

Abstract
Purpose:
The natural history and prognosis of appendiceal adenocarcinomas (AA) differ from those of adenocarcinomas arising in other large bowel sites. We aimed to compare the molecular profiles exhibited by AAs and CRCs, or between the histopathological subtypes of AA.
Experimental Design:
A total of 183 samples from AA (46 adenocarcinoma, not otherwise specified (NOS), 66 pseudomyxoma peritonei (PMP), 44 mucinous adenocarcinoma (MU), and 27 signet ring cell carcinoma (SR)), 994 from right-sided colorectal cancer (R-CRC), and 1080 from left-sided CRC (L-CRC) were analyzed by next-generation sequencing (NGS) and immunohistochemical (IHC) markers. Microsatellite instability (MSI) and tumor mutational burden (TMB) were tested by NGS, and PD-L1 by IHC.
Results:
We observed high mutation rates in AA samples for KRAS (55%), TP53 (40%), GNAS (31%), SMAD4 (16%), and APC (10%). AA exhibited higher mutation rates in KRAS and GNAS, and lower mutation rates in TP53, APC, and PIK3CA (6%) than CRCs. PMP exhibited much higher mutation rates in KRAS (74%) and GNAS (63%), and much lower mutation rates in TP53 (23%), APC (2%), and PIK3CA (2%) than NOS. Alterations associated with immune checkpoint inhibitor response (MSI-high, TMB-high, PD-L1 expression) showed similar frequency in AA compared to L-CRC, but not R-CRC, and those of NOS were higher than other subtypes of AA and L-CRC.
Conclusion:
Molecular profiling of AA revealed different molecular characteristics than noted in R-CRC and L-CRC, and molecular heterogeneity between the histopathological subtypes of AA. Our findings may be critical to develop individualized approach for AA treatment.
Keywords: appendiceal adenocarcinoma, right-sided colorectal cancer, left-sided colorectal cancer, molecular profiling, mutation
Introduction
The natural history and prognosis of appendiceal adenocarcinomas (AA) differs from those of adenocarcinomas arising in other large bowel sites (1, 2). Compared to colorectal adenocarcinoma, AA more commonly are associated with peritoneal dissemination and increased mucin production, and are distinguished by the diagnostic classification as pseudomyxoma peritonei (PMP) (3, 4). Histological variants of appendiceal epithelial neoplasia include low- and high-grade mucinous neoplasms, goblet cell tumors, neuroendocrine neoplasms, adenoma, and adenocarcinomas (common colonic type, mucinous type, and signet ring cell carcinoma) (2, 5). The primary treatment for these neoplasms is surgical resection (6). Also in PMP, cytoreductive surgery (CRS) and hyperthermic intraperitoneal chemotherapy (HIPEC) are associated with favorable outcome when conducted in specialized centers of excellance (6-8). However, both patient selection and the expertise of the treating team are critical to best outcomes. Furthermore, in patients with metastatic AA or recurrent PMP, the most effective chemotherapy and molecular targeted therapy is controversial. At present, AA patients typically receive therapies approved for colorectal cancer (CRC) (3, 9, 10), although their efficacy, especially in low grade tumors, suggests lower vulnerability of these slow growing tumors to conventional therapies as compared to CRCs (11, 12).
Molecular profiling has been used effectively to identify novel treatment options for malignant diseases. In CRC, genetic profiling, such as RAS and BRAF alterations, has suggested phenotypic clustering with profiles that are prognostic as well as predictive of different susceptibilities to molecularly targeted therapeutics (13). In addition, assessment of the presence or absence of microsatellite instability (MSI), tumor mutational burden (TMB), and programmed death-ligand 1 (PD-L1) expression can be predictive of the likelihood that an individual patient will see a tumor response to treatment with immune checkpoint inhibitors (14). With molecular profiling, differences in the genetic and immune characteristics between the right-sided (R-CRC) and left-sided CRC (L-CRC) have been reported and have developed a personalized treatment strategy in CRC (15). On the contrary, studies on molecular profiling of AA have been handicapped due to the rarity of AA and assay failures in available PMP samples because of low cellularity (2), and these analyses have provided limited genetic data (16, 17). Although the prognosis varies according to the histopathological subtypes of AA (18), studies are lacking that correlate molecular profiles with AA subtypes (19). Identification of molecular alterations of AA is critical for the development and selection of more effective therapeutic strategies.
We performed molecular profiling of AA and compared it with those of R-CRC and L-CRC, using integrated data within a total of 183 samples from AA, 994 from R-CRC, and 1,080 from L-CRC. Our analysis demonstrates that histopathological and molecular classification of AA could be a key step towards personalized treatment strategies of AA.
Materials and Methods
Tumor Samples
Figure 1 summarizes the workflow of this study. Consecutive appendiceal cancer (N = 224) and CRC (N = 4,600) cases submitted to a commercial CLIA-certified laboratory (Caris Life Sciences, Phoenix, AZ) from April, 2015 to January, 2018 were retrospectively analyzed for their molecular alterations. Formalin fixed paraffin-embedded (FFPE) samples were sent for analysis from treating physicians around the world. The tissue diagnoses were submitted based on pathologic assessment of physicians who requested the assays and were further verified by a board-certified oncological pathologist at the Caris laboratory. A total of 183 AAs were analyzed and 41 tumors of neuroendocrine/goblet histology were excluded from their analysis. Included in the AA cohort were 66 PMP, 44 mucinous adenocarcinoma (MU), and 27 signet ring cell carcinoma (SR). Fourty-six tumors were determined to be adenocarcinoma, not otherwise specified (NOS) as no additional detailed histological features were noted (Supplementary Figure S1). R-CRC defined as tumors arising from the cecum to the hepatic flexure and transverse colon (N = 994) and L-CRC defined as those arising from the splenic flexure to the rectosigmoidal colon (N = 1,080) were analyzed; while tumors with origin unclearly annotated (N = 2,526) were excluded. Samples taken from original tumor sites were considered primary tumors and samples taken from organs other than the primary were considered metastases. Tissues were profiled by next-generation sequencing (NGS) and immunohistochemical (IHC) analysis using Caris Molecular Intelligence. Human subjects were anonymized prior to analysis. This study was conducted in accordance with guidelines of the Declaration of Helsinki, Belmont report and U.S. Common rule. In keeping with 45 CFR 46.101(b)(4), this study was performed utilizing retrospective, de-identified clinical data. Therefore this study is considered IRB exempt and no patient consent was necessary from the subject.
Figure 1. CONSORT diagram.
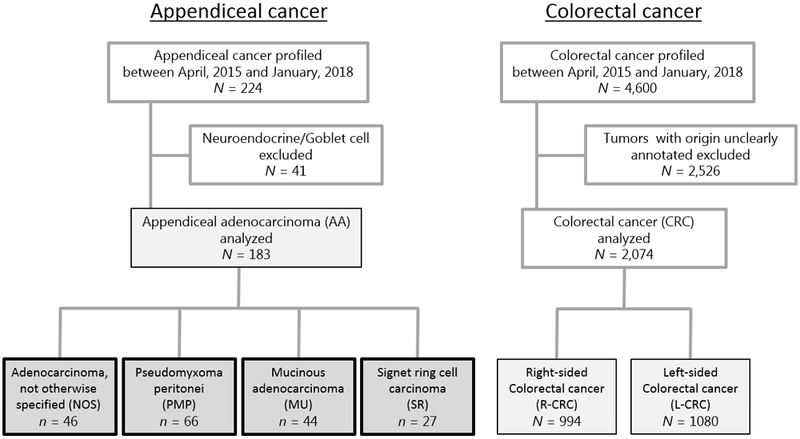
Flow chart showing the inclusion/exclusion criteria in this study.
Next-generation sequencing (NGS)
NGS was performed on genomic DNA isolated from FFPE tumor samples using the NextSeq platform (Illumina, Inc., San Diego, CA). A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with > 99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of 750 and an analytic sensitivity of 5%. Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques. Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as ‘pathogenic,’ ‘presumed pathogenic,’ ‘variant of unknown significance,’ ‘presumed benign,’ or ‘benign,’ according to the American College of Medical Genetics and Genomics (ACMG) standards. When assessing mutation frequencies of individual genes, ‘pathogenic,’ and ‘presumed pathogenic’ were counted as mutations while ‘benign’, ‘presumed benign’ variants and ‘variants of unknown significance’ were excluded.
Microsatellite instability (MSI)
MSI was examined using over 7,000 target microsatellite loci and compared to the reference genome hg19 from the University of California, Santa Cruz (UCSC) Genome Browser database, and the status was defined as MSI-high (MSI-H) or MSI-low/ microsatellite stable (MSS). The number of microsatellite loci that were altered by somatic insertion or deletion were counted for each sample. Only insertions or deletions that increased or decreased the number of repeats were considered. Genomic variants in the microsatellite loci were detected using the same depth and frequency criteria as used for mutation detection. MSI-NGS results were compared with results from over 2,000 matching clinical cases analyzed with traditional PCR-based methods. The threshold to determine MSI by NGS was determined to be 46 or more loci with insertions or deletions to generate a sensitivity of >95% and specificity of >90%.
Tumor mutation burden (TMB)
TMB was measured by counting all non-synonymous missense mutations found per tumor that had not been previously described as germline alterations (592 genes and 1.4 megabases [MB] sequenced per tumor). The threshold to define TMB-high (TMB-H) was greater than or equal to 17 mutations/MB and was established by comparing TMB with MSI by fragment analysis in CRC cases, based on reports of TMB having high concordance with MSI-H in CRC.
Immunohistochemical (IHC) analysis
IHC analysis was performed on full slides of FFPE tumor specimens using automated staining techniques (Benchmark XT, Ventana, and Autostainer Link 48, Dako). The primary antibody and details of evaluation for analysis are shown in Supplementary Table S1. Staining was scored for intensity (0 = no staining; 1+ = weak staining; 2+ = moderate staining; 3+ = strong staining) and staining percentage (0–100%). Results were categorized as positive or negative by defined thresholds specific to each marker, based on published clinical literature.
Statistical analysis
All statistical analyses were performed with SPSS v23 (IBM SPSS Statistics, Cary, USA), and all tests were two-sided at a significant level of 0.05. The comparison of age was analyzed using Student’s t-test and that of molecular profile between groups were analyzed using Fisher’s exact test. Cases with missing information in any of the categorical data were not included in the analysis.
Results
Patient and tumor characteristics
Baseline patient and tumor characteristics in regard to age, gender, and location of tumor sampling are shown in Supplementary Table S2. Patients with AA were significantly younger than patients with R-CRC (P < 0.001) and were more likely to be female than patients with L-CRC (P = 0.011). In addition, more AA samples were collected from metastatic sites than R-CRC (P < 0.001) and L-CRC (P < 0.001) samples. Between histopathological subtypes of AA, there is no significant difference in regard to age, gender, and location of tumor sampling.
Common gene mutations in AA, and comparison with R-CRC and L-CRC
The observed patterns of common gene mutations were totally different between AA, R-CRC, and L-CRC. (Table 1) The most prevalent mutations seen in AA were KRAS (55%), TP53 (40%), GNAS (31%), SMAD4 (16%), APC (10%), ARID1A (8%), RNF43 (7%), PIK3CA (6%) and BRAF (5%). Compared to both R- and L- CRCs, AA had significantly higher mutation rates in GNAS (31% vs 2% vs 1%) and SMAD4 (16% vs 11% vs 10%), and lower mutation rates in TP53 (40% vs 66% vs 75%), APC (10% vs 70% vs 83%), PIK3CA (6% vs 22% vs 17%), FBXW7 (3% vs 11% vs 9%), NRAS (1% vs 3% vs 5%), and AMER1 (0% vs 9% vs 2%). In addition, compared to R-CRC, AA had significantly lower mutation rates in ARID1A (8% vs 26%), BRAF (5% vs 17%), ATM (2% vs 7%), KMT2D (2% vs 7%), PTEN (1% vs 8%), MSH6 (1% vs 5%), HNF1A (1% vs 5%), PTCH1 (1% vs 5%), and CTNNB1 (0% vs 4%); and compared to L-CRC, AA had significantly higher mutation rates in KRAS (55% vs 43%) and RNF43 (7% vs 2%). Moreover, the mutation rates in TP53, GNAS, APC, PIK3CA, and AMER1 had significant differences between AA, R-CRC, and L-CRC. BRAF mutation rate was the highest in R-CRC (AA, R-CRC, L-CRC: 5%, 17%, 5%), and KRAS mutation rate was the lowest in L-CRC (AA, R-CRC, L-CRC: 55%, 56%, 43%).
Table 1.
Frequency of gene mutations in AA, R-CRC, and L-CRC.
| Gene | AA N = 183 |
R-CRC N = 994 |
L-CRC N = 1080 |
P value a | P value a | P value a |
|---|---|---|---|---|---|---|
| % | % | % | AA vs. R-CRC | AA vs. L-CRC | R-CRC vs. L-CRC | |
| TP53 | 40 | 66* | 75* | <0.001 | <0.001 | <0.001 |
| GNAS | 31 | 2* | 1* | <0.001 | <0.001 | 0.049 |
| SMAD4 | 16 | 11* | 10* | 0.042 | 0.029 | |
| APC | 10 | 70* | 83* | <0.001 | <0.001 | <0.001 |
| PIK3CA | 6 | 22* | 17* | <0.001 | <0.001 | 0.008 |
| FBXW7 | 3 | 11* | 9* | 0.002 | 0.015 | |
| NRAS | 1 | 3* | 5* | 0.048 | 0.004 | |
| AMER1 | 0 | 9* | 2* | <0.001 | 0.024 | <0.001 |
| ARID1A | 8 | 26* | 19 | 0.006 | 0.034 | |
| BRAF | 5 | 17* | 5 | <0.001 | <0.001 | |
| ATM | 2 | 7* | 5 | 0.012 | 0.041 | |
| KMT2D | 2 | 7* | 2 | 0.006 | <0.001 | |
| PTEN | 1 | 8* | 3 | <0.001 | <0.001 | |
| MSH6 | 1 | 5* | 2 | 0.016 | <0.001 | |
| HNF1A | 1 | 5* | 1 | 0.024 | <0.001 | |
| PTCH1 | 1 | 5* | 1 | 0.009 | <0.001 | |
| CTNNB1 | 0 | 4* | 1 | 0.004 | 0.001 | |
| KRAS | 55 | 56 | 43* | 0.004 | <0.001 | |
| RNF43 | 7 | 11 | 2* | 0.004 | <0.001 |
P value was based on Fisher’s exact test. Blanks are P > 0.05.
P < 0.05 compared to AA.
AA, appendiceal adenocarcinoma; L-CRC, left-sided colorectal cancer; R-CRC, right-sided colorectal cancer.
Common gene mutations in the histopathological subtypes of AA
We further evaluated the pattern of common gene mutations in the histopathological subtypes of AA (NOS, PMP, MU, and SR). (Table 2) Compared to NOS, PMP exhibited much higher mutation rates in KRAS (74% vs 44%) and GNAS (63% vs 7%), and much lower mutation rates in TP53 (23% vs 51%), APC (2% vs 22%), and PIK3CA (2% vs 15%); MU exhibited higher mutation rate in KRAS (64% vs 44%) and GNAS (25% vs 7%); and SR exhibited lower mutation rate in KRAS (15% vs 44%), GNAS (4% vs 7%), TP53 (33% vs 51%), APC (0% vs 22%), and PIK3CA (0% vs 15%). Notably, BRAF mutations were identified in NOS (7%), MU (9%), and SR (7%), but not identified in PMP.
Table 2.
Frequency of gene mutations in the histopathological subtypes of AA.
| Gene | NOS n = 46 |
PMP n = 66 |
MU n = 44 |
SR n = 27 |
P value a | P value a | P value a |
|---|---|---|---|---|---|---|---|
| % | % | % | % | NOS vs. PMP | NOS vs. MU | NOS vs. SR | |
| KRAS | 44 | 74* | 64 | 15* | 0.002 | 0.019 | |
| GNAS | 7 | 63* | 25* | 4 | <0.001 | 0.020 | |
| TP53 | 51 | 23* | 57 | 33 | 0.004 | ||
| SMAD4 | 15 | 15 | 20 | 11 | |||
| RNF43 | 9 | 6 | 7 | 4 | |||
| APC | 22 | 2* | 16 | 0* | <0.001 | 0.011 | |
| PIK3CA | 15 | 2* | 7 | 0* | 0.009 | 0.042 | |
| BRAF | 7 | 0 | 9 | 7 | |||
| ARID1A | 11 | 0 | 15 | 11 |
P value was based on Fisher’s exact test. Blanks are P > 0.05.
P < 0.05 compared to NOS.
AA, appendiceal adenocarcinoma; L-CRC, left-sided colorectal cancer; MU, mucinous adenocarcinoma; NOS, adenocarcinoma, not otherwise specified; PMP, pseudomyxoma peritonei; R-CRC, right-sided colorectal cancer; SR, signet ring cell carcinoma.
Immune profiling of AA, comparison with R-CRC and L-CRC, and differences between the histopathological subtypes of AA
The immune profile of AA was similar to that of L-CRC but not that of R-CRC. (Figure 2) MSI-H, TMB-H, and PD-L1 high expression rates were much lower in AA (MSI-H, TMB-H, PD-L1 high: 2.2%, 2.2%, 2.8%) than in R-CRC (MSI-H, TMB-H, PD-L1 high: 14.5%, 14.9%, 6.7%), but similar to L-CRC (MSI-H, TMB-H, PD-L1 high: 3.4%, 4.6%, 2.7%). The positive ratio of any of immune check point inhibitor markers (MSI-H, TMB-H, PD-L1 high) is much higher in R-CRC (19.8%) than in AA (5.1%) and in L-CRC (6.7%). Notably, some differences were observed between the histopathological subtypes of AA: PMP had no MSI-H or TMB-H case; and the positive ratio of any one marker (MSI-H, TMB-H, PD-L1 high) was higher in NOS (11.4%) than in PMP (1.6%), in MU (4.8%), and in SR (3.9%).
Figure 2. Immune profiling (the status of MSI, TMB, and PD-L1 expression) of AA, R-CRC and L-CRC, and the histopathological subtypes of AA.
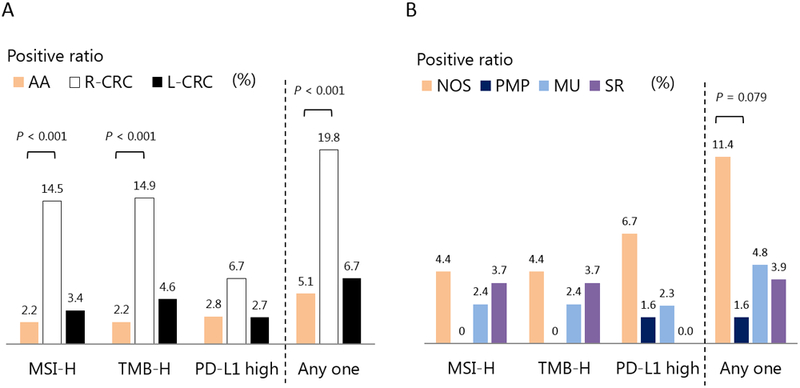
A: The characteristics of immune profiles in AA, R-CRC, and L-CRC. B: The characteristics of immune profiles in the histopathological subtypes of AA.
Abbreviations: AA, appendiceal adenocarcinoma; L-CRC, left-sided colorectal cancer; MSI, microsatellite instability; PD-L1, programmed death-ligand 1; R-CRC, right-sided colorectal cancer; TMB, tumor mutational burden.
Protein expression of chemotherapeutic sensitivity markers in AA, comparison with R-CRC and L-CRC, and differences between the histopathological subtypes of AA
As shown in Table 3, expression status of protein markers for chemotherapeutic sensitivity varied between AA, R-CRC, and L-CRC, and between the histopathological subtypes of AA. The positive ratios of ERCC1, TOPO1, PTEN, and MGMT were higher in AA than in R-CRC and L-CRC. Further, TS was overexpressed most frequently in R-CRC. Between the histopathological subtypes of AA, although the protein expressions were similar in NOS, MU and SR, those expressions were significantly different between NOS and PMP: ERCC1, TOPO1, PTEN, and MGMT were overexpressed in PMP, and TS was suppressed in PMP.
Table 3.
Frequency of protein expressions in AA, R-CRC, L-CRC, and the histopathological subtypes of AA.
| Protein | AA N = 183 |
R-CRC N = 994 |
L-CRC N = 1080 |
P value a | P value a | P value a |
|---|---|---|---|---|---|---|
| % | % | % | AA vs. R-CRC | AA vs. L-CRC | R-CRC vs. L-CRC | |
| TS | 19 | 31* | 18 | <0.001 | <0.001 | |
| ERCC1 | 32 | 16* | 15* | <0.001 | <0.001 | |
| TOPO1 | 65 | 52* | 53* | 0.001 | 0.003 | |
| PTEN | 88 | 65* | 66* | <0.001 | <0.001 | |
| MGMT | 69 | 52* | 56 | 0.047 |
| Protein | NOS n = 46 |
PMP n = 66 |
MU n = 44 |
SR n = 27 |
P value a | P value a | P value a |
|---|---|---|---|---|---|---|---|
| % | % | % | % | NOS vs. PMP | NOS vs. MU | NOS vs. SR | |
| TS | 26 | 8* | 23 | 27 | 0.015 | ||
| ERCC1 | 22 | 44* | 27 | 27 | 0.034 | ||
| TOPO1 | 53 | 76* | 70 | 52 | 0.022 | ||
| PTEN | 83 | 98* | 88 | 76 | 0.010 | ||
| MGMT | 50 | 95* | 60 | 38 | 0.006 |
P value was based on Fisher’s exact test. Blanks are P > 0.05.
P < 0.05 compared to AA or NOS.
AA, appendiceal adenocarcinoma; L-CRC, left-sided colorectal cancer; MU, mucinous adenocarcinoma; NOS, adenocarcinoma, not otherwise specified; PMP, pseudomyxoma peritonei; R-CRC, right-sided colorectal cancer; SR, signet ring cell carcinoma.
Comparison of molecular characteristics between the locations of tumor sampling
Finally, we analyzed the association of molecular characteristics with the location of tumor sampling (primary or metastatic sites). (Supplementary Table S3) In AA, the molecular characteristics had no difference between the locations of tumor sampling. On the other hand, in both R-CRC and L-CRC, the positive ratios of ARID1A mutation and MSI-H were significantly lower in metastatic sites.
Discussion
To the best of our knowledge, we performed so far the largest study to determine molecular profiling of AA and to compare it with R-CRC and L-CRC, using integrated data within a total of 183 samples from AA, 994 from R-CRC, and 1,080 from L-CRC. We found that AA had higher mutation rates in GNAS and KRAS, and lower mutation rates in TP53, APC, and PIK3CA than R-CRC and L-CRC; gene mutation rates differed between the histopathological subtypes of AA; and although the immune profile results (MSI, TMB, and PD-L1 expression status) of AA were similar to those of L-CRC (not to those of R-CRC), those of NOS had higher likelihood of overexpression than did other subtypes of AA and L-CRC. Our results support developing a personalized treatment strategy in patients with AA that is tailored to the individual’s histopathological subtypes.
The molecular feature of AA is different from CRC. As in the past reports (16, 20-22), our current study showed that AA had higher GNAS and KRAS mutation rates in comparison with CRC. The mutations of GNAS, a member of the G-protein family, cause constitutive activation of the protein kinase A (PKA) pathway via elevated levels of cyclic AMP (cAMP), and affected the MAPK and Wnt signaling pathways (20). GNAS mutation is common in benign tumors such as villous adenomas of the stomach (23) and colon (24), and intraductal papillary mucinous neoplasm (IPMN) of the pancreas (25, 26) and bile duct (27). Although the cAMP-PKA pathway can stimulate cell proliferation via MAPK or Wnt signalings, exogenous GNAS mutation in CRC (28) and pancreatic ductal adenocarcinoma (29) did not promote cell proliferation but increased expressions of MUC2 and MUC5AC (which induce mucin production). Additionally, Taki et al. demonstrated that transgenic mice with pancreas-specific GNAS and KRAS mutations developed a cystic pancreatic tumor (30). However, intraperitoneal injection of CRC cells with exogenous GNAS mutation in mice did not produce PMP but resulted in the formation of solid tumors (28). These data suggest that both GNAS and KRAS may contribute to the oncogenesis of PMP. Further, the mutation rates in GNAS and KRAS increased from NOS to MU and to PMP in our data, implying a functional interaction of these two oncogenes on mucin production. Moreover, several studies showed that GNAS and KRAS mutations were independent from pathological grade which is related to PMP activity (31-35). In support of these findings, both GNAS and KRAS mutations were not reported to be prognostic in patients with PMP (32, 36). GNAS and KRAS mutations might be a genetic feature of PMP but not be easy therapeutic targets.
Our data for the first time demonstrated that all the subtypes of AA had much lower TP53, APC, and PIK3CA mutation rates in comparison with CRC, and the mutation rates were further much lower in PMP and SR than in NOS and MU. In CRC, TP53, APC, and PIK3CA are key driver genes for “adenoma-to-carcinoma sequence”, and the mutation rates are high in any stage or tumor status (37, 38). Our findings suggest that carcinogenic mechanisms of AA may be different from that of CRC. Wilson et al. showed that GNAS mutation cooperated with inactivation of APC leading to colorectal tumorigenesis, but not carcinogenesis (39). In addition, Noguchi et al. demonstrated that TP53, PIK3CA, and AKT1 mutations were detected in peritoneal mucinous adenocarcinoma but not in PMP (21). Although GNAS mutation might contribute to APC-driven tumorigenesis, mutations in TP53 and/or genes related to the PI3K-AKT pathway may be necessary for malignant transformation in PMP. On the other hand, SR exhibited lower mutation rates not only in TP53, APC, and PIK3CA, but also in KRAS and GNAS. In addition, the mutation rate of BRAF was much lower (7%) compared to SR of CRC (about 40%) (40, 41), suggesting that SR might differ from both other histopathological subtypes of AA and SR of CRC in both development and treatment strategies.
Recently, immune checkpoint based therapy has demonstrated better survival and tolerance in subsets of patients with both solid and hematological malignancies. Studies further showed benefit in immune checkpoint blockade in patients whose tumors with MSI-H, TMB-H, and PD-L1 high expression (14). There is a need for molecular markers which can identify patients who are likely to benefit from immune checkpoint inhibitors. Therefore, overall immune profiling is an important emerging biomarker for cancer treatment. The appendix originated from the cecum, has many lymphoid clusters, and regulates IgA production in the large bowel sites (42), suggesting that AA may be subject to lymphocytic regulation more than R-CRC and L-CRC. However, interestingly, our study showed that immune profile of AA was similar to L-CRC but not R-CRC. In addition, NOS had a higher positive likelihood of expressing any of the evaluated immune check point inhibitor markers (11.4%) than other subtypes of AA (PMP: 1.6%, MU: 4.8%, SR: 3.9%) and L-CRC (6.7%). Although the carcinogenic mechanism of NOS is different from that of R-CRC and L-CRC, the immune characteristics might be closer to R-CRC. Further experimental studies are necessary to better understand this mechanism.
The efficacy of systematic chemotherapy and molecular targeted therapy for AA has not been well studied so far. Although a phase II trial in unresectable PMP suggested capecitabine combined with mitomycin C, the combination of fluorouracil (5-FU) and oxaliplatin/irinotecan is commonly used for the treatment of metastatic AA and PMP despite the paucity of efficacy data (3, 43). Our data showed that the positive ratios of ERCC1 and TOPO1 were higher in AA, especially in PMP, than in R-CRC and L-CRC, and that of TS was lower in AA, especially in PMP. ERCC1, a protein of the nucleotide excision repair (NER) complex, is essential for repairing platinum-DNA adducts and is involved in drug resistance to oxaliplatin (44); TOPO1, a molecular target of SN38, is a plausible positive predictive marker for irinotecan (45); and TS, a rate-limiting enzyme in the synthesis of pyrimidine nucleotides, is required for DNA synthesis and the activity is a negative predictive marker for 5-FU (44, 46). Thus, compared to CRCs, combination therapy with 5-FU and irinotecan (due to lower TS and higher ERCC1 and TOPO1) may be more effective for AA, especially for PMP treatment. However, the predictive protein expressions for chemosensitivities are still controversial even in CRC. As increased mucin production is responsible for major complications and fatal outcome in patients with PMP, GNAS-related pathways (cAMP-PKA, MAPK, and Wnt signaling pathways) might be a potential therapeutic target: PKA inhibitor (28), BIM-46174 (inhibitor of heterotrimeric G-protein complex) (47), and a MEK-inhibitor (48) were reported to cause mucin production reduction in tumors with GNAS mutation. Biomarker studies according to the histopathological and molecular subtypes are needed to determine individualized treatment strategies for AA.
We also compared the molecular profiles of samples from primary and metastatic tumors to identify features that are associated with distant metastasis in AA. In consensus with the data from past reports (38), a high level of genomic concordance was detected. However, ARID1A mutation and MSI-H were specifically enriched in primary tumors compared with distant metastases in both R-CRC and L-CRC, suggesting their potential protective effects. Recently, ARID1A, a subunit of the chromatin remodeling complex SWI/SNF, mutation was reported to contribute to impaired mismatch repair and mutator phenotype in cancers (49). ARID1A could be a promising target for novel treatment strategies for immune checkpoint based therapy.
Limitations in our study need to be mentioned. First, the retrospective study design could not exclude selection bias. Second, due to the loss of clinical data, such as precise TNM stage, treatment, and patient outcome, the direct effect of our findings in clinical perspectives is unclear, as well as the protective effect of ARID1A mutation and MSI-H for CRC patients. However, using the biggest dataset (183 samples from AA, 994 from R-CRC, and 1,080 from L-CRC), our results may support the past findings and compare molecular profiles between AA, R-CRC, and L-CRC, and also between minor histopathological subtypes of AA. In addition, our dataset included immune profile and protein expressions, which could lead to selection of treatment strategies. Further large-scale prospective studies with detailed clinical data may be warranted to validate our findings and provide us more information for treatment strategies of AA.
In conclusion, molecular profiling of AA revealed different characteristics from R-CRC and L-CRC, and heterogeneity between the histopathological subtypes of AA. Our data suggests that these molecular differences should be recognized in treating the patients with AA. Upon validation with clinical features, our findings may provide novel insight to develop individualized approach for AA treatment.
Supplementary Material
Translational relevance:
Compared to colorectal cancer (CRC), appendiceal adenocarcinoma (AA) is more associated with peritoneal dissemination and increased mucin production. However, AA patients typically receive therapies approved for CRC in spite of lower vulnerability. The molecular profiling of AA and to compare it with those of right-sided CRC (R-CRC) and left-sided CRC (L-CRC) is needed for personalized treatment strategy. We found that AA had higher mutation rates in GNAS and KRAS, and lower mutation rates in TP53, APC, and PIK3CA than CRC, and gene mutation rates differed between the histopathological subtypes. Although the immune profile results (microsatellite instability, tumor mutational burden, and PD-L1 expression status) of AA were similar to those of L-CRC (not to those of R-CRC), not otherwise specified AA (NOS) had higher overexpression of the markers than did other subtypes of AA and L-CRC. This molecular profiling may support developing a personalized treatment strategy in patients with AA.
Acknowledgments:
We thank all the specimen donors and research groups of data sets.
Funding: This work was supported by the Uehara Memorial Foundation, the National Cancer Institute (grant number P30CA014089), Dhont Family Foundation, San Pedro Peninsula Cancer Guild, and Daniel Butler Research Fund.
Abbreviations:
- AA
appendiceal adenocarcinoma
- CRC
colorectal cancer
- CRS
cytoreductive surgery
- FFPE
formalin fixed paraffin-embedded
- HIPEC
hyperthermic intraperitoneal chemotherapy
- IHC
immunohistochemistry
- IPMN
intraductal papillary mucinous neoplasm
- L-CRC
left-sided colorectal cancer
- MSI
microsatellite instability
- MSI-H
MSI-high
- MU
mucinous adenocarcinoma
- NGS
next-generation sequencing
- NOS
adenocarcinoma, not otherwise specified
- PD-L1
programmed death-ligand 1
- PMP
pseudomyxoma peritonei
- R-CRC
riight-sided colorectal cancer
- SR
signet ring cell carcinoma
- TMB
tumor mutational burden
- TMB-H
TMB-high
Footnotes
Conflict of interests: Dr. Xiu, Dr. Johnston, and Dr. Korn are employed by Caris Life Sciences. Dr. Marshall and Dr. Seeber are consultants for Caris Life Sciences. The other authors declare that they have no competing interests. The content is solely the responsibility of the authors.
Use of standardized official symbols:
In this study, HUGO (Human Genome Organisation)-approved official symbols for genes and gene products were used: all of which are described at www.genenames.org.
References
- 1.Compton C, Fenoglio-Preiser CM, Pettigrew N, Fielding LP. American Joint Committee on Cancer Prognostic Factors Consensus Conference: Colorectal Working Group. Cancer. 2000;88:1739–57. [DOI] [PubMed] [Google Scholar]
- 2.Turaga KK, Pappas SG, Gamblin T. Importance of histologic subtype in the staging of appendiceal tumors. Ann Surg Oncol. 2012;19:1379–85. [DOI] [PubMed] [Google Scholar]
- 3.Pietrantonio F, Maggi C, Fanetti G, Iacovelli R, Di Bartolomeo M, Ricchini F, et al. FOLFOX-4 chemotherapy for patients with unresectable or relapsed peritoneal pseudomyxoma. Oncologist. 2014;19:845–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carr NJ, Finch J, Ilesley IC, Chandrakumaran K, Mohamed F, Mirnezami A, et al. Pathology and prognosis in pseudomyxoma peritonei: a review of 274 cases. J Clin Pathol. 2012;65:919–23. [DOI] [PubMed] [Google Scholar]
- 5.Carr NJ, Bibeau F, Bradley RF, Dartigues P, Feakins RM, Geisinger KR, et al. The histopathological classification, diagnosis and differential diagnosis of mucinous appendiceal neoplasms, appendiceal adenocarcinomas and pseudomyxoma peritonei. Histopathology. 2017;71:847–58. [DOI] [PubMed] [Google Scholar]
- 6.Chua TC, Moran BJ, Sugarbaker PH, Levine EA, Glehen O, Gilly FN, et al. Early- and long-term outcome data of patients with pseudomyxoma peritonei from appendiceal origin treated by a strategy of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. J Clin Oncol. 2012;30:2449–56. [DOI] [PubMed] [Google Scholar]
- 7.Sugarbaker PH. New standard of care for appendiceal epithelial neoplasms and pseudomyxoma peritonei syndrome? Lancet Oncol. 2006;7:69–76. [DOI] [PubMed] [Google Scholar]
- 8.Bijelic L, Kumar AS, Stuart OA, Sugarbaker PH. Systemic Chemotherapy prior to Cytoreductive Surgery and HIPEC for Carcinomatosis from Appendix Cancer: Impact on Perioperative Outcomes and Short-Term Survival. Gastroenterol Res Pract. 2012;2012:163284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugarbaker PH, Bijelic L, Chang D, Yoo D. Neoadjuvant FOLFOX chemotherapy in 34 consecutive patients with mucinous peritoneal carcinomatosis of appendiceal origin. J Surg Oncol. 2010;102:576–81. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro JF, Chase JL, Wolff RA, Lambert LA, Mansfield PF, Overman MJ, et al. Modern systemic chemotherapy in surgically unresectable neoplasms of appendiceal origin: a single-institution experience. Cancer. 2010;116:316–22. [DOI] [PubMed] [Google Scholar]
- 11.Pietrantonio F, Berenato R, Maggi C, Caporale M, Milione M, Perrone F, et al. GNAS mutations as prognostic biomarker in patients with relapsed peritoneal pseudomyxoma receiving metronomic capecitabine and bevacizumab: a clinical and translational study. J Transl Med. 2016;14:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farquharson AL, Pranesh N, Witham G, Swindell R, Taylor MB, Renehan AG, et al. A phase II study evaluating the use of concurrent mitomycin C and capecitabine in patients with advanced unresectable pseudomyxoma peritonei. Br J Cancer. 2008;99:591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tosi F, Magni E, Amatu A, Mauri G, Bencardino K, Truini M, et al. Effect of KRAS and BRAF Mutations on Survival of Metastatic Colorectal Cancer After Liver Resection: A Systematic Review and Meta-Analysis. Clin Colorectal Cancer. 2017;16:e153–e63. [DOI] [PubMed] [Google Scholar]
- 14.Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stintzing S, Tejpar S, Gibbs P, Thiebach L, Lenz HJ. Understanding the role of primary tumour localisation in colorectal cancer treatment and outcomes. Eur J Cancer. 2017;84:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Mody K, de Abreu FB, Pipas JM, Peterson JD, Gallagher TL, et al. Molecular profiling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin Chem. 2014;60:1004–11. [DOI] [PubMed] [Google Scholar]
- 17.Raghav KP, Shetty AV, Kazmi SM, Zhang N, Morris J, Taggart M, et al. Impact of molecular alterations and targeted therapy in appendiceal adenocarcinomas. Oncologist. 2013;18:1270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Overman MJ, Fournier K, Hu CY, Eng C, Taggart M, Royal R, et al. Improving the AJCC/TNM staging for adenocarcinomas of the appendix: the prognostic impact of histological grade. Ann Surg. 2013;257:1072–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levine EA, Votanopoulos KI, Qasem SA, Philip J, Cummins KA, Chou JW, et al. Prognostic Molecular Subtypes of Low-Grade Cancer of the Appendix. J Am Coll Surg. 2016;222:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alakus H, Babicky ML, Ghosh P, Yost S, Jepsen K, Dai Y, et al. Genome-wide mutational landscape of mucinous carcinomatosis peritonei of appendiceal origin. Genome Med. 2014;6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noguchi R, Yano H, Gohda Y, Suda R, Igari T, Ohta Y, et al. Molecular profiles of high-grade and low-grade pseudomyxoma peritonei. Cancer Med. 2015;4:1809–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ang CS-P, Shen JP, Hardy-Abeloos CJ, Huang JK, Ross JS, Miller VA, et al. Genomic Landscape of Appendiceal Neoplasms. JCO Precision Oncology. 2018;2:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsubara A, Sekine S, Kushima R, Ogawa R, Taniguchi H, Tsuda H, et al. Frequent GNAS and KRAS mutations in pyloric gland adenoma of the stomach and duodenum. J Pathol. 2013;229:579–87. [DOI] [PubMed] [Google Scholar]
- 24.Yamada M, Sekine S, Ogawa R, Taniguchi H, Kushima R, Tsuda H, et al. Frequent activating GNAS mutations in villous adenoma of the colorectum. J Pathol. 2012;228:113–8. [DOI] [PubMed] [Google Scholar]
- 25.Furukawa T, Kuboki Y, Tanji E, Yoshida S, Hatori T, Yamamoto M, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai JH, Yuan RH, Chen YL, Liau JY, Jeng YM. GNAS Is frequently mutated in a specific subgroup of intraductal papillary neoplasms of the bile duct. Am J Surg Pathol. 2013;37:1862–70. [DOI] [PubMed] [Google Scholar]
- 28.Nishikawa G, Sekine S, Ogawa R, Matsubara A, Mori T, Taniguchi H, et al. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br J Cancer. 2013;108:951–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komatsu H, Tanji E, Sakata N, Aoki T, Motoi F, Naitoh T, et al. A GNAS mutation found in pancreatic intraductal papillary mucinous neoplasms induces drastic alterations of gene expression profiles with upregulation of mucin genes. PLoS One. 2014;9:e87875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taki K, Ohmuraya M, Tanji E, Komatsu H, Hashimoto D, Semba K, et al. GNAS(R201H) and Kras(G12D) cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm. Oncogene. 2016;35:2407–12. [DOI] [PubMed] [Google Scholar]
- 31.Bazan V, Migliavacca M, Zanna I, Tubiolo C, Grassi N, Latteri MA, et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann Oncol. 2002;13:1438–46. [DOI] [PubMed] [Google Scholar]
- 32.Singhi AD, Davison JM, Choudry HA, Pingpank JF, Ahrendt SA, Holtzman MP, et al. GNAS is frequently mutated in both low-grade and high-grade disseminated appendiceal mucinous neoplasms but does not affect survival. Hum Pathol. 2014;45:1737–43. [DOI] [PubMed] [Google Scholar]
- 33.Hosoda W, Sasaki E, Murakami Y, Yamao K, Shimizu Y, Yatabe Y. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows Arch. 2015;466:665–74. [DOI] [PubMed] [Google Scholar]
- 34.Nummela P, Saarinen L, Thiel A, Jarvinen P, Lehtonen R, Lepisto A, et al. Genomic profile of pseudomyxoma peritonei analyzed using next-generation sequencing and immunohistochemistry. Int J Cancer. 2015;136:E282–9. [DOI] [PubMed] [Google Scholar]
- 35.Pietrantonio F, Perrone F, Mennitto A, Gleeson EM, Milione M, Tamborini E, et al. Toward the molecular dissection of peritoneal pseudomyxoma. Ann Oncol. 2016;27:2097–103. [DOI] [PubMed] [Google Scholar]
- 36.Austin F, Mavanur A, Sathaiah M, Steel J, Lenzner D, Ramalingam L, et al. Aggressive management of peritoneal carcinomatosis from mucinous appendiceal neoplasms. Ann Surg Oncol. 2012;19:1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hadac JN, Leystra AA, Paul Olson TJ, Maher ME, Payne SN, Yueh AE, et al. Colon Tumors with the Simultaneous Induction of Driver Mutations in APC, KRAS, and PIK3CA Still Progress through the Adenoma-to-carcinoma Sequence. Cancer Prev Res (Phila). 2015;8:952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018;33:125–36 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson CH, McIntyre RE, Arends MJ, Adams DJ. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene. 2010;29:4567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvi MA, Loughrey MB, Dunne P, McQuaid S, Turkington R, Fuchs MA, et al. Molecular profiling of signet ring cell colorectal cancer provides a strong rationale for genomic targeted and immune checkpoint inhibitor therapies. Br J Cancer. 2017;117:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yalcin S, Onguru O. BRAF mutation in colorectal carcinomas with signet ring cell component. Cancer Biol Med. 2017;14:287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masahata K, Umemoto E, Kayama H, Kotani M, Nakamura S, Kurakawa T, et al. Generation of colonic IgA-secreting cells in the caecal patch. Nat Commun. 2014;5:3704. [DOI] [PubMed] [Google Scholar]
- 43.Tejani MA, ter Veer A, Milne D, Ottesen R, Bekaii-Saab T, Benson AB 3rd, et al. Systemic therapy for advanced appendiceal adenocarcinoma: an analysis from the NCCN Oncology Outcomes Database for colorectal cancer. J Natl Compr Canc Netw. 2014;12:1123–30. [DOI] [PubMed] [Google Scholar]
- 44.Shirota Y, Stoehlmacher J, Brabender J, Xiong YP, Uetake H, Danenberg KD, et al. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J Clin Oncol. 2001;19:4298–304. [DOI] [PubMed] [Google Scholar]
- 45.Braun MS, Richman SD, Quirke P, Daly C, Adlard JW, Elliott F, et al. Predictive biomarkers of chemotherapy efficacy in colorectal cancer: results from the UK MRC FOCUS trial. J Clin Oncol. 2008;26:2690–8. [DOI] [PubMed] [Google Scholar]
- 46.Bendardaf R, Lamlum H, Elzagheid A, Ristamaki R, Pyrhonen S. Thymidylate synthase expression levels: a prognostic and predictive role in advanced colorectal cancer. Oncol Rep. 2005;14:657–62. [DOI] [PubMed] [Google Scholar]
- 47.Prevost GP, Lonchampt MO, Holbeck S, Attoub S, Zaharevitz D, Alley M, et al. Anticancer activity of BIM-46174, a new inhibitor of the heterotrimeric Galpha/Gbetagamma protein complex. Cancer Res. 2006;66:9227–34. [DOI] [PubMed] [Google Scholar]
- 48.Ang C, Stollman A, Zhu H, Sarpel U, Scarborough B, Sahni G, et al. Clinical Benefit from Trametinib in a Patient with Appendiceal Adenocarcinoma with a GNAS R201H Mutation. Case Rep Oncol. 2017;10:548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.