands in the coordinate sphere. Figure was prepared using PyMOL (56).
Table 2.
Distances between metal ions and protein ligands in chain A of the apo structure
| Ligand (Å) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Mn-1 | ND1His-407 (2.10) | NE2His-448 (2.14) | OD1Asp-425 (2.26) | w1 (2.29) | w5 (2.40) | w6 (2.31) | ||
| Ca-1 | OD1Asp-409 (2.75) | OD2Asp-409 (2.62) | OE1Glu-287 (2.64) | OE2Glu-287 (2.64) | OHis-407 (2.57) | OE2Glu-401 (2.63) | w10 (2.63) | w11 (2.64) |
| Ca-2 | OE1Gln-513 (2.58) | OD1Asp-479 (2.63) | OD2Asp-479 (2.65) | OD1Asp-474 (2.60) | OThr-475 (2.61) | w96 (2.65) | w477 (2.61) | |
| Mg-1 | OE2Glu-133 (2.36) | OTrp-180 (1.97) | w160 (2.28) | w440 (2.25) | w443 (2.24) | w598 (2.07) |
Substrate binding and mechanism of Dp0100
The catalytic site of TM5 was initially identified following co-crystallization with substrate M5, and the structure was solved by molecular replacement at a resolution of 2.76 Å (Table 3). Consistent with biochemical data that indicate that the full-length enzyme and TM5 can cleave small oligosaccharides, examination of the resultant electron density map provided clear evidence for the binding of a trisaccharide rather than a pentasaccharide (Fig. 5). This suggests that the binding site is occupied by a product, ΔMM, with an unsaturated uronic acid at the nonreducing end. Thus, the three sugars are presumed to be bound at subsites +1, +2, and +3 with the general location of the active site being adjacent to the unsaturated sugar at +1 subsite. Examination of this region suggested that His-187 might play a pivotal role in the reaction mechanism, and subsequently, an H187A mutant was shown to be catalytically inactive. Co-crystallization of M5 with the TM5 H187A mutant led to a structure with clear density for all the sugars with no sign of any cleavage by the enzyme.
Table 3.
X-ray crystallographic data
| apo-Native | SeMet | ΔMM-bound structure | M5-bound structure | |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 0.97179 | 0.97928 | 0.97625 | 0.97625 |
| Resolution range (Å) | 98.70–2.07 (2.11–2.07) | 108.99–2.38 (2.42–2.38) | 130.96–2.76 (2.81–2.76) | 62.78–2.85 (2.90–2.85) |
| Space group | C 2 2 21 | C 2 2 21 | P 3 2 1 | P 3 2 1 |
| Unit cell (a, b, c) (Å) | 261.3, 394.8, 112.2 | 261.2, 395.4, 111.6 | 261.9, 261.9, 58.3 | 261.4, 261.4, 58.4 |
| (α, β, γ) (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.00, 120.0 | 90.0, 90.0, 120.0 |
| Total reflections | 2,551,319 (104,358) | 2,551,319 (144,764) | 649,310 (32,281) | 594,163 (26,053) |
| Unique reflections | 348,350 (16,738) | 226,731 (11,049) | 58,914 (2878) | 53,459 (2668) |
| Multiplicity | 7.3 (6.2) | 13.6 (13.1) | 11.0 (11.2) | 11.1 (9.8) |
| Completeness (%) | 99.5 (96.3) | 98.5 (96.9) | 100.0 (99.2) | 100.0 (100.0) |
| Mean I/σ | 11.0 (1.8) | 7.4 (2.4) | 6.2 (0.9) | 11.5 (1.0) |
| Wilson B-factor (Å2) | 32 | 18 | 56 | 67 |
| Rmerge | 0.099 (0.699) | 0.325 (0.909) | 0.265 (2.319) | 0.217 (1.827) |
| Rpim | 0.059 (0459) | 0.132 (0.381) | 0.122 (1.062) | 0.101 (0.910) |
| CC½ | 0.992 (0.541) | 0.971 (0.396) | 0.986 (0.516) | 0.996 (0.524) |
| Anomalous completeness (%) | 98.7 (97.1) | |||
| Anomalous multiplicity | 7.0 (6.7) | |||
| Anomalous correlation | 0.229 (0.318) | |||
| Anomalous slope | 1.068 | |||
| Refinement | ||||
| Rfactor | 0.222 | 0.199 | 0.209 | |
| Rfree | 0.240 | 0.226 | 0.223 | |
| Atoms | ||||
| Protein | 21,800 | 6240 | 6235 | |
| Ligands | 61 | 40 | 61 | |
| Ions | 12 | 4 | 4 | |
| Water | 832 | 144 | 37 | |
| Protein residues | 3080 | 770 | 770 | |
| RMSD (bonds) (Å) | 0.0077 | 0.0034 | 0.0040 | |
| RMSD (angles) (°) | 1.515 | 1.283 | 1.326 | |
| Ramachandran favored (%) | 95.4 | 94.7 | 93.8 | |
| Ramachandran outliers (%) | 0.2 | 0.1 | 0.5 | |
| Favored rotamers (%) | 94.8 | 95.8 | 94.3 | |
| Poor rotamers (%) | 0.75 | 0.3 | 0.6 | |
| Molprobity score | 1.28 (99th percentile, 2.07 ± 0.25 Å) | 1.4 (100th percentile, 2.76 ± 0.25 Å) | 1.51 (100th percentile, 2.85 ± 0.25 Å) | |
| Average B-factors | ||||
| Main chain (Å2) | 59 | 64 | 74 | |
| Side chains (Å2) | 51 | 68 | 75 | |
| Ligands (Å2) | 63 | 77 | 89 | |
| Ions (Å2) | 43 | 49 | 58 | |
| Water (Å2) | 44 | 47 | 45 | |
| PDB codes | 6JP4 | 6JPH | 6JPN | |
Figure 5.

Substrate-binding groove of TM5. A, surface representation to show the binding site of M5 (shown as sticks and drawn in atom colors (oxygen, red; carbon, cyan)) in the catalytic domain of Dp0100. B, schematic diagrams to show the binding site of M5 and the surrounding elements of secondary structure (colored as in Fig. 3). C and D, electron density surrounding the substrates (shown as in A) in the (2Fo − Fc) omit map (contoured at 1.0σ) in complexes of the H187A mutant with M5 and the WT enzyme with ΔMM, respectively. The sugar-binding subsites are numbered in C and D. Key residues interacting with the substrates are highlighted (drawn in atom colors (oxygen, red; carbon, gray; and nitrogen, blue)) with the remainder of the enzyme shown transparently in cartoon format. Figure was prepared using PyMOL (56).
Analysis of the structure of the complex with M5 shows that the oligosaccharide binds in a long cleft formed between the N-terminal domain and the central domain with its five sugar molecules in what are presumed to be subsites −2, −1, +1, +2, and +3 (Fig. 5 and Table 4) and superposing well with equivalent parts of the structure of ΔMM. One wall of the substrate-binding groove is formed from residues close to the start of α5 (Ser-127 and Tyr-135), the loop between α6 and α7 (Arg-183, His-185, Asn-186, and His-187), the loop between α13 and α14 (Gly-340, Ser-342, and Tyr-343), and residues from α9 (His-238, Tyr-239, and Tyr-242) (Fig. 4 and Fig. S3). The other wall is formed from residues associated with the loop leading to α15 (Tyr-433), and the loop connecting β3 to β4 (SV40 and His-405) (Fig. 4 and Fig. S3). We note that the substrate-binding cleft is open at both ends such that longer chain polysaccharides can be accommodated by the enzyme (Fig. 5). Critical interactions between the enzyme and the substrate involve the recognition of the C5 carboxyl moiety on each of the uronic acid residues, which alternately point to opposite walls of the oligosaccharide-binding site (−2 (Ser-342), −1 (Tyr-135), +1 (Asn-186, His-187, and His-405), +2 (Ser-127), and +3 (Arg-183 and His-185)) (Fig. 5C and Table 4).
Table 4.
Distances between the substrates and key binding residues in Dp0100
| Binding residue | Target ligand (subsite) | Distance in ΔMM-bound structure | Distance in M5-bound structure |
|---|---|---|---|
| Å | Å | ||
| OGSer-342 | O6 carboxyl (−2) | 2.83 | |
| OGly-340 | O6 carboxyl (−2) | 3.27 | |
| OHTyr-135 | O6 carboxyl (−1) | 3.41 | |
| NE2His-238 | O3 (−1) | 2.86 | |
| OHTyr-239 | O2 (−1) | 2.82 | |
| OHTyr-239 | O4 (+1) | 3.21 | |
| NE2His-187 | O6 carboxyl (+1) | 2.88 | NDa |
| NE2His-405 | O6 carboxyl (+1) | 2.48 | 2.65 |
| ND2Asp-186 | O6 carboxyl (+1) | 2.84 | 3.01 |
| OD1Asp-186 | O6 carboxyl (+1) | 2.52 | 2.81 |
| OD1Asn-404 | O2 (+2) | 2.81 | 2.31 |
| OGSer-127 | O6 carboxyl (+2) | 3.85 | 3.11 |
| NE2His-185 | O6 carboxyl (+3) | 2.72 | 2.83 |
| NH2Arg-183 | O4 (+3) | 3.33 | 3.27 |
| NEArg-183 | O5 (+3) | 3.36 | 3.57 |
a ND means not determined. The side chain of His-187 is absent in the structure of the H187A mutant with M5.
The identification of the unsaturated sugar at the +1 subsite, consideration of its environment, and comparison with the equivalent sugar in M5 strongly implicated His-405 as the catalytic base in the first step of the β-elimination reaction, as this residue is ideally placed to remove the C5 proton of the +1 M sugar, which is acidic as a result of the adjacent carboxyl group (Fig. 6). This would lead to the formation of a C5 aci-carboxylate, which could be stabilized by Asn-186, His-187, and His-405 (Fig. 6B). Subsequent collapse of the aci-carboxylate would then lead to the formation of the double bond between C5 and C4 with the concomitant cleavage of the glycosidic bond between the −1 and +1 uronic acids and the protonation of the O4 leaving group by Tyr-239 to complete the reaction (Fig. 6). The importance of His-187, Tyr-239, and His-405 to catalysis was investigated by mutation with preliminary activity data showing that H187A and H405A are totally inactive, whereas H187F, H405F, Y239A, and Y239F exhibit a dramatic decrease in activity relative to TM5 (Table S4) consistent with the proposed mechanism. The finding that the oligosaccharide-binding site at the nonreducing end of uronic acid at the +1 subsite is open and unhindered, together with the observed binding of sugars in the −1 and −2 subsites with M5, provides a clear explanation of the endolytic activity of the enzyme. Interestingly, we note no major conformational changes or domain rearrangements on substrate binding.
Figure 6.

Schematic diagram of the active site of Dp0100 and the proposed mechanism. A, relative orientation of key functional groups on the enzyme with respect to the substrate is shown. The coordinates and the associated molecular surface displayed are those of the WT enzyme combined with M5 oligosaccharide. The position of the latter is modeled based on the superposition of the WT enzyme onto the structure of the complex of the inactive mutant, H187A, with M5. The sugar-binding subsites are numbered. The proximity of the residues proposed to be involved in the acid/base catalysis (His-405 and Tyr-239) can be seen. Figure was prepared using PyMOL (56). The color scheme is the same as that for Fig. 4. B, proposal for the catalytic mechanism of depolymerization of polyM by Dp0100.
The finding that Dp0100 is capable of depolymerizing both polyM and polyG raises the question as to how this is possible given the stereochemical differences between the two sugars that differ in the chirality of the critical C5 carbon atom. Thus, compared with polyM, the proton removed by enzyme in the depolymerization of polyG would lie on the opposite face of the sugar. We note the existence of the additional histidine residue in the active site, His-187, which lies in a suitable position to possibly fulfill this role.
Mechanistic comparison with other PL family enzymes
Examples of both exotype and endotype alginate lyases belonging to PL family 7 have been identified (15). These single domain enzymes are highly divergent in their amino acid sequence with similarities of the order of 16% identity, yet having a highly-related jelly-roll fold (15). Comparison of these enzymes has shown that they conserve key residues involved the reaction mechanism (15). However, the architecture of the substrate-binding site is radically different such that in the endotype lyases the polysaccharide-binding site is formed from a long groove on the enzyme surface that is open at both ends, and in the exotype enzyme, the pocket is sterically hindered at the nonreducing end sugar such that only the terminal saccharide residue at the −1 position can be cleaved in any one catalytic cycle to leave a saccharide with a Δ unit at the +1 position (15).
Although the fold of Dp0100 is totally unrelated to that of the PL7 enzymes, comparison of the more complex multidomain exotype alginate lyases and Dp0100, the first endotype multidomain alginate lyase to have its structure determined, reveals a striking parallel in how the exo- and endo-specificity is controlled. Thus, despite this difference in specificity, many of the residues implicated in the catalytic mechanism of Dp0100 (e.g. His-187, Tyr-239, and His-405) are conserved in the PL15 and PL17 enzymes (Fig. 7) in an analogous manner to the pattern seen in the PL7 family (30). Moreover, in the PL7, PL15, and PL17 enzymes, as well as in Dp0100, the recognition of the C5 carboxyl group of the polysaccharide substrate is a key determinant of specificity.
In addition, the broad oligosaccharide-binding groove in Dp0100, which is open at both ends (Fig. 8), mirrors the strategy for endolytic activity used by the equivalent PL7 enzyme (AlyA1) (15). Similarly, in a pattern closely related to that in the PL7 enzymes, comparison of Dp0100 and the exotype PL15 and PL17 alginate lyases (Atu3025 from A. tumefaciens and Alg17c from S. degradans, respectively) shows that despite their common architecture, the structure of the exotype lyases is severely sterically hindered so as to allow the binding of only a single sugar at a −1 position to the cleavage site (Fig. 8). In the case of the PL15 enzyme, a major determinant of the steric hindrance arises from a short helix, H3, which blocks the end of the polysaccharide-binding groove (13). In the PL17 enzyme, residues belonging to a β-turn between two strands in the C-terminal domain of a 2-fold related subunit in the dimer seal off the pocket restricting the enzyme to exolytic activity alone (14). Moreover, compared with these enzymes, where significant conformational changes on substrate binding have been reported and proposed as a common feature of the multidomain lyases belonging to the (α/α)n toroid structural class (13, 14), the apparent absence of such changes in Dp0100 would point to this not necessarily being a universal feature of the wider enzyme family. Structural comparisons further show that in addition to the similarity of the overall fold, the active site of Dp0100 is closely related to that of the endotype PL21 heparinase II that can cleave both heparin and heparan sulfate (Fig. 7) (22). This similarity extends to the conservation of the residues that have been implicated in the β-elimination chemistry, including these involved in aspects of acid/base catalysis (Tyr-239 and His-405 (Dp0100 numbering)), in the stabilization of the aci-carboxylate intermediate (Asn-186 and His-187) (Figs. 6B and 7C), and in the open architecture of the substrate-binding cleft (Fig. 8D) (31).
Figure 8.

Surface representations of the substrate-binding site in Dp0100 and related enzymes. A, H187A mutant in Dp0100 in complex with M5. B, H531A mutant of the PL15 alginate lyase from A. tumefaciens in complex with ΔGGG. C, Y258A mutant of the PL17 alginate lyase from S. degradans in complex with ΔMMG. D, heparinase II of PL21 from P. heparinus in complex with a disaccharide product from heparin. Selected residues proposed to be important in catalysis and the oligosaccharides are shown as sticks and drawn in atom colors (oxygen, red; carbon, cyan/gray for the oligosaccharide/protein, respectively, sulfur, yellow; and nitrogen, blue). Figure was prepared using PyMOL (56).
Conclusions
Alginate is one of the major components of the matrix that is responsible for organizing the macrocellular assembly of algal cells in brown seaweed, and this polymer and the small oligosaccharides derived from it have many applications in areas of food, pharmaceutical, and biomaterial industries. The discovery and detailed analysis of Dp0100 from a marine thermophile D. phaphyphila, the first member alginate lyase of a new family of PL39, have provided new insights into the strategies that can be used to bring about the depolymerization of this important polymer. The results presented here not only provide a clear molecular basis for the endotype activity of the enzyme, but also reveal the generic strategies used by unrelated multidomain and single domain enzymes catalyzing this chemistry.
Experimental procedures
Cloning, overexpression, and purification
Isolation and characterization of the D. phaphyphila Alg1 strain have been previously reported (19, 20). Heterologous expression and purification of Dp0100, its truncated derivatives, and the site-directed mutants were made following the methods described by Zhang et al. (32). To facilitate heterologous expression of Dp0100, the N-terminal 26-residue signal peptide was not included in any of the expression constructs. For cloning, the genes were amplified by PCR from genomic DNA, purified, and ligated into pEASY-E1 (TransGen Biotech Inc., Beijing, China) through either Gibson Assembly or TA cloning as described in Table S1. The plasmids containing the genes were then transformed into Escherichia coli DH5α cells. Single colonies were picked from the plate and cultured in LB medium supplemented with 100 μg ml−1 ampicillin. The verified recombinant plasmid was transformed into E. coli BL21 (DE3), incubated overnight at 37 °C on ampicillin LB agar plates from which single colonies were isolated, inoculated in fresh ampicillin LB medium and cultured with aeration overnight at 37 °C, and then transferred into 500 ml of ampicillin LB medium under vigorous shaking (200 rpm) at 37 °C. After growth to an OD600 of 0.5–0.6, isopropyl β-d-thiogalactopyranoside was added to a final concentration of 1 mm; the temperature was decreased to 25 °C, and the culture was incubated for an additional 12 h. The cells were harvested by centrifugation (10,000 × g for 10 min), resuspended in a binding buffer (50 mm Tris-HCl, 500 mm NaCl (pH 8.0)), and ruptured by the cell disruptor. Cell debris was removed by centrifugation at 72,000 × g for 10 min and then heated at 60 °C for 10 min, and the precipitated material was removed by centrifugation as above, and cell-free extract was applied on a 5-ml HisTrap cartridge (GE Healthcare). Proteins were eluted by a 50-ml gradient of imidazole concentration from 0 to 0.3 m at flow rate 5 ml/min. A volume of 3-ml fractions was collected and analyzed by SDS-PAGE. Combined fractions with the target protein were reduced in volume to 1–2 ml using VivaSpin concentration device 30,000 MWCO and further purified by size-exclusion chromatography using 1.6 × 60 HiLoad Superdex 200 column (GE Healthcare) in 50 mm Tris-HCl, 500 mm NaCl (pH 8.0). Chromatography was performed on an ÄKTA purifier system (GE Healthcare).
Enzymes activity assay and kinetic analysis
Alginate lyase activity was assayed by measuring the increase in absorbance at 235 nm (A235) of the reaction products (unsaturated oligosaccharides) for 1 min at 65 °C in a quartz cuvette containing 2 ml of 0.2% alginate in 100 mm acetic acid/sodium acetate buffer (pH 5.8) and 0.5 μg (in the specific activities measurement of the mutant TM5, 12 μg) of purified enzymes. One unit of activity was defined as an increase of 0.1 in A235 per min. Protein concentrations were determined with the Bradford assay kit (Bio-Rad) with BSA as the standard (33). Thin layer chromatography (TLC) assay was performed on a Silica Gel 60 F254 plate (Merck, Germany) with a solvent system of 1-butanol/formic acid/water (4:6:1, v/v). Products of the cleavage of alginate were visualized either by heating TLC plates at 110 °C for 5 min after spraying with 10% (v/v) sulfuric acid in ethanol to detect saturated or unsaturated oligosaccharides (not including unsaturated monosaccharide) (34) or by the thiobarbituric acid (TBA) method to detect unsaturated mono- or oligosaccharides (35).
The kinetic parameters of Dp0100 and its truncation mutants for the depolymerization of alginate were determined by adding 0.5 μg of enzyme to 1 ml of mixture containing 100 mm acetic acid/sodium acetate buffer (pH 5.8) and varying concentrations of substrate (1–20 mg/ml). The mixture was incubated at 65 °C for 1 min, and the A235 was recorded to quantify the amount of oligoalginate with an unsaturated end produced using a molar extinction coefficient of ϵ = 6150 m−1 cm−1 for the reaction products (15). The initial rates of product formation were plotted against substrate concentration, and the apparent kinetic parameters were estimated by the Michaelis-Menten equation using the software GraphPad Prism 5.01 (GraphPad Software Inc., San Diego). Sodium alginate from brown algae (>90% purity) was purchased from Sangon Biotech (Shanghai) Co., Ltd. PolyM (>90% purity with a number-average degree of polymerization of 13) and polyG (>91% purity with a number-average degree of polymerization of 16) were prepared from the alginate according to the method of Haug et al. (36).
Determination of pH and temperature optimum of Dp0100
Three buffers were used for pH profiling of Dp0100 and TM1–5: 100 mm acetic acid/sodium acetate buffer (pH 4.0–5.8), 100 mm Na2HPO4·NaH2PO4 (pH 6.0–7.0), and 100 mm Tris-HCl buffer (pH 7.0–9.0). To determine the optimal pH of the enzymes on alginate, 0.5 μg of Dp0100 or one of its truncation mutants was incubated with 0.2 mg/ml alginate in each buffer at a given pH at 65 °C and assayed as described above. For determination of optimal temperature, each enzyme sample was incubated with 0.2 mg/ml alginate at its optimal pH at various temperatures (35, 45, 55, 65, 70, and 75 °C). The thermostability of Dp0100 and its truncation mutants were determined by incubating the enzymes at 65 °C in a water bath. At different time points, aliquots were taken from the tubes, and residual enzymatic activity was determined.
Affinity gel electrophoresis and substrate specificity
The binding ability of Dp0100 to soluble polysaccharides was evaluated by affinity gel electrophoresis (37–39). Alginate was added at a concentration of 0.1% (w/v) into the separation gel. Electrophoresis was carried out at room temperature in native 10% (w/v) polyacrylamide gels. After electrophoresis, proteins were visualized through staining with Coomassie Blue. For investigation of substrate specificity, 0.5 μg of purified Dp0100 and 0.2% (w/v) alginate, polyM, or polyG as a final concentration in 100 mm acetate/sodium acetate buffer (pH 5.8) was incubated in 500 μl of reaction mixture for 1 min at 65 °C, and the OD at 235 nm was recorded to evaluate the substrate preference.
Electron microscopy
Purified Dp0100 (∼5 μl at a concentration of ∼1 mg/ml) and ∼5 μl of alginate at ∼1 mg/ml were loaded onto carbon-coated grids and incubated for 1 min. Excess protein sample was removed by blotting. The grid was then dipped in negative stain (0.75% uranyl formate) (w/v) for 1 s and washed twice in distilled water. After a final staining for 20 s, the excess staining was removed by blotting. The grids were vacuum-dried and imaged on a Philips CM100 transmission electron microscope at an accelerating voltage of 100 kV. Digital images were collected on a Gatan Multiscan 794 1k × 1k charge-coupled device.
Phylogenetic analyses
Representative enzymes belonging to PL8, PL15, PL17, and PL21 families were identified in the Carbohydrate-Active enZYmes (CAZy) database (11), and their amino acid sequences and that of Dp0100 (NCBI accession QDD67358) were retrieved from the NCBI Protein Database. The sequences were aligned by ClustalW, and phylogenetic analysis was performed using the software package MEGA version 7.0 using the Neighbor-joining method (40–42).
Crystallization and data collection
Initial crystallization conditions were determined by automated screening (Nextal, Qiagen Inc.) using a Matrix Hydra II crystallization robot. Crystals of TM5 construct and SeMet-labeled and the H187A mutant enzyme were optimized by hanging-drop vapor diffusion using a 1:1 ratio of protein to precipitant. In detail, 12 mg/ml protein in 10 mm Tris-HCl (pH 8.0) was added to the same volume of precipitant. For the TM5 construct, a precipitant containing 0.1 m magnesium acetate, 0.1 m sodium cacodylate (pH 6.5), 15% polyethylene glycol (PEG) 6000 was used, and for the SeMet-labeled protein and the H187A mutant, the precipitant contained 0.1 m magnesium chloride, 0.1 m Na HEPES (pH 7.5), 10% w/v PEG 4000. For the co-crystallization with substrate, M5 (Qingdao BZ Oligo Biotech Co., Ltd., China) at a final concentration of 5 mm was added to solutions containing either H187A mutant or the construct-active TM5. Crystals were formed after equilibrating against a 1-ml reservoir of the same precipitant over the course of 1 day at 16 °C. All crystals were cryoprotected in the crystallization solution to which 25% ethylene glycol had been added, prior to flash-cooling in liquid nitrogen, and data sets were collected on the MX beamlines at the Diamond Light Source.
Phasing, structure determination, refinement, and substrate restraints
Crystallographic phases were determined using SAD data collected from a crystal of SeMet-labeled protein. Native data on crystals of the TM5 construct were collected on beamline I03 at the Diamond Light Source. Data were processed in xia2-DIALS (43–46) to 2.07 Å and belong to space group C2221. SeMet peak SAD data were collected at 0.97928 Å wavelength on beamline I04 at the Diamond Light Source. Data were processed in xia2-DIALS to 2.38 Å, in space group C2221. The SHELX program suite (47) was used to identify heavy atom sites and produce an initial electron density map in which three subunits could be readily identified. A preliminary model was built using Coot and Buccaneer (48, 49), and subsequently subjected to rounds of building in Coot. During refinement, weaker electron density for a fourth subunit was identified and confirmed by analysis of the difference map comparing the SeMet and sulfur methionine dataset (apo-native) using the calculated phases to reveal the positions of the selenium atoms in all the subunits. Coordinates for subunit A were then superimposed onto the position of the fourth subunit (D), and although the electron density was at a much lower level, the conformation of subunit D appeared to be the same. In one region of this subunit, a loop between β16 and β17, a severe clash with a neighboring molecule was noted indicating that, locally, the conformation was incorrect. The coordinates in this region were not altered prior to refinement to act as a bias check (50). In the resultant electron density map calculated after refinement, the error in the position of this local region was unambiguous, and the new position was clear. Nevertheless, following the final round of refinement, the average B-factors for subunit D remained high (96 Å2) indicating its somewhat disordered nature in the crystal (51). The final model includes residues from 1 to 770 of the expected 789 residues (772 from the protein and 17 from the N-terminal His-tag) for the three subunits A, B, and C and coordinates for the Cα atoms alone of the fourth subunit D to indicate its position in the cell. Some additional weak electron density could be seen for each subunit at the N terminus arising from residues from the His-tag. These were not modeled in the structure.
Crystals of TM5 and H187A complexes with M5 oligosaccharide were morphologically distinct to these of the apo-enzyme. Data were collected on beamline I03 at the Diamond Light Source and processed in xia2-DIALS, revealing that they belong to space group P321 with one molecule in the asymmetric unit. The co-crystal structures were determined by molecular replacement using PHASER (52) with the refined unliganded TM5 coordinates as a search model to resolutions of 2.76 and 2.85 Å, respectively. The electron density for the TM5 complex substrate confirmed the conversion of M5 to ΔMM by the enzyme during the crystallization experiment. Model building and refinement were carried out in Coot and Refmac5. A single chemical dictionary was generated for each of the polysaccharides in each structure. Fitting into the electron density maps was initially done without activating torsion restraints to allow for the two different chair conformations (4C1 and 1C4) to arise, and as the coordinates in the dictionary had not been subjected to energy minimization, all M5 sugars in the structure are 4C1 chairs. Torsion restraints were activated once the polysaccharides were well-aligned with the electron density and with all rings in conformations that were close to low-energy chairs. Ring conformation was then further restrained by the activation of harmonic torsion restraints. Dictionaries, including these, were generated using the Acedrg program from the CCP4 suite, as this has been shown to create geometric restraints that are comparable (53) with those obtained by Grade (Global Phasing Ltd.) and Elbow (PHENIX) in combination with CSD Mogul. Carbohydrate validation was then done with a development version of Privateer MKIV (54). The final model includes residues from 1 to 770 of the expected 789 residues (772 from the protein and 17 from the His-tag) together with coordinates for the oligosaccharides for the subunit. Residues from the His-tag were not modeled in the structure. Refinement statistics are summarized in Table 3. All models were validated using MolProbity (55), and diagrams were generated using PyMOL (56).
MCA spectrum and ICP-MS
An X-ray fluorescence spectrum (MCA) was collected on a crystal of TM5 on beamline I02 at the Diamond Synchrotron. For ICP-MS analysis, protein samples were purified and dialyzed against 10 mm Tris-HCl (pH 8.0) for 24 h with two changes. Finally, 100 μl of 15 mg/ml protein was introduced to the instrument (Agilent 7500 ICP-MS (Agilent Technologies, Inc.)) for analysis after hydrolysis.
Site-directed mutagenesis
Site-directed mutagenesis was conducted by designing a pair of complementary mutagenic primers to amplify the entire plasmid in a thermocycling reaction with a high-fidelity Pfu polymerase (New England Biolabs). The nucleotide sequences of the mutagenic primers used for mutagenesis are given in Table S1.
The PCR product was digested with DpnI (New England Biolabs) at 37 °C for 1 h to degrade the parental plasmid DNA. The product from the DpnI digestion was transformed into E. coli BL21 (DE3)-competent cells. The E. coli cells were spread on LB plates containing 100 μg/ml ampicillin and incubated at 37 °C overnight. Single colonies were inoculated in 5 ml of LB medium supplemented with 100 μg/ml ampicillin and cultured for 12 h. The plasmids were extracted from the recombinant E. coli cells, and the inserts were sequenced to confirm the presence of the desired mutation. The truncated protein was produced and purified in the same way as described above.
Accession numbers
The X-ray crystal structures for the catalytic domain of Dp0100 and the associated X-ray data have been deposited in the Protein Data Bank under the ID codes 6JP4 (apo) and 6JPH (ΔMM bound), 6JPN (M5 bound), respectively.
Author contributions
S. J., F.-L. L., and D. W. R. conceptualization; S. J., P. J. B., J. B. R., P. A. B., F.-L. L., and D. W. R. resources; S. J., S. R. D., and D. W. R. data curation; S. J., S. R. D., A. A. A., P. J. B., S. B. T., J. A., and D. W. R. software; S. J. supervision; S. J., P. J. B., J. B. R., and D. W. R. funding acquisition; S. J., S. R. D., J. A., and D. W. R. validation; S. J., A. A. A., and S. E. S. investigation; S. J. and D. W. R. visualization; S. J., S. E. S., P. A. B., S. B. T., and J. A. methodology; S. J. and D. W. R. writing-original draft; S. J. and D. W. R. project administration.
Supplementary Material
Acknowledgments
EM was performed in the Faculty of Science Electron Microscopy Facility at the University of Sheffield. We thank the Diamond Light Source for access to beamlines I03 and I04 that contributed to this work and the beamline scientists for their assistance.
This work was supported by National Natural Science Foundation of China (NSFC) Grants 31670001 and 41506155, by Qingdao Municipal Science and Technology Bureau of China Grant 17-1-1-55-jch (to S. J.), by the Royal Society under the International Exchanges 2017 Cost Share (China) Programme Grant IEC\NSFC\170392 (to D. W. R., P. J. B., J. B. R., and S. J.) and under Royal Society University Research Fellow Award UF160039 (to J. A.)., and the China Scholarship Council (CSC) for Visiting Scholarship Award 201704910020 (to S. J.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S4 and Figs. S1–S5.
The atomic coordinates and structure factors (codes 6JP4, 6JPH, and 6JPN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The amino acid sequence of this protein can be accessed through NCBI Protein Database under NCBI Accession No. QDD67358.
- G
- α-l-guluronate
- M
- β-d-mannuronate
- PL
- polysaccharide lyase
- CBM
- carbohydrate-binding module
- SeMet
- selenomethionine
- TBA
- thiobarbituric acid
- TLC
- thin layer chromatography
- SAD
- single-wavelength anomalous diffraction
- RMSD
- root mean square deviation
- ICP-MS
- inductively-coupled mass spectrometry
- PDB
- Protein Data Bank.
References
- 1. Chung I. K., Beardall J., Mehta S., Sahoo D., and Stojkovic S. (2011) Using marine macroalgae for carbon sequestration: a critical appraisal. J. Appl. Phycol. 23, 877–886 10.1007/s10811-010-9604-9 [DOI] [Google Scholar]
- 2. Smith S. V. (1981) Marine macrophytes as a global carbon sink. Science 211, 838–840 10.1126/science.211.4484.838 [DOI] [PubMed] [Google Scholar]
- 3. Enquist-Newman M., Faust A. M., Bravo D. D., Santos C. N., Raisner R. M., Hanel A., Sarvabhowman P., Le C., Regitsky D. D., Cooper S. R., Peereboom L., Clark A., Martinez Y., Goldsmith J., Cho M. Y., et al. (2014) Efficient ethanol production from brown macroalgae sugars by a synthetic yeast platform. Nature 505, 239–243 10.1038/nature12771 [DOI] [PubMed] [Google Scholar]
- 4. Wargacki A. J., Leonard E., Win M. N., Regitsky D. D., Santos C. N., Kim P. B., Cooper S. R., Raisner R. M., Herman A., Sivitz A. B., Lakshmanaswamy A., Kashiyama Y., Baker D., and Yoshikuni Y. (2012) An engineered microbial platform for direct biofuel production from brown macroalgae. Science 335, 308–313 10.1126/science.1214547 [DOI] [PubMed] [Google Scholar]
- 5. Gacesa P. (1988) Alginates. Carbohyd. Polym. 8, 161–182 10.1016/0144-8617(88)90001-X [DOI] [Google Scholar]
- 6. Sellimi S., Younes I., Ayed H. B., Maalej H., Montero V., Rinaudo M., Dahia M., Mechichi T., Hajji M., and Nasri M. (2015) Structural, physicochemical and antioxidant properties of sodium alginate isolated from a Tunisian brown seaweed. Int. J. Biol. Macromol 72, 1358–1367 10.1016/j.ijbiomac.2014.10.016 [DOI] [PubMed] [Google Scholar]
- 7. Grant G. T., Morris E. R., Rees D. A., Smith P. J., and Thom D. (1973) Biological interactions between polysaccharides and divalent cations: the egg-box model. FEBS Lett. 32, 195–198 10.1016/0014-5793(73)80770-7 [DOI] [Google Scholar]
- 8. Lee K. Y., and Mooney D. J. (2012) Alginate: properties and biomedical applications. Prog. Polym. Sci. 37, 106–126 10.1016/j.progpolymsci.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Preiss J., and Ashwell G. (1962) Alginic acid metabolism in bacteria. 1. Enzymatic formation of unsaturated oligosaccharides and 4-deoxy-l-erythro-5-hexoseulose uronic acid. J. Biol. Chem. 237, 309–316 [PubMed] [Google Scholar]
- 10. Preiss J., and Ashwell G. (1962) Alginic acid metabolism in bacteria. 2. Enzymatic reduction of 4-deoxy-l-erythro-5-hexoseulose uronic acid to 2-keto-3-deoxy-d-gluconic acid. J. Biol. Chem. 237, 317–321 [PubMed] [Google Scholar]
- 11. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garron M. L., and Cygler M. (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20, 1547–1573 10.1093/glycob/cwq122 [DOI] [PubMed] [Google Scholar]
- 13. Ochiai A., Yamasaki M., Mikami B., Hashimoto W., and Murata K. (2010) Crystal structure of exotype alginate lyase Atu3025 from Agrobacterium tumefaciens. J. Biol. Chem. 285, 24519–24528 10.1074/jbc.M110.125450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park D., Jagtap S., and Nair S. K. (2014) Structure of a PL17 family alginate lyase demonstrates functional similarities among exotype depolymerases. J. Biol. Chem. 289, 8645–8655 10.1074/jbc.M113.531111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thomas F., Lundqvist L. C., Jam M., Jeudy A., Barbeyron T., Sandström C., Michel G., and Czjzek M. (2013) Comparative characterization of two marine alginate lyases from Zobellia galactanivorans reveals distinct modes of action and exquisite adaptation to their natural substrate. J. Biol. Chem. 288, 23021–23037 10.1074/jbc.M113.467217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davies G. J., Wilson K. S., and Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 10.1042/bj3210557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gacesa P. (1987) Alginate-modifying enzymes–a proposed unified mechanism of action for the lyases and epimerases. FEBS Lett. 212, 199–202 10.1016/0014-5793(87)81344-3 [DOI] [Google Scholar]
- 18. Garron M. L., and Cygler M. (2014) Uronic polysaccharide degrading enzymes. Curr. Opin. Struct. Biol. 28, 87–95 10.1016/j.sbi.2014.07.012 [DOI] [PubMed] [Google Scholar]
- 19. Ji S. Q., Wang B., Lu M., and Li F. L. (2016) Defluviitalea phaphyphila sp. nov., a novel thermophilic bacterium that degrades brown algae. Appl. Environ. Microb. 82, 868–877 10.1128/AEM.03297-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ji S. Q., Wang B., Lu M., and Li F. L. (2016) Direct bioconversion of brown algae into ethanol by thermophilic bacterium Defluviitalea phaphyphila. Biotechnol. Biofuels 9, 81 10.1186/s13068-016-0494-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang B., Ji S. Q., Ma X. Q., Lu M., Wang L. S., and Li F. L. (2018) Substitution of one calcium-binding amino acid strengthens substrate binding in a thermophilic alginate lyase. FEBS Lett. 592, 369–379 10.1002/1873-3468.12965 [DOI] [PubMed] [Google Scholar]
- 22. Shaya D., Tocilj A., Li Y., Myette J., Venkataraman G., Sasisekharan R., and Cygler M. (2006) Crystal structure of heparinase II from Pedobacter heparinus and its complex with a disaccharide product. J. Biol. Chem. 281, 15525–15535 10.1074/jbc.M512055200 [DOI] [PubMed] [Google Scholar]
- 23. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 10.1042/BJ20040892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li S., Yang X., Bao M., Wu Y., Yu W., and Han F. (2015) Family 13 carbohydrate-binding module of alginate lyase from Agarivorans sp. L11 enhances its catalytic efficiency and thermostability, and alters its substrate preference and product distribution. FEMS Microbiol. Lett. 362, fnv054 10.1093/femsle/fnv054 [DOI] [PubMed] [Google Scholar]
- 25. Lyu Q. Q., Zhang K. K., Zhu Q. Y., Li Z. J., Liu Y. J., Fitzek E., Yohe T., Zhao L. M., Li W. H., Liu T., Yin Y. B., and Liu W. Z. (2018) Structural and biochemical characterization of a multidomain alginate lyase reveals a novel role of CBM32 in CAZymes. Biochem. Biophys. Acta Gen. Subj. 1862, 1862–1869 10.1016/j.bbagen.2018.05.024 [DOI] [PubMed] [Google Scholar]
- 26. Han W., Gu J., Cheng Y., Liu H., Li Y., and Li F. (2016) Novel alginate lyase (Aly5) from a polysaccharide-degrading marine bacterium, Flammeovirga sp. strain MY04: effects of module truncation on biochemical characteristics, alginate degradation patterns, and oligosaccharide-yielding properties. Appl. Environ. Microbiol. 82, 364–374 10.1128/AEM.03022-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sim P. F., Furusawa G., and Teh A. H. (2017) Functional and structural studies of a multidomain alginate lyase from Persicobacter sp. CCB-QB2. Sci. Rep. 7, 13656 10.1038/s41598-017-13288-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holm L., and Laakso L. M. (2016) Dali server update. Nucleic Acids Res. 44, W351–W355 10.1093/nar/gkw357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Müller P., Köpke S., and Sheldrick G. M. (2003) Is the bond-valence method able to identify metal atoms in protein structures? Acta Crystallogr. D Biol. Crystallogr. 59, 32–37 10.1107/S0907444902018000 [DOI] [PubMed] [Google Scholar]
- 30. Ogura K., Yamasaki M., Mikami B., Hashimoto W., and Murata K. (2008) Substrate recognition by family 7 alginate lyase from Sphingomonas sp. A1. J. Mol. Biol. 380, 373–385 10.1016/j.jmb.2008.05.008 [DOI] [PubMed] [Google Scholar]
- 31. Shaya D., Zhao W., Garron M. L., Xiao Z., Cui Q., Zhang Z., Sulea T., Linhardt R. J., and Cygler M. (2010) Catalytic mechanism of heparinase II investigated by site-directed mutagenesis and the crystal structure with its substrate. J. Biol. Chem. 285, 20051–20061 10.1074/jbc.M110.101071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang K., Chen X., Schwarz W. H., and Li F. (2014) Synergism of glycoside hydrolase secretomes from two thermophilic bacteria cocultivated on lignocellulose. Appl. Environ. Microbiol. 80, 2592–2601 10.1128/AEM.00295-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 34. Shimokawa T., Yoshida S., Kusakabe I., Takeuchi T., Murata K., and Kobayashi H. (1997) Some properties and action mode of (1 → 4)-α-l-guluronan lyase from Enterobacter cloacae M-1. Carbohydr. Res. 304, 125–132 10.1016/S0008-6215(97)00230-9 [DOI] [PubMed] [Google Scholar]
- 35. Warren L. (1960) Thiobarbituric acid spray reagent for deoxy sugars and sialic acids. Nature 186, 237–237 10.1038/186237a0 [DOI] [PubMed] [Google Scholar]
- 36. Haug A., Larsen B., and Smidsrod O. (1967) Studies on the sequence of uronic acid residues in alginic acid. Acta Chem. Scand. 21, 691–704 10.3891/acta.chem.scand.21-0691 [DOI] [Google Scholar]
- 37. Horejsí V. (1981) Affinity electrophoresis–review. Anal. Biochem. 112, 1–8 10.1016/0003-2697(81)90252-9 [DOI] [PubMed] [Google Scholar]
- 38. Tomme P., Boraston A., Kormos J. M., Warren R. A., and Kilburn D. G. (2000) Affinity electrophoresis for the identification and characterization of soluble sugar binding by carbohydrate-binding modules. Enzyme Microb. Technol. 27, 453–458 10.1016/S0141-0229(00)00246-5 [DOI] [PubMed] [Google Scholar]
- 39. Abbott D. W., Hrynuik S., and Boraston A. B. (2007) Identification and characterization of a novel periplasmic polygalacturonic acid binding protein from Yersinia enterolitica. J. Mol. Biol. 367, 1023–1033 10.1016/j.jmb.2007.01.030 [DOI] [PubMed] [Google Scholar]
- 40. Kumar S., Stecher G., and Tamura K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol 33, 1870–1874 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 42. Saitou N., and Nei M. (1987) The neighbor-joining method–a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
- 43. Winter G. (2010) Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 43, 186–190 10.1107/S0021889809045701 [DOI] [Google Scholar]
- 44. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 46. Collaborative Computational Project, No. 4. (1994) The ccp4 suite–programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 47. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 10.1107/S0108767307043930 [DOI] [PubMed] [Google Scholar]
- 48. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cowtan K. (2008) Fitting molecular fragments into electron density. Acta Crystallogr. D Biol. Crystallogr. 64, 83–89 10.1107/S0907444907033938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rice D. W. (1981) The use of phase combination in the refinement of phosphoglycerate kinase at 2.5 Å resolution. Acta Crystallogr. A 37, 491–500 10.1107/S0567739481001186 [DOI] [Google Scholar]
- 51. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Agirre J. (2017) Strategies for carbohydrate model building, refinement and validation. Acta Crystallogr D. Struct. Biol. 73, 171–186 10.1107/S2059798316016910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Agirre J., Iglesias-Fernández J., Rovira C., Davies G. J., Wilson K. S., and Cowtan K. D. (2015) Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 10.1038/nsmb.3115 [DOI] [PubMed] [Google Scholar]
- 55. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B., Snoeyink J., Adams P. D., et al. (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schrödinger LLC. (2015) The PyMOL molecular graphics system, version 1.8.x, Schrödinger, LLC, New York [Google Scholar]
- 57. Felsenstein J. (1985) Confidence-limits on phylogenies–an approach using the bootstrap. Evolution 39, 783–791 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 58. Shimada E., and Matsumura G. (1984) Thin-layer chromatography of hyaluronate oligosaccharides. J. Biochem. 96, 721–725 10.1093/oxfordjournals.jbchem.a134890 [DOI] [PubMed] [Google Scholar]
- 59. Suzuki H., Suzuki K., Inoue A., and Ojima T. (2006) A novel oligoalginate lyase from abalone, Haliotis discus hannai, that releases disaccharide from alginate polymer in an exolytic manner. Carbohydr. Res. 341, 1809–1819 10.1016/j.carres.2006.04.032 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.