

Abstract
5-Formylcytosine (5fC) is an endogenous epigenetic DNA mark introduced via enzymatic oxidation of 5-methyl-dC in DNA. We and others recently reported that 5fC can form reversible DNA–protein conjugates with histone proteins, likely contributing to regulation of nucleosomal organization and gene expression. The protein component of DNA–protein cross-links can be proteolytically degraded, resulting in smaller DNA–peptide cross-links. Unlike full-size DNA–protein cross-links that completely block replication and transcription, DNA–peptide cross-links can be bypassed by DNA and RNA polymerases and can potentially be repaired via the nucleotide excision repair (NER) pathway. In the present work, we constructed plasmid molecules containing reductively stabilized, site-specific 5fC–polypeptide lesions and employed a quantitative MS-based assay to assess their effects on transcription in cells. Our results revealed that the presence of DNA–peptide cross-link significantly inhibits transcription in human HEK293T cells but does not induce transcription errors. Furthermore, transcription efficiency was similar in WT and NER-deficient human cell lines, suggesting that the 5fC–polypeptide lesion is a weak substrate for NER. This finding was confirmed by in vitro NER assays in cell-free extracts from human HeLa cells, suggesting that another mechanism is required for 5fC–polypeptide lesion removal. In summary, our findings indicate that 5fC-mediated DNA–peptide cross-links dramatically reduce transcription efficiency, are poor NER substrates, and do not cause transcription errors.
Keywords: DNA transcription, epigenetics, DNA damage, protein cross-linking, RNA polymerase II, transcription, gene regulation, nucleotide excision repair, 5-formylcytosine, bulky DNA lesion, DNA–peptide cross-link, epigenetic mark, human cells
Introduction
In living cells, genomic DNA dynamically interacts with a range of architectural and regulatory proteins, allowing for chromatin packaging, DNA repair, gene expression, and propagation of genetic information. Exposure to free radicals, UV radiation, heavy metals, and common antitumor drugs can lead to irreversible cross-linking of proteins to DNA strands to form DNA–protein cross-links (DPCs)2 (1–3). In addition, topoisomerases, DNA polymerases, glycosylases, and meiotic recombinases can be trapped on their DNA substrates (4, 5). DPCs are among the most common DNA lesions in living cells and are known to accumulate in tissues with age, likely contributing to human disease (1).
We recently discovered that 5-formylcytosine (5fC) residues in DNA and lysine or arginine side chains of histone proteins form reversible DPCs that can be stabilized by reduction (6). 5fC is an epigenetic DNA mark generated via oxidation of 5-methylcytosine by ten eleven translocation dioxygenases (Tet) (7, 8). Li et al. (9) independently reported 5fC–histone DPC formation in chromosomal core particles. Although the resulting DNA–histone cross-links are reversible (t½ = ∼1.8 h), they are likely to have a large effect on chromatin structure and gene expression (6). A recent report suggests that 5fC-mediated covalent DNA–histone interactions play a role in controlling nucleosome positioning, establishing distinct regulatory regions for fine-tuning the levels of gene expression (10).
The presence of endogenous 5fC-mediated DNA–protein cross-links in living cells raises a question of how such bulky lesions are tolerated by cells and whether they influence the efficiency and the fidelity of DNA replication and transcription. We recently investigated the effects of reductively stabilized 5fC-mediated DPC on DNA synthesis and found that TLS DNA polymerase η and κ were completely blocked by covalent DNA–histone lesions, whereas smaller DNA–polypeptide lesions were readily bypassed (11). We have also shown that large DPCs completely inhibit T7 RNA polymerase (35).
It has been suggested that in vivo, the protein component of DPCs is proteolytically degraded by specialized metalloproteases (SPRTN/Wss1) and/or the ubiquitin/proteasome pathway to smaller polypeptide conjugates (DpCs) (12–16), which could be removed via the nucleotide excision repair (NER) pathway (17, 18). In Xenopus extracts, DPC proteolysis is replication-dependent (12, 14). However, evidence is accumulating for additional mechanisms of DPC recognition and repair in human cells. For example, Chesner and Campbell (19) examined the kinetics of DPC repair in human cells and reported that hOGG1-abasic site lesions present on nonreplicating plasmids are rapidly repaired. This suggests that DPC recognition and repair in human cells can take place in the absence of DNA replication. Interestingly, DPC lesions on the transcribed strand of DNA were removed faster than the same DPC lesions on the nontranscribed strands (19), suggesting that DPC-mediated blockage of transcription complex may facilitate their repair.
In the present work, reductively stabilized, site-specific 5fC–polypeptide lesions were incorporated into the coding region of pTGFP-T7-Hha10 plasmids under the control of the cytomegalovirus promoter. The resulting constructs were transfected in WT or NER-deficient human cells, and the effects of DpC lesions on transcription were evaluated using the new competitive transcription and bypass assay developed and fully validated by You and Wang et al. (20). The potential role of NER in repair of 5fC–polypeptide lesions was further investigated using in vitro assays employing cell-free extracts from human HeLa cells.
Results
Construction and characterization of plasmid vectors containing site-specific 5fC–polypeptide and 5-formylC-Lys cross-links
A gapped plasmid strategy (21) was employed to create plasmid vectors containing site-specific 5fC–polypeptide cross-links. Nonreplicative pTGFP-T7-Hha10 plasmids containing cytomegalovirus promoter were used for this purpose. First, 5fC-containing DNA 12-mers (5′-ATGGCGGGXTAT-3′, where X = 5fC) were conjugated to an 11-mer peptide (RPKPQQFFGLM-CONH2) using reductive amination in the presence of NaCNBH3, generating covalent DpCs (Fig. 1A) (6, 11). DpCs were purified by denaturing PAGE (Fig. 1B), characterized by MALDI-TOF-MS (Fig. 1C), and ligated into gapped plasmids (Fig. 2 and Fig. S1).
Figure 1.

A, formation of DNA–protein and DNA–peptide cross-links via reductive amination reactions between 5-formyl-dC in DNA and the -NH2 group of Lys. B, denaturing PAGE analysis of reaction mixtures after reductive amination reaction between 5′-ATGGCGGGXTAT-3′, where X = 5fC and RPKPQQFFGLM-CONH2. The 5′ ends of oligonucleotides were radiolabeled with [γ-32P]ATP and T4 PNK. C, representative MALDI-MS spectrum of synthetic DNA–polypeptide conjugate.
Figure 2.
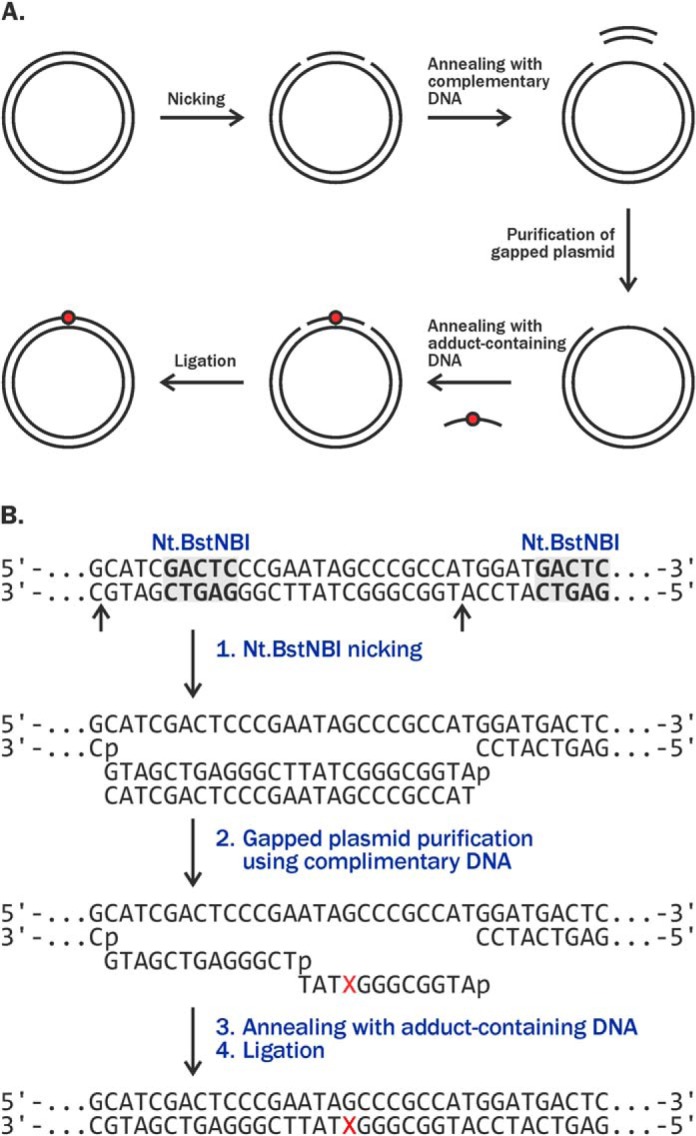
Construction of plasmids containing site-specific incorporated 5fC conjugated to 11-mer peptide or one lysine residue using gapped strategy: overall strategy (A) and restriction sites used for incorporation of DpC (B).
The corresponding cross-links to single Lys were generated analogously using l-lysine, purified by HPLC, and characterized by MS (Fig. S2) prior to incorporation into gapped plasmid. In the resulting plasmid constructs, DNA lesions were placed on the transcribed strand of DNA 56 nucleotides downstream of the transcription start site (21). In addition to control plasmid containing standard dC in place of DpC lesion, a competitor plasmid was prepared containing three additional bp near the modification site (Fig. 3B). Co-transfection with competitor plasmid makes it possible to account for any differences in transfection efficiency by normalizing transcript amounts from test plasmid relative to transcripts amounts from competitor plasmid (21). The use of DNA sequence containing three additional bp in the competitor plasmid allows for distinguishing between transcription products originating from lesion-bearing vectors and competitor plasmids by HPLC-ESI-MS/MS because of their distinct molecular weight (21).
Figure 3.

A and B, NcoI/SfaNI-mediated restriction digestion and post-labeling assays for characterization of the plasmid vectors containing unmodified dC or DNA lesions (A) and the competitor plasmids (B). C, denaturing PAGE analysis of restriction-digested undamaged control plasmids (lane 1), the competitor plasmids (lane 2), 1:1 mixture of the control and competitor plasmids (lane 3), and plasmids containing site-specific 5fC conjugated to 11-mer peptide (lane 4).
Plasmids containing site-specific DNA–peptide and DNA–lysine conjugates were characterized by NcoI/SfaNI-mediated double-restriction digestion and post-labeling assays (Fig. 3). Briefly, plasmids were cleaved with restriction endonucleases, and the excision products were radiolabeled with 32P (Fig. 3, A and B), generating 13-mer and 16-mer oligonucleotides from DpC-containing and competitor vectors, respectively (Fig. 3, A and B, and lanes 1–3 in C). Restriction sites were selected to encompass the modification site (Fig. 3A). Plasmids containing 5fC–11-mer polypeptide conjugates yielded a band with reduced mobility on denaturing PAGE (lane 4 in Fig. 3C), confirming the presence of site-specific DpC. Similar results were observed for plasmids containing 5fC–Lys conjugates, with the exception that the gel shift was less pronounced (Fig. S2B). These experiments confirmed the presence of DNA–peptide and DNA–Lys conjugates at the specified site within the plasmid vectors.
Competitive transcription and bypass assay (CTAB) assay
Human embryonic kidney (HEK293T) cells were transfected with a mixture of DpC-containing vectors and competitor plasmids. In control experiments, unmodified plasmids containing unmodified dC instead of the DpC lesion were employed. 24 h post-transfection, RNA transcripts were isolated and amplified using RT-PCR (Fig. S3). The resulting RT-PCR products were digested with SfaNI/NcoI to release short 13-mer oligonucleotides containing the region of interest from lesion-bearing or control plasmids while at the same time generating 16-mer oligonucleotides from competitor plasmids (Figs. 3 and 4A and Fig. S3). HPLC-ESI-HRMS/MS on an Orbitrap Velos mass spectrometer was employed to quantify and sequence RT-PCR products. We found that the use of high-resolution MS on an Orbitrap MS allows for accurate and sensitive detection of RT-PCR products with minimal background from the sample matrix. The use of competitor plasmid as an internal control accounts for any variations in transfection efficiency or plasmid degradation (19).
Figure 4.

A, CTAB assay to investigate the effects of DpC lesions on transcription in human cells. B, relative bypass efficiencies of unmodified dC, 5fC conjugated to Lys, and 5fC–11-mer peptide conjugates (RPKPQQFFGLM-CONH2) in human embryonic kidney cells (HEK293T), human NER-deficient cells (GM04429, XPA), and XPA-complemented cells (XPA-corrected). Each value represents the average of at least three independent experiments. ns, not significant. ****, p < 0.0001.
Capillary HPLC-ESI-HRMS/MS analysis of RT-PCR–amplified transcript products from in vivo transcription experiments revealed that 5fC–polypeptide conjugates significantly inhibited transcription in HEK293T cells. By normalizing the amounts of transcripts generated from lesion-bearing plasmids (13-mer oligonucleotides) to the amounts of transcription products from the competitor plasmids (16-mer oligonucleotides), we found that the relative transcription efficiency of the templates containing DNA–peptide conjugates was only ∼10% as compared with unmodified dC in the control experiments (Fig. 4B). Transcription levels of probes containing 5fC–lysine conjugates were ∼40%, indicating that these smaller lesions are less blocking as compared with the 11-mer peptide conjugates (Fig. 4B).
Despite their blocking effects on human RNAP II, DpC conjugates did not induce transcription errors. Only error-free products (5′-CCGAATAGCCCGC, [M-4H]4− = 974.7), where the correct base (G) was inserted opposite the modified dC during the RNA synthesis, were observed. We tried but failed to detect any transcription products containing nucleobase substitutions, deletions, insertions, or post-lesion synthesis errors (Fig. 5A). MS/MS spectrum of the error-free product (5′-CCGAATAGCCCGC) was consistent with the theoretical spectrum predicted from Mongo Oligo Mass Calculator (Fig. 5B). Similar results were observed for 5fC conjugated to Lys residue, indicating that the fidelity of in vivo transcription was unaffected.
Figure 5.

A, capillary HPLC-ESI-HRMS/MS analysis of in vivo transcription products generated from site-specific 5fC-conjugated to 11-mer peptide (RPKPQQFFGLM-CONH2). 13-mer error-free products (5′-CCGAATAGCCCGC-3′, [M-4H]4− = 974.665); C to T (5′-CCGAATAACCCGC-3′, [M-4H]4− = 970.667); C to A (5′-CCGAATATCCCGC-3′, [M-4H]4− = 968.414); C to G (5′-CCGAATACCCCGC, [M-4H]4− = 964.664); deletion (5′-CCGAATA_CCCGC-3′, [M-4H]4− = 892.402); and 16-mer products from competitor plasmids (5′-CACAATAGCATATCGC-3′, [M-4H]4− = 1206.957). B, CID MS2 spectrum of the 13-mer error-free products.
Potential role of NER in 5fC DpC removal in human cells
To investigate potential role of nucleotide excision repair in the removal of DpC lesions conjugated to 5fC in DNA, in vivo transcription assays were repeated using XPA-deficient cells and XPA-complemented human fibroblast cells. NER deficiency in XPA cells and competency in complemented cells was confirmed by the strand-specific primer extension-quantitative polymerase chain reaction assay using a known NER substrate (cholesterol-dR) (19), UV sensitivity (Fig. S8), and in vitro assays with known NER substrates (22). Our CTAB experiments revealed that the relative transcription efficiencies of 5fC-conjugated 11-mer peptide cross-links in XPA-deficient cells and XPA-complemented cells 24 h post-transfection were ∼12% and ∼18%, respectively (Fig. 4B). This difference was not statistically significant (p > 0.1, n = 3). Similarly, the transcription efficiency of 5fC–Lys–containing templates was ∼49 and 63% relative to control in XPA-deficient and XPA-complemented cells, respectively, and this difference was not statistically significant (Fig. 4B, p > 0.1, n = 3). Analogous results were obtained when RNA was isolated from transfected cells at a longer time point (48 h after transfection; Fig. S4). These results indicate that unlike other DpC lesions reported in the literature (17, 18), 5fC-conjugated peptide cross-links are poor substrates of nucleotide excision repair.
In vitro NER assay for DpC lesions using HeLa cell extracts
To confirm our results of the repair of 5fC conjugates in human cells, in vitro NER assays in cell-free extracts were performed (Fig. 6). Briefly, DNA–peptide and DNA–lysine cross-links were site-specifically incorporated into 135-mer DNA duplexes with an internal 32P-radiolabel near the lesion (Fig. 6A). The resulting 135-mer DNA duplexes were incubated with NER-competent nuclear extracts from human HeLa cells (23). The time-dependent appearance of characteristic 24–30-mer dual-incision products resulting from lesion-bearing templates indicates successful NER activity (Fig. 6B). In positive control experiments performed with a known NER substrate, (+)-cis-BPDE-dG, time-dependent formation of the characteristic NER products at ∼26–28 nt in length was observed (lanes 10 and 11 in Fig. 6C). In the corresponding NER experiments employing 5fC–polypeptide templates, a series of weak bands ∼35 nt in length were detected (lanes 6 and 7 in Fig. 6C) that were not observed in negative controls employing unmodified dC (lanes 12 and 13 in Fig. 6C). These NER products moved slower as compared with typical NER products (lane 11 in Fig. 6C), probably because of the presence of a covalently attached 11-mer peptide. When treated with proteinase K to digest the polypeptides to single amino acids, the mobility of these bands was increased, but they were still slightly shifted as compared with BPDE control because of the presence of 5fC–Lys lesions in their structure (lanes 1–5 in Fig. 6C).
Figure 6.

In vitro NER assays. A, construction of 32P-labeled 135-mer oligonucleotide duplexes containing site-specific cross-links to 11-mer peptide and single lysine. B, scheme of in vitro NER assays in HeLa cell extracts. C, denaturing PAGE analysis of in vitro NER assays of 5fC-mediated 11-mer peptide cross-links in HeLa cell extracts. Left panel, original gel image. Right panel, gel image with enhanced contrast. Lanes 1–3 and 10–13, standard sample preparation with phenol:chloroform extraction and protease K treatment before loading onto denaturing PAGE gel (lanes 1–3, 5fC conjugated to 11-mer peptide; lanes 10 and 11, (+)-cis-BPDE-dG as positive control; lanes 12 and 13, dC as negative control). Lanes 4–9, preparation of DNA–peptide cross-links samples without phenol: chloroform extraction (lanes 4 and 5), without proteinase K digestion (lanes 6 and 7), or with trypsin digestion of peptides conjugates before the in vitro NER (lanes 8 and 9). D, densitometry tracings of individual lanes with ×5 vertical scale amplification (left panel) or no amplification (right panel). E, example of time-dependent formation of NER products (left panel) and the zoomed in view (right panel); the NER dual excision products were resolved from the 135-mer unreacted DNA oligonucleotides by denaturing 12% PAGE (23). The relative yields of dual incision products were determined from densitometry tracings of the gel autoradiographs. F, the relative yields of 24–26 oligonucleotide excision repair products containing single 5fC DNA–peptide/Lys cross-links after incubation of the 135-mer oligonucleotide substrates in human cell extracts for 45 min as described in more detail elsewhere (23). The averages of four independent experiments are shown.
The fractions of excised 24–30-nt NER products were determined from the gel autoradiographs by comparing the relative intensities of the bands corresponding to intact and incised products. Quantitative assessment of individual gel lanes revealed that 5fC conjugated to 11-mer peptide is a poor NER substrate (<5% of the (+)-cis-BPDE-dG used as a positive control; Fig. 6, D–F). Furthermore, in vitro NER assays using the 5fC conjugated to a single lysine residue did not generate any detectable NER products (Fig. 6E and Fig. S5). Overall, our in vitro NER experiment confirmed that DpCs conjugated to the C5 position of cytosine are strongly resistant to NER in human cell extracts.
Discussion
The primary goal of this study was to elucidate the effects of 5fC-mediated DNA–polypeptide lesions on transcription in human cells. Although the global levels of 5fC in living cells are relatively low (0.002–0.02% of all cytosine) (7, 8), these epigenetic marks are preferentially found in the regulatory elements of genes and are thought to influence gene expression levels (24, 25). We and others recently reported that 5fC can form reversible DNA–protein conjugates with histone proteins in nucleosome core particles and in human cells (6, 9). Because of their endogenous formation, it is of critical importance to establish how 5fC-mediated DPCs influence the efficiency and the fidelity of transcription in human cells. Other types of DPCs have been reported to be processed by specialized proteases, giving rise to smaller DNA–peptide cross-links, which may be better tolerated by DNA replication and transcription machineries and are ultimately repaired by NER (12, 13, 17, 18).
Our earlier in vitro study revealed that T7 RNA polymerase is blocked by 5fC-conjugated protein lesions but that smaller polypeptide lesions were readily bypassed (35). However, T7 RNA polymerase is a small, single subunit enzyme that differs greatly from RNA polymerase II employed by human cells and lacks the proofreading activity (26, 27).
In the present work, we for the first time addressed the effects of 5fC–polypeptide conjugates on transcription in human cells. For this purpose, we engineered plasmid vectors containing DpC or DNA–Lys lesions 56 nucleotides downstream from transcription start site. We found that presence of reductively stabilized 5fC–polypeptide conjugates on DNA significantly inhibited transcription in human cells (Fig. 4B). Bulky peptide lesions likely represent a physical impediment to transcription during RNA elongation, resulting in the stalling of RNA polymerase II at the lesion site. 5fC conjugated to a single lysine is less blocking to RNA polymerases as compared with an 11-mer peptide (Fig. 4B). However, it is not known whether the DNA–peptide lesions can be cleaved to single amino acid lesions in cells.
In vivo transcription of DNA containing 5fC-mediated DNA–peptide/lysine cross-links predominately generated error-free transcription products (Fig. 5). This is different from our in vitro T7 RNA polymerase bypass assays, where we observed preferential incorporation of A opposite modified dC (35). This is probably due to the lack of proofreading activity in viral T7 RNAP as compared with RNAP II, because the latter can detect and remove incorporated incorrectly added nucleotides via “backtracking” (27–29). Our results differ from a report by Nakano et al. (30) showing that in vitro T7 RNAP bypass of oxanine-induced DPCs produced large amounts of substitution and deletion transcriptional mutations. Collectively, these results suggest that the cross-linking site within DNA and the identity of the RNA polymerase significantly affect transcription fidelity.
In the case of transcription coupled repair, the arrest of transcription by bulky DNA lesions is the initiation step that subsequently leads to the recruitment of the NER proteins that lead to the excision and generation of the characteristic 24–30-nt excision repair products. There are previous data showing that the NER-deficient cells are hypersensitive to DPC-inducing agents such as formaldehyde (31) and that NER is capable of repairing certain types of small DPCs and DNA–polypeptide remaining after proteolytic processing (19). XPA proteins serve as the scaffold for other repair proteins at the damage site and play an key role for the appropriate excision of DNA damage during NER (32).
To elucidate the potential role of NER in DpC removal, we performed in vivo transcription assays (20) in human XPA-deficient cells and XPA-complemented cells. Although transcription of templates containing DNA–peptide and DNA–lysine conjugates was less efficient in NER-deficient cells, the difference between XPA and XPA-corrected cells was not statistically significant (p > 0.1, n = 3) (Fig. 4B), suggesting that 5fC DpCs are not efficiently repaired by NER. This is in contrast to known NER substrates, where a 2–4-fold decrease of the bypass efficiency was observed in XPA-deficient cells using the CTAB assay (21).
To further examine the role of NER in 5fC DpC removal, we performed in vitro NER assays using cell extracts from human HeLa cells. When incubated with NER-competent cell extracts, characteristic dual incision NER products were observed, and protease K digestion confirmed that the peptides were still attached at the adduct site after the NER excision from the templates (Fig. 6, C and D). However, 5fC conjugated to 11-mer peptides were poor NER substrates in vitro, with repair efficiency of only 0.5% after 45 min of incubation (Fig. 6, C–E). In contrast, ∼3.5% repair was observed in the case of the (+)-cis-BPDE-dG adduct, which is known as a good NER substrate (Fig. 6, D and E). When performing in vitro NER assays with 5fC conjugated to one lysine residue, no NER activity was observed (Fig. 6F and Fig. S5).
The inefficient recognition and repair of C5-dC–conjugated polypeptide lesions by NER can be explained by relatively minor effects of such lesions on DNA structure. Molecular modeling of 5fC–polypeptide DpCs indicates that the polypeptide is readily accommodated in the DNA major groove, with minimal effects on DNA structure (Fig. 7). Therefore, despite their significant size, such DpC lesions may not be readily detected by the NER machinery. In contrast, Reardon et al. (33) reported that tetra- and dodecapeptides conjugated at AP sites can be removed very efficiently by NER. The observed differences in NER efficiency here may be attributed to different structures of DpCs employed in the two studies. The DpC model employed in this study has peptide attached to C5 position of dC, which preserves Watson–Crick hydrogen bonding and minimally disrupts the structure of the DNA duplex (Fig. 7). In contrast, Reardon et al. (33) used a polypeptide attached to an abasic site polypeptide that was attached to an abasic site, creating a lesion with missing nucleobase and significant structural perturbations, which could elicit a NER response.
Figure 7.
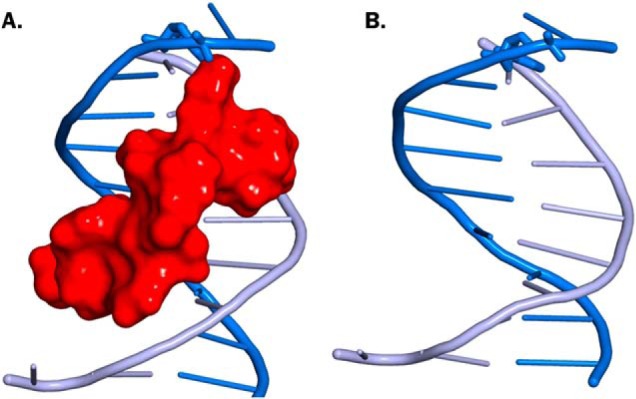
B-DNA duplex containing 11-mer peptide cross-linked to the C5-dC (A) or containing unmodified dC (B). The 11-mer peptide is shown in red surface. See “Experimental procedures.”
In summary, this study for the first time investigated the effects of 5fC-mediated DNA–peptide cross-links on transcription in human cells and found that these lesions dramatically reduce transcription efficiency but do not cause transcription errors. Because reversible 5fC–polypeptide conjugates naturally form in cells (6), it is not surprising that human RNA polymerases have evolved to allow for error-free bypass of such lesions by the transcription machinery. Furthermore, because the 5fC–histone conjugates formed in cells are reversible (6), it is possible that other mechanisms exist for the removal of transiently attached proteins. However, if present in a coding region of a gene, such structures are expected to reduce the levels of gene expression.
Experimental procedures
Synthesis and characterization of 5fC-containing oligonucleotide
DNA oligodeoxynucleotides (5′-ATGGCGGGXTAT-3′, where X = 5fC) were synthesized using solid-phase synthesis on an ABI 394 DNA synthesizer as previously described (6). Nucleoside phosphoramidites and other reagents for solid-phase synthesis were purchased from Glen Research (Sterling, VA). 5fC-containing synthetic oligodeoxynucleotides were cleaved off the solid support using 30% ammonium hydroxide at room temperature overnight and deprotected in 80% acetic acid at room temperature for 6 h. The resulting DNA strands were purified by semipreparative HPLC and characterized by MS as described previously (11). Standard unmodified oligonucleotides were purchased from IDT (Coralville, IA) and purified by semipreparative HPLC or denaturing PAGE before use.
Synthesis, purification, and characterization of DNA conjugated to 11-mer peptides or lysine residues
DNA–polypeptide cross-links were synthesized and characterized by gel electrophoresis and MS as described in earlier publications (Fig. 1A) (6, 11). Briefly, 5fC-containing DNA 12-mers (5′-ATGGCGGGXTAT-3′, 300 pmol) were incubated with 20-fold molar excess of 11-mer peptides (RPKPQQFFGLM-CONH2) in 16 μl of sodium phosphate buffer (4.5 mm, pH 7.4) for 3 h at 37 °C. To stabilize DNA–peptide linkages, Schiff base conjugates were reduced by adding 4 μl of 100 mm NaCNBH3 and incubated at 37 °C overnight. The reaction mixtures were heated at 90 °C for 15 min and purified by 20% denaturing PAGE containing 7 m urea (Fig. 1B). Gel-purified DNA–peptide cross-links were desalted by solid-phase extraction on Sep-Pak C18 cartridges and characterized by MALDI-TOF-MS using 3-HPA matrix as previously described (Fig. 1C) (6).
DNA strands containing 5fC–Lys cross-links were synthesized analogously, with the exception that 25 mm synthetic l-lysine was used in the reaction. The reaction mixture was purified by HPLC on an Xbridge BEH C18 column (2.5 μm) with a gradient of 100 mm TEAA at pH 7.0 (A) and acetonitrile (B). Solvent composition was changed linearly from 6% to 15% over 40 min, increased to 20% in 10 min, further to 75% in 1 min, and held at 75% for 5 min. HPLC-purified oligodeoxynucleotides containing 5fC–Lys conjugates were characterized by HPLC-ESI−-MS on a Zorbax 300SB C18 column with a gradient of 15 mm ammonium acetate (A) and acetonitrile (B) (Fig. S2A).
Construction of plasmids containing site-specific 5fC–11-mer peptide and 5fC–Lys conjugates
pTGFP-T7-Hha10 (control) and pTGFP-T7-Hha10comp (competitor) plasmids were a kind gift from Prof. Yinsheng Wang at the University of California–Riverside (20). As compared with control plasmid, competitor plasmid contains three additional bp near the modification site. Plasmids were amplified in TOP10F′ chemically competent Escherichia coli, characterized by gel electrophoresis, and sequenced prior to use. Plasmid vectors containing site-specific DpC lesions were prepared using the gapped vector strategy described elsewhere (20, 21, 34) as shown in Fig. 2. Briefly, pTGFP-T7-Hha10 plasmids were treated with Nt.BstNBI to introduce two nicks 25 nucleotides apart, and the resulting 25-mer oligonucleotides (5′-pATGGCGGGCTATTCGGGAGTCGATG-3′) were removed from the plasmid by incubating with ∼70 molar excess of the complementary strand (5′-CATCGACTCCCGAATAGCCCGCCAT-3′). The resulting gapped plasmids were purified via agarose gel and ligated with the 13-mer oligodeoxynucleotide, 5′-pTCGGGAGTCGATG-3, and the 12-mer oligodeoxynucleotide containing site-specific DpC lesions (5′-ATGGCGGGXTAT-3′, where X = 5fC conjugated to 11-mer peptide RPKPQQFFGLM-CONH2 or single Lys) (Fig. 2). Lesion-bearing supercoiled plasmids were purified by agarose gel prior to use as shown in Figs. S1 and S6.
The presence of site-specific DpC lesions on engineered plasmid vectors was confirmed via NcoI/SfaNI-mediated restriction digestion and post-labeling assays (Fig. 3). In brief, lesion-bearing plasmids (40 ng) were treated with NcoI (5 units) and shrimp alkaline phosphatase (0.5 unit). The 5′ ends of dephosphorylated fragments were radiolabeled with [γ-32P]ATP and T4 PNK (5 units). Treatment with SfaNI (2 units) released radiolabeled DpC-containing 13-mers. The reaction mixtures were analyzed by 20% denaturing PAGE. The presence of a DpC lesion was detected via a characteristic gel shift (Fig. 3C and Fig. S2B). The competitor vector was characterized similarly and produced DNA 16-mers because of the presence of three additional bases in its sequence (Fig. 3).
CTAB assay
In vivo CTAB assays were performed as described elsewhere (20). Briefly, human embryonic kidney cells (HEK293T), human XPA-deficient fibroblast cells (GM04429), and XPA-complemented fibroblasts (1.25 × 105) obtained from the NIGMS Human Genetic Cell Repository (Camden, NJ) were seeded into 24-well plates in Dulbecco's modified Eagle's medium supplemented with 9% fetal bovine serum and cultured in a humidified atmosphere of 5% carbon dioxide and 95% air at 37 °C overnight. NER deficiency in XPA cells was confirmed by the strand-specific primer extension-quantitative polymerase chain reaction assay using a known NER substrate (cholesterol-dR) (19), UV sensitivity (Fig. S8), and an in vitro NER assay (22). Lesion-bearing or unmodified control plasmids were mixed with the competitor plasmids at a 3:1 molar ratio, and 50 ng of the plasmid mixture was transfected into cells using Lipofectamine 2000 (Thermo Fisher Scientific). The cells are harvested after 24 h, and RNA was extracted with total RNA extraction kit (Omega BioTek, Norcross, GA). The purified RNA was treated twice with Ambion DNA-free kit (Thermo Fisher Scientific). RT-PCR was used to confirm that there was no residual DNA contamination (Fig. S7). cDNA was synthesized using a specific primer (5′-TCGGTGTTGCTGTGAT-3′) and Moloney murine leukemia virus reverse transcriptase. The resulting cDNA was amplified via PCR with Phusion DNA polymerases (New England Biolabs, Ipswich, MA) using primers 5′-CTAGCGGATGCATCGACTC-3′ and 5′-TGCTGCGGATGATCTTGTCG-3′. PCR amplification started with incubation at 98 °C for 3 min, followed by 37 cycles at 98 °C for 20 s, 58 °C for 30 s, 72 °C for 20 s, and a final extension at 72 °C for 5 min. PCR products were purified by E.Z.N.A. Cycle Pure kit (Omega BioTek, Norcross, GA), followed by treatment with SfaNI (30 units), shrimp alkaline phosphatase (15 units), and NcoI (50 units; New England Biolabs) (Fig. S3). Proteins were extracted with an equal volume of phenol:chloroform:isoamyl alcohol solution (25:24:1), and the DNA was precipitated with 2.5 volumes of ethanol and 0.1 volumes of 3 m sodium acetate at −80 °C overnight. DNA was reconstituted in LC-MS grade water for the HPLC-ESI-MS/MS analysis.
UV sensitivity of XPA and XPA corrected cells
Human XPA-deficient fibroblasts (GM04429) and the corresponding XPA-complemented fibroblasts obtained from NIGMS Human Genetic Cell Repository (Camden, NJ) were grown in Dulbecco's modified Eagle's medium containing 15% FBS and 1× antibiotic. The cells were trypsinized and counted. 100,000–200,000 cells were plated in 35-mm dishes. The cells were incubated at 37 °C overnight or until they reached 60% confluence. To perform UV dosing experiments, the dishes were removed from the incubator, the medium was removed, and the dishes were immediately placed in a UVP HybriLinker HL-2000 apparatus. Samples (in triplicate) were irradiated with specified UV dosage (0–8 J/cm2). Immediately after irradiation, fresh medium was added to the dishes, and they were returned to the 37 °C incubator. 20 h after UV dosing, the cells were trypsinized and counted.
HPLC-ESI-MS/MS analysis of RT-PCR products
RT-PCR products generated from in vivo transcription were analyzed and quantified using a Dionex UltiMate 3000 HPLC coupled to a LTQ Orbitrap Velos mass spectrometer. A Zorbax 300SB-C18 column (150 × 0.5 mm, 5 μm) was eluted at a flow rate of 15.0 μl/min using 15 mm ammonium acetate in water (A) and acetonitrile (B). Solvent composition was linearly changed from 2 to 20% B in 25 min. Mass spectrometry analyses were performed at a resolution of 60,000 and a scan range of m/z 300–2000. The mass spectrometer was set to monitor 13-mer oligonucleotides corresponding to transcription products (5′-CCGAATAXCCCGC-3′, where X = A, T, C, or G) and 16-mer oligonucleotides originating from competitor plasmids (5′-CACAATAGCATATCGC-3′) as an internal standard for quantification. Relative quantification was performed by comparing HPLC-ESI-MS peak areas in extracted ion chromatograms corresponding to each RT-PCR product relative to the internal standard. MS/MS spectra were used to confirm the sequence of each oligodeoxynucleotide product.
In vitro NER experiments in HeLa cell extracts
DNA 135-mer duplexes containing site-specific DpC lesions were constructed as described in detail by Kropachev et al. (23) Briefly, gel-purified 12-mer oligonucleotides containing reductively stabilized 5fC–polypeptide or 5fC–Lys conjugates were radiolabeled with [γ-32P]ATP and T4 PNK and ligated to produce 135-mer oligonucleotide duplexes (see Fig. 6A for DNA sequence). Nuclear extracts were prepared using human HeLa cells and utilized for in vitro NER assays as described earlier (23). Internally 32P-labeled 135-mer oligonucleotide duplex containing site-specific DNA lesion at the 70th nucleotide from the 5′ end (1 pmol; Fig. 6A) was mixed with 17.5 μl of 1 m KCl, 20 μl of Tris-ATP (10 mm at pH 7.9), 10 μl of freshly prepared HeLa cell extracts, and sufficient dialysis buffer containing 12 mm MgCl2, 25 mm HEPES-KOH, pH 7.9, 2.5 mm DTT, 1 mm EDTA, and 10% glycerol in a total volume of 50 μl. After preselected incubation times and appropriate preparation, aliquots of the repair reaction mixtures were quenched with gel loading buffer and loaded onto 12% denaturing PAGE for analysis.
Model of the DpC-containing B-DNA duplex
The structure of the 11-mer peptide (RPKPQQFFGLM-CONH2) cross-linked to the C5-dC in B-DNA (5′-CATGACGCT-3′) was extracted from the most representative structure of our previous MD simulation for this DpC bound to human DNA polymerase η (11). Similarly, the unmodified B-DNA structure was extracted from our previous simulation of DNA polymerase η without DpC (11).
Author contributions
S. J. formal analysis; S. J., D. P., K. K., M. K., and I. F. investigation; S. J. methodology; S. J. writing-original draft; I. F. software; S. B. data curation; S. B., N. E. G., and N. Y. T. supervision; S. B., N. E. G., and N. Y. T. writing-review and editing; M.E. validation; N. Y. T. conceptualization; N. Y. T. funding acquisition; N. Y. T. project administration.
Supplementary Material
Acknowledgments
We gratefully acknowledge Prof. Yinsheng Wang at the University of California at Riverside for providing the pTGFP-T7-Hha10 and pTGFP-T7-Hha10comp plasmids as kind gifts. We thank Xun Ming and Dr. Peter W. Villalta (University of Minnesota) for help with MS analyses, Dr. Colin Campbell for advice with viability essays, and Robert Carlson (University of Minnesota) for help with figure preparation. We thank Dr. Lihua Wang for dedicated support of computational infrastructure in the Broyde laboratory.
This work was supported by the NIEHS, National Institutes of Health Grants R01-ES-023350 (to N. Y. T.), R01-ES-025987 (to S. B.), and R01-ES-024050 (to N. E. G.). This work was also supported in part by a Wayland E. Noland graduate student fellowship (to S. J.) and a doctoral dissertation fellowship from the University of Minnesota (to S. J.). Computational resources were provided by Extreme Science and Engineering Discovery through Grant MCB060037 (to S. B.) and by New York University IT High Performance Computer Resources and Services. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8.
- DPC
- DNA–protein cross-link
- DpC
- DNA–peptide cross-links
- 5fC
- 5-formylcytosine
- NER
- nucleotide excision repair
- ESI
- electrospray ionization
- CTAB
- competitive transcription and bypass assay
- HRMS
- high-resolution MS
- XPA
- group A xeroderma pigmentosum.
References
- 1. Barker S., Weinfeld M., and Murray D. (2005) DNA–protein crosslinks: their induction, repair, and biological consequences. Mutat. Res. 589, 111–135 10.1016/j.mrrev.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 2. Tretyakova N. Y., Groehler A. 4th, and Ji S. (2015) DNA–protein cross-links: formation, structural identities, and biological outcomes. Acc. Chem. Res. 48, 1631–1644 10.1021/acs.accounts.5b00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wickramaratne S., Ji S., Mukherjee S., Su Y., Pence M. G., Lior-Hoffmann L., Fu I., Broyde S., Guengerich F. P., Distefano M., Schärer O. D., Sham Y. Y., and Tretyakova N. (2016) Bypass of DNA–protein cross-links conjugated to the 7-deazaguanine position of DNA by translesion synthesis polymerases. J. Biol. Chem. 291, 23589–23603 10.1074/jbc.M116.745257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neale M. J., Pan J., and Keeney S. (2005) Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057 10.1038/nature03872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang S. W., Burgin A. B. Jr., Huizenga B. N., Robertson C. A., Yao K. C., and Nash H. A. (1996) A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. U.S.A. 93, 11534–11539 10.1073/pnas.93.21.11534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ji S., Shao H., Han Q., Seiler C. L., and Tretyakova N. Y. (2017) Reversible DNA–protein cross-linking at epigenetic DNA marks. Angew. Chem. Int. Ed. Engl. 56, 14130–14134 10.1002/anie.201708286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., and Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pfaffeneder T., Hackner B., Truss M., Münzel M., Müller M., Deiml C. A., Hagemeier C., and Carell T. (2011) The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 50, 7008–7012 10.1002/anie.201103899 [DOI] [PubMed] [Google Scholar]
- 9. Li F., Zhang Y., Bai J., Greenberg M. M., Xi Z., and Zhou C. (2017) 5-Formylcytosine yields DNA–protein cross-links in nucleosome core particles. J. Am. Chem. Soc. 139, 10617–10620 10.1021/jacs.7b05495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raiber E. A., Portella G., Martínez Cuesta S., Hardisty R., Murat P., Li Z., Iurlaro M., Dean W., Spindel J., Beraldi D., Liu Z., Dawson M. A., Reik W., and Balasubramanian S. (2018) 5-Formylcytosine organizes nucleosomes and forms Schiff base interactions with histones in mouse embryonic stem cells. Nat. Chem. 10, 1258–1266 10.1038/s41557-018-0149-x [DOI] [PubMed] [Google Scholar]
- 11. Ji S., Fu I., Naldiga S., Shao H., Basu A. K., Broyde S., and Tretyakova N. Y. (2018) 5-Formylcytosine mediated DNA–protein cross-links block DNA replication and induce mutations in human cells. Nucleic Acids Res. 46, 6455–6469 10.1093/nar/gky444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vaz B., Popovic M., Newman J. A., Fielden J., Aitkenhead H., Halder S., Singh A. N., Vendrell I., Fischer R., Torrecilla I., Drobnitzky N., Freire R., Amor D. J., Lockhart P. J., Kessler B. M., et al. (2016) Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA–protein crosslink repair. Mol. Cell 64, 704–719 10.1016/j.molcel.2016.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vaz B., Popovic M., and Ramadan K. (2017) DNA–protein crosslink proteolysis repair. Trends Biochem. Sci. 42, 483–495 10.1016/j.tibs.2017.03.005 [DOI] [PubMed] [Google Scholar]
- 14. Duxin J. P., Dewar J. M., Yardimci H., and Walter J. C. (2014) Repair of a DNA–protein crosslink by replication-coupled proteolysis. Cell 159, 346–357 10.1016/j.cell.2014.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maskey R. S., Flatten K. S., Sieben C. J., Peterson K. L., Baker D. J., Nam H. J., Kim M. S., Smyrk T. C., Kojima Y., Machida Y., Santiago A., van Deursen J. M., Kaufmann S. H., and Machida Y. J. (2017) Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 45, 4564–4576 10.1093/nar/gkx107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Larsen N. B., Gao A. O., Sparks J. L., Gallina I., Wu R. A., Mann M., Räschle M., Walter J. C., and Duxin J. P. (2019) Replication-coupled DNA–protein crosslink repair by SPRTN and the proteasome in xenopus egg extracts. Mol. Cell 73, 574–588.e7 10.1016/j.molcel.2018.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baker D. J., Wuenschell G., Xia L., Termini J., Bates S. E., Riggs A. D., and O'Connor T. R. (2007) Nucleotide excision repair eliminates unique DNA–protein cross-links from mammalian cells. J. Biol. Chem. 282, 22592–22604 10.1074/jbc.M702856200 [DOI] [PubMed] [Google Scholar]
- 18. Minko I. G., Zou Y., and Lloyd R. S. (2002) Incision of DNA–protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. Proc. Natl. Acad. Sci. U.S.A. 99, 1905–1909 10.1073/pnas.042700399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chesner L. N., and Campbell C. (2018) A quantitative PCR-based assay reveals that nucleotide excision repair plays a predominant role in the removal of DNA–protein crosslinks from plasmids transfected into mammalian cells. DNA Repair (Amst.) 62, 18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. You C., and Wang Y. (2015) Quantitative measurement of transcriptional inhibition and mutagenesis induced by site-specifically incorporated DNA lesions in vitro and in vivo. Nat. Protoc. 10, 1389–1406 10.1038/nprot.2015.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. You C., Dai X., Yuan B., Wang J., Wang J., Brooks P. J., Niedernhofer L. J., and Wang Y. (2012) A quantitative assay for assessing the effects of DNA lesions on transcription. Nat. Chem. Biol. 8, 817–822 10.1038/nchembio.1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shafirovich V., Kropachev K., Kolbanovskiy M., and Geacintov N. E. (2019) Excision of oxidatively generated guanine lesions by competing base and nucleotide excision repair mechanisms in human cells. Chem. Res. Toxicol. 32, 753–761 10.1021/acs.chemrestox.8b00411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kropachev K., Kolbanovskii M., Cai Y., Rodríguez F., Kolbanovskii A., Liu Y., Zhang L., Amin S., Patel D., Broyde S., and Geacintov N. E. (2009) The sequence dependence of human nucleotide excision repair efficiencies of benzo[a]pyrene-derived DNA lesions: insights into the structural factors that favor dual incisions. J. Mol. Biol. 386, 1193–1203 10.1016/j.jmb.2008.12.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song C. X., Szulwach K. E., Dai Q., Fu Y., Mao S. Q., Lin L., Street C., Li Y., Poidevin M., Wu H., Gao J., Liu P., Li L., Xu G. L., Jin P., and He C. (2013) Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153, 678–691 10.1016/j.cell.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spruijt C. G., Gnerlich F., Smits A. H., Pfaffeneder T., Jansen P. W., Bauer C., Münzel M., Wagner M., Müller M., Khan F., Eberl H. C., Mensinga A., Brinkman A. B., Lephikov K., Müller U., et al. (2013) Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159 10.1016/j.cell.2013.02.004 [DOI] [PubMed] [Google Scholar]
- 26. Ebright R. H. (2000) RNA polymerase: structural similarities between bacterial RNA polymerase and eukaryotic RNA polymerase II. J. Mol. Biol. 304, 687–698 10.1006/jmbi.2000.4309 [DOI] [PubMed] [Google Scholar]
- 27. Cramer P. (2002) Multisubunit RNA polymerases. Curr. Opin. Struct. Biol. 12, 89–97 10.1016/S0959-440X(02)00294-4 [DOI] [PubMed] [Google Scholar]
- 28. Xu L., Wang W., Chong J., Shin J. H., Xu J., and Wang D. (2015) RNA polymerase II transcriptional fidelity control and its functional interplay with DNA modifications. Crit. Rev. Biochem. Mol. Biol. 50, 503–519 10.3109/10409238.2015.1087960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. E C., Duan B., and Yu J. (2017) Nucleotide selectivity at a preinsertion checkpoint of T7 RNA polymerase transcription elongation. J. Phys. Chem. B 121, 3777–3786 10.1021/acs.jpcb.6b11668 [DOI] [PubMed] [Google Scholar]
- 30. Nakano T., Ouchi R., Kawazoe J., Pack S. P., Makino K., and Ide H. (2012) T7 RNA polymerases backed up by covalently trapped proteins catalyze highly error prone transcription. J. Biol. Chem. 287, 6562–6572 10.1074/jbc.M111.318410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Graaf B., Clore A., and McCullough A. K. (2009) Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA–protein crosslinks. DNA Repair (Amst.) 8, 1207–1214 10.1016/j.dnarep.2009.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Laat W. L., Jaspers N. G., and Hoeijmakers J. H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev. 13, 768–785 10.1101/gad.13.7.768 [DOI] [PubMed] [Google Scholar]
- 33. Reardon J. T., and Sancar A. (2006) Repair of DNA–polypeptide crosslinks by human excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 103, 4056–4061 10.1073/pnas.0600538103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. You C., Wang P., Dai X., and Wang Y. (2014) Transcriptional bypass of regioisomeric ethylated thymidine lesions by T7 RNA polymerase and human RNA polymerase II. Nucleic Acids Res. 42, 13706–13713 10.1093/nar/gku1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ji S., Thomforde J., Rogers C., Fu I., Broyde S., Tretyakova N. Y. (2019) Transcriptional bypass of DNA-protein and DNA-peptide conjugates by T7 RNA polymerase. ACS Chem Biol. 10.1021/acschembio.9b00365 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.