

Abstract
Cellular senescence has been shown to contribute to skin ageing. However, the role of melanocytes in the process is understudied. Our data show that melanocytes are the only epidermal cell type to express the senescence marker p16INK4A during human skin ageing. Aged melanocytes also display additional markers of senescence such as reduced HMGB1 and dysfunctional telomeres, without detectable telomere shortening. Additionally, senescent melanocyte SASP induces telomere dysfunction in paracrine manner and limits proliferation of surrounding cells via activation of CXCR3‐dependent mitochondrial ROS. Finally, senescent melanocytes impair basal keratinocyte proliferation and contribute to epidermal atrophy in vitro using 3D human epidermal equivalents. Crucially, clearance of senescent melanocytes using the senolytic drug ABT737 or treatment with mitochondria‐targeted antioxidant MitoQ suppressed this effect. In conclusion, our study provides proof‐of‐concept evidence that senescent melanocytes affect keratinocyte function and act as drivers of human skin ageing.
Keywords: melanocytes, SASP, senescence, skin ageing, telomeres
Subject Categories: Ageing; Cell Cycle; DNA Replication, Repair & Recombination
Senescence in melanocytes of the ageing epidermis induces telomere damage and reduced proliferation of surrounding cells, which can be rescued by a senolytic drug.
Introduction
Cellular senescence is characterised by an irreversible cell‐cycle arrest, which is accompanied by a number of phenotypic changes such as the development of a senescence‐associated secretory phenotype (SASP) (Coppé et al, 2008). Senescent cells have been shown to play beneficial roles in vivo, such as tissue regeneration, wound healing and embryonic development (Munoz‐Espin et al, 2013; Demaria et al, 2014; Ritschka et al, 2017). Paradoxically, accumulation of senescent cells has also been causally implicated in age‐associated tissue dysfunction and pathologies (Baker et al, 2011, 2016; Xu et al, 2015). The detrimental effects of senescent cells can be largely attributed to the SASP, which comprises a number of pro‐inflammatory cytokines, chemokines and growth factors, allowing extracellular communication. Indeed, senescent cells can induce paracrine senescence in normal neighbouring cells via secretion of SASP factors (Acosta et al, 2013), and chronic exposure to the SASP has been shown to impair the regenerative capacity of keratinocytes in mice in vivo (Ritschka et al, 2017).
The senescence programme can be triggered by a number of stressors, such as telomere dysfunction. Telomeres are specialised structures present at the ends of linear chromosomes, which are critical in maintaining genomic stability (Blackburn, 1991). They consist of tandem TTAGGG repeats and are stabilised by a group of proteins known as the shelterin complex. The latter are important for the formation of the T‐loop, a lariat‐like structure which physically protects the ends of chromosomes from being recognised as double‐stranded breaks (DSBs) (Griffith et al, 1999). It is believed that telomere shortening that occurs due to extensive rounds of cell division results in loss of shelterin components (i.e. telomere uncapping), destabilising the T‐loop, and subsequently triggering a DNA damage response (DDR) similar to double‐stranded breaks, which initiates the senescence programme (d'Adda di Fagagna et al, 2003; Takai et al, 2003). However, it is now becoming increasingly evident that a DDR can also be activated at telomeres independently of length, and such length‐independent persistent DNA damage signalling has been reported in senescent cells in vitro and in many mammalian tissues with age (Herbig et al, 2006; Kaul et al, 2011; Fumagalli et al, 2012; Hewitt et al, 2012; Birch et al, 2015; Anderson et al, 2019).
Skin ageing arises from a combination of both intrinsic and extrinsic factors, which ultimately compromise the structural integrity and physiological function of the skin (Kammeyer & Luiten, 2015). In general, aged skin is characterised by profound phenotypic changes such as dermal and epidermal atrophy, flattening of the epidermal–dermal junction, a loss of collagen and a gain of elastic fibres (Farage et al, 2007). Although senescent cells have been shown to accumulate in human skin with age (Dimri et al, 1995; Ressler et al, 2006), the majority of studies on cellular senescence in the skin have thus far focused on dermal fibroblasts, and very little is known about the impact of melanocyte senescence in skin ageing. Recent reports have demonstrated that melanocytes expressing senescence markers accumulate in human skin, and this was significantly associated with increased facial wrinkles, higher perceived age and age‐associated elastin morphology in the papillary dermis (Waaijer et al, 2012b, 2016). Interestingly, expression of p16INK4A, a known marker of senescence, was shown to co‐localise almost exclusively to melanocytes in the epidermis, suggesting that melanocytes represent the major p16INK4A senescent cell population in the epidermis of human skin (Waaijer et al, 2012b, 2016; Pawlikowski et al, 2013). This observation is surprising given that fully differentiated melanocytes have an extremely low replicative capacity (Jimbow et al, 1975; Taylor et al, 2011). However, the mechanisms by which melanocytes become senescent and whether they contribute causally to skin ageing phenotypes are still unknown. Therefore, in our study, we investigated the link between senescent melanocytes with age‐related skin changes and examined whether these cells could act in a cell non‐autonomous manner, impacting on the regenerative capacity of surrounding cells.
Results
Senescent melanocytes accumulate in human skin with age
In order to investigate melanocyte senescence in vivo, we obtained skin punch biopsies taken from the sun‐protected part of the upper inner arm of young and older human donors (Table 1). Firstly, we performed immunohistochemistry to analyse the expression of p16, a cyclin‐dependent kinase inhibitor which is highly expressed in senescent cells (Serrano et al, 1996). Melanocytes were identified by immunohistochemistry using antibodies against S100 and Melan‐A. We found a significant increase in the frequency of p16‐positive melanocytes in human skin with age (Fig 1A and B). Interestingly, all p16‐positive cells detected in the epidermis were also positive for melanocyte markers (Fig 1A), supporting previous observations that melanocytes are the main senescent cell population in the epidermis of human skin (Waaijer et al, 2016). To determine whether melanocytes expressed other markers of senescence with age, we conducted immunofluorescence for HMGB1, which has been shown to migrate from the nucleus to the extracellular milieu in senescent cells (Davalos et al, 2013) and cyclin‐dependent kinase inhibitor p21 in combination with Melan‐A. We observed a decline in expression of HMGB1 in melanocytes from older subjects (Fig 1A and C); however, we did not observe a significant age‐dependent change in p21‐positive melanocytes (Fig 1A and D).
Table 1.
Age and gender of subjects involved in this study
| Young donors | Older donors | |
|---|---|---|
| n | 20 | 41 |
| Age range, year | 22–31.8 | 54.9–81.3a |
| Mean age, year | 26.5 ± 2.8 | 66.5 ± 6.6 |
| Gender (male/female) | 10/10 | 25/16 |
n = number of subjects.
Mean age = mean ± SD.
P < 0.0001 compared with young donors.
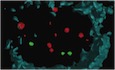
Figure 1. Senescent melanocytes accumulate in human skin with age.
-
ARepresentative immunofluorescence images for p16, HMGB1 and p21 together with Melan‐A in young and older skin. Arrows indicate melanocytes, which are amplified on the right. Scale bar is 30 μm. Bottom panel shows representative immuno‐FISH images for γH2AX (green) and telomeres (red) in melanocytes in young and older skin sections. Arrows indicate co‐localisation between γH2AX foci and telomeres (TAF), which are amplified on the right. Images are 3D reconstructions of immuno‐FISH using confocal microscopy and taken with a 100× oil objective. Scale bar is 10 μm.
-
B–GDot plots show (B) percentage of p16‐positive melanocytes, (C) mean HMGB1 intensity in melanocytes, (D) percentage of p21‐positive melanocytes, (E) mean number of γH2AX foci, (F) mean number of TAF in melanocytes and (G) percentage of melanocytes containing ≥ 3 TAF for each individual subject (n = 10–14 young and 10–12 old donors). The horizontal line represents the mean for each group ± SEM.
-
HHistograms showing distribution of telomere signal intensities in melanocytes analysed by Q‐FISH in young (n = 8) and old (n = 7) skin biopsies. Dotted lines indicate median intensity.
-
IHistograms showing distribution of intensities of γH2AX‐negative (non‐TAF) and γH2AX‐positive (TAF) telomeres in melanocytes in older skin (n = 8 donors). Dotted lines indicate median intensity.
-
JRepresentative immuno‐FISH image (red: telomeres; green: γH2AX) of a melanocyte in the skin of an older donor, showing a longer telomere co‐localising with γH2AX (a) and a shorter one which is γH2AX‐negative (b). Arrows indicate telomeres measured in (K), and dashed lines show the area quantified in (K). Images are Huygen (SVI) deconvolved Z projections and were taken using a 100× oil objective.
-
KGraphs showing the quantification of telomere intensity of γH2AX‐positive (a) and γH2AX‐negative (b) telomeres. Red line indicates telomere intensity, and green shows γH2AX intensity.
-
LGraph showing FISH intensities for individual telomeres co‐localising (TAF; red) or not co‐localising (non‐TAF; black) with γH2AX foci in individual melanocytes (n = 18). Green horizontal line indicates the mean FISH intensity.
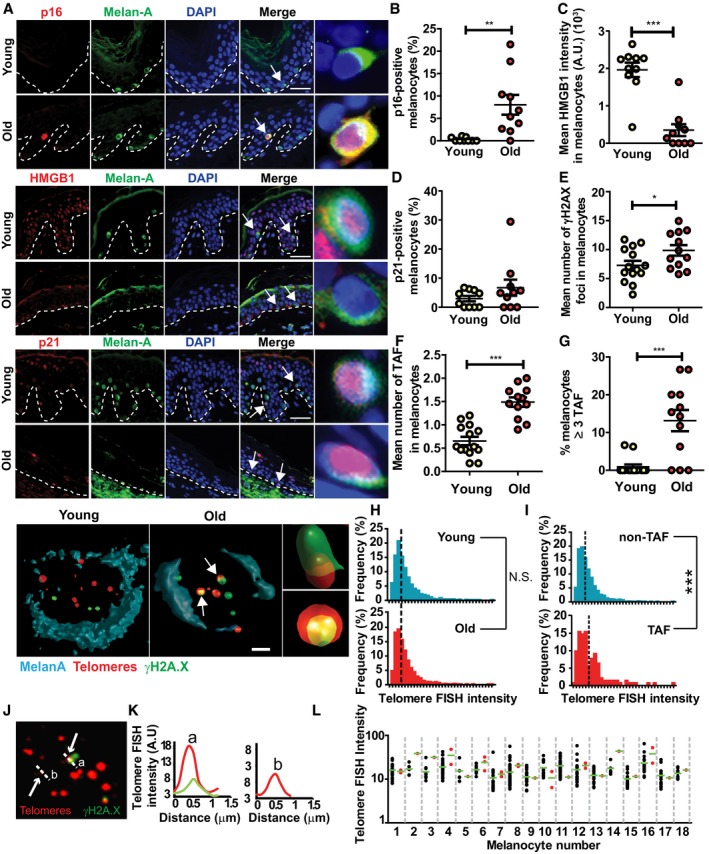
A decline in SIRT1 (a NAD+‐dependent protein deacetylase) has been associated with ageing and senescence (Sasaki et al, 2006; Imai & Guarente, 2014; Birch et al, 2015). Accordingly, we observed a significant decrease in SIRT1 expression in melanocytes in the skin of older donors (Fig EV1A and B).
Figure EV1. Melanocytes express increased markers of senescence in human skin with age.
-
ARepresentative images of SIRT1 immunofluorescence in combination with the melanocyte marker Melan‐A in young and older skin. Arrow indicates a melanocyte, which is amplified on the right.
-
BDot plot showing percentage of SIRT1‐positive melanocytes for each individual donor. The horizontal line represents the group mean (n = 12 donors for each group). Data are shown as mean ± SEM.
-
C–EGraphs showing (C) mean number of 53BP1 foci in melanocytes, (D) mean number of 53BP1 foci co‐localising with telomeres in melanocytes and (E) percentage of melanocytes containing ≥ 3 53BP1 foci co‐localising with telomeres for each individual subject. The horizontal line represents the mean for each group (n = 10 young donors; n = 8 old donors). Data are shown as mean ± SEM.
Telomere dysfunction, characterised by the association of DNA damage response (DDR) proteins with telomeres, has also been shown to drive senescence and increase in multiple mammalian tissues during ageing (Kaul et al, 2011; Fumagalli et al, 2012; Hewitt et al, 2012). Therefore, in order to investigate whether telomere dysfunction was associated with melanocyte senescence, we performed immuno‐FISH combining immunofluorescence against DNA damage response proteins γH2AX and 53BP1 and in situ hybridisation using a telomere‐specific PNA probe. The analysis of Melan‐A‐positive cells revealed a small but significant increase in the total number of γH2AX foci in melanocytes with age (Fig 1E). By quantifying the co‐localisation of γH2AX and telomeres, we found that the mean number of telomere‐associated foci (TAF) significantly increased in melanocytes in aged skin (Fig 1F). Similar results were obtained when quantifying co‐localisation between 53BP1 and telomeres (Fig EV1C and D). Moreover, we observed a significant increase in the percentage of melanocytes containing ≥ 3 TAF in the skin of older donors (Figs 1G and EV1E), which was rarely seen in younger skin. It is still unknown how many TAF are required to induce senescence; however, previous work had shown that % of cells containing ≥ 3 TAF was quantitatively comparable to the frequency of other senescent markers (Ogrodnik et al, 2017; Anderson et al, 2019).
Since a DNA damage response can be activated at short telomeres as a result of attrition driven by cell division (d'Adda di Fagagna et al, 2003), we next investigated whether telomere shortening occurs during ageing of melanocytes. By performing telomere quantitative FISH (Q‐FISH) in skin sections from human donors and analysing individual telomere signal intensity, we found no significant difference in median telomere FISH signal intensity between melanocytes from young and older skin (Fig 1H), suggesting that these cells do not undergo significant telomere shortening with age in vivo. This is consistent with the low replicative potential of differentiated melanocytes previously reported (Jimbow et al, 1975; Taylor et al, 2011). Interestingly, by comparing the distribution of FISH intensities of telomeres that did not co‐localise with γH2AX (non‐TAF) with those co‐localising with γH2AX (TAF) in melanocytes, we found a slight shift towards stronger telomere FISH signal intensities in the latter (Fig 1I). Further analysis of telomere FISH intensity in individual melanocytes showed that telomeres co‐localising with γH2AX were not preferentially short (Fig 1J–L). Together, these data suggest that telomere dysfunction in aged melanocytes is not driven by reduced telomere length.
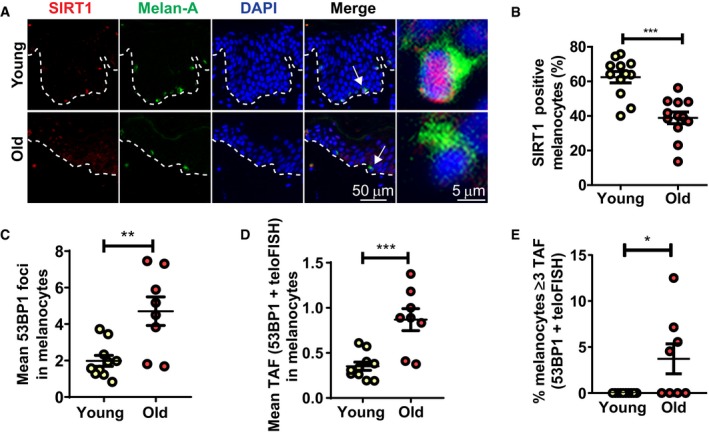
Interestingly, we observed in human skin that both the mean number of TAF in melanocytes and the percentage of melanocytes containing ≥ 2 dysfunctional telomeres positively correlated with a flatter epidermal curvature, which is an important feature of skin ageing (Fig EV2A and B), and this was still significant even when correcting for subject age (Fig EV2C and D). These findings and previous published data (Waaijer et al, 2016) led us to hypothesise that senescent melanocytes may contribute to the decreased epidermal turnover observed during ageing (Farage et al, 2007).
Figure EV2. Telomere dysfunction in melanocytes correlates with age‐associated loss of epidermal curvature.
-
AGraph showing the correlation between epidermal curvature and age for each individual donor (n = 22 donors) accounting for subject age. A curvature value closer to 1 denotes a flatter epidermal–dermal junction (EDJ).
-
BImages illustrating the loss in epidermal curvature (red dotted lines) with age. Red dotted lines represent the EDJ.
-
C, DGraphs showing correlations between (C) mean number of TAF in melanocytes and (D) percentage of melanocytes containing ≥ 2 TAF and epidermal curvature for each individual donor. When accounting for subject age, the statistical significance for these correlations was (C) P = 0.007 and (D) P = 0.02.
Senescent melanocytes induce paracrine telomere damage and senescence in surrounding cells
Senescent cells have been shown to induce paracrine DNA damage and senescence in neighbouring cells, and this has been suggested to be a mechanism by which they contribute to age‐related tissue dysfunction (Nelson et al, 2012; Acosta et al, 2013). Therefore, we hypothesised that accumulation of senescent melanocytes could contribute to skin ageing phenotypes by compromising surrounding epidermal cells. Firstly, we investigated whether there was evidence of paracrine telomere damage exerted by senescent melanocytes on neighbouring keratinocytes in human skin. To that end, melanocytes containing 0 to 3 TAF in aged human skin were separated into groups, and the number of TAF in keratinocytes located in the immediate vicinity of each melanocyte was quantified. We found that keratinocytes surrounding melanocytes with 3 TAF have a significantly higher number of dysfunctional telomeres when compared to cells neighbouring melanocytes with 0 TAF (Fig 2A and B). In order to ensure that the differences in the number of TAF in keratinocytes were not due to inter‐subject variability (such as age differences), we carried out within‐subject analysis in the older subjects. A positive correlation between TAF in melanocytes and surrounding keratinocytes was present in 6 of the 7 older subjects, and 3 of the 6 correlations were statistically significant, suggesting that the correlation between the number of TAF in melanocytes and in the surrounding keratinocytes was not just due to inter‐subject differences (Appendix Fig S1A and B). Additionally, we investigated whether TAF in keratinocytes changed in function of the distance from senescent melanocytes. We found that the mean number of TAF was the highest in keratinocytes located in the immediate vicinity of senescent melanocytes and that this number significantly declined the farther they were located (Appendix Fig S1C).
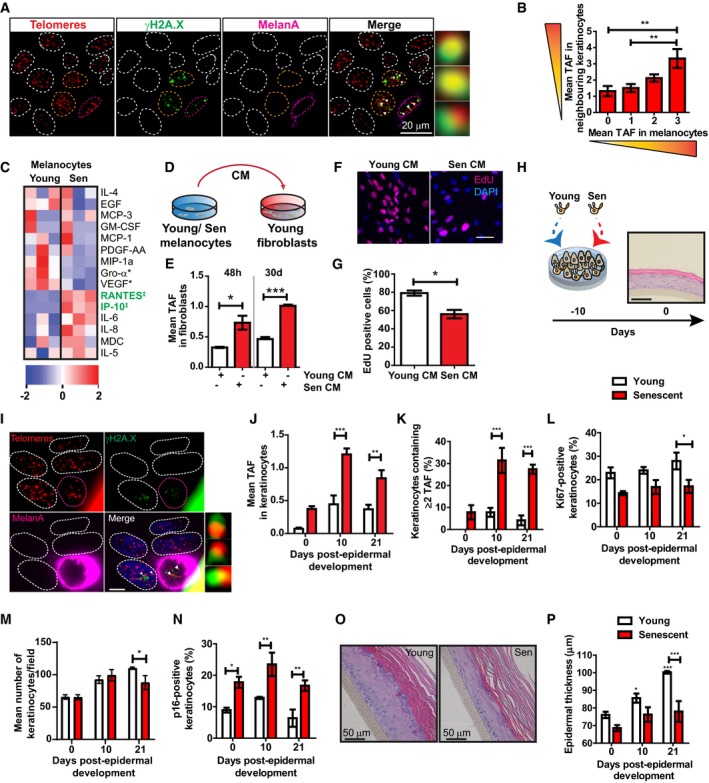
Figure 2. Senescent melanocytes induce paracrine senescence and contribute to epidermal atrophy in a 3D epidermal equivalent.
-
ARepresentative immuno‐FISH images of keratinocytes around melanocytes with 3 TAF in skin from older donors. Melanocytes are circled in pink, and keratinocytes analysed are circled in orange. Areas encircled with white dashed lines represent other keratinocytes not included in this analysis. Arrows represent co‐localisation between γH2AX and telomeres (TAF), which are amplified on the right. Images are Z projections taken using a 100× oil objective.
-
BGraph showing mean number of TAF in keratinocytes adjacent to melanocytes containing 0–3 TAF in skin biopsies of older donors (n = 7). Data are shown as mean ± SEM.
-
CHeatmap showing cytokines detected in conditioned medium from young and senescent melanocytes. Blue represents low expression, whereas red denotes high expression. Each column represents one independent experiment for the corresponding condition (n = 3). Heatmap was generated using Ingenuity Pathway Analysis (IPA). ‡ = cytokines significantly up‐regulated; * = cytokines significantly down‐regulated.
-
DExperimental scheme. Young dermal fibroblasts were cultured with conditioned medium collected from either young or senescent melanocyte.
-
EGraph showing mean number of TAF in fibroblasts cultured with young or senescent melanocyte CM for 48 h and 30 days. Data are shown as mean ± SEM (n = 3 independent experiments).
-
FRepresentative EdU immunofluorescence images of fibroblasts cultured in young or senescent melanocyte CM for 20 days. Images were taken using a 20× objective. Scale bar is 50 μm.
-
GGraph showing percentage of EdU‐positive fibroblasts at the indicated culture conditions. Data are shown as mean ± SEM (n = 3 independent experiments).
-
HScheme representing the development of 3D human epidermal equivalents. Proliferating or senescent melanocytes are co‐cultured with keratinocytes for 10 days before fully differentiating into an epidermal equivalent. Day 0 indicates the day of complete epidermal differentiation. Scale bar is 50 μm.
-
IRepresentative immuno‐FISH image of a melanoderm containing senescent melanocytes. Telomeres are shown in red, γH2AX is shown in green, and Melan‐A is shown in pink. Arrow indicates co‐localisation between γH2AX and telomeres (TAF), which is amplified on the right. Images are Z projections taken with a 63× oil objective. Scale bar is 10 μm.
-
J–NGraphs showing (J) mean number of TAF in keratinocytes, (K) percentage of keratinocytes containing ≥ 2 TAF, (L) percentage of Ki‐67‐positive keratinocytes, (M) mean number of keratinocytes and (N) percentage of p16‐positive keratinocytes in melanoderms containing either young (white bars) or senescent (red bars) melanocyte at the time points indicated. Data are shown as mean ± SEM of n = 3–4 (proliferating) and n = 3 (senescent) melanoderms.
-
ORepresentative H&E images showing epidermal thickness of melanoderms containing young or senescent melanocytes. Images were taken using a 20× objective.
-
PGraph showing epidermal thickness of melanoderms containing either young (white bars) or senescent (red bars) melanocyte at the time points indicated. Data are shown as mean ± SEM of n = 4 (proliferating) or n = 3 (senescent) melanoderms.
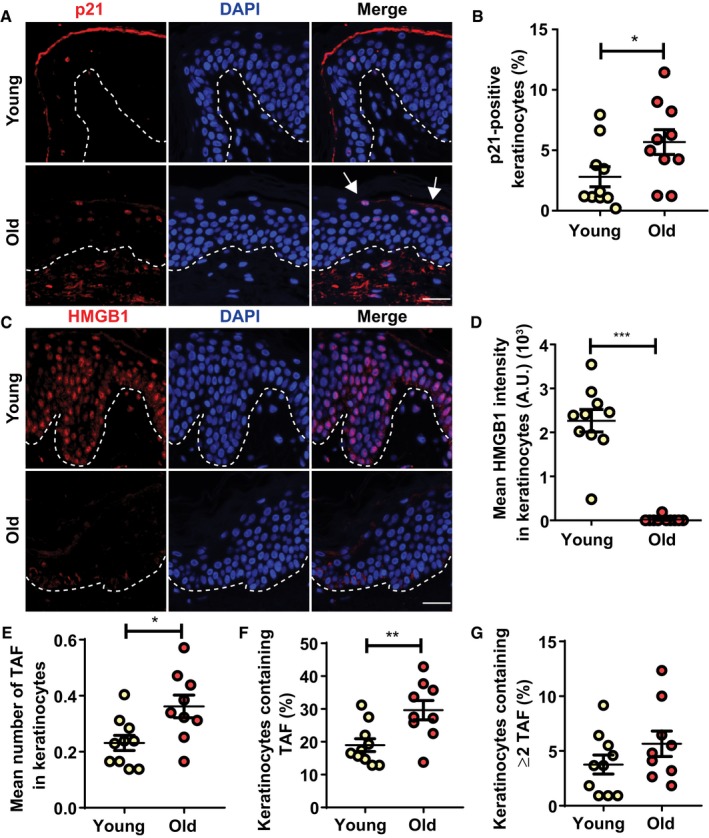
In order to investigate whether keratinocytes also acquired senescent markers with age, we evaluated markers of senescence p16, p21, TAF and absence of HMGB1 specifically in keratinocytes from young and old subjects. We did not detect any p16 positivity in keratinocytes from any age group, however, we found an age‐dependent increase in p21‐positive cells located predominantly in the granular layer of the epidermis (Fig EV3A and B). We also found a significant decrease in expression of HMGB1 (Fig EV3C and D). Finally, we observed that both the mean number and % of keratinocytes positive for TAF increased in older subjects (Fig EV3E–G).
Figure EV3. Keratinocytes express senescence markers in human skin with age.
-
A–DRepresentative immunofluorescence images of (A) p21 and (C) HMGB1 in skin from young and old donors. Images were taken using a 20× objective. Arrows indicate p21‐positive keratinocytes in the uppermost layer of the epidermis. Scale is 30 μm. Graphs showing (B) percentage of p21‐positive keratinocytes per donor and (D) HMGB1 intensity in keratinocytes. The horizontal line represents the mean for each group (n = 10 donors per group). Data are shown as mean ± SEM.
-
E–G(E) Mean number of TAF in keratinocytes, (F) percentage of keratinocytes containing TAF and (G) percentage of keratinocytes containing ≥ 2 TAF in skin sections from young and old donors. The mean for each group is represented by the horizontal line (n = 10 young donors; n = 9 old donors). Data are shown as ± SEM.
Next, in order to assess whether senescent melanocytes are capable of inducing paracrine DNA damage in vitro, we induced senescence in human epidermal melanocytes using X‐ray irradiation, which is a widely used model of stress‐induced cellular senescence (Passos et al, 2010). We found that the majority of melanocytes became senescent 10 days following irradiation, which was confirmed by significantly decreased proliferation, increased senescence‐associated β‐galactosidase activity (Sen‐β‐Gal) and increased p16 expression (Appendix Fig S2A–F). X‐ray‐induced senescent melanocytes also showed increased numbers of γH2AX foci, as well as a higher frequency of TAF when compared to controls (Appendix Fig S2G–I). An important phenotype of senescent cells is the development of the SASP. In order to characterise the SASP of senescent melanocytes, we carried out a cytokine array in conditioned media (CM) collected from these cells. From the panel of soluble proteins analysed, we observed that the secretion of RANTES and IP‐10 was significantly increased in senescent melanocytes, whereas Gro‐α and VEGF were significantly down‐regulated compared to young, proliferating controls (Fig 2C). The majority of other cytokines and chemokines analysed were secreted below detection level. Although secretion of the classical SASP factors, IL‐6 and IL‐8, was increased in senescent melanocytes, it did not reach statistical significance. This is consistent with previous reports showing that the composition of the SASP is highly dependent on the cell type and on the senescence‐inducing stimulus (Coppe et al, 2008, 2010).
To determine whether the SASP of senescent melanocytes could induce telomere damage in neighbouring cells in vitro, human dermal fibroblasts were cultured with CM from either young or senescent melanocytes (Fig 2D). We were not able to co‐culture senescent melanocytes and keratinocytes since the melanocyte culture medium inadvertently induced differentiation of keratinocytes which impacted on our analysis; therefore, we chose to carry out our experiments using dermal fibroblasts as a proof‐of‐concept. The analysis of telomere dysfunction by immuno‐FISH revealed that CM from senescent melanocytes was effective at triggering a DDR at telomeres in dermal fibroblasts which was present both at 2 and 30 days following CM exposure (Fig 2E). We also examined whether fibroblasts exposed to senescent CM expressed additional markers of senescence. Although p16 and Sen‐β‐Gal expression were not affected at the time points analysed, we observed a significant decrease in EdU incorporation when fibroblasts were cultured in senescent melanocyte CM for 20 days (Fig 2F and G). These results suggest that SASP factors secreted from senescent melanocytes can induce paracrine telomere dysfunction and impair proliferation of surrounding cells.
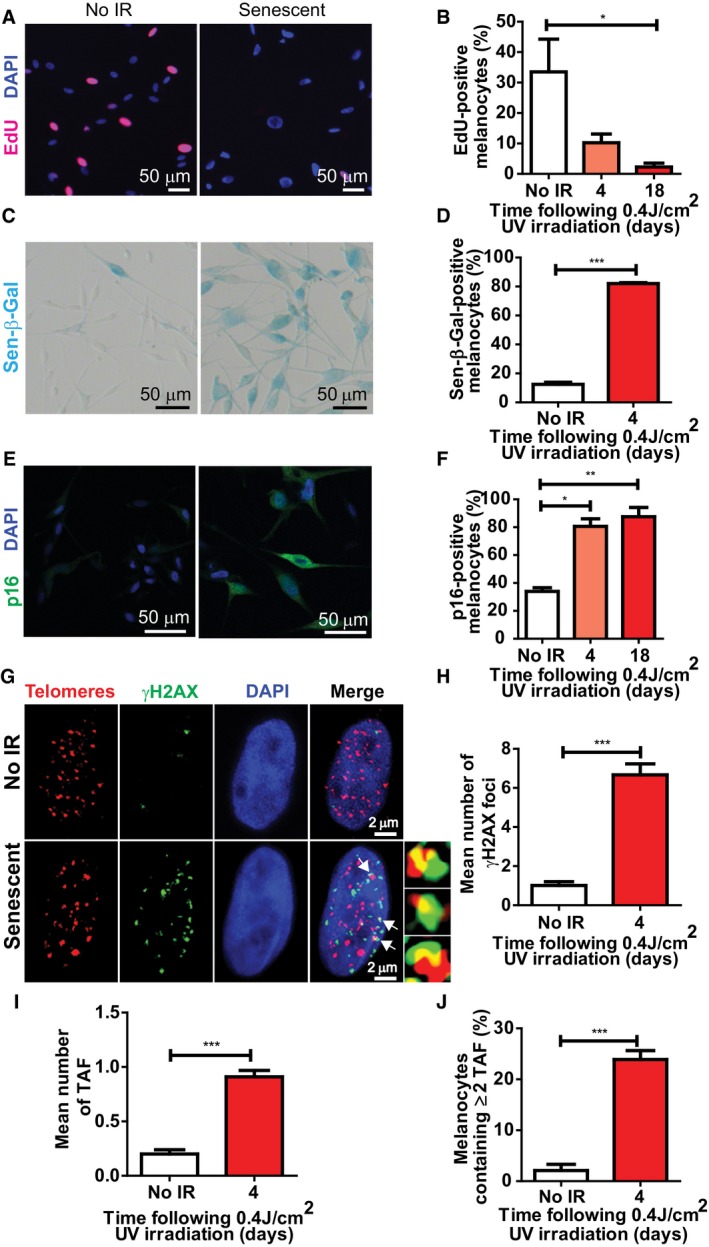
Given that melanocytes are in direct contact with keratinocytes in human skin, and that many age‐associated epidermal changes can be attributed to the loss of replicative capacity of keratinocytes (Gilhar et al, 2004), we next examined whether senescent melanocytes could also exert adverse paracrine effects in these cells. In order to investigate this, we utilised 3D living epidermal equivalents (also known as melanoderms), which consisted of either proliferating or senescent melanocytes and young keratinocytes cultured at a ratio of 1:10 melanocytes: keratinocytes. During a period of 10 days, these cells develop into a multi‐layered, highly differentiated human epidermis equivalent (Fig 2H). Similar models have been widely used to study age‐associated morphological skin changes (Adamus et al, 2014; Dos Santos et al, 2015; Diekmann et al, 2016; Lammermann et al, 2018). With the purpose of achieving a more physiologically relevant model, we used melanocytes which were induced to become senescent by repeated UVA+B exposure. Senescence was achieved by exposing melanocytes to UV irradiation for 5 consecutive days and was confirmed by reduced EdU incorporation, increased Sen‐β‐Gal activity and increased p16 expression 4 days following the last exposure (Fig EV4A–F). The percentage of proliferating melanocytes remained significantly lower at 18 days following UV exposure, whilst p16 expression was significantly higher compared to controls, indicating that repeated UVA+B exposure is sufficient to induce a stable proliferation arrest and melanocyte senescence in vitro (Fig EV4E and F). Similarly to X‐ray‐induced senescence, UV‐induced senescent melanocytes showed significantly increased numbers of γH2AX foci and TAF (Fig EV4G–J).
Figure EV4. Repeated UVA+B exposure induces senescence in human epidermal melanocytes in vitro. Melanocytes were exposed to 0.4 J/cm2 UVA and UVB radiation once a day for five consecutive days. Cells were harvested 4 and 18 days following the last exposure.
-
ARepresentative EdU immunofluorescence images of proliferating and irradiated melanocytes (18 days post‐IR) (red: EdU; blue: DAPI). Images were taken using a 20× objective.
-
BGraph showing the percentage of EdU‐positive melanocytes at the time points indicated. Data are shown as mean ± SEM of n = 3 independent experiments.
-
CRepresentative images of Sen‐β‐Gal (blue) of proliferating and UV‐irradiated melanocytes. Images were taken using a 20× objective.
-
DGraph showing the percentage of control and irradiated (4 days after the last exposure) Sen‐β‐Gal‐positive melanocytes. Data are shown as mean ± SEM of n = 3 independent experiments.
-
ERepresentative p16 immunofluorescence images of proliferating and senescent melanocytes (18 days following the last UV exposure). Images were taken using a 20× objective.
-
FGraph showing the percentage of p16‐positive melanocytes at the time points indicated following the last UV exposure. Data are shown as mean ± SEM of n = 3 independent experiments.
-
GRepresentative immuno‐FISH images of proliferating (top) and senescent (bottom) melanocytes. Telomeres are shown in red, γH2AX is depicted in green, and DAPI is shown in blue. Arrows indicate co‐localisation between γH2AX and telomeres, which are amplified on the right. Images are Z projections and were taken using a 63× oil objective.
-
H–JGraphs showing (H) mean number of γH2AX foci, (I) mean number of TAF, (J) percentage of melanocytes containing ≥ 2 TAF. Data are shown as mean ± SEM of n = 3 independent experiments.
To determine whether senescent melanocytes were capable of inducing paracrine telomere damage in keratinocytes, we performed immuno‐FISH in melanoderms which were obtained at 0, 10 and 21 days following epidermal development. We found that keratinocytes in melanoderms containing senescent melanocytes exhibited a significantly higher number of TAF when compared to those cultured with young melanocytes, both at 10 and 21 days post‐epidermal development (Fig 2I and J). Furthermore, melanoderms containing senescent melanocytes showed a significantly higher percentage of keratinocytes that contained 2 or more TAF, and this difference was significant at days 10 and 21 post‐development (Fig 2K).
Next, to investigate whether senescent melanocytes could also induce paracrine senescence in keratinocytes, we assessed expression of senescence markers in melanoderms. We found that melanoderms containing senescent melanocytes have a consistently lower proportion of proliferating keratinocytes, as demonstrated by a decreased percentage of Ki‐67‐positive keratinocytes in these models (Fig 2L). Consistent with decreased proliferation, we found a significant reduction in the total number of keratinocytes at day 21 in melanoderms containing senescent melanocytes (Fig 2M). Additionally, p16 expression was significantly increased in keratinocytes from melanoderms containing senescent melanocytes at all time points investigated (Fig 2N). Interestingly, we also observed that melanoderms containing senescent melanocytes have a significantly thinner epidermis at day 21 compared to their proliferating counterparts (Fig 2O and P) and this corresponded to a significantly decreased number of epidermal cell layers positive for keratin 10—a marker of keratinocyte differentiation (Appendix Fig S3A). Whilst numbers of senescent melanocytes remained unchanged, the total number of young melanocytes present in melanoderms doubled 21 days post‐epidermal development. However, given the relatively low number of melanocytes present in melanoderms, this cannot explain per se the differences in epidermal thickness (Appendix Fig S3B).
It is also possible that the observed changes in epidermal thickness induced by senescent melanocytes are affected by alterations in extracellular matrix composition. Recently, it was reported that the hemidesmosome component collagen XVII was significantly decreased during ageing and that its increased expression impacted positively on skin regeneration (Liu et al, 2019). Consistently, we found that collagen XVII expression was significantly reduced in melanoderms containing senescent melanocytes (Appendix Fig S3C and D). Together, these results suggest that senescent melanocytes induce paracrine telomere damage and senescence in surrounding keratinocytes. This in turn may compromise the proliferative potential of keratinocytes, resulting in age‐associated epidermal atrophy.
Paracrine telomere dysfunction is mediated by CXCR3 activation and increased ROS production in bystander cells
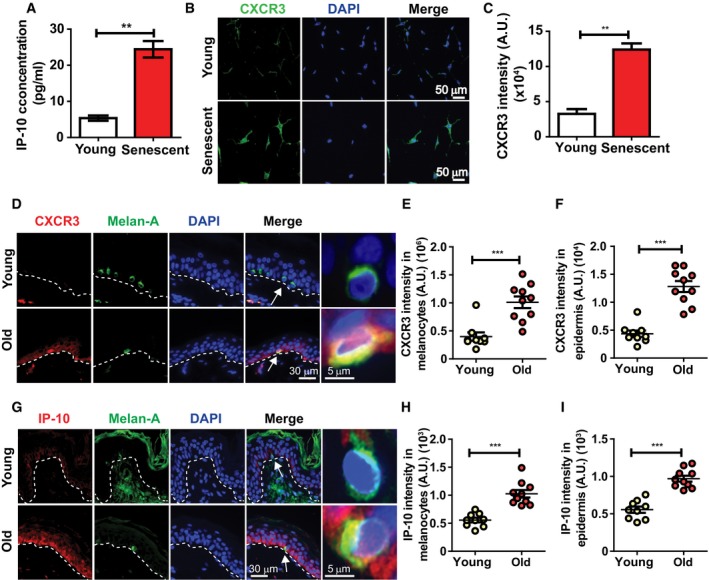
Following the observation that soluble factors secreted by senescent melanocytes can induce paracrine telomere dysfunction and senescence in neighbouring cells, we investigated whether specific components of the SASP are responsible for mediating this process. Cytokine array analysis done in CM obtained from X‐ray‐induced senescent melanocytes in vitro revealed that IP‐10 was one of the cytokines which was highly secreted by these cells (Fig 3A). We also observed that expression of CXCR3, a cell‐surface receptor for IP‐10, was up‐regulated in senescent melanocytes in vitro (Fig 3B and C), and chemical inhibition of CXCR3 was sufficient to reduce a number of senescence markers during X‐ray‐induced senescence (Fig EV5), suggesting that CXCR3 is involved in autocrine signalling important for the establishment of melanocyte senescence. Moreover, by performing immunofluorescence in the skin of young and older human donors, we also found that CXCR3 expression was significantly higher both in melanocytes specifically (Fig 3D and E) and, more generally, across the basal layer of the epidermis in older human skin (Fig 3D and F). Similarly, expression of IP‐10 was significantly increased in melanocytes (Fig 3G and H) and in the basal layer of older human skin (Fig 3G and I). Therefore, we hypothesised that IP‐10 secreted by senescent melanocytes could be involved in mediating the paracrine effects observed thus far.
Figure 3. Expression levels of CXCR3 and IP‐10 increase in senescent melanocytes in vitro and in human skin with age.
-
AGraph showing the concentration of IP‐10 secreted by senescent melanocytes in culture. Data are shown as mean ± SEM of n = 3 independent experiments.
-
BRepresentative immunofluorescence images of CXCR3 (green) in young and stress‐induced senescent melanocytes. Images were taken using a 20× objective.
-
CGraph showing CXCR3 fluorescence signal intensity in young and senescent melanocytes. Data are shown as mean ± SEM of n = 3 independent experiments.
-
DRepresentative immunofluorescence images of CXCR3 (red) in skin sections from young and older donors. Images were taken using a 20× objective. Arrows indicate melanocytes, which are amplified on the right.
-
E, FDot plots showing CXCR3 fluorescence intensity in (E) melanocytes specifically and (F) whole epidermis of young (n = 9) and older (n = 10) human skin for each individual donor. The horizontal line represents the mean for each group. Data are shown as mean ± SEM.
-
GRepresentative immunofluorescence images of IP‐10 (red) in skin sections from young and older donors. Images were taken using a 20× objective. Arrows indicate melanocytes, which are amplified on the right.
-
H, IDot plots showing IP‐10 fluorescence intensity in (H) melanocytes specifically and (I) whole epidermis of young (n = 9) and older (n = 10) human skin for each individual donor. The horizontal line represents the mean for each group. Data are shown as mean ± SEM.
Figure EV5. CXCR3 signalling is involved in the initiation of melanocyte senescence. Senescence was induced in melanocytes by 10 Gy X‐ray irradiation, and cells were treated with either AMG487 or vehicle DMSO (control) for 10 days.
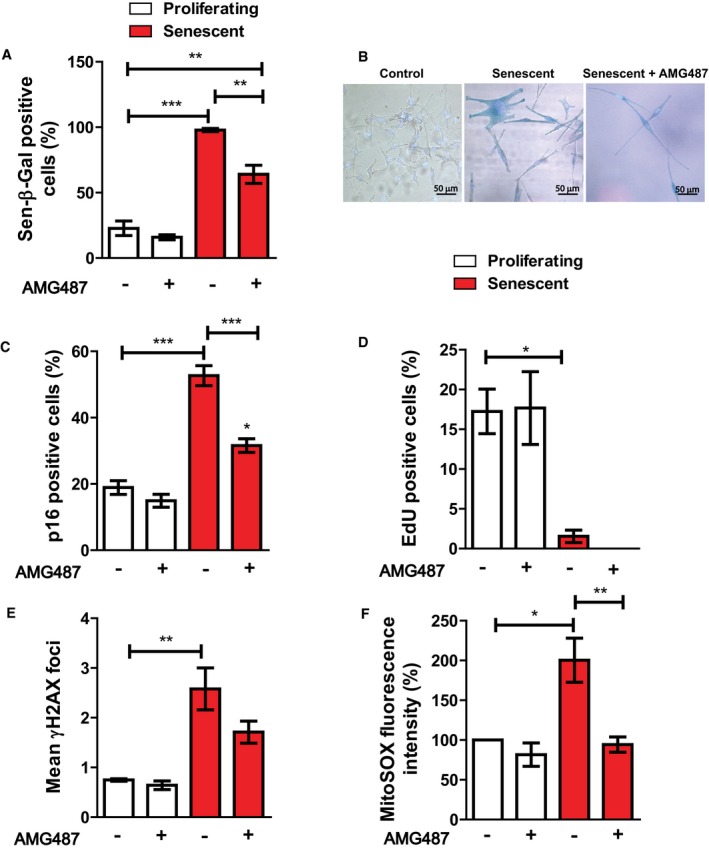
-
AGraph showing the percentage of Sen‐β‐Gal‐positive melanocytes at the conditions indicated (white: proliferating melanocytes; red: senescent melanocytes). Data are shown as ± SEM (n = 3 independent experiments).
-
BRepresentative Sen‐β‐Gal images of proliferating and senescent melanocytes with or without AMG487 treatment. Images were taken using a 20× objective.
-
C–FGraphs showing the percentage of (C) p16‐positive and (D) EdU‐positive melanocytes at the conditions indicated. (E) Mean number of γH2AX foci in proliferating and senescent melanocytes with or without AMG487 treatment. (F) MitoSOX fluorescence intensity of melanocytes at the conditions indicated. Values are a percentage fold change normalised to proliferating controls (untreated). ROS measurements were performed at 3 days following irradiation. Data are shown as mean ± SEM of N = 3 independent experiments.
To determine whether CXCR3 activation was necessary for paracrine telomere damage induction, dermal fibroblasts were cultured with either proliferating or senescent melanocyte CM, and were simultaneously treated with the CXCR3 inhibitor, AMG487, or with siRNAs against CXCR3, for 48 h (Fig 4A). In accordance with our previous observations, CM from senescent melanocytes induced a threefold increase in the mean number of TAF in fibroblasts, which was prevented by treatment with CXCR3 inhibitor AMG487 (Fig 4B). Similarly, knockdown of CXCR3 (Fig 4C) prevented induction of TAF by senescent melanocyte CM (Fig 4D).
Figure 4. Senescent melanocytes transmit paracrine telomere dysfunction to neighbouring cells via mechanisms involving IP‐10‐CXCR3‐ROS signalling.
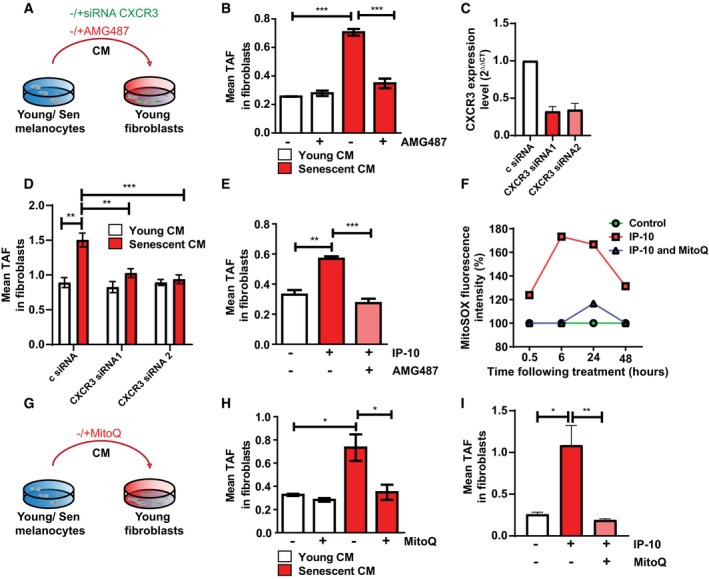
- Dermal fibroblasts were cultured with either young or senescent melanocyte conditioned medium. CXCR3 inhibition was achieved either by siRNA knockdown of CXCR3 or by treating fibroblasts with the CXCR3 inhibitor, AMG487.
- Graph showing the mean number of TAF in fibroblasts cultured with young or senescent melanocyte CM with or without AMG487. Data are shown as mean ± SEM of n = 3 independent experiments.
- Knockdown efficiency of two different siRNAs against CXCR3. Data are shown as mean ± SD of n = 2 independent experiments.
- Mean number of TAF in fibroblasts cultured with young or senescent melanocyte CM after CXCR3 knockdown. Data are shown as mean ± SEM of n = 3 independent experiments.
- Mean number of TAF in fibroblasts cultured with IP‐10 with or without AMG487. Data are shown as mean ± SEM of n = 3 independent experiments.
- Graph showing MitoSOX fluorescence intensity of fibroblasts treated with IP‐10 with or without MitoQ at the time points indicated. Values are a percentage fold change normalised to controls. Graph is representative of one out of n = 3 independent experiments.
- Dermal fibroblasts were cultured with either young or senescent melanocyte conditioned medium with or without MitoQ.
- Mean number of TAF in fibroblasts cultured with young or senescent melanocyte CM with or without MitoQ. Data are shown as mean ± SEM of n = 3 independent experiments.
- Mean number of TAF in fibroblasts treated with IP‐10 with or without MitoQ. Data are shown as mean ± SEM of n = 3 independent experiments.
Next, to assess whether telomere dysfunction could be triggered as a consequence of direct CXCR3 stimulation by IP‐10, we treated dermal fibroblasts in culture with human recombinant IP‐10 for 48 h. We found that IP‐10 effectively induces TAF in fibroblasts, and this is completely abolished by AMG487 treatment (Fig 4E), suggesting that IP‐10 secreted by senescent melanocytes activates CXCR3 signalling in surrounding cells, in turn leading to telomere dysfunction.
Mechanistically, it is unclear how CXCR3 activation by IP‐10 contributes to paracrine telomere dysfunction. It is known that oxidative stress is an important contributor to activation of a DDR, with telomeres being particularly sensitive to imbalances in ROS homeostasis (Kruk et al, 1995; Petersen et al, 1998; Rochette & Brash, 2010). In fact, mitochondria are a major source of ROS generation and have been implicated as key components for the generation and replenishment of DNA damage foci, an important effector of senescence (Passos et al, 2007; Correia‐Melo et al, 2016, 2018). Moreover, stimulation of the CXCR3 receptor has been shown to increase oxidative stress in human kidney cells (Bek et al, 2003). Therefore, we hypothesised that CXCR3 activation in bystander cells could lead to an increase in intracellular ROS, thus generating TAF. To that end, we treated dermal fibroblasts with human recombinant IP‐10 and measured mitochondrial ROS production using MitoSOX fluorescent dye. We found that IP‐10 triggered a significant increase in MitoSOX fluorescence intensity, which peaked at 6 h and was suppressed by treatment with the mitochondria‐targeted antioxidant, MitoQ (Fig 4F and Appendix Fig S4A–D). To assess whether paracrine TAF induction by senescent melanocytes was mediated by increased mitochondrial ROS in bystander cells, we next treated dermal fibroblasts with CM from either young or senescent melanocytes in the presence of MitoQ (Fig 4G). Consistent with our previous observations, immuno‐FISH revealed that senescent melanocyte CM triggered a significant increase in the mean number of TAF in fibroblasts, which was prevented by treatment with MitoQ (Fig 4H). Similarly, IP‐10‐induced telomere dysfunction was also abolished by treating cells with MitoQ (Fig 4I). Altogether, these results suggest that CXCR3 activation by IP‐10 leads to an increase in mitochondrial ROS generation, which in turn induces telomere‐associated foci in neighbouring cells.
The signalling pathways that contribute to ROS production downstream of CXCR3 activation have not been fully elucidated. It has been shown that CXCR3 signalling leads to activation of the PI3K/Akt signalling pathway in human airway epithelial cells, mediating chemotaxis (Bonacchi et al, 2001; Shahabuddin et al, 2006). In fact, Akt has been involved in signalling cascades that converge signals from the DDR into PGC‐1β‐dependent mitochondrial biogenesis, contributing to increased ROS generation (Correia‐Melo et al, 2016). Furthermore, ERK phosphorylation has also been reported to occur as result of CXCR3 activation (Xia et al, 2000; Shahabuddin et al, 2006; Jenkins et al, 2015), although the links between the MEK/ERK pathway and ROS production are less clear. Indeed, we observed an increase in both Akt and ERK(1/2) phosphorylation upon treating dermal fibroblasts with IP‐10, which was prevented by inhibiting CXCR3 with AMG487 (Appendix Fig S4E).
Clearance of senescent melanocytes rescues epidermal atrophy in 3D epidermal equivalents
Epidermal atrophy is feature of ageing skin which is normally attributed to a decrease in the number of epidermal cells as well as reduced basal keratinocyte proliferation (Gilhar et al, 2004; Rinnerthaler et al, 2015). We have shown that senescent melanocytes can impair keratinocyte proliferation and contribute to epidermal atrophy in 3D epidermal equivalents. In order to establish a causal relationship between melanocyte senescence and epidermal atrophy, we treated melanoderms containing senescent melanocytes with ABT‐737, a small‐molecule inhibitor of the BCL‐2 family of anti‐apoptotic proteins, which has been shown to induce apoptosis preferentially in senescent cells (Yosef et al, 2016). Melanoderms were treated with ABT‐737 upon establishment of an epidermal stratum corneum (which occurs 10 days after co‐culture of melanocytes and keratinocytes, here referred to as day 0) for 48 h, and the tissues were cultured without any treatment thereafter (Fig 5A). Such treatment effectively eliminated senescent melanocytes from melanoderms, as shown by loss of p16‐positive melanocytes from these tissues (Fig 5B) and absence of Melan‐A expression (Appendix Fig S5A). Clearance of senescent melanocytes was sufficient to prevent significant TAF induction in keratinocytes (Fig 5C and D). Moreover, elimination of senescent melanocytes resulted in a decrease in the percentage of p16‐positive keratinocytes, which was comparable to control levels (Fig 5E and F). Therefore, these data implicate senescent melanocytes as causal in the induction of paracrine telomere damage and senescence in neighbouring epidermal cells. Consistent with a role for senescent melanocytes in contributing to age‐associated skin changes, we found that ABT‐737 treatment rescued the epidermal atrophy induced by senescent melanocytes (Fig 5G and H) and restored keratinocyte numbers (Appendix Fig S5B).
Figure 5. Clearance of senescent melanocytes or reducing mitochondrial ROS production rescues epidermal atrophy in 3D human epidermal equivalents.
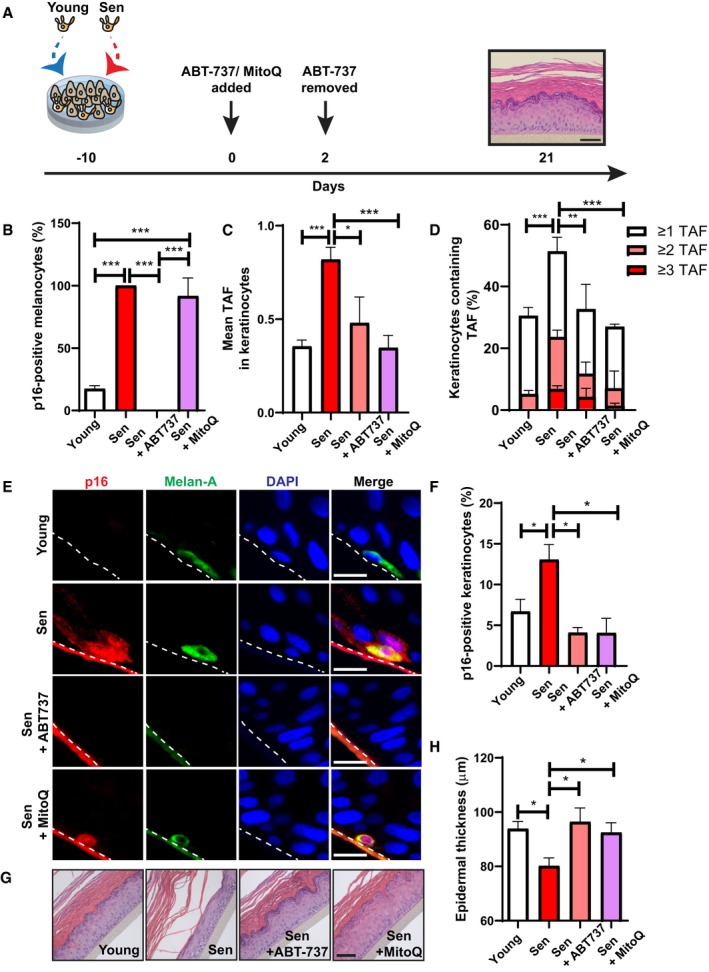
-
AMelanoderms comprised young keratinocytes with either young or senescent melanocytes. ABT737 or MitoQ was added at day 0 (day of full epidermal differentiation). ABT737 was removed at day 2, and melanoderms were cultured under normal culture conditions for the remainder of the experiment. MitoQ was refreshed every 2 days until the end of the experiment at day 21. Tissues were analysed at day 21. Scale bar is 50 μm.
-
BGraph showing the percentage of p16‐positive melanocytes in melanoderms with or without ABT737 treatment. Data are shown as mean ± SEM of n = 4 (young) and n = 3 (senescent control and treated) melanoderms.
-
C, DGraphs showing (C) the mean number of TAF in keratinocytes and (D) the percentage of keratinocytes containing ≥ 1, ≥ 2 and ≥ 3 TAF in melanoderms in the conditions indicated. Data are shown as mean ± SEM of n = 3–6 melanoderms (statistical significance is indicated for ≥ 2 TAF).
-
ERepresentative immunofluorescence images of p16 (red) in melanoderms containing young or senescent melanocytes with or without ABT737 or MitoQ treatment. Images were taken using a 20× objective. Scale bar = 50 μm.
-
FGraph showing the percentage of p16‐positive keratinocytes in melanoderms in the conditions indicated. Data are shown as mean ± SEM of n = 3–7 melanoderms.
-
GRepresentative H&E images showing epidermal thickness of melanoderms containing young or senescent melanocytes with and without ABT737 or MitoQ at 21 days following epidermal development. Images were taken using a 20× objective. Scale bar = 50 μm.
-
HGraph showing epidermal thickness of melanoderms containing either young or senescent melanocytes with or without ABT737 or MitoQ at the time points indicated. Data are shown as mean ± SEM of n = 3–8 melanoderms.
Given that our previous data showed that paracrine telomere damage is mediated by mitochondrial ROS, melanoderms were also treated with the mitochondria‐targeted antioxidant, MitoQ. Unlike ABT737, MitoQ did not affect the number of p16‐positive melanocytes following treatment (Fig 5B). Consistent with our previous results, MitoQ prevented paracrine telomere damage induction by senescent melanocytes (Fig 5C and D), and it also inhibited the accumulation of p16‐positive keratinocytes in melanoderms containing senescent melanocytes (Fig 5E and F). Furthermore, a rescue in epidermal thickness was also observed with MitoQ treatment (Fig 5G and H). These data support our hypothesis that the paracrine effects of senescent melanocytes are mediated by mitochondrial ROS.
Discussion
Senescent cells have been shown to accumulate in human skin with age (Dimri et al, 1995; Ressler et al, 2006), and evidence suggests that accumulation of these cells contributes to age‐associated skin changes by compromising regeneration and structure (Adamus et al, 2014; Velarde et al, 2015). However, the majority of human studies so far have focused on dermal fibroblasts, whilst the contribution of senescent melanocytes to the skin ageing process remains poorly understood.
We have shown that p16 expression is significantly increased in melanocytes in the epidermis of older donors, in agreement with previous reports (Waaijer et al, 2012b, 2016; Pawlikowski et al, 2013). In the epidermis, we found that p16 expression could only be detected in cells expressing melanocyte markers, indicating that melanocytes represent the main population of p16‐positive senescent cells. This observation prompted our investigation into the mechanisms by which melanocytes acquire a senescent phenotype, since differentiated melanocytes have an extremely low proliferative capacity in vivo and are thus unlikely to undergo sufficient telomere attrition to induce replicative senescence (Jimbow et al, 1975). It is possible that in post‐mitotic and/or slow‐dividing cells, such as melanocytes, senescence occurs as a result of length‐independent telomere damage caused by oxidative stress (Victorelli & Passos, 2017). Consistent with this hypothesis, we observed increased co‐localisation between γH2A.X and telomeres in aged melanocytes, which denotes telomere dysfunction, without significant shortening. The accumulation of DDR proteins at telomeres that are not critically short has been previously reported in non‐dividing cells, such as neurons (Fumagalli et al, 2012; Ogrodnik et al, 2019), cardiomyocytes (Anderson et al, 2019) and hepatocytes (Hewitt et al, 2012). Consistent with a role for the DDR in melanocyte senescence and in accordance with our results, a previous study reported an age‐dependent increase in 53BP1‐positive melanocytes (Suram et al, 2012). We should note that for technical reasons, we have not been able to determine whether p16‐positive melanocytes are also positive for TAF.
It has been proposed that long telomeres may provide a more abundant target for DNA damage to occur (Fumagalli et al, 2012). Another mechanism by which telomeres can activate a DNA damage response irrespectively of length is via the loss of shelterin components (Takai et al, 2003). Given that our data suggest that telomeres do not shorten significantly in senescent melanocytes in vivo, it is unlikely that telomere uncapping would occur as a result of extensive loss of telomeric repeats. However, it has been shown that oxidative damage at telomeres can disrupt the binding of certain shelterin proteins (Opresko et al, 2005), possibly providing a length‐independent mechanism for telomere uncapping. Our data suggest that telomere length is not a limiting factor in melanocyte senescence; however, whilst interphase telomere FISH is the only method that will allow us to obtain information regarding telomere length in specific cells within a tissue, the methodology does not allow the recognition of telomere‐free ends. Thus, one of the limitations of our study is that we may be failing to detect critically short telomeres. Additionally, it is also possible that clusters of telomere FISH signals may contribute to artefacts in our analysis as previously discussed (Anderson et al, 2019); however, we did not find any indication that the presence of telomere clusters occurs preferentially in old melanocytes.
Evidence suggests that persistent DDR signalling at telomeres plays an important role in the establishment of senescence. Studies have shown that DNA damage at telomeres is less efficiently repaired compared to non‐telomeric damage (Kruk et al, 1995; Fumagalli et al, 2012; Hewitt et al, 2012; Anderson et al, 2019). Consistently, live‐cell imaging studies have demonstrated that the majority of long‐lived DNA damage foci co‐localise with telomeres in stress‐induced senescence (Hewitt et al, 2012). Such irreparable DNA damage generates persistent DDR signalling at telomeres, which is believed to be important for the initiation and maintenance of the senescent state (Fumagalli et al, 2012; Hewitt et al, 2012).
Skin ageing is accompanied by many morphological changes, including loss of epidermal curvature (Waaijer et al, 2012a). In accordance with this, we observed significant age‐dependent epidermal flattening in the older donor cohort used in this study. Since accumulation of senescent cells has been shown to contribute to tissue dysfunction with age (Baker et al, 2011, 2016; Xu et al, 2015), we investigated whether increased TAF in melanocytes was associated with age‐related skin characteristics. We observed a highly significant association between increased TAF in melanocytes and flattening of the epidermal–dermal junction (EDJ), suggesting that senescent melanocytes contribute to this skin ageing phenotype. Flattening of the EDJ occurs primarily as a result of the retraction of the rete ridges, which consist of epidermal invaginations into the dermis. Physiologically, a flattened dermal–epidermal interface means that the epidermis is less resistant to shearing forces and reduces the exchange surface between the epidermis and dermis, compromising nutrient supply to the epidermis. This has been suggested to contribute to reduced keratinocyte proliferation with ageing (Briggaman & Wheeler, 1975; Lavker, 1979; Lavker et al, 1989; Gilhar et al, 2004). Although it is not known whether flattening of the EDJ is a direct consequence of senescent melanocyte accumulation, our results implicate TAF as a potential marker for epidermal flattening that occurs during skin ageing.
Senescent cells have been shown to induce paracrine DNA damage and senescence in healthy neighbouring cells via mechanisms involving secretion of SASP factors and increased ROS (Nelson et al, 2012; Acosta et al, 2013; Razdan et al, 2018). In agreement with this, our data showed that keratinocytes surrounding senescent melanocytes also exhibited significantly more telomere damage, providing evidence for a possible bystander effect of senescent melanocytes in human skin in vivo. Moreover, we showed that conditioned media from senescent melanocytes in vitro induced TAF in dermal fibroblasts, suggesting that paracrine TAF induction is mediated by soluble factors released by senescence melanocytes. Senescent melanocytes were also shown to induce paracrine telomere dysfunction in surrounding keratinocytes in 3D epidermal equivalents. Dysfunctional telomeres provide a source of persistent DDR that limits the replicative capacity of cells (Fumagalli et al, 2012, 2014; Hewitt et al, 2012). In support of this, we observed that senescent melanocytes impaired replication of keratinocytes in 3D epidermal equivalents, resulting in epidermal atrophy, which is a skin ageing phenotype. Importantly, epidermal thickness was rescued by eliminating senescent melanocytes using the senolytic drug ABT737 (Yosef et al, 2016), suggesting that senescent melanocytes directly contribute to epidermal atrophy through limiting proliferation of keratinocytes. Previous studies have shown that elimination of senescent cells by ABT737 rescued proliferation of hair follicle stem cells in a transgenic mouse model where senescence is specifically induced in the basal layer of the epidermis (Yosef et al, 2016). Given that proliferation of stem and progenitor cells is also crucial for epidermal regeneration and homeostasis (Blanpain & Fuchs, 2009), it would also be important to assess the bystander effects of senescent melanocytes on epidermal stem cells. However, since our in vitro models do not comprise stem cells, we postulate that keratinocyte cell‐cycle arrest triggered by senescent melanocytes is sufficient to induce epidermal atrophy. We acknowledge that our results using melanoderms have a number of limitations, such as the lack of dermis, subcutaneous fat and a number of cell types, including immune cells, which have been previously implicated in the skin ageing process. Nonetheless, the model used is valuable since it allows the investigation of the specific interactions between senescent melanocytes and keratinocytes as well as test pharmacological interventions without any additional confounding factors.
We demonstrate that paracrine TAF induction is mediated by mechanisms involving the cell‐surface receptor CXCR3. Interestingly, we found that IP‐10 and its receptor, CXCR3, were up‐regulated both in senescent melanocytes in vitro and in human skin with age. Since telomeres are particularly susceptible to oxidative stress due to their high content of guanine triplets (Oikawa & Kawanishi, 1999), we hypothesised that paracrine TAF was mediated by increased intracellular ROS. Indeed, our results show that IP‐10 released by senescent melanocytes activates CXCR3 signalling in surrounding cells, triggering an increase in mitochondrial ROS production and consequently inducing telomere dysfunction. Consistent with our data, previous studies have shown that stimulation of the CXCR3 receptor results in increased ROS generation (Bek et al, 2003). Moreover, others have also demonstrated that SASP components, notably TGF‐β1, induce paracrine telomere dysfunction in a ROS‐dependent manner (Razdan et al, 2018). Although the mechanisms that result in enhanced mitochondrial ROS production downstream of CXCR3 signalling have not been fully elucidated, we and others have shown that Akt phosphorylation occurs as a result of CXCR3 activation (Bonacchi et al, 2001; Shahabuddin et al, 2006). In fact, Akt has been involved in signalling cascades that enhance mitochondrial ROS generation in senescence (Correia‐Melo et al, 2016).
Our results support a role for mitochondrial ROS in the development of skin ageing phenotypes, as epidermal atrophy induced by senescent melanocytes is rescued by MitoQ in 3D epidermal equivalents. Increased ROS generation has been shown to contribute to skin ageing in laboratory mice. Deletion of Sod2, a mitochondrial antioxidant enzyme, increased mitochondrial ROS and induced cellular senescence in mouse skin, resulting in epidermal atrophy (Velarde et al, 2012).
In summary, our results indicate that senescent melanocytes accumulate in human skin with age. We show that senescent melanocytes induce paracrine telomere damage in surrounding cells. Moreover, we demonstrate that senescent melanocytes contribute causally to epidermal atrophy in 3D epidermal equivalents, likely by inhibiting basal keratinocyte proliferation, and thus impairing epidermal turnover.
Materials and Methods
Cell culture and treatments
Primary human lightly pigmented epidermal neonatal melanocytes were obtained from Thermo Fisher Scientific (HEMn‐LP, C0025C) and Lonza (NHEM‐Neo, CC‐2504). Melanocytes were cultured in MBM‐4 basal medium supplemented with MGM‐4 SingleQuot Supplements and Growth Factors (Lonza), and maintained at atmospheric conditions (air plus 5% CO2). Primary human dermal fibroblasts were obtained from Lonza (NHDF, CC‐2509) and were cultured in DMEM (Sigma) supplemented with 10% foetal bovine serum (FBS), 2 mM l‐glutamine and penicillin/streptomycin 100 U/ml, and maintained at atmospheric conditions (air plus 5% CO2).
Stress‐induced senescence was induced in melanocytes by either 10 Gy X‐ray irradiation or by 5 consecutive daily exposures to 0.4 J/cm2 of UVA and UVB irradiation. The UVA and UVB sources consisted of a Dr Honle SOL 500s lamp (Dr. Honle AG UV Technology) equipped with a UVA+UVB filter glass (Eurobond Adhesives), and emitting in the spectral range of 320–400 nm for UVA and 295–315 nm for UVB. No residual UVC is emitted.
To assess paracrine telomere damage induction, conditioned medium from control (proliferating) and stress‐induced senescent melanocytes was collected 11 days following X‐ray irradiation. CM was centrifuged at 481 g for 5 min to eliminate any cell debris, and it was then added to fibroblasts at a ratio of 1:1 DMEM: conditioned medium. Fibroblasts were cultured in this mixture of DMEM and CM for the duration of the experiment. For longer‐term culture, medium was replenished every 3 days.
For CXCR3 inhibition, X‐ray‐irradiated melanocytes were treated with 10 μM AMG487 (Tocris) immediately following irradiation, and treatment was refreshed every 2 days for 10 days. Dermal fibroblasts were also treated with 10 μM AMG487 for 48 h in the presence of melanocyte CM, as described above.
For scavenging of mitochondrial ROS, dermal fibroblasts in melanocyte CM were treated with 20 nM MitoQ (provided and developed by Prof Mike Murphy) for the duration of the experiment. Prior to the addition of melanocyte CM, fibroblasts were pre‐treated with MitoQ for 6 h in normal culture conditions.
Recombinant human IP‐10 protein (R&D System) was added to dermal fibroblasts at a concentration of 200 ng/ml for the duration of each experiment.
For EdU incorporation assays, cells were incubated in 10 μM EdU for 24 h in normal growth medium. All subsequent EdU detection steps were carried out using Click‐iT EdU Alexa Fluor Imaging Kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
Knockdown by small interfering RNA (siRNA)
Dermal fibroblasts were transiently transfected with siRNAs using HiPerFect Transfection Reagent (Qiagen, 301705). Cells were transfected with 20 nM siRNA (negative control siRNA, Qiagen, SI03650318; Hs_CXCR3_2 FlexiTube siRNA, Qiagen, SI00357336; Hs_CXCR3_3 FlexiTube siRNA, Qiagen, SI00357343) following the HiPerFect Transfection Reagent manufacturer's protocol. Cells were transfected 24 h prior to addition of CM and harvested 72 h after transfection (2 days after addition of CM). siRNA transfection efficiency was assessed by qPCR.
Human tissue collection and ethics
Young subjects consisted of white British subjects. Skin punch biopsies of 4 mm were obtained from the sun‐protected site of the inner upper arm of each donor and were taken by a qualified dermatologist in 2007. The study was approved by the Unilever Colworth Ethics Committee (UCR2007‐07), and subjects gave informed written consent prior to inclusion in this study. Data from 20 randomly selected Colworth donors were used in this study.
Skin samples from older subjects were obtained from the Leiden Longevity Study (LLS). Study design and methodologies of the LLS have been previously described (Schoenmaker et al, 2006). Briefly, the LLS consisted of men and women aged above 89–91, respectively, with at least one sibling who matches the same age criterion. The offspring of either long‐lived sibling and the partners of the offspring were recruited to participate in the study. Skin punch biopsies of 4 mm were taken from the sun‐protected part of the inner upper arm of each donor. The study protocol was approved by the Medical Ethics Committee of the Leiden University Medical Centre (following the Declaration of Helsinki), and participants gave informed written consent. Skin biopsies from 41 randomly selected LLS participants were used in this study.
Skin biopsies were formalin‐fixed, paraffin‐embedded (FFPE) and cut into 4‐μm sections for subsequent immunohistochemical analysis.
Human epidermal equivalents (melanoderms)
Melanoderms were produced by MatTek Corporation (Ashland, MA) and contained neonatal human keratinocytes and either proliferating or UV‐induced senescent melanocytes. Briefly, melanocytes and keratinocytes are co‐cultured for a period of 10 days before developing into a multi‐layered, highly differentiated model of human epidermis (also referred to as “day 0”). For the purposes of this study, tissues were collected either 0, 10 and 21 days post‐epidermal development or at days 7 and 21 post‐epidermal development. Tissues were FFPE and cut into 4‐μm sections for subsequent immunohistochemical analysis.
Melanoderms were treated with 2.5 μM ABT‐737 at day 0 (post‐epidermal development) for 48 h. Treatment was then removed, and tissues were subsequently cultured without the drug for the remainder of the experiment. MitoQ (20 nM) was added to tissues at day 0 (post‐development), and treatment was refreshed every 48 h for the duration of the experiment.
Flow cytometry
Analysis of mitochondrial ROS production was carried out by measuring MitoSOX fluorescence intensity by flow cytometry. MitoSOX is a red fluorescent dye, which is used as a mitochondrial superoxide indicator. MitoSOX permeates the membrane of live cells and is selectively targeted to mitochondria, where it is oxidised by superoxide anions, exhibiting red fluorescence.
Cells were harvested and centrifuged at 1,600 rpm for 2 min. Supernatant was discarded, and approximately 2.5 × 105 live cells were incubated with 5 μM MitoSOX Red (Invitrogen) in serum‐free culture medium for 10 min at 37°C in the dark. Following another centrifugation at 1,600 rpm for 2 min, supernatant was discarded, and the cell pellet was resuspended in 2 ml of serum‐free culture medium. MitoSOX median fluorescence intensity was determined by flow cytometry using the red fluorescence channel (FL3 channel). An FSC/SSC dot plot was used to define the population of live cells and apoptotic cells, and debris was excluded by gating.
Quantitative real‐time PCR
RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, 74106). Complementary DNAs were synthesised using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher, 4368814) following the manufacturer's instructions. Quantitative real‐time PCR was carried out using Power SYBR® Green PCR Master Mix (Invitrogen, 4367659) in a C100TM Thermal Cycler, CFX96TM Real‐Time PCR System (Bio‐Rad) and Bio‐Rad CFX Manager software. Gene expression was normalised to 18S. Primers used were as follows: Hs_CXCR3_va.1_SG (Qiagen, QT02410212) and Hs_RRN18S_1_SG (Qiagen, QT00199367).
Immunofluorescence
Cells grown on coverslips were fixed in 2% paraformaldehyde in PBS for 5 min. Cells were then permeabilised in PBG‐Triton (PBS, 0.4% Fish‐skin gelatin, 0.5% BSA, 0.5% Triton X‐100) for 45 min and incubated with primary antibody overnight at 4°C. Following PBS washes, cells were incubated with secondary antibody for 45 min and mounted onto glass microscope slides with ProLong Gold Antifade Mountant with DAPI (Invitrogen).
Primary antibodies used were as follows: mouse monoclonal anti‐p16 (dilution as provided by manufacturer, 9511; CINTec Histology, Roche) and mouse monoclonal anti‐CXCR3 (1:400, ab64714; Abcam).
Secondary antibodies used were as follows: goat anti‐mouse Fluorescein‐conjugated secondary antibody Alexa Fluor 488 (1:4,000, A21042; Invitrogen) and goat anti‐mouse Fluorescein‐conjugated secondary antibody Alexa Fluor 594 (1:4,000, A11005; Invitrogen).
Immunohistochemistry
For FFPE tissues, sections were deparaffinised in 100% Histoclear and hydrated in 100, 90 and 70% ethanol (twice for 5 min each), and incubated twice for 5 min in distilled water. Antigen retrieval was performed by incubating sections in 0.01 M citrate buffer (pH 6.0) and heated until boiling for 10 min. Sections were allowed to cool down to room temperature followed by two washes in distilled water for 5 min. Next, sections were blocked in normal goat serum (1:60) in BSA/PBS for 30 min and incubated with primary antibody overnight at 4°C. Following three PBS washes, sections were then incubated with secondary antibody for 1 h, followed by PBS washes and mounted using ProLong Gold Antifade Mountant with DAPI (Invitrogen).
Primary antibodies used were as follows: rabbit polyclonal anti‐Ki67 (1:250, ab15580; Abcam), rabbit polyclonal anti‐SIRT1 (1:100, ab13749; Abcam), mouse monoclonal anti‐Melan‐A (1:20, M7196; Dako), rabbit polyclonal anti‐Melan‐A (1:50, sc‐28871; Santa Cruz), mouse monoclonal anti‐p16 (dilution as provided by manufacturer, 9511; CINTec Histology, Roche), mouse monoclonal anti‐CXCR3 (1:400, ab64714; Abcam), rabbit polyclonal anti‐IP10 (1:200, ab9807; Abcam), rabbit polyclonal anti‐53BP1 (1:200, 4937S, Cell Signaling), rabbit polyclonal anti‐HMGB1 (1:200, 3935S, Cell Signaling), rabbit monoclonal anti‐p21 (1:200, 2947S, Cell Signaling), mouse monoclonal anti‐cytokeratin 10 (1:100, ab9026, Abcam), rabbit monoclonal anti‐collagen XVII (1:100, ab186415, Abcam) and rabbit monoclonal anti‐collagen XVII (1:100, ab186415, Abcam).
Secondary antibodies used were as follows: goat anti‐rabbit Fluorescein‐conjugated secondary antibody Alexa Fluor 488 (1:1,000, A11008; Invitrogen), goat anti‐rabbit Fluorescein‐conjugated secondary antibody Alexa Fluor 594 (1:1,000, A11012; Invitrogen), goat anti‐mouse Fluorescein‐conjugated secondary antibody Alexa Fluor 488 (1:1,000, A21042; Invitrogen), goat anti‐mouse Fluorescein‐conjugated secondary antibody Alexa Fluor 594 (1:1,000, A44005; Invitrogen) and goat anti‐mouse Fluorescein‐conjugated secondary antibody Alexa Fluor 647 (1:1,000, A21235; Invitrogen).
Immuno‐FISH and Q‐FISH
For cells grown on coverslips, immunocytochemistry was performed as described above using mouse monoclonal anti‐γH2AX (1:200, 05‐636; Millipore). Following secondary antibody incubation, cells were fixed in methanol: acetic acid (3:1 ratio) for 30 min followed by dehydration in graded cold ethanol solutions (70, 90, 100%) for 2 min each. Cells were incubated in PBS at 37°C for 5 min and fixed in 4% paraformaldehyde at 37°C for 2 min. Following a PBS wash, cells were dehydrated again with cold ethanol solutions (70, 90, 100%) for 2 min each and left to air‐dry. Coverslips were then placed onto glass slides containing 10 μl of PNA hybridisation mix (70% deionised formamide (Sigma), 25 mM MgCl2, 1 M Tris pH 7.2, 5% blocking reagent (Roche) containing 2.5 μg/ml Cy‐3‐labelled telomere‐specific (CCCTAA) peptide nucleic acid probe (PANAGENE)), and samples were denatured for 10 min at 80°C. Coverslips were incubated in a humidified chamber protected from light for 2 h at room temperature to allow hybridisation to occur. Cells were then washed with 70% formamide in 2× SSC (3 × 10 min each) followed by three washes in 0.05% TBS‐Tween‐20 for 5 min. Cells were then dehydrated in graded cold ethanol solutions (70, 90, 100%) and left to air‐dry before they were mounted onto glass microscope slides with ProLong Gold Antifade Mountant with DAPI (Invitrogen). Cells were images using in‐depth Z stacking (a minimum of 35 optical slices with 63× objective).
For FFPE tissues, immunohistochemistry was carried out as described above. Following an overnight incubation with rabbit monoclonal anti‐γH2AX (1:400, 9718; Cell Signalling), sections were then incubated with a goat anti‐rabbit biotinylated secondary antibody (1:200, PK‐6101; Vector Labs) for 30 min at room temperature. Following three PBS washes, tissues were incubated with fluorescein avidin DCS (1:500, A‐2011; Vector Labs) for another 30 min at room temperature. Sections were then washed three times in PBS and cross‐linked by incubation in 4% paraformaldehyde in PBS for 20 min. Sections were washed in PBS three times and then dehydrated in graded cold ethanol solutions (70, 90, 100%) for 3 min each. Tissues were then allowed to air‐dry prior to being denatured in 10 μl of hybridisation mix (as described above) for 10 min at 80°C and then incubated for 2 h at room temperature in a dark humidified chamber to allow hybridisation to occur. Sections were washed in 70% formamide in 2× SCC for 10 min, followed by a wash in 2× SSC for 10 min, and a PBS wash for 10 min. Tissues were then mounted using ProLong Gold Antifade Mountant with DAPI (Invitrogen). Sections were imaged using in‐depth Z stacking (a minimum of 40 optical slices with 63× objective) followed by Huygens (SVI) deconvolution.
Quantitative FISH (Q‐FISH) analysis of telomere FISH intensity was performed on fixed cells and FFPE tissue following immuno‐FISH staining (as described above). Image analysis was performed using ImageJ software, where Z projections were created for each individual image and the oval tool was used to measure the integrated density of each individual telomere signal.
Senescence‐associated β‐galactosidase activity assay
Cells grown on coverslips were fixed in 0.2% glutaraldehyde in 2% PFA in PBS for 5 min. Cells were incubated in Sen‐β‐Gal staining solution (150 mM sodium chloride, 2 mM magnesium chloride, 40 mM citric acid, 12 mM sodium phosphate pH 6.0, 1 mg/ml 5‐bromo‐4‐chloro‐3‐inolyl‐β‐d‐galactosidase (X‐Gal), 5 mM potassium hexacyanoferrate (II) trihydrate, 5 mM potassium hexacyanoferrate (III) trihydrate) (pH 6.0) overnight at 37°C in the dark, followed by three PBS washes for 5 min each, and were then mounted onto glass microscope slides using ProLong Gold Antifade Mountant with DAPI (Invitrogen).
Luminex assay
Detection of cytokines and chemokines in cell culture supernatants was performed using the commercially available kit MILLIPLEX Human Cytokine/Chemokine Magnetic Bead Panel—Premixed 41 Plex (Merck Millipore, HCYTMAG‐60K‐PX41). Protocol was followed according to the manufacturer's instructions. Assays were analysed using a Luminex 200 reader.
Western blotting analysis
Cells (0.5 × 106) were lysed in 50 μl of lysis buffer (150 mM NaCl, 1% NP40, 0.5% NaDoC, 0.1% SDS, 50 mM Tris pH 7.4, 1× phosphatase and protease inhibitors cocktail in H2O). Equal amounts of protein (30 μg) from each sample were resolved on a 10% SDS–PAGE gel. Proteins were blotted onto a 0.45 μm polyvinylidene difluoride (PVDF) membrane (Millipore) using Trans‐Blot SD Semi‐Dry Transfer Cells (Bio‐Rad). Following blocking with PBS‐Tween blocking buffer (5% milk, 0.05% Tween‐20 in PBS), the membranes were incubated with primary antibodies at 4°C overnight. The next day, membranes were washed with H2O and incubated with a peroxidase‐conjugated secondary antibody for 1 h at room temperature. Membranes were washed three times in distilled H2O, once in 0.05% PBS‐Tween for 5 min, and then again in distilled H2O to remove excess Tween. Membranes were incubated with chemiluminescence agent Clarity Western ECL substrate (Bio‐Rad, 170‐5060) and visualised using Fuji Film Intelligent Dark Box II and Image Reader Las‐100 software. Signal intensity of protein bands was analysed using ImageJ analysis software. Background subtraction was applied, and signal intensity of the protein of interest was normalised to a loading control.
Primary antibodies used were as follows: rabbit polyclonal anti‐Akt (1:1,000, 9272; Cell Signaling), rabbit polyclonal anti‐phospho Akt (T308) (1:1,000, 2965S; Cell Signaling), rabbit polyclonal anti‐Erk1/2 (p44/42 MAPK) (1:1,000, 9102; Cell Signaling), rabbit polyclonal anti‐phospho ERK1/2 (p‐44/42 MAPK) (Thr202/Tyr204) (1:1,000, 9101; Cell Signaling) and rabbit monoclonal anti‐GAPDH (1:5,000, 5174; Cell Signaling).
Secondary antibody used was as follows: goat anti‐rabbit IgG‐HRP‐conjugated (1:5,000, A0545; Sigma‐Aldrich).
Heatmaps
Heatmaps were generated using Ingenuity Pathway Analysis (IPA).
Morphometric skin measurements
For human skin, the measurements of morphological characteristics, such as epidermal curvature, have been previously described (Waaijer et al, 2012a).
For melanoderms, haematoxylin and eosin staining was performed in human epidermal equivalent sections, which were then imaged (20× objectives) using a Nikon E800 widefield microscope and captured with a Leica DFC420 camera using LASAF software (Leica). For each section, the full length of the epidermis was imaged. Epidermal thickness was measured throughout the section using five randomly selected areas for each field by applying a randomly offset grid overlay on ImageJ. Mean thickness per field and then mean thickness per section were calculated. Data are plotted as mean thickness (μm) per section ± SEM.
Quantification of immunohistochemical stainings
Quantifications were carried out in a blinded manner and were performed by 3 independent analysts. All melanocytes present in the micrographs were analysed (an average of 80 nuclei), and the majority of keratinocytes were counted in each section (an average of 860 keratinocytes per donor). For p16 and p21 analysis, nuclei with strong staining were considered positive cells. For SIRT1, a threshold was set for the fluorescence intensity, which was kept constant through all micrographs, and only nuclei with SIRT1 fluorescence intensity above the threshold were considered positive. For HMGB1 fluorescence intensity analysis, the background intensity for each section was measured from 6 random sites without nuclei. Then, the average background intensity was subtracted from the nuclear HMGB1 intensity for each cell type. For this analysis, all melanocytes present in each section and 50–100 nuclei from keratinocytes were analysed per donor. For immuno‐FISH analysis, an average of 20 melanocytes and 100 keratinocytes were analysed per donor.
Microscopy
For fluorescence microscopy, the following microscopes were used: Leica DM5500B widefield fluorescence microscope, Zeiss AxioObserver Spinning Disk confocal microscope, Leica SP8 confocal and Digital Light Sheet (DLS) microscope.
Statistical analysis
All statistical analyses were carried out using GraphPad Prism 7.01, and a P <0.05 was considered as statistically significant. For comparisons between 2 groups, a two‐tailed unpaired t‐test was used, and where the data were not normally distributed, a Mann–Whitney U‐test was performed. For multi‐group comparison, a one‐way ANOVA with Tukey's or Holm–Sidak's post hoc test was used. A two‐way ANOVA with Bonferroni post hoc test was performed for data sets containing two independent factors. Correlations were analysed using Pearson's rank correlation test.
Author contributions
SV performed the majority of the experiments; AL, JH, WM, DT, KB, JC, JB, MO and JSP performed and evaluated individual experiments; AM, DJ, PDA, DVH, MB and PES designed and supervised individual experiments; DAG and JFP designed and supervised the study; and SV and JFP wrote the manuscript with the contributions from all the authors.
Conflict of interest
DG, DT and KB are Unilever employees. Although no products were tested, this work could potentially promote the use of anti‐ageing products and lead to financial gain for Unilever.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was funded by Unilever and by BBSRC grants: [BB/L502066/1; BB/K017314/1]. M.O. is supported by the Glenn Foundation for Medical Research Postdoctoral Fellowship PD1921. D.J. was funded by the Academy of Medical Sciences (SBF003_1179). AL, SV and JFP would like to thank the Ted Nash Foundation, Mayo Clinic CTSA grant number UL1TR002377 from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH) and the Kogod Center on Aging Career Development Award. We would like to thank Matthew Rowson and Filimon Meichanetzidis for their work analysing the in vivo TAF data, to Andrea Maier for her work and advice in the collection of the LLS biopsies and Barry Monk for the acquisition of the biopsies from the young subjects.
The EMBO Journal (2019) 38: e101982
References
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M et al (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15: 978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamus J, Aho S, Meldrum H, Bosko C, Lee JM (2014) p16INK4A influences the aging phenotype in the living skin equivalent. J Invest Dermatol 134: 1131–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere‐initiated senescence. Nature 426: 194–198 [DOI] [PubMed] [Google Scholar]
- Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D et al (2019) Length‐independent telomere damage drives post‐mitotic cardiomyocyte senescence. EMBO J 38: e100492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature 479: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A et al (2016) Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature 530: 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bek MJ, Reinhardt HC, Fischer KG, Hirsch JR, Hupfer C, Dayal E, Pavenstadt H (2003) Up‐regulation of early growth response gene‐1 via the CXCR6 receptor induces reactive oxygen species and inhibits Na+/K+‐ATPase activity in an immortalized human proximal tubule cell line. J Immunol 170: 931–940 [DOI] [PubMed] [Google Scholar]
- Birch J, Anderson RK, Correia‐Melo C, Jurk D, Hewitt G, Marques FM, Green NJ, Moisey E, Birrell MA, Belvisi MG et al (2015) DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 309: L1124–L1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH (1991) Structure and function of telomeres. Nature 350: 569–573 [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E (2009) Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol 10: 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonacchi A, Romagnani P, Romanelli RG, Efsen E, Annunziato F, Lasagni L, Francalanci M, Serio M, Laffi G, Pinzani M et al (2001) Signal transduction by the chemokine receptor CXCR10: activation of Ras/ERK, Src, and phosphatidylinositol 3‐kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem 276: 9945–9954 [DOI] [PubMed] [Google Scholar]
- Briggaman RA, Wheeler CE Jr (1975) The epidermal‐dermal junction. J Invest Dermatol 65: 71–84 [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S, Hodgson JG, Chin K, Desprez PY, Campisi J (2010) A human‐like senescence‐associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE 5: e9188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J‐P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez P‐Y, Campisi J (2008) Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: e301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia‐Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A et al (2016) Mitochondria are required for pro‐ageing features of the senescent phenotype. EMBO J 35: 724–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia‐Melo C, Birch J, Fielder E, Rahmatika D, Taylor J, Chapman J, Lagnado A, Carroll BM, Miwa S, Richardson G et al (2018) Rapamycin improves healthspan but not inflammaging in nfkappab1(‐/‐) mice. Aging Cell 18: e12882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe J‐P, Rodier F, Campisi J (2013) p53‐dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201: 613–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME et al (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell 31: 722–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekmann J, Alili L, Scholz O, Giesen M, Holtkotter O, Brenneisen P (2016) A three‐dimensional skin equivalent reflecting some aspects of in vivo aged skin. Exp Dermatol 25: 56–61 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira‐Smith O et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo . Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos M, Metral E, Boher A, Rousselle P, Thepot A, Damour O (2015) In vitro 3‐D model based on extending time of culture for studying chronological epidermis aging. Matrix Biol 47: 85–97 [DOI] [PubMed] [Google Scholar]
- Farage MA, Miller KW, Elsner P, Maibach HI (2007) Structural characteristics of the aging skin: a review. Cutan Ocul Toxicol 26: 343–357 [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM et al (2012) Telomeric DNA damage is irreparable and causes persistent DNA‐damage‐response activation. Nat Cell Biol 14: 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Rossiello F, Mondello C, d'Adda di Fagagna F (2014) Stable cellular senescence is associated with persistent DDR activation. PLoS ONE 9: e110969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhar A, Ullmann Y, Karry R, Shalaginov R, Assy B, Serafimovich S, Kalish RS (2004) Aging of human epidermis: reversal of aging changes correlates with reversal of keratinocyte fas expression and apoptosis. J Gerontol A Biol Sci Med Sci 59: 411–415 [DOI] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999) Mammalian telomeres end in a large duplex loop. Cell 97: 503–514 [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM (2006) Cellular senescence in aging primates. Science 311: 1257 [DOI] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques FD, Correia‐Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress‐induced senescence. Nat Commun 3: 708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol 24: 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MH, Brinckerhoff CE, Mullins DW (2015) CXCR31 signaling in BRAFWT melanoma increases IL‐8 expression and tumorigenicity. PLoS ONE 10: e0121140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbow K, Roth SI, Fitzpatrick TB, Szabo G (1975) Mitotic activity in non‐neoplastic melanocytes in vivo as determined by histochemical, autoradiographic, and electron microscope studies. J Cell Biol 66: 663–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammeyer A, Luiten RM (2015) Oxidation events and skin aging. Ageing Res Rev 21: 16–29 [DOI] [PubMed] [Google Scholar]
- Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR (2011) Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep 13: 52–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruk PA, Rampino NJ, Bohr VA (1995) DNA damage and repair in telomeres: relation to aging. Proc Natl Acad Sci USA 92: 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammermann I, Terlecki‐Zaniewicz L, Weinmullner R, Schosserer M, Dellago H, de Matos Branco AD, Autheried D, Sevcnikar B, Kleissl L, Berlin I et al (2018) Blocking negative effects of senescence in human skin fibroblasts with a plant extract. NPJ Aging Mech Dis 4: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavker RM (1979) Structural alterations in exposed and unexposed aged skin. J Invest Dermatol 73: 59–66 [DOI] [PubMed] [Google Scholar]
- Lavker RM, Zheng PS, Dong G (1989) Morphology of aged skin. Clin Geriatr Med 5: 53–67 [PubMed] [Google Scholar]
- Liu N, Matsumura H, Kato T, Ichinose S, Takada A, Namiki T, Asakawa K, Morinaga H, Mohri Y, De Arcangelis A et al (2019) Stem cell competition orchestrates skin homeostasis and ageing. Nature 568: 344–350 [DOI] [PubMed] [Google Scholar]
- Munoz‐Espin D, Canamero M, Maraver A, Gomez‐Lopez G, Contreras J, Murillo‐Cuesta S, Rodriguez‐Baeza A, Varela‐Nieto I, Ruberte J, Collado M et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118 [DOI] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin‐Ruiz C, von Zglinicki T (2012) A senescent cell bystander effect: senescence‐induced senescence. Aging Cell 11: 345–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM et al (2017) Cellular senescence drives age‐dependent hepatic steatosis. Nat Commun 8: 15691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Krüger P, Fielder E, Victorelli S, Ruswhandi RA, Giorgadze N, Pirtskhalava T et al (2019) Obesity‐induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab 29: 1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa S, Kawanishi S (1999) Site‐specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett 453: 365–368 [DOI] [PubMed] [Google Scholar]
- Opresko PL, Fan J, Danzy S, Wilson DM III, Bohr VA (2005) Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res 33: 1230–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K et al (2007) Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere‐dependent senescence. PLoS Biol 5: e110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A et al (2010) Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 6: 347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowski JS, McBryan T, van Tuyn J, Drotar ME, Hewitt RN, Maier AB, King A, Blyth K, Wu H, Adams PD (2013) Wnt signaling potentiates nevogenesis. Proc Natl Acad Sci USA 110: 16009–16014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen S, Saretzki G, von Zglinicki T (1998) Preferential accumulation of single‐stranded regions in telomeres of human fibroblasts. Exp Cell Res 239: 152–160 [DOI] [PubMed] [Google Scholar]
- Razdan N, Vasilopoulos T, Herbig U (2018) Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell 17: e12838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter‐Kochanek K, Jansen‐Durr P, Wlaschek M (2006) p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 5: 379–389 [DOI] [PubMed] [Google Scholar]
- Rinnerthaler M, Streubel MK, Bischof J, Richter K (2015) Skin aging, gene expression and calcium. Exp Gerontol 68: 59–65 [DOI] [PubMed] [Google Scholar]
- Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM (2017) The senescence‐associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31: 172–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette PJ, Brash DE (2010) Human telomeres are hypersensitive to UV‐induced DNA damage and refractory to repair. PLoS Genet 6: e1000926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Maier B, Bartke A, Scrable H (2006) Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell 5: 413–422 [DOI] [PubMed] [Google Scholar]
- Schoenmaker M, de Craen AJ, de Meijer PH, Beekman M, Blauw GJ, Slagboom PE, Westendorp RG (2006) Evidence of genetic enrichment for exceptional survival using a family approach: the Leiden Longevity Study. Eur J Hum Genet 14: 79–84 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon‐Cardo C, Beach D, DePinho RA (1996) Role of the INK4a locus in tumor suppression and cell mortality. Cell 85: 27–37 [DOI] [PubMed] [Google Scholar]
- Shahabuddin S, Ji R, Wang P, Brailoiu E, Dun N, Yang Y, Aksoy MO, Kelsen SG (2006) CXCR58 chemokine receptor‐induced chemotaxis in human airway epithelial cells: role of p38 MAPK and PI3K signaling pathways. Am J Physiol Cell Physiol 291: C34–C39 [DOI] [PubMed] [Google Scholar]
- Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung RL et al (2012) Oncogene‐induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J 31: 2839–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556 [DOI] [PubMed] [Google Scholar]
- Taylor KL, Lister JA, Zeng Z, Ishizaki H, Anderson C, Kelsh RN, Jackson IJ, Patton EE (2011) Differentiated melanocyte cell division occurs in vivo and is promoted by mutations in Mitf. Development 138: 3579–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde MC, Flynn JM, Day NU, Melov S, Campisi J (2012) Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 4: 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde MC, Demaria M, Melov S, Campisi J (2015) Pleiotropic age‐dependent effects of mitochondrial dysfunction on epidermal stem cells. Proc Natl Acad Sci USA 112: 10407–10412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victorelli S, Passos JF (2017) Telomeres and cell senescence ‐ size matters not. EBioMedicine 21: 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaijer ME, Gunn DA, Catt SD, van Ginkel M, de Craen AJ, Hudson NM, van Heemst D, Slagboom PE, Westendorp RG, Maier AB (2012a) Morphometric skin characteristics dependent on chronological and biological age: the Leiden Longevity Study. Age 34: 1543–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaijer ME, Parish WE, Strongitharm BH, van Heemst D, Slagboom PE, de Craen AJ, Sedivy JM, Westendorp RG, Gunn DA, Maier AB (2012b) The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 11: 722–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaijer ME, Gunn DA, Adams PD, Pawlikowski JS, Griffiths CE, van Heemst D, Slagboom PE, Westendorp RG, Maier AB (2016) P16INK4a positive cells in human skin are indicative of local elastic fiber morphology, facial wrinkling, and perceived age. J Gerontol A Biol Sci Med Sci 71: 1022–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT (2000) Expression of the chemokine receptor CXCR68 on neurons and the elevated expression of its ligand IP‐10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer's disease. J Neuroimmunol 108: 227–235 [DOI] [PubMed] [Google Scholar]
- Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, Sepe A, Johnson KO, Stout MB, Giorgadze N et al (2015) Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 4: e12997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef R, Pilpel N, Tokarsky‐Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R et al (2016) Directed elimination of senescent cells by inhibition of BCL‐W and BCL‐XL. Nat Commun 7: 11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File