

Abstract
Tunneling nanotubes (TNTs) are actin‐based transient tubular connections that allow direct communication between distant cells. TNTs play an important role in several physiological (development, immunity, and tissue regeneration) and pathological (cancer, neurodegeneration, and pathogens transmission) processes. Here, we report that the Wnt/Ca2+ pathway, an intracellular cascade that is involved in actin cytoskeleton remodeling, has a role in TNT formation and TNT‐mediated transfer of cargoes. Specifically, we found that Ca2+/calmodulin‐dependent protein kinase II (CaMKII), a transducer of the Wnt/Ca2+ pathway, regulates TNTs in a neuronal cell line and in primary neurons. We identified the β isoform of CaMKII as a key molecule in modulating TNT formation and transfer, showing that this depends on the actin‐binding activity of the protein. Finally, we found that the transfer of vesicles and aggregated α‐synuclein between primary neurons can be regulated by the activation of the Wnt/Ca2+ pathway. Our findings suggest that Wnt/Ca2+ pathway could be a novel promising target for therapies designed to impair TNT‐mediated propagation of pathogens.
Keywords: CaMKII, intercellular communication, tunneling nanotubes, Wnt pathway, α‐synuclein
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Development & Differentiation; Signal Transduction
Wnt‐mediated activation of actin‐binding CaMKII regulates tunneling nanotube formation and transfer of vesicles and pathological protein aggregates between neurons.
Introduction
Tunneling nanotubes (TNTs) are actin‐rich membranous connections that act as intercellular bridges allowing direct communication between distant cells (Rustom et al, 2004). Recent studies have implicated TNTs in key processes, such as development, immunity, and tissue regeneration, but also in the transmission of several pathogens (Ariazi et al, 2017). In cultured cells, TNTs appear as straight intercellular connections with a diameter of 20–500 nm and a length up to 100 μm, which hover freely in the medium without touching the substrate (Abounit & Zurzolo, 2012). Many different cellular components such as organelles (including mitochondria and lysosomes), endocytic vesicles, proteins, ions, and even pathogens can be transported inside TNTs and transferred between TNT‐connected cells (Marzo et al, 2012; Gerdes et al, 2013).
Of interest, TNTs can mediate the transfer of infectious prion particles (Gousset et al, 2009) and other prion‐like proteins (Victoria & Zurzolo, 2017). Specifically, we have shown that fibrils of α‐synuclein (α‐syn), a prion‐like protein involved in Parkinson's disease (PD) pathology, can be transferred between neuronal cells (CAD cells) in culture through TNTs (Abounit et al, 2016a). Furthermore, we reported that transfer of α‐syn fibrils takes place both between primary neurons and astrocytes in culture, and occurs with a higher efficiency between neurons when cellular contacts are allowed (Abounit et al, 2016a; Loria et al, 2017), suggesting that primary neurons also communicate through a cell contact‐mediated mechanism, possibly by TNTs. In fact, the role of TNTs in neurons as a mean for the spreading of aggregated proteins has been previously proposed (Costanzo et al, 2013; Abounit et al, 2016a; Tardivel et al, 2016); however, their existence in these cells has not been proven yet.
TNTs can arise from the extension of filopodia‐like protrusions toward neighboring cells, a process in which actin polymerization plays an important role (Abounit & Zurzolo, 2012). Because of their morphological resemblance to filopodia, we hypothesized that factors influencing filopodia formation could also be involved in TNT formation. Our previous data showed that TNTs and filopodia use the same actin modulators and Rab proteins for their formation (Gousset et al, 2013; Delage et al, 2016; Zhu et al, 2018). However, these two cellular structures appear to be differently regulated by the same molecular players (Delage et al, 2016).
Here, we investigated the role of Wnt pathway in TNT formation. Wnt ligands are secreted glycoproteins that act as signaling molecules in diverse cellular processes, such as cell adhesion, morphology, proliferation, and migration (Mikels & Nusse, 2006). Wnt ligands bind to different receptors at the plasma membrane and can signal through several branches that can be classified according to specific effectors of the signaling cascade: Wnt/β‐catenin, Wnt/JNK, and Wnt/Ca2+ pathways (Widelitz, 2005; Grainger & Willert, 2018). Of interest, filopodia formation in various cell types can be regulated, among other factors, by the Wnt/β‐catenin‐independent signaling (Nishita et al, 2006). Both Wnt/JNK and Wnt/Ca2+ pathways have been shown to play an important role in regulating the cytoskeleton organization (Mezzacappa et al, 2012). In particular, the activation of Ca2+/calmodulin‐dependent protein kinase II (CaMKII), a transducer of the Wnt/Ca2+ pathway, is involved in the actin cytoskeleton remodeling (Wang et al, 2010). Specifically, the β isoform of CaMKII interacts with filamentous actin (F‐actin) and functions as an actin‐bundling protein (Shen & Meyer, 1999). In addition, βCaMKII inhibits actin polymerization by binding actin monomers and reducing the amount of freely available monomers (G‐actin) for nucleate polymer assembly (Sanabria et al, 2009). By controlling actin dynamics, Wnt pathway can regulate the formation of cellular protrusions, such as filopodia and lamellipodia, thus influencing dendritic spine outgrowth, as well as axonal spreading and enlargement of the axonal growth cone in developing neurons (Ciani et al, 2011; Stamatakou et al, 2015; Ferrari et al, 2018).
We report here for the first time that TNT‐like structures can be identified in primary neurons at early developmental stages and that the activation of the Wnt/Ca2+ pathway upregulates TNT formation and TNT‐mediated vesicle transfer in CAD cells and in neurons. We demonstrate that CaMKII is involved in the basal establishment of TNT connections in CAD cells. CaMKII, through the actin‐binding activity of its β‐isoform, contributes to stabilize TNTs. Importantly, the interneuronal transfer of α‐syn fibrils through TNT‐like protrusions can be modulated by the activation of CaMKII and is significantly reduced in primary neurons from a βCaMKII knock‐out mouse.
Results
Wnt7a increases TNT formation and TNT‐mediated transfer of vesicles in CAD cells
In our previous reports, we have used CAD cells as a neuronal cellular model to study TNTs in vitro. When cultured at an appropriate density (60–70% confluency), these cells form numerous TNTs, resulting in about 40% of TNT‐connected cells (Gousset et al, 2009).
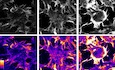
In order to test a possible role of Wnt signaling in TNT formation, we first determined whether these cells respond to Wnt ligands by evaluating the expression of Wnt receptors, Frizzled proteins (Fzd). Using qPCR analysis, we found that CAD cells express Fzd2, Fzd3, Fzd4, and Fzd6, suggesting that these cells could respond to Wnt ligands (Fig EV1A).
Figure EV1. CAD cells are responsive to Wnt signals.
- mRNA expression levels of 10 Fzd receptors in CAD cells were determined by qPCR analysis. RPLP0 mRNA expression was used for normalization. Efficiency (eff) was calculated for each set of primers and used for the calculation of the expression by −ΔΔC T method.
- Representing confocal images of CAD cells treated for 4 h with 200 μM H2O2, 200 ng/ml Wnt7a, or 200 ng/ml Wnt5a. Cells were fixed and then stained with WGA (green) and DAPI (blue).
- Percentage of TNT‐connected CAD cells with each of the indicated treatments.
- Average transferred vesicle per acceptor cell with indicated treatments.
- Immunoblot of activated β‐catenin in CAD cells cultured in control conditions or treated for 4 h with 200 μM H2O2; 10 mM LiCl; or 50, 100, and 200 ng/ml Wnt7a. GAPDH was used as loading control.
- Immunoblot of activated β‐catenin and phosphorylated GSK3β (S9) in CAD cells treated in the presence or absence of Wnt7a for the indicated time points. Immunoblots show that 200 ng/ml Wnt7a could not activate β‐catenin, while 10 mM LiCl effectively activated β‐catenin after 60 min of incubation. GAPDH was used as loading control.
- Representative confocal images of control CAD cells and cells treated with 200 μM H2O2, LiCl, or NaCl for 4 h. Cells were stained with WGA (green) and Hoechst (blue).
- Percentage of TNT‐connected CAD cells with each of the indicated treatments.
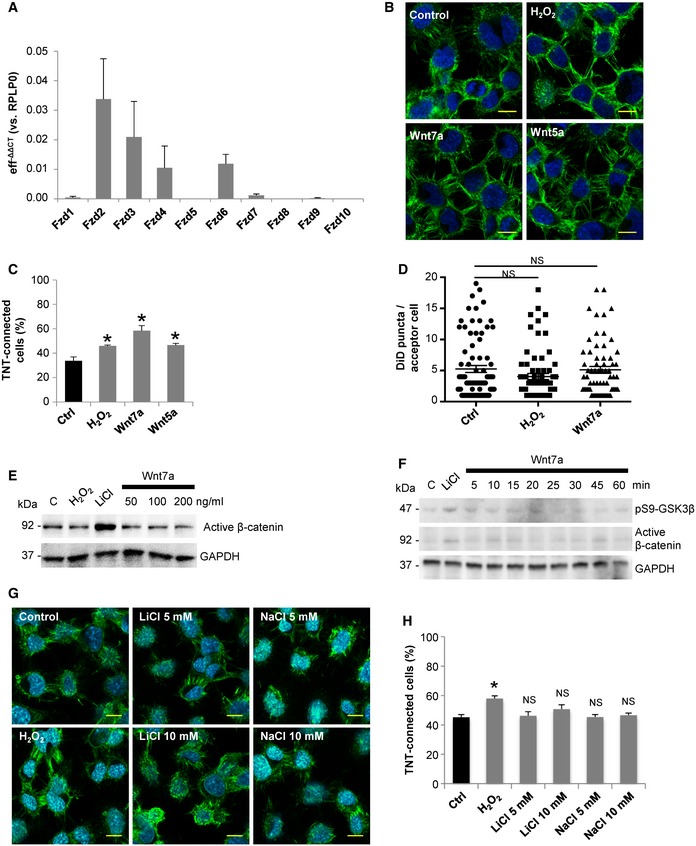
Next, among the Wnt ligands, we tested the effect on TNTs of Wnt5a and Wnt7a, for two main reasons: (i) Both Wnt5a and Wn7a have been involved in the formation of filopodia‐like protrusions in both neuronal and non‐neuronal cells (Nishita et al, 2006; He et al, 2008; Yang et al, 2012; Bentzinger et al, 2014), and (ii) they are expressed in a variety of tissues in vertebrates, including the central nervous system where the Wnt pathway modulates neuronal development (Oliva et al, 2013). We found that both Wnt5a and Wnt7a significantly increase TNT connections in CAD cells (Fig EV1B and C; for detailed criteria used to identify and count TNTs, please refer to the Materials and Methods section). However, Wnt7a produced a stronger effect, and therefore, we decided to use Wnt7a for further assays in CAD cells. To test the effect of Wnt7a on TNT formation, CAD cells were treated with increasing concentrations of Wnt7a for 4 h (Fig 1A). In these experiments, 200 μM of H2O2 treatment was used as a positive control for TNT induction (Gousset et al, 2013; Zhang & Zhang, 2015). We found that Wnt7a is able to increase the percentage of TNT‐connected cells in a dose‐dependent manner (Fig 1A and B). To confirm that cellular protrusions induced by Wnt7a exposure are in fact functional TNTs, we analyzed the intercellular transfer of vesicles in the presence or absence of Wnt7a. Since filopodia have closed ends and do not appear to allow vesicle transfer in CAD cells (Sartori‐Rupp et al, 2019), an effect on the transfer of vesicles should occur only in the case that TNTs are formed. Thus, if Wnt7a promotes the formation of TNTs rather than filopodia, we expected to observe an increase in vesicle transfer when donor cells (containing fluorescently labeled vesicles) are co‐cultured with acceptor cells (labeled with a different fluorescent dye) (see schematic in Fig 1C). To test this, donor cells were stained with the non‐specific membrane dye DiD to label all internal vesicles, while acceptor cells were stained with CellTracker™ Green (CTG). Cell contact‐dependent transfer of vesicles was assessed by seeding donor and acceptor cells at a 1:1 ratio and treating them or not with H2O2 (as a positive control for TNT increase), and Wnt7a for 4 h (Fig 1C). Compared to control cells, Wnt7a significantly increased the percentage of acceptor cells that received vesicles from donor cells (Fig 1C bottom, and D), while the average transferred vesicle number per acceptor cell did not change between the conditions (Fig EV1D). These results suggest that Wnt7a is likely affecting TNT formation, but not vesicle transfer per se. Furthermore, to exclude that Wnt treatment induced vesicle transfer through a secretory mechanism, we cultured acceptor cells with the conditioned medium from Wnt7a‐treated cells (Fig 1E). Interestingly, no significant changes in the percentage of acceptor cells containing DiD‐labeled vesicles were observed after treating the cells with the conditioned media from donor cells, for any of the assayed treatments (Fig 1F). Although basal secretion of vesicles occurs in these cells, the transfer to acceptor cells mediated by secretion is significantly less efficient than the cell‐to‐cell contact‐mediated transfer. Thus, altogether these data suggest that Wnt7a can increase the number of functional TNTs in CAD cells.
Figure 1. Wnt7a increases TNTs and vesicle transfer in CAD cells.
- Representative confocal images of CAD cells exposed for 4 h to the indicated treatments. Cells were fixed and then stained with WGA (green) and DAPI (blue).
- Percentage of TNT‐connected CAD cells with each of the indicated treatments.
- Top: Schematic of the co‐culture system used to assess vesicle transfer from donor (labeled with DiD) to acceptor (labeled with CTG) cells. After labeling, cells were detached, replated at 1:1 ratio, and cultured in control conditions or treated with 200 μM H2O2 or 200 ng/ml Wnt7a for 4 h. Middle: Representative confocal images of co‐cultured donor and acceptor CAD cells. Each image corresponds to the projection of the entire Z‐stack. Bottom: The insets correspond to the projection of selected Z‐stacks of the area marked with a yellow square in each condition to better illustrate the transferred vesicles in acceptor cells. Yellow arrows point to DiD‐labeled vesicles inside acceptor cells.
- Percentage of acceptor cells containing DiD‐labeled vesicles in the co‐culture systems with the indicated treatments.
- Top: Schematic of the culture system used to assess vesicle transfer by secretion. Donor cells (labeled with DiD) were cultured in control conditions or treated with 200 μM H2O2 or 200 ng/ml Wnt7a for 4 h. Then acceptor cells (labeled with CTG) were treated for 4 h with the conditioned media from donor cells. Bottom: Representative confocal images of acceptor CAD cells treated with the conditioned media from donor cells. Yellow arrows point to DiD‐labeled vesicles inside acceptor cells.
- Percentage of acceptor cells containing DiD‐labeled vesicles after being cultured for 4 h with the conditioned media of treated donor cells.
Wnt7a‐induced effect on TNTs is mediated by the activation of Wnt/Ca2+ pathway
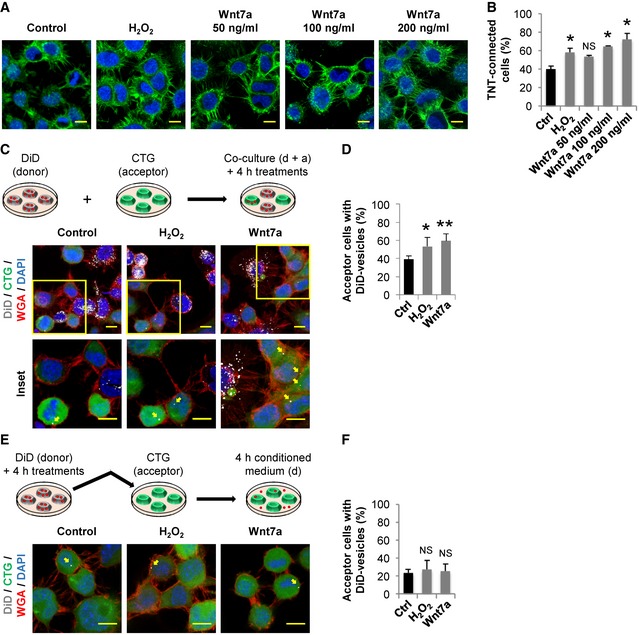
Depending on the cell type, Wnt7a has been shown to activate both β‐catenin‐dependent and β‐catenin‐independent pathways (Caricasole et al, 2003; Ciani et al, 2011; Yang et al, 2012). To identify which signaling cascade is activated by Wnt7a in CAD cells, resulting in TNT formation, we measured the activation of downstream proteins involved in the different Wnt signaling branches following Wnt7a treatment. First, we analyzed the β‐catenin‐dependent pathway. After 4‐h exposure to increasing concentrations of Wnt7a, we found no activation of β‐catenin compared with the positive control LiCl, a known inducer of β‐catenin accumulation in various cell types (Caricasole et al, 2003; Sutton et al, 2007) (Fig EV1E). In addition, to rule out if we missed the activation peak of β‐catenin after Wnt7a treatment, we performed a time course assay. However, also in this case, Wnt7a failed to induce the activation of Wnt/β‐catenin, because the levels of the inhibitory phosphorylation of the glycogen synthase kinase 3β (pS9‐GSK3β) and active β‐catenin, remained unchanged upon Wnt7a treatment across all time points assayed (Fig EV1F). Moreover, we found that LiCl and NaCl (negative control) failed to induce a significant increase in the percentage of TNT‐connected cells as compared to the H2O2 treatment (Fig EV1G and H). These results indicate that Wnt7a does not activate Wnt/β‐catenin pathway in CAD cells and that TNT increase does not depend on β‐catenin signal transduction. Instead, we found that Wnt7a rapidly induces an increase in the phosphorylation levels of CaMKII (Fig EV2A) and JNK (Fig EV2B). This indicates that, in CAD cells, Wnt7a activates the β‐catenin‐independent pathways: Wnt/Ca2+ and Wnt/JNK pathways.
Figure EV2. Wnt7a induces the activation of Wnt/β‐catenin‐independent pathways in CAD cells.
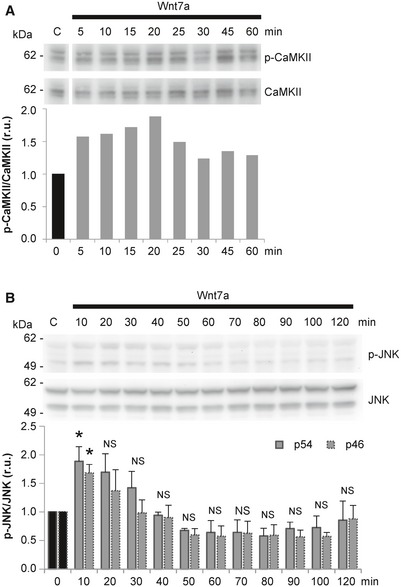
- Western blot analysis of CaMKII and p‐CaMKII in CAD cells treated in the presence or absence of Wnt7a for the indicated time points. Immunoblot and bar graph show that 200 ng/ml Wnt7a activated CaMKII in CAD cells. The graph shows p‐CaMKII/CaMKII values (r.u., relative units) for each time point normalized to time 0.
- Western blot analysis of JNK and p‐JNK in CAD cells treated in the presence or absence of Wnt7a for the indicated time points. Immunoblot and bar graph show that 200 ng/ml Wnt7a activated JNK in CAD cells. The graph shows p‐JNK/JNK values (r.u., relative units) for each time point normalized to time 0. The data are presented as the average of three independent experiments ± SEM. Statistical significance was calculated with respect to control (Ctrl); *P ≤ 0.05, NS = not significant (one‐way ANOVA).
Source data are available online for this figure.
In order to determine which of these pathways contribute to the formation and function of TNTs, we tested the effect on TNT formation and TNT‐mediated vesicle transfer of the pharmacological inhibitors KN‐93 and TAT‐TI‐JIP (TAT), which block the activation of CaMKII and JNK, respectively. When cells were co‐incubated with Wnt7a and KN‐93, we observed that Wnt7a‐induced increase in TNT connections was blocked (Fig 2A and B). Interestingly, KN‐93 alone, but not TAT alone, significantly decreased the percentage of TNT‐connected cells compared to control (Fig 2B), which suggests that Wnt/Ca2+ pathway plays a role in the basal TNT formation. Consistently with these results, we found a reduction in vesicle transfer upon treatment with KN‐93, but not with TAT alone (Fig 2C and D). This result suggests that CaMKII, but not JNK, might be required for Wnt7a‐induced formation of functional TNTs that mediate the intercellular transport of vesicles. Moreover, in order to rule out a potential effect of the Wnt/Ca2+ pathway on filopodia, we assessed the effect of Wnt7a on the formation of filopodia by staining the cells with vinculin, a frequently used marker for attached filopodia (He et al, 2017; Huang et al, 2017). We found that upon Wnt7a treatment, CAD cells exhibit a significant increase in vinculin‐positive puncta (Fig 2E and F). However, in line with our previous data indicating that TNTs and filopodia are regulated differently, we found that KN‐93 does not affect the average number of attached filopodia, while TAT significantly reduced it (Fig 2F). This indicates that Wnt7a‐induced increase in filopodia is dependent on the Wnt/JNK pathway and not on the Wnt/Ca2+ pathway. Overall, our results suggest that Wnt7a activates both Wnt/Ca2+ and Wnt/JNK pathways in CAD cells, which specifically modulate TNTs and filopodia, respectively.
Figure 2. Pharmacological inhibition of CaMKII decreases TNT's formation and function.
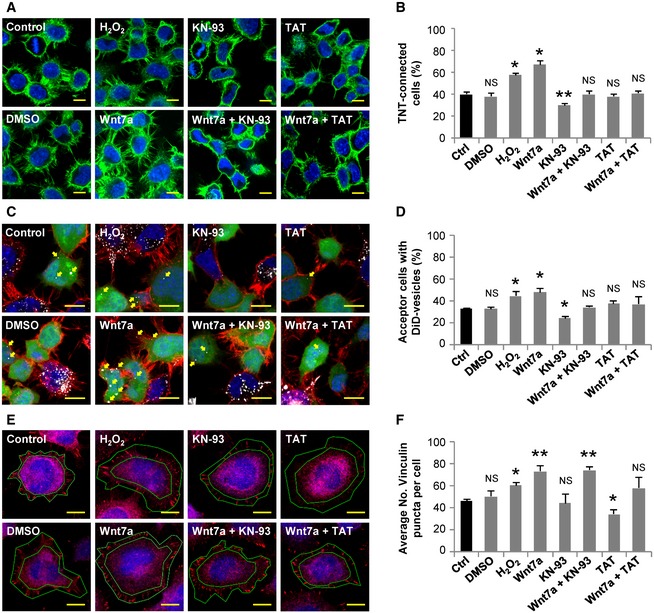
- Representative confocal images of CAD cells cultured in control conditions or treated with 0.2% DMSO, 200 μM H2O2, 200 ng/ml Wnt7a, 10 μM KN‐93, 200 ng/ml Wnt7a plus 10 μM KN‐93, 1 μM TAT, or 200 ng/ml Wnt7a plus 1 μM TAT for 4 h. Cells were fixed and stained with WGA (green) and DAPI (blue).
- Percentage of TNT‐connected CAD cells with each of the indicated treatments.
- Representative confocal images of co‐cultured donor and acceptor CAD cells. Cells were co‐cultured in control conditions or treated with 0.2% DMSO, 200 μM H2O2, 200 ng/ml Wnt7a, 10 μM KN‐93, 200 ng/ml Wnt7a plus 10 μM KN‐93, 1 μM TAT, or 200 ng/ml Wnt7a plus 1 μM TAT for 4 h. Yellow arrows point to DiD‐labeled vesicles inside acceptor cells.
- Percentage of acceptor cells containing DiD‐labeled vesicles in the co‐culture systems with the indicated treatments.
- Representative confocal images of CAD cells stained for vinculin (red) and HCS CellMask™ Blue after being treated with 0.2% DMSO, 200 μM H2O2, 200 ng/ml Wnt7a, 10 μM KN‐93, 200 ng/ml Wnt7a plus 10 μM KN‐93, 1 μM TAT, or 200 ng/ml Wnt7a plus 1 μM TAT for 4 h. The ring circumscribed between the green lines mark the area where the vinculin puncta were counted for each cell. This area is defined by the board of the HCS CellMask™ Blue staining and the vinculin staining around it.
- Average number of vinculin puncta per cell with the indicated treatments.
Actin‐binding activity of βCaMKII is required for TNT stabilization
Four different isoforms of CaMKII (α, β, γ and δ) are expressed in mammals (Gaertner et al, 2004). In particular, the α and β subunits are the most abundant in the brain (Tobimatsu & Fujisawa, 1989). Here, we decided to further investigate βCaMKII because of its ability to interact with F‐actin, observed both in cell culture models and in neurons (Shen & Meyer, 1999). Previous reports have proposed that βCaMKII can bundle F‐actin through its actin‐binding and association domains, thus impacting the actin turnover (Okamoto et al, 2007; Khan et al, 2016). Of interest, by controlling the turnover of actin molecules in the dendritic spines of neurons, βCaMKII can modulate the outgrow of dendritic filopodia, a precursor form of mature spines (Fink et al, 2003; Okamoto et al, 2007).
We hypothesized that βCaMKII could be modulating TNTs by controlling the remodeling of the actin cytoskeleton, thus influencing their formation and/or stabilization. One approach used to study the interaction between βCaMKII and F‐actin is to overexpress the βCaMKII mutants: kinase deficient (K43R), calmodulin‐binding deficient (A303R), and a constitutively active form (T287D). By using this approach, it was reported that the actin‐binding activity of βCaMKII is regulated by the phosphorylation state of the protein (Shen & Meyer, 1999). In fact, in rat basophilic leukemia (RBL) cells upon an increase in the intracellular Ca2+ concentration, it was observed that the K43R mutant can dissociate from actin, while the A303R mutant cannot detach from actin and the T287D mutant does not bind to actin at all (Shen & Meyer, 1999). To test whether βCaMKII is involved in TNT formation, we transfected CAD cells with GFP‐βCaMKII wild‐type (WT) or mutant plasmids and compared the resultant percentage of TNT‐connected cells to control cells expressing a GFP‐vector construct (Fig 3A and B). Of interest, the GFP‐βCaMKII WT, the kinase‐inactive GFP‐βCaMKII K43R mutant and the calmodulin‐binding deficient GFP‐βCaMKII A303R mutant exhibited a punctuated pattern when expressed in CAD cells (Fig 3A), similar to what was previously shown in RBL cells (Shen & Meyer, 1999) and different from the diffuse fluorescence of control GFP‐vector (Fig 3A). In addition, all three constructs significantly increased the percentage of TNT‐connected cells in comparison with control (Fig 3B). On the contrary, the phosphomimetic GFP‐βCaMKII T287D mutant showed a diffused fluorescent pattern in the cytoplasm of transfected cells (Fig 3A), and no change was observed in the percentage of TNT‐connected cells when compared to GFP‐vector‐transfected cells (Fig 3B).
Figure 3. TNT's formation and function depend on βCaMKII phosphorylation status.
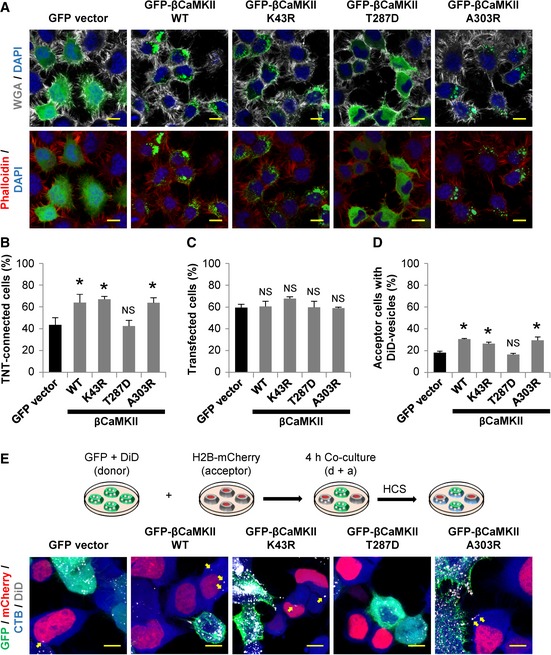
- Representative confocal images of CAD cells expressing the indicated GFP‐fusion proteins. Top: Cells showing WGA (white) and DAPI (blue) staining. Bottom: Cells showing phalloidin (red) and DAPI (blue) staining. Note that the expression pattern of βCaMKII WT and the mutants K43R and A303R is rather punctate than diffused.
- Percentage of TNT‐connected CAD cells 24 h after transfection with indicated GFP‐fusion proteins.
- Percentage of transfection rate of indicated GFP‐fusion proteins in CAD cells.
- Percentage of acceptor cells containing DiD‐labeled vesicles when acceptor cells were co‐cultured with donor cells expressing the indicated GFP‐fusion proteins.
- Top: Schematic of the co‐culture system used to assess vesicle transfer in transfected cells. Donor cell was transfected with the indicated GFP‐fusion proteins and then labeled with DiD. Acceptor cells were transfected with a H2B‐mCherry plasmid. After 24 h, the cells were detached, replated at 1:1 ratio, and cultured for 4 h. Cells were then fixed and stained with HCS CellMask™ Blue. Bottom: Representative confocal images of co‐cultured donor and acceptor CAD cells. Yellow arrows point to DiD‐labeled vesicles inside acceptor cells.
When we analyzed the transfection efficiency of the different βCaMKII plasmids, we found no significant differences among them (Fig 3C), ruling out that the observed effects on TNTs might be due to different expression levels. To substantiate the effect of βCaMKII on TNTs, next we measured vesicle transfer in cells transfected with the aforementioned constructs (Fig 3D and E). To this aim, donor cells were transfected with GFP‐vector or GFP‐βCaMKII constructs, while acceptor cells were transfected with a H2B‐mCherry plasmid (Fig 3E). Consistent with the changes observed on the percentage of TNT‐connected cells, we found that the expression of GFP‐βCaMKII WT, GFP‐βCaMKII K43R, and GFP‐βCaMKII A303R, but not the GFP‐βCaMKII T287D mutant, significantly increased vesicle transfer in comparison with control (Fig 3D).
One possible explanation for these results is that GFP‐βCaMKII T287D mutant fails to bind F‐actin filaments (Shen & Meyer, 1999; Okamoto et al, 2007) and that the binding activity of βCaMKII is required for the effect on TNT formation. We speculated that by binding to actin, βCaMKII could stabilize TNTs, thus increasing the number of TNT‐connected cells and TNT‐mediated vesicle transfer. To test this hypothesis, we measured by live imaging, the average lifetime of TNTs in cells transfected either with GFP‐vector, βCaMKII WT, or GFP‐βCaMKII T287D plasmid (Fig 4A and B, and Movie EV1). We found that the overexpression of βCaMKII WT increased the lifetime of TNTs compared to control, while the expression of the GFP‐βCaMKII T287D mutant did not (Fig 4B). These results suggest that βCaMKII contributes to stabilize the very dynamic TNT structures by increasing their lifetime, most likely by stabilizing the actin cytoskeleton. Consistently, this effect was dependent on the actin‐binding activity of βCaMKII, but not on its kinase activity.
Figure 4. Actin‐binding activity of βCaMKII affects the stability of TNTs.
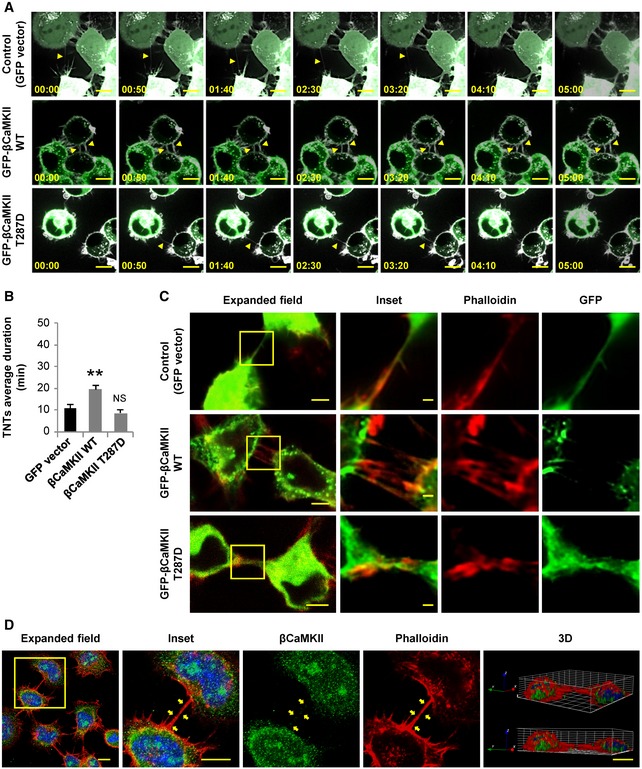
- Time lapses showing a TNT connecting two transfected cells. Top: GFP‐vector‐transfected cells. Middle: GFP‐βCaMKII WT‐transfected cells. Bottom: GFP‐βCaMKII T287D‐transfected cells. Cells were stained with WGA (white). Yellow arrowheads point out a TNT in each condition. Scale bars represent 10 μm.
- TNT's average duration between GFP‐vector, GFP‐βCaMKII WT, and GFP‐βCaMKII T287D‐transfected cells. Graph represents the average lifetime of 15‐20 TNTs per condition ± SEM. Statistical significance was calculated with respect to control (Ctrl); **P ≤ 0.01, NS = not significant (one‐way ANOVA).
- CODIM super‐resolution images (three right‐side panels) of TNTs shown in the confocal expanded field images (left panels) of transfected cells showing expression of indicated GFP‐tagged proteins (green). Cells were stained for F‐actin with phalloidin (red). Scale bars in the expanded fields represent 5 μm and 1 μm in the insets.
- Confocal images and 3D reconstruction showing the intracellular localization of endogenous βCaMKII (green) in CAD cells. Cells were stained with DAPI (blue) and phalloidin (red). Yellow arrows point to βCaMKII‐positive puncta inside a TNT. Scale bars in the expanded fields represent 10 μm in the micrographs and 1 μm in the 3D projection.
In order to further investigate how βCaMKII could stabilize the F‐actin cytoskeleton in TNTs, we studied the localization of βCaMKII in TNTs labeled with phalloidin. We used the conical diffraction microscopy (CODIM) super‐resolution technique, which allows the visualization of TNTs with a higher resolution than confocal microscopy, as it can be compared in Fig EV3A–C. Using this technique, we observed that in cells transfected with GFP‐βCaMKII WT plasmid, the GFP signal was highly enriched at the base of the TNTs, but almost absent inside of them (Fig 4C). Conversely, we found that the GFP signal in cells transfected with the GFP‐βCaMKII T287D mutant plasmid was distributed along the TNTs, and a similar pattern was observed in cells transfected with the control plasmid, GFP‐vector (Fig 4C). Interestingly, we observed that endogenous βCaMKII in CAD cells is also found forming protein clusters similar to overexpressed GFP‐βCaMKII WT (Fig 4D). Moreover, puncta of endogenous βCaMKII can be found inside TNTs and in association with F‐actin (Fig 4D). These data suggest that βCaMKII could stabilize TNTs by binding to F‐actin filaments at the base of the protrusion.
Figure EV3. Actin‐binding activity of βCaMKII modulates actin dynamics.
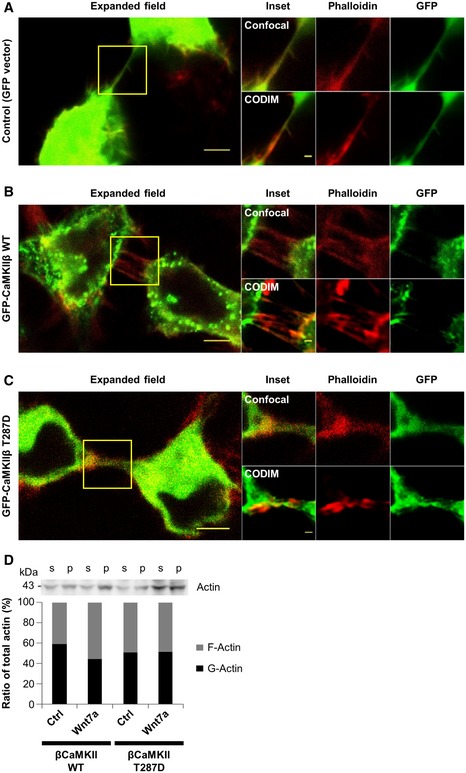
-
A–CImage resolution comparison between images obtained with CODIM super‐resolution and confocal microscopy techniques. Confocal (3 upper panels at the right) and CODIM super‐resolution images (3 bottom panels at the right) of the area depicted in the confocal expanded field images (left panels) of transfected cells (green) showing expression of GFP‐vector (A), GFP‐βCaMKII WT (B), or GFP‐βCaMKII T287D (C). Cells were stained for F‐actin with phalloidin (red). Note: CODIM images presented here are the same as those in Fig 4C. Scale bars in the expanded field represent 5 μm and 1 μm in the inset.
-
DAnti‐actin immunoblot and bar graph showing changes in the ratio between G‐actin (in supernatant fraction, s) and F‐actin (in the pellet fraction, p) of CAD cells transfected with either GFP‐βCaMKII WT or GFP‐ βCaMKII T287D plasmids and treated or not with 200 ng/ml Wnt7a.
Source data are available online for this figure.
Wnt7a‐induced effect on TNTs is due to changes in the interaction between βCaMKII and actin
Previously, it was shown that inhibition of CaMKII activity causes a reduction in the interaction between F‐actin and βCaMKII, producing an actin‐destabilizing effect that can induce the retraction of cellular processes (Waggener et al, 2013). This suggests that modulation of CaMKII activity could affect the number of TNT‐connected cells by influencing the formation of new TNTs and/or by changing the stability of pre‐existing ones. To evaluate these possibilities, we compared F‐actin accumulation and distribution in cells transfected with GFP‐βCaMKII WT and treated or not with Wnt7a (Fig 5A). We found that the overexpression of βCaMKII did not drastically affect F‐actin levels in comparison with non‐transfected cells (Fig 5A). However, Wnt7a treatment markedly increased the F‐actin signal (white and yellow in pseudocolor images), and this increment seemed to be localized in protrusions and adjacent areas (Fig 5A).
Figure 5. Wnt7a regulates actin dynamics in CAD cells overexpressing βCaMKII .
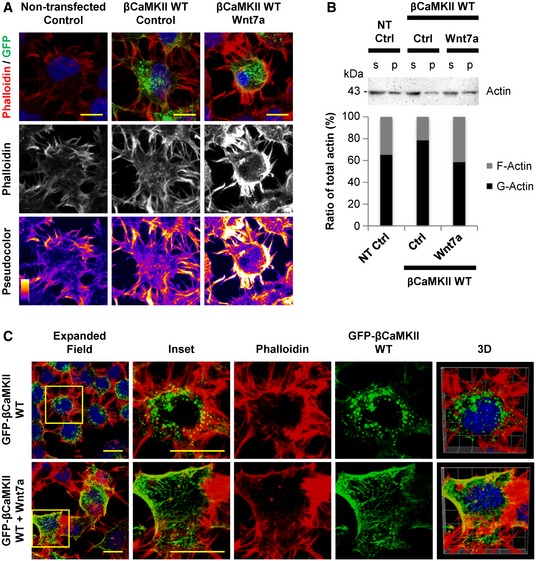
- Confocal images of untransfected and GFP‐βCaMKII WT‐transfected CAD cells (green), treated or not with Wnt7a. Top: Cells were stained with phalloidin (red) and DAPI (blue). Middle: Phalloidin staining (white) of the cells is shown. Bottom: Pseudocolor images of cells stained with phalloidin highlight the differences in F‐actin levels among the treatments. Scale bars represent 5 μm.
- Anti‐actin immunoblot and bar graph showing changes in the ratio between G‐actin (in supernatant fraction, s) and F‐actin (in the pellet fraction, p) of CAD cells untransfected or transfected with GFP‐βCaMKII WT plasmid and treated or not with Wnt7a.
- Confocal images of CAD cells transfected with GFP‐βCaMKII WT plasmid (green) and treated or not with Wnt7a. Cells were fixed and then stained with phalloidin (red) and DAPI (blue). The insets (right panels) show a magnified image of the area depicted in the expanded field images (left). 3D reconstruction shows colocalization of GFP/phalloidin signals. Scale bars represent 10 μm.
Source data are available online for this figure.
Since it has been proposed that actin‐binding activity of βCaMKII alters actin dynamics and impacts actin polymerization (Sanabria et al, 2009), we then studied whether the activation of βCaMKII by Wnt7a treatment could modulate actin dynamics in CAD cells. To test this, we measured the G‐actin/F‐actin ratio in cells that were either untransfected or transfected with GFP‐βCaMKII WT and treated with or without Wnt7a. We found that the overexpression of GFP‐βCaMKII WT increased the percentage of total G‐actin in relation to total F‐actin when compared to untransfected cells (Fig 5B). Interestingly, a marked reduction in G‐actin percentage with a concomitant increase in F‐actin percentage was observed in cells expressing GFP‐βCaMKII WT treated with Wnt7a, in comparison with control cells (Fig 5B). On the contrary, overexpressing GFP‐βCaMKII T287D in cells did not influence the G‐actin/F‐actin ratio upon Wnt7a treatment (Fig EV3D). These results suggest that Wnt7a could affect the G‐actin/F‐actin ratio by controlling the interaction between βCaMKII and actin. Thus, in conditions where the actin‐binding activity of βCaMKII is impaired (i.e., T287D mutant), Wnt7a is unable to modulate actin dynamics. In support to this hypothesis, qualitative image analysis indicated that upon exposure to Wnt7a the colocalization of βCaMKII with F‐actin increased (Fig 5C). Overall, these results suggest that Wnt7a could induce the stabilization of TNTs by promoting the interaction of βCaMKII with the actin cytoskeleton.
Wnt/Ca2+ pathway regulates vesicle transfer between primary neurons
In order to validate the results obtained with the neuronal CAD cell line in a more physiological model, we decided to use primary mouse cortical neurons. First, we corroborated that mouse cortical neurons express βCaMKII protein (Fig EV4A), and that this isoform can be detected as early as 1‐day in vitro (1 DIV), as previously shown in the literature (Lin & Redmond, 2008). Interestingly, we observed that endogenous βCaMKII displays a similar expression pattern in cortical neurons as that in CAD cells, with a marked accumulation in F‐actin enriched neuronal structures (Fig EV4A). Previous reports indicate that differently from CAD cells, in cortical neurons, Wnt7a activates the Wnt/β‐catenin pathway (Hirabayashi et al, 2004). On the other hand, Wnt5a activates the β‐catenin‐independent branches, Wnt/JNK and Wnt/Ca2+, in cortical neurons (Zhou et al, 2017). In agreement with those reports, we found that in cortical neurons Wnt5a induced an increase in the phosphorylation levels of CaMKII (Fig EV4B), while Wnt7a did not activate CaMKII (Fig EV4C).
Figure EV4. Wnt5a, but not Wnt7a, activates CaMKII in cortical neurons.
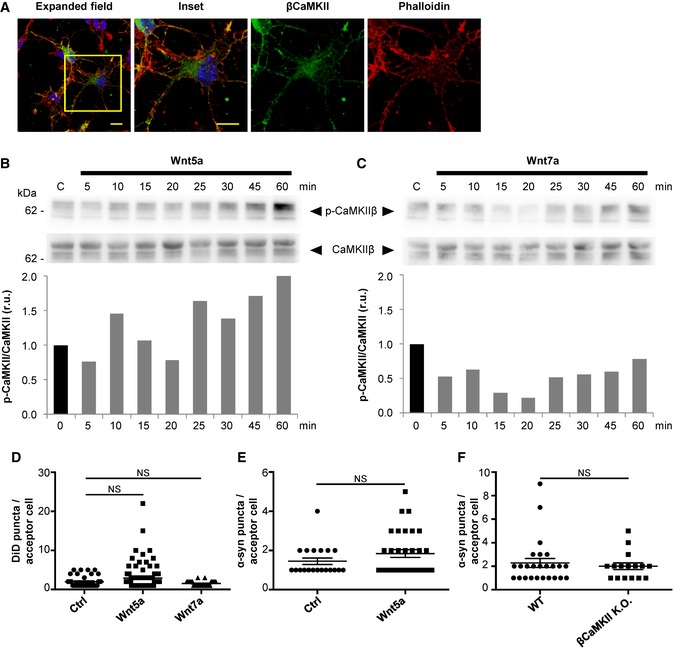
-
AConfocal images representing the intracellular localization of endogenous βCaMKII in primary mouse cortical neurons of 1 DIV. Cells were also stained with DAPI (blue) and phalloidin (red). Scale bars represent 10 μm.
-
B, CWestern blot analysis of CaMKII and p‐CaMKII in CAD cells treated with Wnt5a (B) or Wnt7a (C) for the indicated time points. The graphs show p‐CaMKII/CaMKII values (r.u., relative units) for each time point normalized to time 0.
-
D–FAverage number of transferred vesicles or α‐syn puncta per acceptor neurons for the indicated treatment or genotypes. Graphs represent the average of three independent experiments ± SEM. Statistical significance was calculated with respect to control (Ctrl); NS = not significant (one‐way ANOVA for D and Student's t‐test for E, F).
Source data are available online for this figure.
To test a possible role of the Wnt/Ca2+ pathway in TNT‐mediated communication in primary neurons, we investigated the effect of both Wnt7a and Wnt5a on vesicle transfer. To this aim, cortical neurons were labeled in suspension either with DiI (donor) or with CTG (acceptor) after dissection. Donor and acceptor neurons were seeded at 1:1 ratio and cultured for 20 h. Co‐cultures were then treated for 4 h with either Wnt5a or Wnt7a. In these conditions, we found that Wnt5a, but not Wnt7a, significantly increased vesicle transfer in comparison with control conditions (Fig 6A and B), while the average transferred vesicle number per acceptor neuron was not significantly changed among conditions (Fig EV4D). As control for cell contact‐dependent transfer, we looked at the effect of Wnt ligands on transfer mediated by secretion. We observed that when the acceptor neurons were incubated for 24 h with the conditioned media of donor neurons previously treated for 4 h with either Wnt5a or Wnt7a, neither Wnt5a nor Wnt7a treatments were able to induce changes in the percentage of vesicle transfer (Fig 6C; transfer ratio of cell–cell contact vs. secretion mechanisms for control: 80% vs. 20%, Wnt5a: 92% vs. 8% and Wnt7a 80% vs. 20%, respectively). This indicates that the activation of Wnt pathway is not affecting the transfer mediated by secretion, similar to what we found in CAD cells (Fig 1D and F). Interestingly, we found that donor and acceptor neurons when cultured together can form connections that resemble the TNTs found in CAD cells (Fig 6D).
Figure 6. Wnt/Ca2+ pathway modulates vesicle transfer in primary neurons.
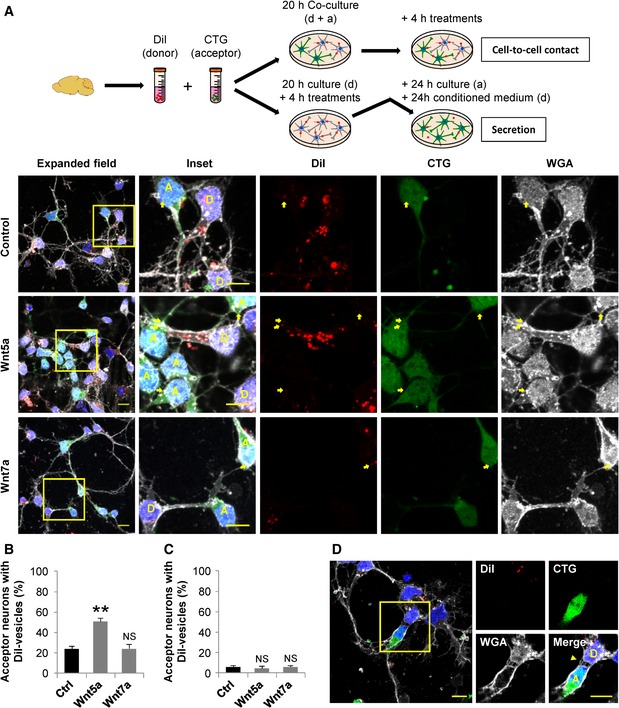
- Top: Schematic of the culture systems used to assess cell‐to‐cell contact‐dependent or secretion‐mediated transfer of vesicles in primary neurons. After dissection, neurons were labeled in suspension with DiI (donor, D) or CTG (acceptor, A). In the co‐culture system, neurons were plated at 1:1 ratio and cultured for 20 h and then treated with 200 ng/ml of Wnt5a or Wnt7a for 4 h. In the secretion‐based system, donor neurons cultured alone for 20 h were treated with 200 ng/ml of Wnt5a or Wnt7a for 4 h, and the medium was then collected and replaced for the medium of acceptor neurons cultured separately. Acceptor neurons were further incubated with the conditioned media of donor neurons for 24 h. Bottom: Representative confocal images of co‐cultured donor and acceptor neurons. The insets (right panels) show a magnification of the area depicted in the expanded field images (left). Yellow arrows point to DiI‐labeled vesicles inside acceptor neurons.
- Percentage of acceptor neurons containing DiI‐labeled vesicles after being co‐cultured with donor neurons and incubated for 4 h with the indicated treatments.
- Percentage of acceptor neurons containing DiI‐labeled vesicles after being cultured for 4 h with the conditioned media of treated donor neurons.
- Confocal image showing a donor (D) and an acceptor (A) neuron connected by TNT‐like structures (yellow arrowhead in the merged panel). Neurons were stained with WGA (white) and DAPI (blue). The insets (right panels) show a magnified image of the area depicted in the expanded field (left).
To further support the contribution of Wnt pathway to the TNT‐mediated transport of vesicles between primary neurons, we used the secreted frizzled‐related protein 2 (sFRP‐2), a Wnt ligand scavenger that has been reported to inhibit Wnt5a effect in neurons (Godoy et al, 2014). Thus, we co‐cultured donor (DiI) and acceptor (CTG) neurons for 20 h, and 4 h before fixation, we treated the cells with either Wnt5a, sFRP‐2 or Wnt5a plus sFRP‐2. Our results show that the increase in vesicle transfer induced by Wnt5a was completely abolished in the presence of sFRP‐2 (Fig 7A and B). More importantly, the treatment with sFRP‐2 alone resulted in a small but significant reduction of vesicle transfer in comparison with control (Fig 7B), suggesting that the activity of the endogenous Wnt pathway is required for a small proportion of basal vesicle transfer to take place in neurons. This small effect could be explained in part, because sFRP‐2, which is known to sequester several Wnt ligands, including Wnt5a (Galli et al, 2006; Wolf et al, 2008; Godoy et al, 2014), does not bind all Wnt ligands. Therefore, the small reduction on vesicle transfer observed in the presence of sFRP‐2 alone could be caused by a compensatory effect of unbound endogenous Wnt ligands. On the other hand, the effect of sFRP‐2 has been shown to be dependent on its concentration and on cell context. In fact, Kele et al (2012) demonstrated that high concentrations of sFRP‐2 activate the Wnt/JNK pathway in dopaminergic neurons. This further supports our findings that the Wnt/JNK pathway is not involved in TNT formation.
Figure 7. Neurons form functional TNT‐like structures.
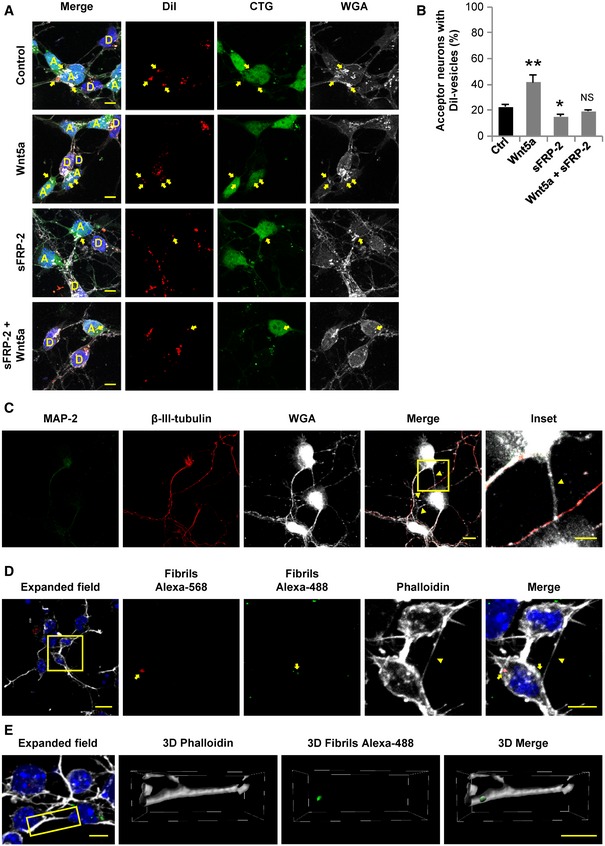
- Representative confocal images of co‐cultured donor (D, labeled with DiI) and acceptor (A, labeled with CTG) neurons treated or not with 200 ng/ml of Wnt5a, 200 ng/ml sFRP‐2 or 200 ng/ml Wnt5a plus 200 ng/ml sFRP‐2 for 4 h. Yellow arrows point to DiI‐labeled vesicles inside acceptor neurons.
- Percentage of acceptor neurons containing DiI‐labeled vesicles after being co‐cultured with donor neurons and incubated for 4 h with the indicated treatments.
- Primary cortical neurons at 1 DIV were fixed and labeled with MAP‐2 (green), β‐III‐tubulin (red), and WGA (white). The inset shows a magnified image of the area depicted in the merged panel. Yellow arrowheads point out TNT‐like structures.
- Transfer of α‐syn fibrils is shown in neurons at 1 DIV. Cells were loaded in suspension with either Alexa‐568 α‐syn fibrils (red) or Alexa‐488 α‐syn fibrils (green) and cultured together. The insets (right panels) show a magnification of the area depicted in the expanded field image (left). Yellow arrows point to red and green α‐syn puncta contained in the soma of a neuron. Yellow arrowhead points to a TNT‐like connection.
- A TNT‐like connection containing Alexa‐488 α‐syn‐positive puncta is found in neurons at 1 DIV stained with phalloidin (white) and DAPI (blue). The insets (right panels) show the 3D reconstruction of the area depicted in the expanded field image (left).
Primary neurons form TNT‐like structures that mediate the transfer of α‐syn aggregates
Although the results presented above suggest that primary neurons can be connected through TNTs, such connections have not been shown before and are indeed very difficult to identify because of the lack of a specific TNT marker. Previously, it was shown that primary neurons cultured with astrocytes can connect through TNT‐like structures, which allow them to establish electrical coupling and share calcium signals (Wang et al, 2012). However, it is not known whether this kind of connections can be formed between neurons and whether they would allow the exchange of cargoes, other than ions. Here, we could recognize TNT‐like structures formed between neurons at an early stage of development (Fig 7C). These membranous connections were labeled by fluorescent WGA, but not with the axonal marker β‐III‐tubulin, or by the dendrite marker MAP‐2, and can be observed between two somas or between a soma and an axon (Fig 7C). These observations together with the fact that neurons can transfer vesicles mainly by a cell‐to‐cell contact‐dependent mechanism (Fig 6B and C) support the hypothesis that neurons are able to form functional TNT‐like structures.
TNT‐mediated transfer of α‐syn fibrils between primary neurons can be modulated by the activation of Wnt/Ca2+ pathway
Recently, we reported that CAD cells can efficiently transfer α‐syn fibrils through TNTs (Abounit et al, 2016a). Moreover, the transport of α‐syn via TNTs has also been reported in SH‐SY5Y cells and astrocytes (Dieriks et al, 2017; Rostami et al, 2017). Also, the transfer of α‐syn fibrils in mature neurons has been observed to depend on cell‐to‐cell contacts (Abounit et al, 2016a). With the data shown above suggesting the presence of TNT‐like structures in young neurons, we decided to evaluate whether the transfer of α‐syn fibrils from neuron‐to‐neuron can occur at this developmental stage. To this aim, we loaded one population of neurons with Alexa‐568‐labeled α‐syn (red fibrils) and another one with Alexa‐488‐labeled α‐syn (green fibrils). Both populations of loaded neurons were then seeded at 1:1 ratio. After 24 h of co‐culture, we found neurons containing both red and green α‐syn puncta (Fig 7D), suggesting transfer of the fibrils. Interestingly, we found α‐syn puncta in TNT‐like structures (Fig 7E and Movie EV2), supporting the hypothesis that α‐syn can spread among neurons via TNT‐like connections.
Since the exposure to Wnt5a can induce upregulation of vesicle transfer in neurons (Figs 6B and 7B), we hypothesized that the activation of Wnt/Ca2+ pathway can also modulate the transfer from neuron‐to‐neuron of α‐syn fibrils. To address this, we evaluated the effect of Wnt5a on α‐syn transfer in neurons. Donor neurons (loaded with α‐syn fibrils) were co‐cultured with acceptor neurons (labeled with CTG) for 20 h and then treated for 4 h with Wnt5a (Fig 8A). We found that Wnt5a significantly increased α‐syn transfer when compared to control neurons (Fig 8B) but no change in the average number of transferred vesicles per acceptor cell upon treatment was found (Fig EV4E). To gather more insight, we also performed live imaging of neurons loaded with fluorescent α‐syn fibrils and treated or not with Wnt5a. Qualitative analysis of the videos revealed that TNT‐like structures were observed connecting the soma of cultured neurons (Fig 8C and Movie EV3), and transfer of α‐syn puncta from neuron‐to‐neuron was observed when the cells were exposed to Wnt5a (Fig 8C and Movie EV4).
Figure 8. Transfer of α‐syn fibrils between neurons occurs through TNT‐like protrusions, and it can be regulated by the Wnt/Ca2+ pathway.
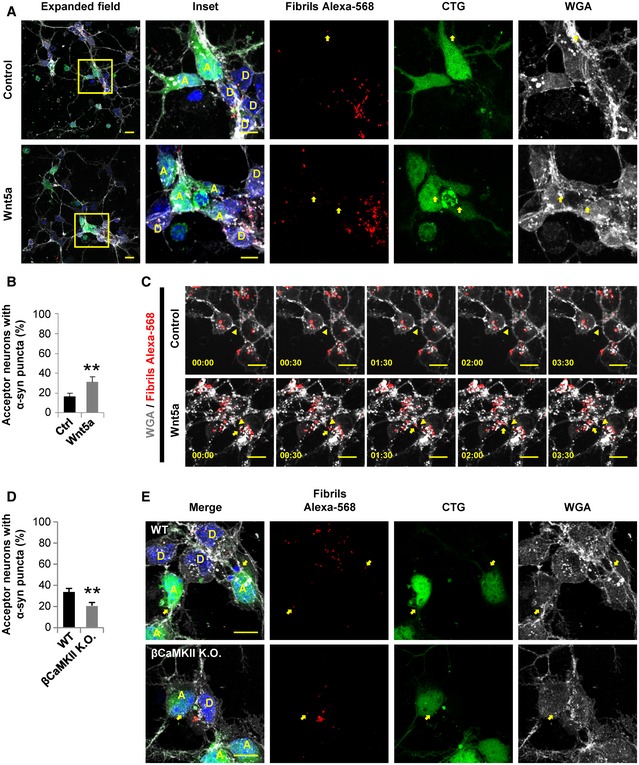
- Representative confocal images of 1 DIV co‐cultured donor (D, loaded with Alexa‐568 α‐syn fibrils) and acceptor (A, labeled with CTG) neurons. After 20 h post‐plating, neurons were treated with 200 ng/ml Wnt5a for 4 h. The insets (right panels) show a magnification of the area depicted in the expanded field (left). Yellow arrows point to α‐syn puncta inside acceptor neurons.
- Percentage of acceptor neurons containing α‐syn puncta for each of the indicated treatments.
- Time lapses of neurons loaded with Alexa‐568 α‐syn fibrils and cultured in control conditions or treated with 200 ng/ml Wnt5a. Neuron‐to‐neuron transfer of α‐syn puncta (yellow arrows) through a TNT‐like structure (yellow arrowheads) is shown upon Wnt5a treatment.
- Percentage of acceptor neurons containing α‐syn puncta in WT or βCaMKII K.O. acceptor neurons.
- Representative confocal images of 1 DIV co‐cultured donor (D, loaded with Alexa‐568 α‐syn fibrils) and acceptor (A, labeled with CTG) neurons. Neurons were obtained from WT or βCaMKII K.O. mice. Yellow arrows point to α‐syn puncta inside acceptor neurons in both conditions.
As Wnt5a induced an increase in the phosphorylation levels of CaMKII (Fig EV4B) and at the same time increased cell‐to‐cell contact‐mediated transfer of vesicles (Fig 6A and B) and α‐syn fibrils (Fig 8A and B), these results suggest that in primary neurons Wnt5a could modulate TNT formation and affect TNT‐mediated transfer by activating the Wnt/Ca2+ pathway, similarly to Wnt7a in CAD cells. To directly test the hypothesis that βCaMKII protein is involved in TNT‐mediated transfer, we evaluated the transfer of α‐syn fibrils in neurons derived from βCaMKII (−/−) mutant mice (Borgesius et al, 2011; Kool et al, 2016). By using the same approach shown to study vesicle transfer in co‐culture experiments (Figure 6A and 7A), we found that in βCaMKII K.O. neurons, the transfer of α‐syn fibrils was significantly reduced in comparison with WT neurons (Fig 8D and E). Yet, the average number of transferred vesicles per acceptor cell remained similar between the genotypes (Fig EV4F). This indicates that βCaMKII expression is affecting the interneuronal transfer of α‐syn, most likely due to an increase in TNT connections between cells. Overall, these findings suggest that neurons can spread and transport α‐syn fibrils through TNTs and this transfer can be regulated by the Wnt/Ca2+ pathway.
Discussion
Increasing evidence suggests that TNTs could be involved in the progression of several neurodegenerative diseases, because of their role in mediating the intercellular spreading of pathogenic protein assemblies (such as α‐syn, amyloid‐β, huntingtin, prion, and tau) involved in the pathogenesis of these diseases (Gousset et al, 2009; Wang et al, 2011; Costanzo et al, 2013; Abounit et al, 2016a,2016b; Tardivel et al, 2016; Victoria et al, 2016; Dieriks et al, 2017; Rostami et al, 2017). It has been proposed that these aggregated proteins propagate throughout the brain along neuronal networks, accumulating intra‐ and/or extracellularly and causing neuronal death (Jucker & Walker, 2018). Although the spreading of these aggregates via TNTs has been documented in several cell lines, the existence of TNTs in neurons and their involvement in the interneuronal propagation of aggregated proteins have not been proved yet. Understanding whether and how neurons can form functional TNTs is key for the design of therapeutic strategies aimed to halt or to slow down disease progression.
To study the formation of TNTs, here we used first a neuronal cell line (CAD cells) and then we corroborated our results in primary neurons. We show that in our in vitro conditions, the activation of Wnt/Ca2+ pathway (by Wnt7a in CAD cells or by Wnt5a in cortical neurons) is involved in the establishment of TNTs and that βCaMKII plays a key role in this event. We also show that by modulating this pathway, the intercellular spreading of α‐syn fibrils can be affected. Whether this mechanism plays a role in the brain during development and/or in the case of α‐synucleinopathies remains to be studied.
Given that one of the main constituents of TNTs is F‐actin, we hypothesized that proteins involved in the regulation of actin dynamics could have a role in the formation of TNTs. Our previous findings in neuronal CAD cells suggest that the same actin modulators affect both TNTs and filopodia, but in a different manner (Delage et al, 2016). Based on these premises, here we focused on Wnt pathway because of its role in controlling the remodeling of the actin cytoskeleton and its participation on filopodia formation, specifically in neurons (Yang et al, 2012; Onishi et al, 2013; Stamatakou et al, 2015). The formation of filopodia protrusions guiding axonal remodeling in developing dorsal root ganglion neurons can be controlled by Wnt3a‐induced regulation of the actin cytoskeleton through the recruitment of Eps8 by disheveled‐1, a scaffold protein crucial for Wnt signaling (Stamatakou et al, 2015). Furthermore, Wnt pathway activation also influences the formation of cytonemes, a specialized type of filopodia that transport signaling proteins between distant cells (Kornberg & Roy, 2014). These filopodia‐like protrusions can be formed in epithelial cells of the somite in chicken embryos and can transport components of the Wnt pathway, such as the Frizzled receptor, Fzd7 (Sagar et al, 2015). Recently, it has been shown that during neural plate formation in zebrafish, Wnt/JNK pathway through the activation of Cdc42, regulates the formation of cytonemes that transport Wnt8a ligands toward Wnt‐responsive cells where the β‐catenin‐dependent signaling is activated (Stanganello et al, 2015; Mattes et al, 2018). Thus, cytonemes do not only use Wnt pathways for their formation, but they can also serve as a mean for distributing Wnt proteins through tissues during the embryonic growth (Stanganello & Scholpp, 2016).
Here, we report that Wnt ligands through the activation of Wnt/Ca2+ pathway can influence the formation of TNTs in primary neurons and neuron‐like cells and increase the transfer of cargoes through them. We also show that Wnt‐mediated activation of JNK and β‐catenin does not affect TNT formation, indicating that TNT and cytonemes are differently regulated. Although we did not analyze particularly the transfer of Wnt pathway components, it is possible that, as cytonemes discussed above, TNTs also transport signaling components that could participate in the establishment and maintenance of TNTs in connected cells. Further studies should explore this hypothesis.
Our findings suggest that activation of Wnt/Ca2+ pathway regulates TNT formation and stability by modulating the interaction between βCaMKII and actin. Phosphorylation state of βCaMKII has been shown to modulate the actin‐binding activity of this protein (Shen & Meyer, 1999). Interestingly, by using different βCaMKII mutants we demonstrated that the actin‐binding activity of βCaMKII is necessary to induce an increase in the number of TNT‐connected neuronal CAD cells. This effect could be due to an upregulation of de novo formation of TNTs, or to an increase in the stabilization of the already formed TNTs. Our data indicate that TNTs have a longer half‐life when the actin‐binding activity of βCaMKII is not impaired. Furthermore, by super‐resolution microscopy we observed that GFP‐βCaMKII WT exhibits a punctuated expression colocalizing with phalloidin at the base of the TNTs. Conversely, the GFP‐βCaMKII T287D mutant, unable to bind actin, showed a diffuse signal throughout the cytosol and was distributed along the TNTs, similar to the pattern of the control GFP. These data suggest that βCaMKII stabilizes TNTs by binding to F‐actin filaments at the base of the protrusion. The increase in the stabilization of TNT could produce an increase in the number of TNT‐connected cells that is not necessarily linked to de novo formation of TNTs, as the increase in the number of connected cells could be due to the greater persistence of pre‐existing TNTs.
In the presence of a Wnt ligand, phosphorylation of CaMKII occurs rapidly and transiently. Thus, several hours after Wnt stimulation, CaMKII gets dephosphorylated and can re‐associate to actin again. Indeed, we observed a redistribution of βCaMKII evidenced by an increase in the colocalization with F‐actin, 4 h after Wnt treatment. Loss of the interaction between βCaMKII and actin could induce actin polymerization by increasing the amount of actin monomers available and allowing the access of actin‐polymerizing factors to the actin filaments (Sanabria et al, 2009). In fact, it has been previously shown that βCaMKII stabilizes F‐actin by limiting the access of actin‐regulatory proteins, and that this effect can be inhibited by autophosphorylation of βCaMKII, causing its dissociation from F‐actin (Kim et al, 2013). Moreover, the inhibition of CaMKII by KN‐93 can also interfere with the actin‐binding/stabilizing activity of βCaMKII (Lin & Redmond, 2008). We speculate that upon Wnt stimulation, detachment of βCaMKII from actin could induce an increase in actin polymerization, as suggested here by the increase in the ratio of F‐actin over G‐actin. This increase in actin polymerization could produce in turn, new TNTs, thus increasing the intercellular connectivity.
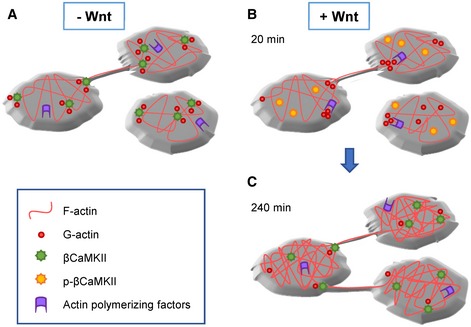
Based on these findings, we propose the following model for the sequence of events underlying TNT formation (Fig 9): (i) Exposure to the appropriate Wnt ligand induces the phosphorylation of CaMKII (all isoforms). (ii) Phosphorylated βCaMKII detaches from actin (both G‐ and F‐actin). (iii) Loss of interaction between βCaMKII and F‐actin could allow the access of actin regulator proteins to the actin filaments, as previously demonstrated (Kim et al, 2013). (iv) These would lead to an increase in actin polymerization events that cause an increment in the formation of TNT structures. (v) After dephosphorylation, βCaMKII reattach to F‐actin, increasing the stability of formed TNTs.
Figure 9. Model of Wnt's mechanism of action on TNT formation.
-
A–CIn the absence of Wnt (A), dephosphorylated βCaMKII is attached to actin (G‐ and F‐actin), helping to stabilize the actin cytoskeleton by impairing the access of actin‐polymerizing factors. In the presence of Wnt (B), βCaMKII gets rapidly phosphorylated and detaches from actin. Actin‐polymerizing factors can now promote the remodeling of the actin cytoskeleton, which could generate new TNT structures. After dephosphorylation of βCaMKII, several hours after Wnt stimulation (C), βCaMKII reattaches to actin and it accumulates at the bases of TNTs, thus contributing to the stabilization of this protrusions. The increase in TNT number and duration could impact the intercellular transfer of cargoes occurring through them.
Consequently, Wnt‐induced increase in TNT formation results in an increment on the intercellular vesicle transfer. Both, TNT formation and TNT‐meditated vesicle transfer can be blocked by the CaMKII inhibitor, KN‐93. The decrease in basal TNT formation induced by KN‐93 exposure suggests that there is a basal level of βCaMKII activity, which is required for the establishment of TNT connections in control conditions. Altogether, this indicates that βCaMKII constitutes a druggable target for the modulation of TNT formation and the regulation of TNT‐mediated transfer. The latter is particularly important for the developing of therapies designed toward impairment of the spreading of pathogens that are transported through TNTs.
To substantiate our findings in a more physiological condition, we analyzed TNT‐mediated vesicle and α‐syn fibrils transfer in primary cortical neurons from wild‐type and βCaMKII K.O. mice. Our data show that the interneuronal transfer of α‐syn fibrils is significantly impaired when βCaMKII is absent. We further provide the first demonstration that neurons can connect via TNT‐like structures and transfer different type of cargoes through them. To identify these connections and distinguish them from other neuronal processes in the absence of a specific TNT marker, we used in vitro cultured neurons at an early stage of development, which are more easily analyzed, as they have less protrusions and branches.
The involvement of Wnt pathway in regulating the formation of lamellar and filopodia protrusions that drive the early neuronal morphogenesis has been well documented (Shafer et al, 2011; Onishi et al, 2013; Stamatakou et al, 2015). Here, we show that in developing neurons Wnt/Ca2+ pathway, through the activation of CaMKII, can modulate the interneuronal TNT‐mediated transfer of vesicles and aggregated proteins. Whether these connections can be formed later in development, when the cells are mature, and whether they are induced in the brain by the presence of amyloid proteins as in the case of neuronal cells (Abounit et al, 2016a,2016b; Victoria et al, 2016) remains to be determined.
Materials and Methods
Reagents
Recombinant Wnt5a, Wnt7a, and sFRP‐2 proteins were obtained from R&D System. Hydrogen peroxide (30% in water) for cell culture was purchased from Thermo Fisher Scientific. Lithium chloride and the cell‐permeable inhibitors KN‐93 and JNK Inhibitor VII (TAT‐TI‐JIP153–163) were obtained from Merck‐Millipore.
Culture, treatments, and transfection of CAD cells
Mouse neuronal CAD (Cath.‐a‐differentiated) cells were kindly given by Hubert Laude (Institut National de la Recherche Agronomique, Jouy‐en‐Josas, France) and were cultured in Opti‐MEM™ (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Confluent CAD cells were mechanically detached and counted, and 200,000 cells were plated for 16 h on Ibidi μ‐dishes (Biovalley). Cells were treated for 4 h with soluble drugs that were added to the medium to reach the indicated concentrations. For transient transfections, 180,000 cells were plated for 6 h on Ibidi μ‐dishes. Transfections were performed with Lipofectamine 2000 (Invitrogen) following manufacturer's instructions. The various GFP‐tagged βCaMKII constructs (pEGFP‐C1‐CAMKIIbeta(T287D), pEGFP‐C1‐CAMKIIbeta(A303R), pEGFP‐C1‐CAMKIIbeta(K43R), and GFP‐C1‐CAMKIIbeta), as well as GFP‐vector and H2B‐mCherry constructs, were obtained from Addgene. After 24 h post‐transfection, cells were directly fixed, or in some cases, and were treated with soluble drugs added into the medium and incubated during 4 h before fixation.
Quantification of TNT‐connected cells in CAD cells
Cells were fixed for 15 min at 37°C in 2% PFA, 0.05% glutaraldehyde, and 0.2 M HEPES in PBS, and then, a second step of fixation with 4% PFA and 0.2 M HEPES in PBS was performed for 15 min at 37°C. Cells were carefully washed in PBS, labeled for 20 min at RT° with a 1:300 (v/v) solution of Wheat Germ Agglutinin (WGA) Alexa Fluor™ conjugate (Thermo Fisher Scientific) in PBS, washed again, and sealed with Aqua‐Poly/Mount (Polysciences) (Abounit et al, 2015). The whole cellular volume was imaged by acquiring Z‐stacks (0.4 μm steps) with an inverted confocal microscope (Zeiss LSM 700) controlled by the Zen acquisition software (Zeiss). After image acquisition, remote cells connected by TNTs were manually counted by using the semi‐automatized TNT counting tool of the ICY software (Quantitative Image Analysis Unit, Institut Pasteur. http://icy.bioimageanalysis.org/), as previously described (Abounit et al, 2015). TNT‐connected cells, i.e., cells connected by straight WGA‐labeled structures were identified by scanning through the Z‐stacks and only those connections that fitted the following criteria were counted as TNTs: (i) not touching the substratum, thus protrusions present above the first 3–4 Z‐stacks were considered; (ii) thin protrusions, thinner than 1 μm; (iii) continuous projections, which clearly start from one cell and uninterruptedly continue toward the other cell to form a “bridge”. TNT counting experiments have been coupled to vesicle transfer experiments, since material transfer is also an important criterion of TNTs which distinguish them from other types of protrusions, such as filopodia that do not allow material transfer between cells (Mattila & Lappalainen, 2008; Gallo, 2013; Jacquemet et al, 2015; Delage et al, 2016; Sartori‐Rupp et al, 2019). Data are presented as percentage of TNT‐connected cells (number of TNT‐connected cells over the total number of cells). Displayed images correspond to stack projections. Only linear corrections were applied using the ICY software.
Quantification of vesicle transfer in CAD cells
Acceptor cells were incubated for 30 min at 37°C with 10 μM CellTracker™ Green (Thermo Fisher Scientific) diluted in serum‐free medium or transfected with H2B‐mCherry plasmid as stated above. Donor cells were transfected or not with the aforementioned GFP‐fusion protein constructs and incubated for 30 min at 37°C with 1:3,000 (v/v) dilution of Vybrant™ DiD cell‐labeling solution (Thermo Fisher Scientific) in complete medium. Cells were carefully washed with warmed PBS, detached, counted, and plated on Ibidi μ‐dishes. Treatments were added as indicated. Then, the cells were fixed and stained with 1:300 (v/v) dilution of WGA‐Alexa Fluor™ conjugate or 1:5,000 (v/v) dilution of HCS CellMask™ Blue Stain (Thermo Fisher Scientific), both in PBS. Image acquisition was performed as detailed above. Transferred vesicles, i.e., acceptor cells containing DiD‐labeled vesicles, were detected with the Spot Detector script developed for ICY software.
Quantification of vinculin‐positive peripheral focal adhesion
Vinculin detection was performed as previously described (Delage et al, 2016; Zhu et al, 2018). Briefly, cells were plated rather sparse for 16 h on Ibidi μ‐dishes and then fixed with 4% PFA in PBS for 15 min at 37°C. Then, cells were permeabilized with 0.01% saponin in PBS containing 2% BSA (w/v) for 20 min at 37°C. After 1‐h incubation with mouse anti‐vinculin antibody (Sigma‐Aldrich) diluted 1:500 in PBS containing 0.01% saponin and 2% BSA (w/v), cells were thoroughly washed and incubated for 40 min with goat anti‐mouse Alexa Fluor©‐488 (ThermoFisher) diluted 1:500 in PBS containing 0.01% saponin and 2% BSA (w/v). Cells were washed and stained for 30 min with a 1:5,000 (v/v) solution of HCS CellMask™ Blue. Samples were imaged with an inverted confocal microscope (Zeiss LSM700), by focusing in the bottom of the cell (in contact with the plastic dish). Displayed images correspond to stack projections. Only linear corrections were applied, using the software ICY software. Vinculin‐positive peripheral focal adhesions were automatically detected and counted using an ICY software plugging.
Confocal imaging
After transfection, cells were fixed as stated above. The cells were labeled for 30 min at RT° with 1:250 (v/v) dilution of Rhodamine‐Phalloidin (Thermo Fisher Scientific) in PBS, washed, and mounted with Aqua‐Poly/Mount. For imaging the samples, the microscope used includes a Nikon C2 confocal system (Nikon Corporation). The confocal scan head is plugged to a Nikon TiE Eclipse inverted microscope. All confocal images were acquired using a 1,024 × 1,024 pixels format with an averaging of 2 frames with a pixel dwell time of 4.9 μs.
Super‐resolution imaging
Super‐resolution fluorescence imaging was performed with Conical Diffraction Microscopy (CODIM; Caron et al, 2014; Fallet et al, 2014). The BioAxial super‐resolution module (CODIM100, BioAxial) is an add‐on integrated to the Nikon confocal system previously described. The CODIM module acts as a powerful beam shaper generating local structured illumination. A sCMOS camera plugged at the back port of the microscope (Orca Flash 4.0, Hamamatsu Photonics) is used for the detection generating individual micro‐images for each scanning point containing independent information. The set of all micro‐images obtained from the scan procedure are processed and reconstructed by CODIM algorithm to generate a super‐resolved image. In both confocal and super‐resolution modalities, a 60 × 1.49NA Oil immersion Nikon Plan Apo TIRF objective was used to focus the laser beam and collect of the emitted fluorescence. The 488 and 561 excitation wavelengths of a multi laser engine (iChrome MLE, Toptica Photonics Inc.) are used for fluorescence excitation. The confocal image captures were performed using Nikon Nis‐Elements software (Nikon Instruments Europe). The laser power was properly chosen to be out of the saturation regime. Confocal and super‐resolution montages were subsequently built in ImageJ (NIH).
G‐Actin/F‐actin measurements
CAD cells seeded into 6‐well plates were transfected and treated as indicated. A G‐Actin/F‐actin in vivo assay kit (Cytoskeleton) was used following the manufacturer's instructions. Briefly, cells were lysed using a buffer from the kit and then homogenized with a 200‐μl pipette tip. Lysates were incubated for 10 min at 37°C and then centrifuged at 700 g for 5 min at RT°. Supernatants were then ultracentrifuged at 100,000 g for 1 h at 37°C, to separate G‐actin (supernatant) from F‐actin (pellet). After separation, a F‐actin depolymerization buffer was added to the pellets for resuspension. Then, pellet and supernatant samples were mixed with SDS sample buffer and 10 μl of each sample were run in Mini‐PROTEAN TGX Precast Gels (Bio‐Rad) and transferred to a PDVF membrane. Immunoblotting of the membrane was performed using a rabbit anti‐actin primary antibody (1:500; Cytoskeleton).
Western immunoblots
For the Western blot analysis, 300,000 cells were seeded for 16 h on 6‐well flat‐bottom plates (Falcon) and then treated with soluble drugs for the time points indicated. Cells were rinsed with PBS and then scraped in RIPA buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS). The resulting soluble fractions were centrifuged at 18,000 g for 5 min at 4°C, and then, the supernatants were taken as the whole cell lysate, mixed with Laemmli sample buffer (1 M Tris–HCl, pH 6.8, 20% glycerol, 4% SDS, 5% β‐mercaptoethanol, and 0.02% bromophenol blue), and boiled for 5 min. For Western blots, equal amounts of proteins (20 μg/lane) were separated on NuPAGE™ 4‐12% Bis‐Tris Protein Gels (Thermo Fisher Scientific) and transferred to PDVF membranes (Thermo Fisher Scientific), following standard procedures. Membranes were then blocked in Tris‐buffered saline containing 0.1% Tween‐20 and 5% non‐fat dried milk for 1 h and probed overnight at 4°C with primary antibodies. The membranes were then washed and exposed for 1 h at RT° to the anti‐rabbit peroxidase‐conjugated antibody (1:50,000; Jackson ImmunoResearch). The specific protein bands were visualized using the ECL‐immunoblotting chemiluminescence system (GE Healthcare Life sciences) and the ImageQuant LAS 500TM camera (GE Healthcare Life sciences). Primary antibodies used in this study were purchased from Cell Signaling Technology: rabbit anti‐non‐phospho‐(Active)‐β‐Catenin (1:1,000), rabbit anti‐(pan)‐CaMKII (1:1,000), rabbit anti‐phospho‐(α‐β‐γ)‐CaMKII (1:1,000), rabbit anti‐SAPK/JNK (1:1,000), rabbit anti‐phospho‐SAPK/JNK (1:1,000), rabbit anti‐phospho‐GSK3β (1:1,000), and rabbit anti‐GAPDH (1:500). To determine the apparent molecular weights of the protein bands, a PageRuler plus prestained protein ladder (Thermo Fisher Scientific) was used.
qPCR analysis
RNA (1 μg) was reverse transcribed using M‐MLV reverse transcriptase (Thermo Fisher Scientific) according to the manufacturer's instructions. cDNA was used for quantitative PCR using SYBR Green mix buffer (LightCycler® 480 SYBR Green I Master) in a total reaction volume of 9 μl. The PCR was carried out as follows: 8 min at 95°C followed by 50 cycles of the following: 15 s at 95°C, 15 s at 60°C, and 15 s at 72°C. Specificity of the PCR products was checked by melting curve analysis using the following program: 65°C increasing by 0.11°C/s to 97°C. The expression level of each mRNA was normalized to that of or murine RPLP0 mRNA (large ribosomal protein, subunit P0) expression. Efficiency for each set of primers was determined by using a dilution curve of cDNA, and expression levels for each gene were calculated according to the −ΔΔC T method using the experimentally calculated efficiency.
Expression, purification, preparation, and labeling of α‐syn fibrils
Human wild‐type α‐syn in pRK172, a construct containing α‐syn that lacks cysteine because of mutagenesis of codon 136 (TAC to TAT) as described previously (Masuda et al, 2006), was transformed into Escherichia coli BL21 (DE3). Expression and purification were performed as described previously (Nonaka et al, 2005, 2010). The protein concentrations of monomeric α‐syn were determined by RP‐HPLC as described previously (Nonaka et al, 2005, 2010). Purified recombinant α‐syn monomers (~5 mg/ml) containing 30 mM Tris–HCl, pH 7.5, 10 mM DTT, and 0.1% sodium azide were incubated at 37 °C with shaking using a horizontal shaker (TAITEC) at 200 rpm. After incubation for 7 days, the samples were ultracentrifuged at 100,000 g for 20 min at room temperature, and the ppt fraction was recovered as α‐syn fibrils. They were resuspended in saline and ultracentrifuged again. The pellets were re‐suspended in saline and sonicated with an ultrasonic homogenizer (VP‐5S, TAITEC). The fibrils were labeled with Alexa Fluor 488 or 568 Protein Labeling Kit (Invitrogen) according to the manufacturer's instructions. After incubation with Alexa Fluor dye, the samples were ultracentrifuged again. The pellets were re‐suspended in 30 mM Tris–HCl, pH 7.5 and ultracentrifuged again. The labeled α‐syn fibrils were re‐suspended in saline containing 0.1% sodium azide. The protein concentration of the fibrils was determined by RP‐HPLC as described previously (Nonaka et al, 2005, 2010). To check the in vitro seeding activity of the labeled fibrils, the fibrils (3 μg) were added to 100 μl of 1 mg/ml α‐syn monomer in 30 μM thioflavin T and 80 mM HEPES, pH 7.5. Amyloid‐like fibril formation was continuously monitored in terms of thioflavin T fluorescence (excitation 442 nm, emission 485 nm) with a plate reader (Varioskan Flash, Thermo Scientific).
Immediately before the experiments, fibrils were diluted in the neuronal medium and sonicated for 5 min at 80% amplitude with a pulse cycle of 5 s on and 2 s off in a Vibra‐Cell 75041 ultrasonic water bath (Bioblocks Scientific).
Animals
C57BL/6 wild‐type mice from Institut Pasteur (Paris, France) in‐house colony were used for all primary neuron experiments, unless otherwise indicated. Homozygous βCaMKII (−/−) exon 2 knock‐out (βCaMKII K.O.) mice and WT littermates were generated and maintained at the facilities of Erasmus Medical Center (Rotterdam, The Netherlands; Kool et al, 2016). All animals were housed in cages with filter tops in a ventilated rack and maintained on food and water ad libitum. Handling of animals was performed in compliance with the guidelines of animal care set by the European Union and approved by the Ethics Committees of Institut Pasteur and Erasmus Medical Center.
Primary neuronal cultures
Primary cortical neurons were prepared from mice at embryonic day 17. Briefly, after removing and decapitating the fetuses, the meninges were stripped off the brain. For C57BL/6 mice, cortices were pooled prior homogenization, while for βCaMKII K.O. (n = 1) and WT (n = 2) mice, cortices were prepared separately. The cortices were digested in a mixture of trypsin (0.25%; Gibco) and DNaseI (1 mg/ml; Roche Diagnostics) for 15 min at 37°C, and then dissociated by trituration with two fire‐polished Pasteur pipettes of different diameter. The dissociated cells were labeled in suspension or directly plated (45,000/cm2) in 4‐well dishes with PLL‐coated (1 mg/ml; Sigma‐Aldrich) coverslips and incubated with Neurobasal medium supplemented with B‐27, Glutamax®, and penicillin/streptomycin (all from Gibco). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Immunocytochemistry assays in neurons
Neurons of 1DIV were rinsed with PBS, fixed with 4% PFA for 20 min, and then permeabilized and blocked for 1 h in blocking solution (PBS with 2% BSA and 0.01% saponin). Then, cells were incubated overnight at 4°C with primary antibodies diluted in blocking solution. After the cells were rinsed with PBS, antibody binding was detected with the respective Alexa‐conjugated secondary antibody for 1 h at RT°. Cells were then rinsed with PBS and stained with 1:300 (v/v) dilution of WGA‐Alexa Fluor™ 647 conjugate (Thermo Fisher Scientific), and the nuclei were counterstained with 1:2,500 (v/v) dilution of DAPI (Sigma‐Aldrich). Samples were mounted using aqua‐poly/mount (Polysciences). The antibodies used were rabbit anti‐MAP‐2 (1:500; Sigma‐Aldrich), mouse anti‐β‐III‐tubulin (1:250; Sigma‐Aldrich), and mouse monoclonal anti‐βCaMKII CB‐beta‐1 (1:500; ThermoFisher). Images were acquired with an inverted confocal microscope (Zeiss LSM 700) and further processed with ICY software.
Quantification of transfer in neurons
In order to obtain two different cell populations, immediately after dissection, neurons were separated to be labeled in suspension. Acceptor neurons were labeled with 10 μM CellTracker™ Green or 1 μM Alexa Fluor 488‐tagged α‐syn fibrils, in neuronal medium. Donor neurons were labeled with 1:3,000 (v/v) dilution of Vybrant™ DiI cell‐labeling solution (Thermo Fisher Scientific) or 1 μM Alexa Fluor 568‐tagged α‐syn fibrils, in neuronal medium. Cells incubated for 30 min at 37°C in rotation using a Tube Revolver/Rotator (Thermo Fisher Scientific) at minimum speed. The cells were centrifuged at 1,000 rpm for 10 min at RT°, and then, the pellets were carefully washed with warmed neuronal medium. Cells were resuspended, counted, and plated on PLL‐coated coverslips (85,000/cm2). Neurons were cultured overnight, and then, treatments were added as indicated. After 24 h of culture, the cells were fixed and stained with 1:300 (v/v) dilution of WGA‐Alexa Fluor™ 647 conjugate or 1:250 (v/v) dilution of Alexa Fluor™ 647 Phalloidin (Thermo Fisher Scientific), both in PBS. Image acquisition was performed using an inverted confocal microscope (Zeiss LSM 700). Transfer analysis was performed using the Fiji software.
Live imaging
For time‐lapse fluorescent microscopy, transfected CAD cells plated on Ibidi μ‐dishes or 1 DIV primary neurons loaded in suspension with 1 μM Alexa Fluor 568‐tagged α‐syn fibrils and plated on 35‐mm glass‐bottom dish with 10‐mm micro‐well (MatTek Corporation) were used. Cells were labeled with 1:300 (v/v) dilution of WGA‐Alexa Fluor™ conjugate in the corresponding media and put in an open chamber equilibrated in 5% CO2 and maintained at 37°C. Time‐lapse sequences were recorded every 25 s up to 50 s apart, using a Nikon Eclipse Ti inverted microscope with a ×60 1.4 NA PL‐APO VC objective controlled by Metamorph software (Universal Imaging). Image montages were built afterward in ImageJ software (NIH).
Statistical analyses
Statistical analyses were performed using the GraphPad Prism version 6 software. All the results are expressed as the mean ± SEM. For comparisons between two groups, Student's t‐test was used. For comparisons between more than two groups, one‐way ANOVA with Tukey's post hoc analysis was employed. Differences were considered significant at *P ≤ 0.05 or **P ≤ 0.01.
Author contributions
JYV designed and performed the experiments, analyzed the data, lead the project, and wrote the manuscript. FL and Y‐JW designed and performed the experiments, analyzed the data, and helped to write the manuscript. GC performed and analyzed qPCR and WB experiments, provided scientific input, and contributed with the manuscript. TN prepared the α‐synuclein tagged fibrils. SB acquired and analyzed the CODIM images. SS gave technical assistance with WB and IF experiments. MH gave feedback on the manuscript. GMW provided the βCaMKII knock‐out mice and gave feedback on the manuscript. CT provided reagents and gave feedback on the manuscript. CZ conceived and coordinated the research, discussed experiments and results, provided financial support, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 5
Acknowledgements
The authors are thankful to Dr. Seng Zhu and Shaarvari Bhat for their help in the acquisition of live imaging experiments. This work was supported by the Agence Nationale de la Recherche [ANR‐16‐CE16‐0019‐01], Fondation pour la Recherche Médicale [FRM‐2016‐DEQ20160334896] and grants from LECMA‐Vaincre Alzheimer and France Alzheimer foundations to CZ. Funding from E‐Rare/ERANET (project SIRD) to CT. Grant‐in‐Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) (JP26117005) from MEXT and Grant‐in‐Aid for Scientific Research on Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) (JP14533254) from AMED and JST CREST (JP18071300) to MH. Grant from Brain Science Foundation to TN. FL was recipient of Marie Skłodowska‐Curie individual fellowships and is funded through the Marie Skłodowska‐Curie Action COFUND 2015 (EU project 713366 – InterTalentum). Y‐JW was recipient of Pasteur‐Roux‐Cantarini fellowships.
The EMBO Journal (2019) 38: e101230
References
- Abounit S, Zurzolo C (2012) Wiring through tunneling nanotubes–from electrical signals to organelle transfer. J Cell Sci 125: 1089–1098 [DOI] [PubMed] [Google Scholar]
- Abounit S, Delage E, Zurzolo C (2015) Identification and characterization of tunneling nanotubes for intercellular trafficking. Curr Protoc Cell Biol 67: 12.10.1‐21 [DOI] [PubMed] [Google Scholar]
- Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo‐Marin J‐C, Melki R, Zurzolo C (2016a) Tunneling nanotubes spread fibrillar α‐synuclein by intercellular trafficking of lysosomes. EMBO J 35: 2120–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abounit S, Wu JW, Duff K, Victoria GS, Zurzolo C (2016b) Tunneling nanotubes: a possible highway in the spreading of tau and other prion‐like proteins in neurodegenerative diseases. Prion 10: 344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariazi J, Benowitz A, De Biasi V, Den Boer ML, Cherqui S, Cui H, Douillet N, Eugenin EA, Favre D, Goodman S et al (2017) Tunneling nanotubes and gap junctions‐their role in long‐range intercellular communication during development, health, and disease conditions. Front Mol Neurosci 10: 333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger CF, von Maltzahn J, Dumont NA, Stark DA, Wang YX, Nhan K, Frenette J, Cornelison DDW, Rudnicki MA (2014) Wnt7a stimulates myogenic stem cell motility and engraftment resulting in improved muscle strength. J Cell Biol 205: 97–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgesius NZ, van Woerden GM, Buitendijk GHS, Keijzer N, Jaarsma D, Hoogenraad CC, Elgersma Y (2011) βCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting αCaMKII to synapses. J Neurosci 31: 10141–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caricasole A, Ferraro T, Iacovelli L, Barletta E, Caruso A, Melchiorri D, Terstappen GC, Nicoletti F (2003) Functional characterization of WNT7A signaling in PC12 cells: interaction with A FZD5 x LRP6 receptor complex and modulation by Dickkopf proteins. J Biol Chem 278: 37024–37031 [DOI] [PubMed] [Google Scholar]
- Caron J, Fallet C, Tinevez J‐Y, Moisan L, Braitbart LPO, Sirat GY, Shorte SL (2014) Conical diffraction illumination opens the way for low phototoxicity super‐resolution imaging. Cell Adh Migr 8: 430–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani L, Boyle KA, Dickins E, Sahores M, Anane D, Lopes DM, Gibb AJ, Salinas PC (2011) Wnt7a signaling promotes dendritic spine growth and synaptic strength through Ca2+/Calmodulin‐dependent protein kinase II. Proc Natl Acad Sci USA 108: 10732–10737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C (2013) Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci 126: 3678–3685 [DOI] [PubMed] [Google Scholar]
- Delage E, Cervantes DC, Pénard E, Schmitt C, Syan S, Disanza A, Scita G, Zurzolo C (2016) Differential identity of Filopodia and Tunneling Nanotubes revealed by the opposite functions of actin regulatory complexes. Sci Rep 6: 39632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieriks BV, Park TI‐H, Fourie C, Faull RLM, Dragunow M, Curtis MA (2017) α‐synuclein transfer through tunneling nanotubes occurs in SH‐SY5Y cells and primary brain pericytes from Parkinson's disease patients. Sci Rep 7: 42984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallet C, Caron J, Oddos S, Tinevez J‐Y, Moisan L, Sirat GY, Braitbart PO, Shorte SL (2014) Conical diffraction as a versatile building block to implement new imaging modalities for superresolution in fluorescence microscopy. In Nanoimaging and Nanospectroscopy II p 916905. International Society for Optics and Photonics Available at: https://www.spiedigitallibrary.org/conference-proceedings-of-spie/9169/916905/Conical-diffraction-as-a-versatile-building-block-to-implement-new/10.1117/12.2061059.short [Accessed June 20, 2018]
- Ferrari ME, Bernis ME, McLeod F, Podpolny M, Coullery RP, Casadei IM, Salinas PC, Rosso SB (2018) Wnt7b signalling through Frizzled‐7 receptor promotes dendrite development by coactivating CaMKII and JNK. J Cell Sci 131: jcs216101 [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer K‐U, Myers JW, Ferrell JE, Schulman H, Meyer T (2003) Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39: 283–297 [DOI] [PubMed] [Google Scholar]
- Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN (2004) Comparative analyses of the three‐dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2 + ‐calmodulin‐dependent protein kinase II. J Biol Chem 279: 12484–12494 [DOI] [PubMed] [Google Scholar]
- Galli LM, Barnes T, Cheng T, Acosta L, Anglade A, Willert K, Nusse R, Burrus LW (2006) Differential inhibition of Wnt‐3a by Sfrp‐1, Sfrp‐2, and Sfrp‐3. Dev Dyn 235: 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G (2013) Mechanisms underlying the initiation and dynamics of neuronal filopodia: from neurite formation to synaptogenesis. Int Rev Cell Mol Biol 301: 95–156 [DOI] [PubMed] [Google Scholar]
- Gerdes H‐H, Rustom A, Wang X (2013) Tunneling nanotubes, an emerging intercellular communication route in development. Mech Dev 130: 381–387 [DOI] [PubMed] [Google Scholar]
- Godoy JA, Arrázola MS, Ordenes D, Silva‐Alvarez C, Braidy N, Inestrosa NC (2014) Wnt‐5a ligand modulates mitochondrial fission‐fusion in rat hippocampal neurons. J Biol Chem 289: 36179–36193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J et al (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11: 328–336 [DOI] [PubMed] [Google Scholar]
- Gousset K, Marzo L, Commere P‐H, Zurzolo C (2013) Myo10 is a key regulator of TNT formation in neuronal cells. J Cell Sci 126: 4424–4435 [DOI] [PubMed] [Google Scholar]
- Grainger S, Willert K (2018) Mechanisms of Wnt signaling and control. Wiley Interdiscip Rev Syst Biol Med 10: e1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Xiong W, Yu X, Espinoza‐Lewis R, Liu C, Gu S, Nishita M, Suzuki K, Yamada G, Minami Y et al (2008) Wnt5a regulates directional cell migration and cell proliferation via Ror2‐mediated noncanonical pathway in mammalian palate development. Development 135: 3871–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Sakai T, Tsukasaki Y, Watanabe TM, Ikebe M (2017) Myosin X is recruited to nascent focal adhesions at the leading edge and induces multi‐cycle filopodial elongation. Sci Rep 7: 13685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y (2004) The Wnt/beta‐catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131: 2791–2801 [DOI] [PubMed] [Google Scholar]
- Huang DL, Bax NA, Buckley CD, Weis WI, Dunn AR (2017) Vinculin forms a directionally asymmetric catch bond with F‐actin. Science 357: 703–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemet G, Hamidi H, Ivaska J (2015) Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr Opin Cell Biol 36: 23–31 [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21: 1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kele J, Andersson ER, Villaescusa JC, Cajanek L, Parish CL, Bonilla S, Toledo EM, Bryja V, Rubin JS, Shimono A et al (2012) SFRP1 and SFRP2 dose‐dependently regulate midbrain dopamine neuron development in vivo and in embryonic stem cells. Stem Cells 30: 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Conte I, Carter T, Bayer KU, Molloy JE (2016) Multiple CaMKII binding modes to the actin cytoskeleton revealed by single‐molecule imaging. Biophys J 111: 395–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ho D‐H, Suk J‐E, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee H‐J et al (2013) Neuron‐released oligomeric α‐synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4: 1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool MJ, van de Bree JE, Bodde HE, Elgersma Y, van Woerden GM (2016) The molecular, temporal and region‐specific requirements of the beta isoform of Calcium/Calmodulin‐dependent protein kinase type 2 (CAMK2B) in mouse locomotion. Sci Rep 6: 26989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg TB, Roy S (2014) Cytonemes as specialized signaling filopodia. Development 141: 729–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y‐C, Redmond L (2008) CaMKIIbeta binding to stable F‐actin in vivo regulates F‐actin filament stability. Proc Natl Acad Sci USA 105: 15791–15796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, Zurzolo C (2017) α‐Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol 134: 789–808 [DOI] [PubMed] [Google Scholar]
- Marzo L, Gousset K, Zurzolo C (2012) Multifaceted roles of tunneling nanotubes in intercellular communication. Front Physiol 3: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M, Dohmae N, Nonaka T, Oikawa T, Hisanaga S, Goedert M, Hasegawa M (2006) Cysteine misincorporation in bacterially expressed human alpha‐synuclein. FEBS Lett 580: 1775–1779 [DOI] [PubMed] [Google Scholar]
- Mattes B, Dang Y, Greicius G, Kaufmann LT, Prunsche B, Rosenbauer J, Stegmaier J, Mikut R, Özbek S, Nienhaus GU et al (2018) Wnt/PCP controls spreading of Wnt/β‐catenin signals by cytonemes in vertebrates. Elife 7: e36953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PK, Lappalainen P (2008) Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 9: 446–454 [DOI] [PubMed] [Google Scholar]
- Mezzacappa C, Komiya Y, Habas R (2012) Activation and function of small GTPases Rho, Rac, and Cdc42 during gastrulation. Methods Mol Biol 839: 119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikels AJ, Nusse R (2006) Wnts as ligands: processing, secretion and reception. Oncogene 25: 7461–7468 [DOI] [PubMed] [Google Scholar]
- Nishita M, Yoo SK, Nomachi A, Kani S, Sougawa N, Ohta Y, Takada S, Kikuchi A, Minami Y (2006) Filopodia formation mediated by receptor tyrosine kinase Ror2 is required for Wnt5a‐induced cell migration. J Cell Biol 175: 555–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, Iwatsubo T, Hasegawa M (2005) Ubiquitination of alpha‐synuclein. Biochemistry 44: 361–368 [DOI] [PubMed] [Google Scholar]
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010) Seeded aggregation and toxicity of {alpha}‐synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem 285: 34885–34898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K‐I, Narayanan R, Lee SH, Murata K, Hayashi Y (2007) The role of CaMKII as an F‐actin‐bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci USA 104: 6418–6423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva CA, Vargas JY, Inestrosa NC (2013) Wnt signaling: role in LTP, neural networks and memory. Ageing Res Rev 12: 786–800 [DOI] [PubMed] [Google Scholar]
- Onishi K, Shafer B, Lo C, Tissir F, Goffinet AM, Zou Y (2013) Antagonistic functions of Dishevelleds regulate Frizzled 3 endocytosis via filopodia tips in Wnt‐mediated growth cone guidance. J Neurosci 33: 19071–19085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami J, Holmqvist S, Lindström V, Sigvardson J, Westermark GT, Ingelsson M, Bergström J, Roybon L, Erlandsson A (2017) Human astrocytes transfer aggregated alpha‐synuclein via tunneling nanotubes. J Neurosci 37: 11835–11853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes H‐H (2004) Nanotubular highways for intercellular organelle transport. Science 303: 1007–1010 [DOI] [PubMed] [Google Scholar]
- Sagar N, Pröls F, Wiegreffe C, Scaal M (2015) Communication between distant epithelial cells by filopodia‐like protrusions during embryonic development. Development 142: 665–671 [DOI] [PubMed] [Google Scholar]
- Sanabria H, Swulius MT, Kolodziej SJ, Liu J, Waxham MN (2009) {beta}CaMKII regulates actin assembly and structure. J Biol Chem 284: 9770–9780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori‐Rupp A, Cordero Cervantes D, Pepe A, Gousset K, Delage E, Corroyer‐Dulmont S, Schmitt C, Krijnse‐Locker J, Zurzolo C (2019) Correlative cryo‐electron microscopy reveals the structure of TNTs in neuronal cells. Nat Commun 10: 342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer B, Onishi K, Lo C, Colakoglu G, Zou Y (2011) Vangl2 promotes Wnt/planar cell polarity‐like signaling by antagonizing Dvl1‐mediated feedback inhibition in growth cone guidance. Dev Cell 20: 177–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284: 162–166 [DOI] [PubMed] [Google Scholar]
- Stamatakou E, Hoyos‐Flight M, Salinas PC (2015) Wnt signalling promotes actin dynamics during axon remodelling through the actin‐binding protein Eps8. PLoS One 10: e0134976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanganello E, Hagemann AIH, Mattes B, Sinner C, Meyen D, Weber S, Schug A, Raz E, Scholpp S (2015) Filopodia‐based Wnt transport during vertebrate tissue patterning. Nat Commun 6: 5846 [DOI] [PubMed] [Google Scholar]
- Stanganello E, Scholpp S (2016) Role of cytonemes in Wnt transport. J Cell Sci 129: 665–672 [DOI] [PubMed] [Google Scholar]
- Sutton LP, Honardoust D, Mouyal J, Rajakumar N, Rushlow WJ (2007) Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled‐3. J Neurochem 102: 153–169 [DOI] [PubMed] [Google Scholar]
- Tardivel M, Bégard S, Bousset L, Dujardin S, Coens A, Melki R, Buée L, Colin M (2016) Tunneling nanotube (TNT)‐mediated neuron‐to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun 4: 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobimatsu T, Fujisawa H (1989) Tissue‐specific expression of four types of rat calmodulin‐dependent protein kinase II mRNAs. J Biol Chem 264: 17907–17912 [PubMed] [Google Scholar]
- Victoria GS, Arkhipenko A, Zhu S, Syan S, Zurzolo C (2016) Astrocyte‐to‐neuron intercellular prion transfer is mediated by cell‐cell contact. Sci Rep 6: 20762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria GS, Zurzolo C (2017) The spread of prion‐like proteins by lysosomes and tunneling nanotubes: implications for neurodegenerative diseases. J Cell Biol 216: 2633–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggener CT, Dupree JL, Elgersma Y, Fuss B (2013) CaMKIIβ regulates oligodendrocyte maturation and CNS myelination. J Neurosci 33: 10453–10458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Symes AJ, Kane CA, Freeman A, Nariculam J, Munson P, Thrasivoulou C, Masters JRW, Ahmed A (2010) A novel role for Wnt/Ca2 + signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS One 5: e10456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cui J, Sun X, Zhang Y (2011) Tunneling‐nanotube development in astrocytes depends on p53 activation. Cell Death Differ 18: 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bukoreshtliev NV, Gerdes H‐H (2012) Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS One 7: e47429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widelitz R (2005) Wnt signaling through canonical and non‐canonical pathways: recent progress. Growth Factors 23: 111–116 [DOI] [PubMed] [Google Scholar]
- Wolf V, Endo Y, Rubin JS (2008) Purification and Wnt‐inhibitory activities of secreted frizzled‐related proteins. Methods Mol Biol 468: 31–44 [DOI] [PubMed] [Google Scholar]
- Yang G, Liu Y, Yang K, Liu R, Zhu S, Coquinco A, Wen W, Kojic L, Jia W, Cynader M (2012) Isoform‐specific palmitoylation of JNK regulates axonal development. Cell Death Differ 19: 553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang Y (2015) Tunneling nanotubes between rat primary astrocytes and C6 glioma cells alter proliferation potential of glioma cells. Neurosci Bulletin 31: 371–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chen D, Huang X‐M, Long F, Cai H, Yao W‐X, Chen Z‐C, Liao Z‐J, Deng Z‐Z, Tan S et al (2017) Wnt5a promotes cortical neuron survival by inhibiting cell‐cycle activation. Front Cell Neurosci 11: 281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Bhat S, Syan S, Kuchitsu Y, Fukuda M, Zurzolo C (2018) Rab11a‐Rab8a cascade regulates the formation of tunneling nanotubes through vesicle recycling. J Cell Sci 131: jcs215889 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 5