

Abstract
Background:
Dry eye disease (DED) affects one third of population worldwide. In prior studies, experimental autoimmune lacrimal keratoconjunctivitis (EALK) induced by desiccating stress in mice has been used as a model of DED. This model is complicated by a requirement for exogenous epithelial cell injury and the administration of anticholinergic agents that have broad immunological effects.
Objective:
We sought to develop a novel mouse model of EALK, and to demonstrate the responsible immunological pathogenic mechanisms.
Methods:
CD4+CD45RBhigh naïve T cells with and without CD4+CD45RBloCD25+ regulatory T cells were adoptively transferred to C57BL/10 RAG2−/− mice. The eyes, draining lymph nodes, lacrimal glands and surrounding tissues of mice that spontaneously developed keratoconjuctivitis were evaluated for histopathological changes, cellular infiltration, and cytokine production in tissues and by isolated cells. Furthermore, the integrity of corneal nerves was evaluated by whole tissue immunofluorescence imaging. Gene-deficient naïve T cells or RAG2-hosts were evaluated to assess the roles of IFN-γ, IL-17A and IL-23 in disease pathogenesis. Finally, cytokine levels were determined in the tears of patients with DED.
Results:
EALK spontaneously developed in C57BL/10 RAG2−/− mice following adoptive of CD4+CD45RBhigh naïve T cells characterized by the infiltration of CD4+ T cells, macrophages, and neutrophils. In addition to lacrimal keratoconjunctivitis, mice also developed damage to the corneal nerve, which connects components of lacrimal functional unit (LFU). Pathogenic T cell differentiation was dependent on IL-23p40 and controlled by co-transferred CD4+CD45RBloCD25+ regulatory T cells (Tregs). Th17 rather than Th1 CD4+ cells were primarily responsible for EALK even though both IL-17 and IFN-γ were increased in inflammatory tissues likely due to their ability to drive the expression of CXC chemokines within the cornea, and the subsequent influx of myeloid cells. Consistent with the findings of this model, the tears of patients with DED had increased levels of inflammatory cytokines including IL-17A and TNFα.
Conclusion:
We describe a novel model of spontaneous EALK that supports a role for Th17 cells in disease pathogenesis, and that will contribute to our understanding of autoimmune lacrimal keratoconjunctivitis in many human eye diseases, including DED.
Keywords: Autoimmune lacrimal keratoconjunctivitis, Th17 cell
Graphical Abstract
Introduction
Desiccating stress is known to induce experimental autoimmune lacrimal keratoconjunctivitis (EALK) in mice model mimicking dry eye disease (DED). Evidence suggests that dry eye disease is an ocular surface inflammatory disease affecting the combined lacrimal functional unit (LFU; cornea, conjunctiva, eyelid and lacrimal glands).1–4 Autoimmune lacrimal keratoconjunctivitis also occurs in patients with a number of underlying autoimmune systemic diseases as rheumatoid arthritis, Sjögren’s syndrome, Wegener’s granulomatosis, systemic lupus erythematosus (SLE), graft versus host disease (GVHD), surgically induced keratoscleritis, inflammatory bowel disease (IBD) and others. Inflammatory damage to the cornea and lacrimal gland results in a number of clinical presentations that can include sight-threatening complications.1–3,5–8
The role of helper T cells in EALK has been studied in some patients and animal models,8–10 however, a direct role for CD4+ cells in the pathogenesis autoimmune keratitis has not been thoroughly investigated. In particular, studies have not excluded a role for B cells and CD8+ cells in models using desiccating stress using scopolamine hydrobromide injection and constant airflow. Furthermore, in desiccating DED experiments, the role of CD4+ Foxp3+ regulatory T cells (Tregs) was suggested by the resistance of Th17 to Treg suppression1 and the role of macrophages by clodronate liposome mediated depletion,2 however, no comprehensive spontaneous model of EALK had been developed that could directly address the contributions of effector T cells, Tregs, and antigen-presenting cells, in particular dendritic cells (DCs) to disease development.
The adoptive T cell transfer model of colitis in mice is well established as model for inflammatory bowel disease (IBD).11 Transfer the CD4+CD45RBhigh naïve T cells to RAG2−/− mice lacking T and B cells make it possible to investigate the role of CD4+ T cells in the development of inflammation in the absence of other lymphocyte populations, and co-transfer of CD4+Foxp3+ regulatory T cells (Foxp3+ Tregs) prevents disease development This model has contributed to our understanding of the characteristics of intestinal inflammation and the role of helper T cells in human IBD.12,13
More recently, studies from our laboratory have demonstrated an enhanced intestinal inflammatory phenotype in RAG−/− mice on a C57BL/10 compared to C57BL/6 background, as well as the development of the development of a psoriasiform dermatitis in this model, both of which could be ameliorated by anti-CR3 antibody treatment.14,15 The enhanced intestinal inflammation in the C57BL10 RAG−/− mice was associated with poor de novo induction of CD4+ Foxp3+ regulatory T cell and enhanced effector T cell expansion in the draining mesenteric lymph nodes following CD4+CD45RBhi (naïve) T cell transfer into the C57BL10 Rag−/− mice, and was likely due to enhanced IL-23 produced by migrating CD103+ DCs.15 Furthermore, on careful observation, a significant number of naïve CD4+ T cell-transferred C57BL/10 mice developed opacified corneas, which was not found following the co-transfer of Tregs, indicating the possibility that a spontaneous autoimmune inflammatory disease was affecting the eyes of these mice.
In the current study, we demonstrate the development of inflammation in the LFU including the cornea and ocular adnexa (eyelids conjunctiva and lacrimal gland) after the transfer the CD4+CD45RBhigh naïve T cells to C57BL/10 RAG2−/− mice. The inflammation is characterized by the infiltration of Gr-1+, CD4+, and F4/80+ cells, and co-transfer of naïve helper T cell cells with regulatory CD4+ ameliorated the development of EALK. Furthermore, IL-12/IL-23p40−/− RAG2−/− double knockout mice were protected from disease development, as were RAG2−/− mice transferred IL-17-deficent, but not IFN-γ-deficient T cells. Remarkably, we also found diffuse damage to the corneal nerves accompanied by an increase in expression of pro-inflammatory cytokines in corneal tissues over time after naïve CD4+ T cell transfer. Therefore, this model has the phenotype of autoimmune lacrimal keratoconjunctivitis and comprehensively shows the role of T cell and APC in the disease process. This spontaneous EALK murine model is important for furthering our understanding of the pathogenesis of autoimmune keratitis and dry eye in human and the precise role of helper T cells and endogenous microbiota in the disease processes. It may also be important for evaluating therapeutic candidates for human autoimmune lacrimal keratoconjunctivitis.
Methods
Mice.
C57BL/10SgSnAi and C57BL/10SgSnAi-(KO) RAG2 (RAG-2−/−) mice were purchased from Taconic Farms. IL-17A-deficient mice were the kind gift of Yoichiro Iwakura, The University of Tokyo. IFN-γ-deficient mice were from Taconic farms, and IL-12p40 x Rag2−/− were kindly provide by Alan Sher, NIAID, NIH. Age-matched female mice were used in all control experiments. All mice were maintained under specific pathogen-free conditions in the National Institute of Allergy and Infectious Diseases animal facilities, and all animal experiments were performed under an animal study proposal approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee.
Flow cytometry.
or flow cytometric analysis, the following antibodies were used. FITC-conjugated rat anti-mouse CD4 (GK1.5), and PE-conjugated rat anti-mouse CD45RB (16A) Abs were used for cell sorting. FITC-conjugated rat-anti–mouse IFN-γ (XMG1.2), PE-conjugated rat anti-mouse IL-17A (TC11–18H10.1), PE-Cy5–conjugated rat anti-mouse CD4, and allophycocyanin-conjugated hamster anti-mouse TCRβ (H57–597) Abs were used for intracellular cytokine staining. Isotype-matched IgG was used as a negative control. These Abs were purchased from eBioscience (San Diego, CA). Gr-1 (Ly6G/C; RB6–8C5) was purchased from BD Biosciences (San Jose, CA). F4/80 Ab was obtained from Serotec. Cells were stained and analysed on a FACSCaliber flow cytometer (BD Biosciences) using FlowJo software (FlowJo; Tree Star, Ashland, OR).
Induction of keratitis by adoptive transfer of CD4+CD45RBhigh cells into C57BL/10 RAG-2−/− mice.
CD4+ T cells were isolated from spleens of 5- to 8-wk-old female C57BL/10SgSnAi mice via negative selection using the MACS CD4 T cell isolation kit (Miltenyi Biotec Inc, Auburn, CA) and the AutoMACS separation system (Miltenyi Biotec). Enriched cells were subsequently stained for CD4+ and CD45RB and sorted by flow cytometry into fractions containing the 15% of CD4+ cells with the brightest CD45RB-staining (CD4+CD45RBhigh) and the 35% with the lowest CD45RB-staining (CD4+CD45RBlow) cells using a FACS Aria cell sorter (BD Biosciences). Sorted cells were >95% pure. A total of 2 × 105 CD45RBhigh cells or 2 × 105 CD45RBhigh plus 2 × 105 CD45RBlow were transferred i.p. to recipient 5- to 8-wk-old female RAG-2−/− mice. Non-transferred RAG-2−/− mice served as controls. Mice were followed for the development of inflammatory changes of eyelids, and haziness of the cornea, which correlates with histological evidence of keratitis.
Histological assessment of ocular tissues.
Mice were euthanized at the indicated time points. Eyeballs, eyelids, and lacrimal and salivary glands were collected and fixed in 10% neutral buffered formaldehyde. Coronal sections were made from each sample and stained with H&E.
RNA isolation and RT-PCR.
Total RNA extraction from four corneal tissues of each group was performed using the RNeasy mini kit (QIAGEN, Valencia, CA). cDNA was reverse-transcribed using Superscript III first-strand synthesis kit (Invitrogen). Quality and quantity of RNA was assessed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Real-time PCR analyses were performed using Applied Biosystems 7900 HT instrument (Applied Biosystems, Foster City, CA). All primers and probes used for gene expression analyses were purchased from Applied Biosystems. Relative target gene expression is expressed as 1/2ΔΔCt with GAPDH (glyceraldehyde 3-phosphate dehydrogenase) as endogenous control and non-transferred mice as reference and Ct is threshold cycle.
Isolation of lymphocytes from lymph nodes.
Lymphocytes were isolated as follows: Lymph nodes were mechanically disrupted in HBSS and filtered through 100-μm cell strainers. RBCs were removed using ACK lysing buffer (Lonza, Walkersville, MD), and lymphocytes were enriched by discontinuous density gradient centrifugation to remove dead cells using Percoll (Sigma-Aldrich, St. Louis, MO ) by isolating cells at the 40%:70% interface as per manufacturer’s instructions. Cells were washed and resuspended in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin and used in further analyses.
Intracellular staining and flow cytometric analysis.
For intracellular cytokine staining, cells were restimulated with PMA and ionomycin (both from Sigma-Aldrich, St. Louis, MO), together with monensin (BD Biosciences. San Jose, California, USA), for 5 h. After restimulation, cells were stained for IFN-γ or IL-17A, using BD Cytofix/Cytoperm Plus Fixation/Permeabilization Kit (BD Biosciences, San Jose, California, USA), according to the manufacturer’s instructions.
Tear Sample Collection and Analysis.
To collect tear samples, 30 μL of phosphate-buffered saline was instilled into the inferior fornix. A total of 20 μL of tear fluid and buffer were collected with a micropipette at the medial and lateral canthus. To minimize ocular surface irritation, we collected the mixture of tear fluid and buffer solution as soon as possible. The fluid was placed into a 1.5-mL Eppendorf tube and stored at −70-degrees C until further analysis by immunoassay. Cytokine concentrations were measured using a multiplex immunobead assay (BDTM Cytometric Bead Array Human Soluble Protein Flex Set; BD Biosciences, San Jose, California, USA) and flow cytometry (BDTM FACS LSR II; BD Biosciences). The lower limits of detection were as follows: IL-2, 2.6 pg/mL; IL-10, 4.5 pg/mL; IL-17α, 18.9 pg/mL; IL-4, 4.9 pg/mL; IFN-γ, 3.7 pg/mL; and TNF-α, 3.8 pg/mL. The value 0 was used for statistical comparison between tear samples with concentrations below the value of the lowest cytokine concentration (detection limit) in the linear portion of the standard curve. Samples were obtained under human protocol approved by the Institutional Review Board (4–2009-0694) of Yonsei University College of Medicine. Human samples were obtained in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. Informed consent was obtained for experimentation with human subjects.
Statistical analysis
Statistical significance of histological scores was determined using the Mann-Whitney U test. The relative amount of mRNA and the results of flow cytometry were analyzed using unpaired two-tailed Student t test or one-way or two-way ANOVA, followed by Bonferroni posthoc test, as indicated in the figure legends. The p values <0.05 were considered statistically significant. All analyses were performed using Prism 5 software (GraphPad, La Jolla, CA).
Results
Development of autoimmune lacrimal keratoconjunctivitis following adoptive transfer of naïve T cells to C57BL/10 RAG2−/− mice.
To induce the eye inflammation, we adoptively transferred CD4+CD45RBhigh T cells from wild type C57BL/10 mice to C57BL/10 RAG2−/− mice (Supplemental Figure 1A). The C57BL/10 strain was chosen because previous studies demonstrated that C57BL/10 mice, while having significant genetic identity with C57/BL6 mice, have a mutation in a locus on chromosome 11, termed tnbs2, that enhances the susceptibility of C57BL/10 mice to colitis following the intra-rectal administration of trinitrobenzene sulfonic acid (TNBS) as well as IL-12p70 production following systemic administration of lipopolysaccharide (LPS).14,16
Furthermore, C57BL/10 RAG−/− mice were found to develop more severe colitis as well as psoriasiform dermatitis, esophageal inflammation, and pancreatic islet inflammation compared to C57BL/6 RAG−/− mice following adoptive T cell transfer, which was associated with higher IL-12p12 and IL23 production by dendritic and enhanced expansion of Th1 and Th17 cells and lower Treg induction in lymphoid tissues.14,15 Therefore, C57BL/10 mice are more genetically susceptible to inflammation at multiple sites of exogenous antigen exposure that C57BL/6 mice.
Consistent with this enhanced susceptibility, 50% of adoptively transferred C57BL/10 RAG2−/− mice developed eye inflammation 6–7 wk. after transfer (Figure 1C) compared to only 20% of C57BL/6-RAG-2−/− mice (data not shown). On the ocular surface, inflammation was dominant in the central cornea (Figure 1A); with cell infiltration in the anterior portion of corneal stroma (Figure 1B). Immunohistochemical staining of tissue sections (Figure 2) revealed that the infiltrating cells were mainly Gr-1+, F4/80+ and CD4+ cells, consistent with PMNs, macrophages and T lymphocytes. In addition, infection was excluded in the affected tissues by negative Gram, Giemsa, and silver nitrate staining (Supplemental Figure 2).
Figure 1. Adoptive transfer of CD4+CD45RBhigh naïve T cells induced the development of corneal lesions in C57BL/10 RAG2−/− mice.

(A) Representative images of corneal lesions in RAG2−/− mice induced by transfer of CD4+CD45RBhigh naïve T cells from three similar experiments. Left: normal cornea. Center: minimal keratitis. Right: severe keratitis. (B) H&E-stained cross sections of eyeballs including the center of cornea. Arrow heads indicate the center of cornea. Left: normal cornea. Center: minimal keratitis. Right: severe keratitis.(X40) (C) The mean of cumulative incidences of corneal lesion in 20 mice of each experiment after transfer of naïve T cells. Three similar experiments were performed to the total 60 mice. Shown are means +/− SD.
Figure 2. Identification of infiltrated cells in cornea after adoptive transfer of CD4+CD45RBhigh naïve T cells.
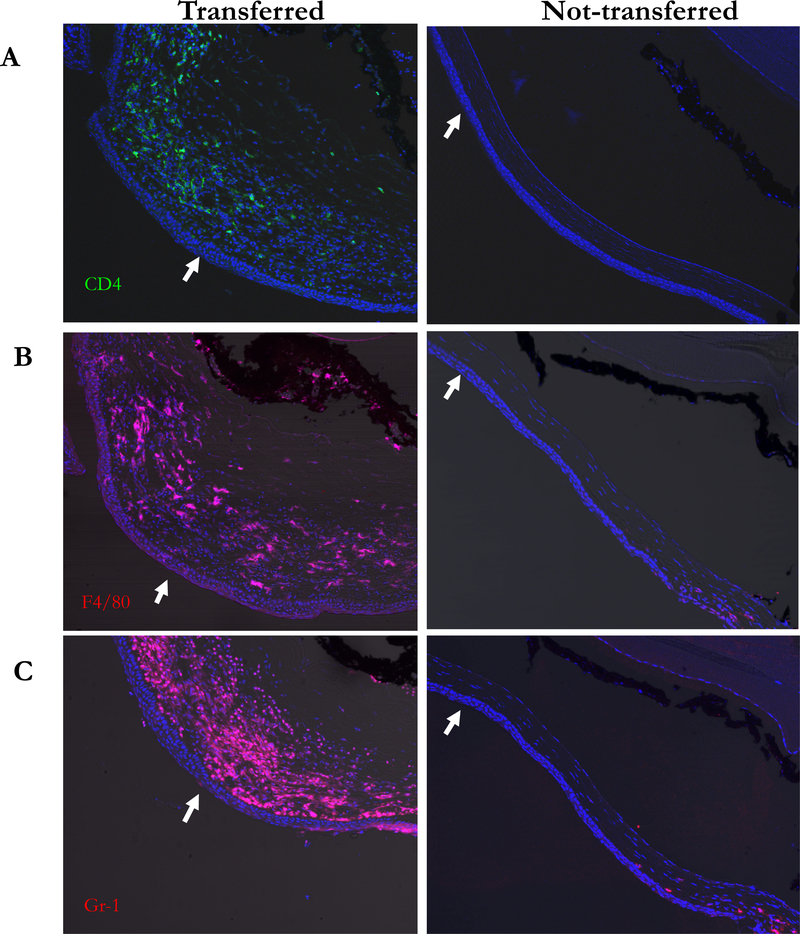
Cornea sections were stained for CD4 (A, green) F4/80 (B, red) and Gr-1 (C, red) and the nuclear dye Hoechst 33258 (blue). Center of each cornea was indicated as arrow. Increase of CD4+ F4/80+ and Gr-1+ cells were present in the corneal stroma of adoptive transferred mice. Original magnification X200.
Inflammatory changes in the lacrimal functional unit (LFU) of adoptively transferred mice
As shown in Figure 3B, the eyelids of transferred mice also showed inflammatory changes with swelling and loss of hair around eyelids. Eyelid histology on day 35 after transfer revealed epidermal acanthosis and hyperparakeratosis, and dermal inflammatory infiltrates with tarsal conjunctival inflammation (Figure 3D). Most of infiltrating cells were Gr-1+, F4/80+ and CD4+ similar to the corneal stroma (Figure 3E-G). Histologic analysis of the lacrimal glands at this time point revealed various degrees of lymphocytic infiltration, destruction of the acinar structures, and hypertrophy of the ductal structures (Figure 4A). However, inflammatory changes were not observed in the salivary glands up to 8 weeks after transfer (Figure 4B).
Figure 3. Eyelid inflammation by CD4+ F4/80+ and Gr-1+ cell population.
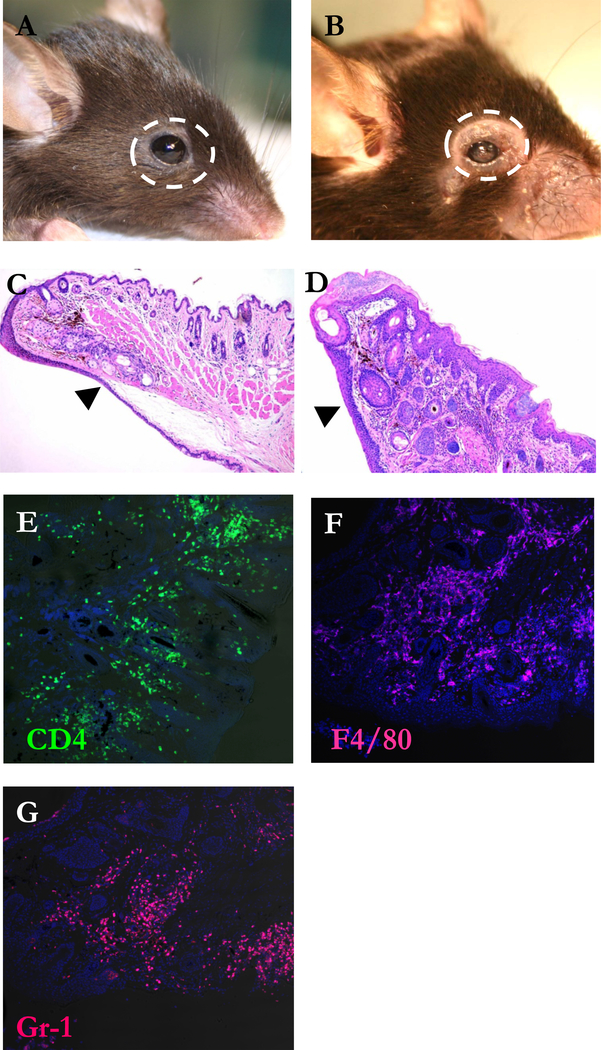
(A) Representative photograph of affected eyelid in C57BL/10 RAG2−/− mouse 6 weeks after CD4+CD45RBhigh T cells reconstitution from 3 similar experiments. H&E-stained cross sections of eyelids of not-transferred (B) and transferred mouse (C). Arrow heads indicate the skin side of eyelids. Original magnification X100. Cornea sections were stained for CD4 (D, green) F4/80 (E, red) and Gr-1 (F, red) and the nuclear dye Hoechst 33258 (blue). Original magnification X200.
Figure 4. Lymphocytic infiltration of the lacrimal glands in adoptive transferred mice.
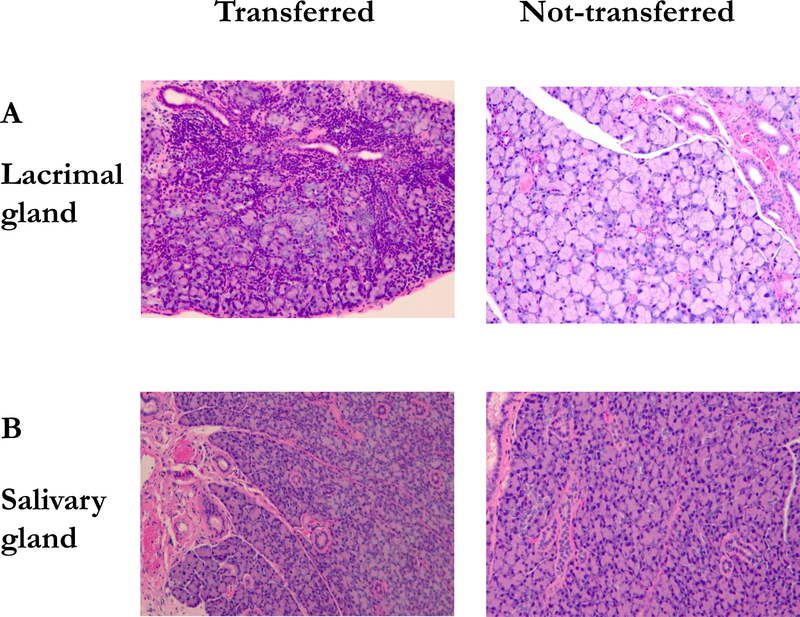
(A) H&E-stained cross sections of lacrimal gland of transferred and not-transferred mouse. (B) H&E-stained cross sections of salivary gland of transferred and not-transferred mouse. Original magnification X200.
Role of regulatory T cell in the development of keratitis
Prior studies have implicated suppressed Treg function contributes to the pathogenesis of DED.1,8 To evaluate the ability of Tregs to control the expansion of effector T cells12 and thus the keratitis and changes in the ocular adnexa seen in our model, we compared the incidence of keratitis, the presence of Th1 and Th17 cells, and the expression of inflammatory cytokines in the corneal tissue between mice transferred CD4+CD45RBhigh alone or together with CD4+Foxp3+ Tregs contained in the CD45RBlow fraction (Supplemental Figure 1B).14 Co-transfer of CD4+CD45RBhigh naïve T cell with Tregs completely prevented the development of keratitis (Figure 5A, B) Also co-transferred mice did not develop eyelid inflammation. Neutrophil dominant cornea infiltration was not seen in the cornea of co-transferred mice (Figure 5B). CD4+ cells from draining lymph nodes 6 weeks after transfer showed decreased expression of IL-17 and IFN- γ compared to mice receiving only CD4+CD45RBhigh cells (Figure 5C). Within the corneal tissue, mRNA for IFN-γ, IL-17, IL-1β, and iNOS were suppressed with co-transfer of Tregs, while levels of TNF-α and TGF-β where similar (Figure 5D).
Figure 5. Co-transfer of CD4+CD45RBhigh and CD4+CD45RBlow T cells prevent the development of keratitis in C57BL/10 RAG2−/− mice.


(A) The mean of cumulative incidences of keratitis in 20 mice in each group after transfer of naïve T cells. (B) Representative photographs and H&E-stained cross sections of cornea. Left: mice transferred by CD4+CD45RBhigh naïve T cells. Right: mice transferred by CD4+ CD45RBhigh and CD4+CD45RBlow T cells (1:1). Arrow heads indicate the center of cornea. (C) TCRβ+CD4+ cells of draining lymph nodes from 6 weeks after transfer of T cells were stained with antibodies recognizing IL-17 and IFN-γ and were analyzed by flow cytometry. Results are representative of 3 similar experiments from 3 individual mice. (D) Total RNA was purified from six corneas of three mice. Mice transferred by CD4+CD45RBhigh naïve T cells (open bars), mice transferred by CD4+ CD45RBhigh with CD4+CD45RBlow T cells (closed bars) mRNAs were quantified by RT-qPCR. Levels of target mRNAs were normalized to GAPDH mRNA as an endogenous control. Shown are means +/− SD of values from 3 similar experiments. * p < 0.05 ** p< 0.01.
Neural damage within the cornea is induced in chronic keratitis
Decreased corneal sensation is common in DED, and the LFU is linked mainly by nerves, suggesting that a disruption in neural signals controlling LFU function may be involved in DED pathogenesis.3,17,18 We hypothesized that the increased activity of iNOS in corneal tissue in our model could induce nerve damage mediated by reactive oxygen species.19 To explore this possibility, we stained corneal neural fibers with anti-β-tubulin III for the confocal imaging. Seven weeks after naïve T cell transfer, staining for corneal nerve fibers was markedly diminished from the periphery to the center of the cornea (Figure 6A) compared to corneas from C57BL/10 RAG2−/− mice without transfer (Figure 6B). Interestingly, different from the damage in the unmyelinated part of corneal nerve20 the myelinated nerve plexus in the limbus around the cornea remained intact after transfer (Arrow head, Figure 6A). To determine whether these changes progressed over time, we examined corneal tissue 0, 4 and 7 weeks after transfer. As early as 4 weeks after transfer, the majority of corneal nerves were already poorly detectable (Figure 6C). In addition, the levels of mRNA for iNOS, IL-1β, and IL-6, but not TNF-α increased in corneal tissue at this time point (Figure 6D), as were levels of the chemokines CXCL1 and CXCL5 that are known to recruit PMN to inflammatory sites (data not shown), consistent with the presence of active inflammation that results in nerve damage, likely through the production of NO.
Figure 6. Induction of neuronal fiber damage in cornea by adoptive transfer of CD4+CD45RBhigh naïve T cells.



(A) Representative nerve staining whole mount cornea from three similar experiments. Corneas from RAG2 −/− mice 7 weeks after naïve T cells transfer were taken and labeled for nerve fibers (red) with anti-β-tubulin III. Arrow head indicates nerve plexus in the limbus surrounding the cornea. (B) Representative nerve staining whole mount of cornea from RAG2 −/− mice as control. (C) Nerve changes in the center of corneas from RAG2 −/− mice after naïve T cells transfer by time course. (D) Total RNA was purified from corneas of three mice in each group of mice not transferred by CD4+CD45RBhigh naïve T cells (open bars), after 4 weeks transferred by CD4+ CD45RBhigh (grey bars), and after 7 weeks (black bars). mRNAs were quantified by RT-qPCR. Levels of target mRNAs were normalized to GAPDH mRNA as an endogenous control. Results are presented as the mean mRNA levels obtained from three similar experiments. Error bars indicate the SD.
Contribution of CD4+ T cell cytokines to keratitis development
An increase in IFN-γ and IL-17 in the affected tissues is common feature of DED in humans and in other animal models.1,10,21 To determine the role of CD4+ T cell-derived IFN-γ and IL-17 in the development of keratitis in the transfer model, we adoptively transferred CD4+CD45RBhigh T cells from IFN-γ KO mice to C57BL/10 RAG2−/− mice by intraperitoneal injection. There was no significant difference in the development of keratitis between the mice that received IFN-γ KO or wild type CD4+ naïve T cells, despite the presence of IFN-γ-producing CD4+ cells in the draining LN of mice given wild type T cells. (Figure 7A-B). Therefore, the production of IFN-γ by CD4+ T cell is not required for the expression of keratitis. We also transferred CD4+CD45RBhigh T cells from IL-17A KO mice to C57BL/10 RAG2−/− mice by intraperitoneal injection (Figures 7C-D). Starting 5 weeks after transfer, IL-17A KO T cell-transferred mice showed a reduced incidence of keratitis compared to the wild type CD4+ naïve T cells transferred mice, however keratitis was not completely eliminated. These data suggest that cytokines produced by the differentiated CD4+ T cells have limited effects on the process of keratitis, which are likely to include the enhanced recruitment of monocyte and leukocyte into the ocular tissues.
Figure 7. Adoptive transfer of IFN-γ−/− or IL-17A−/− CD4+CD45RBhigh naïve T cells to induce corneal lesions in RAG2 −/− mice.


(A) The mean of cumulative incidences of keratitis in mice after transfer of IFN-γ−/− naïve T cells from three similar experiments of 6 or 7 mice in each experiment. (B) CD4+ cells of draining lymph nodes from 8 weeks after transfer of naïve T cells were stained with antibodies recognizing IL-17 and IFN-γ and were analyzed by flow cytometry. (C) The mean of cumulative incidences of corneal lesion in mice after transfer of IL-17A−/− naïve T cells. (D) CD4+ cells of draining lymph nodes 8 weeks after transfer of naïve T cells were stained with antibodies recognizing IL-17 and IFN-γ and were analyzed by flow cytometry. Shown are means +/− SD of values for cells from 3 individual mice, and results are representative of 3 similar experiments. * p < 0.05 ** p< 0.01.
Role of IL-12/IL-23p40 in the recipient mice in disease development
Antigen presenting cells (APCs) have been shown to be important for development of dry eye in the desiccating model of keratitis.2 To evaluate the role of cytokine production by recipient APCs on the incidence of keratitis in our transfer model, we adoptively transferred CD4+CD45RBhigh T cells to RAG2−/− or IL-12/IL-23p40 (p40)−/− x RAG2−/− mice. There was no evidence of eye disease (Figure 8A), and a reduction in the frequency of IL-17 and IFN-γ-producing T cells in the draining LNs (Figure 8B) of p40−/− x RAG2−/− mice compared to RAG2−/− mice. Furthermore, in corneal tissues, the expression of IFN-γ, IL-17A and IL-17F were all decreased compared to RAG2−/− mice. These data indicate that IL-12 and/or IL-23 has an essential role in the differentiation of transferred CD4+ T cell, cytokine production in corneal tissues and the expression of keratitis.
Figure 8. Adoptive transfer of CD4+CD45RBhigh naïve T cells to C57BL/10 IL-12p35−/−RAG2−/− or IL-12/IL-23p40−/−RAG2−/− mice.

(A) The mean of cumulative incidences of keratitis in mice after transfer of naïve T cells from three similar experiments of 6 or 7 mice in each experiment. (B) CD4+ 7AAD- cells of draining lymph nodes from 8 weeks after transfer of naïve T cells were stained with antibodies recognizing IL-17 and IFN-Y and were analyzed by flow cytometry. (C) Representative photos of corneal lesions in IL-12p35−/−RAG2−/− or IL-12/IL-23p40−/−RAG2−/− mice after transfer of CD4+CD45RBhigh naïve T cells. (D) Total RNA was purified from six corneas of three mice. Mice transferred by CD4+CD45RBhigh naïve T cells (open bars), mice transferred by CD4+ CD45RBhigh with CD4+CD45RBlow T cells (closed bars). mRNAs were quantified by RT-qPCR. Levels of target mRNAs were normalized to GAPDH mRNA as an endogenous control. Shown are means +/− SD of values for cells from 3 individual mice, and results are representative of 3 similar experiments. * p < 0.05 ** p< 0.01.
Tear cytokines from patients of autoimmune related dry eye diseases.
To show the relationship between mouse model of autoimmune lacrimal keratoconjunctivitis and human disease, we measured the concentrations of cytokines in tears of 26 patients (26 eyes) from each of the following groups; Sjögren syndrome dry eye disease (SSDE), non-Sjögren syndrome dry eye disease (NSDE) patients, and healthy, sex- and age-matched controls (only females; 56.2±10.6, 55.6±11.8, 51.3±12.1 years old, respectively). SSDE patients showed highest levels of all cytokines in tears, followed by NSDE and controls. Particularly, IL-17A levels in tears of SSDE patients (13.4±8.3 ng/ml) were significantly higher than in controls (1.3±1.9 ng/ml), but also higher than in NSDE patients (4.8±3.3 ng/ml) (p<0.001). Other pro-inflammatory cytokines in SSDE tears, TNF-α, IFN-γ, IL-2, and −4, were also higher than in both NSDE patients and controls. Furthermore, the concentration of anti-inflammatory IL-10 in SSDE tears was significantly two-fold higher compared to controls (2.7±1.6 vs 1.3±0.8 ng/ml, p<0.001). In NSDE patients, TNF-α and IL-17A of tears were significantly increased compared to control (1.8±1.0 vs 0.7±0.7, 4.8±3.3 vs 1.3±1.9 ng/ml, respectively).
Discussion
Following adoptive transfer of naïve CD4+ CD45RBhigh T cells to B10-RAG2 KO, we demonstrated the central role of CD4+ cells in EALK as a model of DED and Sjögren’s syndrome. This spontaneous disease model minimizes environmental damage and therefore accurately models the disease process in humans. In particular, desiccation models of DED cause corneal epithelial damage with consequent trauma-induced inflammation, and the inclusion of the anticholinergic, scopolamine to increase eye dehydration has additional systemic effects, including the inhibition of the cholinergic anti-inflammatory pathway.22 Furthermore, even though CD4+ cells are have been implicated as a main effector in prior models,8–10 the participation of other lymphocytes including CD8+ cells and B cells could not be ruled out. This includes models of enhanced DED after adoptive transfer of CD4+ T cells from mice with desiccating injury. Therefore, the current model in RAG2 KO mice which lack T cells and B cells allows for the definitive evaluation of the contributions of individual CD4+ T cell populations to EALK. Adoptive transfer of naïve CD4+ T cells could induce inflammatory damage of the LFU in mice, thereby demonstrating that CD4+ T cells are sufficient to drive DED.
In prior studies, poor regulatory T cell function induced by Th17 cell responses has been implicated in the pathogenesis of DED.1 Our study confirms that lack of Treg cell function is critical to the development of EALK in that disease occurred following the transfer of naïve CD4+ T cells, but was suppressed by the co-transfer of Tregs. Furthermore, prior studies from our laboratory showed that naïve CD4+ T cells when transferred to C57Bl/10 Rag−/− mice differentiate into Foxp3+ Treg cells less efficiently than when transferred to C57BL/6 RAG−/− mice,15 consistent with a role for induced Tregs in the protection of C57BL/6 RAG−/−, but not C57Bl/10 RAG−/− mice to EALK development. Foxp3+Tregs suppress a variety of immune cells including B cells, NK cells, NKT cells, CD4+, and CD8+ T cells, as well as monocytes and dendritic cells (DCs). In this study, we present that Treg suppress disease phenotype and expression of effector T helper cell without other B or CD8+ lymphocytes in DED, most likely via direct inhibition of DC-mediated CD4+ T cell priming in the draining LNs.23
An increase in IFN-γ is commonly associated with DED and CD4+ cells are a source of IFN-γ in EALK. However, transfer of naïve CD4+ T cells from IFN-γ deficient mice to lymphopenic mice did not result in a lower incidence of EALK. Recently NK cells have been suggested to be a source of IFN-γ in DED. IFN-γ-secreting NK cells were shown to damage the cornea directly and influence the priming phase of the adaptive immune response in a model of DED.24 Furthermore, depletion of NK cells improved corneal damage in desiccating conditions and transfer of CD4+ T cell from NK-depleted mice resulted in less IL-17 produced at the ocular surface.25 Our findings of a decreased incidence of EALK after the transfer of IL-17-deficient naïve CD4+ T cells to lymphopenic mice indicate that Th17 cells play the central role in EALK, and suggest that IFN-γ from NK cells, but not CD4+ T cells, may contribute to the pathology in DED. These data are consistent with a role of Th17 cells in acute and chronic inflammation in the desiccation model,1 however, as mentioned above because of the lack of direct trauma to the corneal epithelium and the systemic effects of scopolamine, we fell that the transfer model described here is highly informative and is likely to prove a better model for DED in humans.
The concept of a lacrimal functional unit (LFU) is important for understanding the pathogenesis DED. Corneal sensation promotes tear secretion and tears protect the ocular surface and increase eyelid hygiene. Inflammation of the lacrimal gland affects both the volume of tears, and the presence of damaging cytokines. The increased cytokines or desiccating stress can result in damaged corneal nerves that then signal to the lacrimal gland to decrease tear production thus contributing to DED in a vicious cycle. The central portion of the corneal nerve is unmyelinated.26 In our model, when inflammation occurs, part of the unmyelinated corneal nerve is lost, in contrast to the intact myelinated limbal nerve. These data indicate that the unmyelinated corneal nerve is susceptible to inflammation, and in the current model this is likely due to increased levels of tissue cytokines produced by macrophage and neutrophils recruited into the corneal tissue (Figure 6).
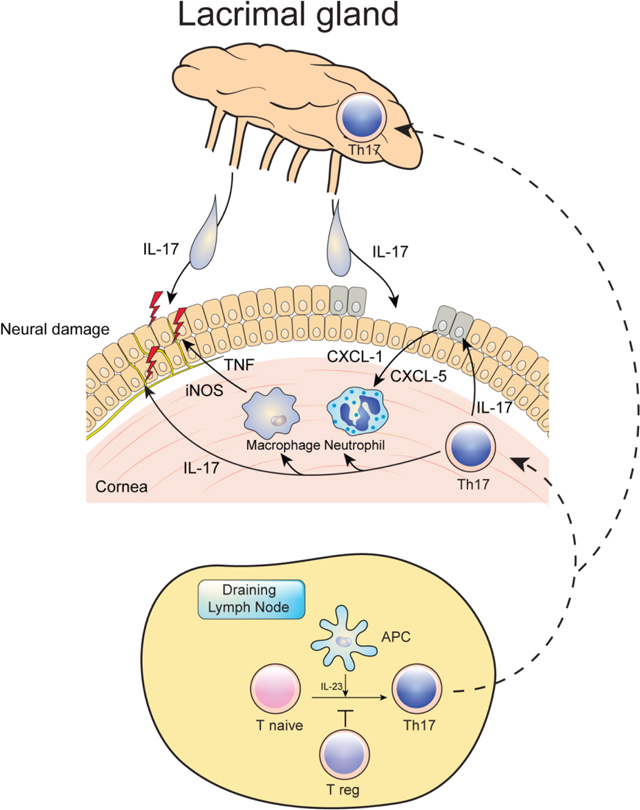
A recent study indicated that antigen presenting myeloid cells are critical for the initiation and development of desiccation induced ocular inflammation and suggested DED is a self-antigen-driven autoimmune-based inflammatory disease.2 In our study, IL-12/IL-23p40 was crucial for the development of EALK. IL-12/IL-23p40 is the common subunit in the heterodimers of IL-12 and IL-23. As expected with the role of Th17 cells in our disease model, lack of IL-23 ameliorates CD4+ differentiation toward Th17 cells and disease induction. Overall, our data consistent with the following pathological process in DED: naïve CD4+ T cells differentiate into Th17 cells in the draining cervical lymph node in response to migrating DCs that produce IL-23, a process that is normally controlled by Tregs. In the relative absence of Tregs in this model, Th17 cells migrate to the cornea and lacrimal gland in EALK. IL-17 from Th17 cells in cornea, conjunctiva and lacrimal gland causes the epithelial damage10 and induces CXCL-1 and CXCL-5 production from that recruits macrophage and PMNs. IL-17 with iNOS also induces the damage of corneal nerve (Figure 8).27–29 Consistent with these findings, we found an increase in inflammatory cytokines in the tears of patients with DED. In particular, IL-17, as well as TNFα is increased in patients with and without Sjögren’s syndrome.
Adoptively transferred C57BL/10 RAG2−/− mouse populations developing keratitis also develop colitis and skin inflammation, but the frequency and severity of any one of these inflammatory manifestations is dependent on the environmental conditions of the mice; with mice in one NIH facility developing predominantly colits and skin inflammation, with one predominating, and in the other facility developing keratoconjunctivitis, but less colitis and skin inflammation. Interestingly, common to all inflammatory conditions in this model is the external exposure to antigens and commensal microbes. Therefore, it seems likely that differing symptoms are due to the development of different microbiomes in the gut, skin, and eyes at the different facilities, or to different PAMPS/DAMPS in routinely autoclaved food or bedding. Current studies are addressing these issues including whether the presence of certain bacterial strains in the eye (e.g., strains of Corynebacterium30) may predispose to keratoconjunctivitis. Furthermore, when OTII Ovalbumin-specific TCR-transgenic T cells were transferred to C57BL/10 RAG2−/− mice in the same facility resulting in a high incidence of keratitis, they did not develop EALK (reference 30, and data not shown), suggesting that the keratitis was due to T cell responses to self or microbial antigens.
In conclusion, our study supports a working animal model for DED expressed as EALK, in which Th17 cells are generated to lacrimal and microbial antigens in the draining lymph node in response to cognate antigen and IL-23, a process that is normally controlled by CD4+ Foxp3+ regulatory T cells. These Th17 cells migrate to the lacrimal gland where they result in inflammation and cytokines, including IL-17, are secreted into the tears that drive initial injury to the cornea, followed by inflammation and further IL-17 mediated damage to the cornea. This includes the influx of Th17 cells, as well as macrophages and neutrophils into the cornea in response to CXC chemokines, and their production of NO and TNFα which enhances destructive inflammation of the corneal layer, and results in nerve damage and further physical injury. This model includes pathologic findings in the whole LFU and is spontaneously induced without the requirement for extreme environmental desiccating conditions. This model also allows for the precise evaluation of CD4+ T cells, Tregs and myeloid cells, and will thus be a valuable for future studies. Furthermore, by studying disease development in different animal facilities, following the treatment with antibiotics, and in germ-free conditions, this spontaneous model should also help in understanding the contribution of commensal bacteria and other exogenous stimuli in driving DED development. Finally, these data support the recent anti-inflammatory therapeutic approach for DED that targets T cell immunity and suggest that cytokines involved in myeloid function could be a new target for disease intervention.
Supplementary Material
Supplemental Figure 1. Adoptive transfer of CD4+CD45RBhigh naïve T cells to RAG2−/− mice. (A) CD4+CD45RBhigh T cells from spleen of C57BL/10 donor mice were sorted by FACS sorting after negative selection by MACS. One to three thousand cells were adoptively transferred to C57BL/10 RAG2−/− mice by i.p. injection. (B) CD4+CD45RBhigh T cells and CD4+CD45RBlow T cells from spleen of C57BL/10 donor mice were isolated by FACS after negative selection by MACS. One to three thousand mixed (1:1) cells were adoptively transferred to C57BL/10 RAG2−/− mice by i.p. injection.
Supplemental Figure 2. No evidence of microbial infection. Section of the corneal tissue were evaluated by Gram (A, B), Giemsa (C, D), and Silver nitrate (E, F) staining for excluding the infection by bacteria or fungi in corneal lesions. Original magnification X100 (A, C, E), X600 (B, D, F).
Acknowledgements
This work was supported by the Division of Intramural Research, NIAID, NIH.
Abbreviations:
- DED
dry eye disease
- EALK
experimental autoimmune lacrimal keratoconjunctivitis
- LFU
lacrimal functional unit
- IBD
inflammatory bowel disease
- Treg
CD4+ Foxp3+ regulatory T cell
- APC
antigen presenting cell
Footnotes
Disclosures
The authors declare no conflict of interest.
References
- 1.Chauhan SK, Dana R. Role of Th17 cells in the immunopathogenesis of dry eye disease. Mucosal Immunol. 2009;2(4):375–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaumburg CS, Siemasko KF, De Paiva CS, Wheeler LA, Niederkorn JY, Pflugfelder SC, et al. Ocular surface APCs are necessary for autoreactive T cell-mediated experimental autoimmune lacrimal keratoconjunctivitis. J Immunol. 2011;187(7):3653–3662. [DOI] [PubMed] [Google Scholar]
- 3.Dartt DA. Neural regulation of lacrimal gland secretory processes: relevance in dry eye diseases. Prog Retin Eye Res. 2009;28(3):155–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern ME, Gao J, Siemasko KF, Beuerman RW, Pflugfelder SC. The role of the lacrimal functional unit in the pathophysiology of dry eye. Exp Eye Res. 2004;78(3):409–416. [DOI] [PubMed] [Google Scholar]
- 5.Mohsenin A, Huang JJ. Ocular manifestations of systemic inflammatory diseases. Conn Med. 2012;76(9):533–544. [PubMed] [Google Scholar]
- 6.Patel SJ, Lundy DC. Ocular manifestations of autoimmune disease. Am Fam Physician. 2002;66(6):991–998. [PubMed] [Google Scholar]
- 7.Stern ME, Schaumburg CS, Dana R, Calonge M, Niederkorn JY, Pflugfelder SC. Autoimmunity at the ocular surface: pathogenesis and regulation. Mucosal Immunol. 2010;3(5):425–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chauhan SK, El Annan J, Ecoiffier T, Goyal S, Zhang Q, Saban DR, et al. Autoimmunity in dry eye is due to resistance of Th17 to Treg suppression. J Immunol. 2009;182(3):1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niederkorn JY, Stern ME, Pflugfelder SC, De Paiva CS, Corrales RM, Gao J, et al. Desiccating stress induces T cell-mediated Sjogren’s Syndrome-like lacrimal keratoconjunctivitis. J Immunol. 2006;176(7):3950–3957. [DOI] [PubMed] [Google Scholar]
- 10.De Paiva CS, Chotikavanich S, Pangelinan SB, Pitcher JD 3rd, Fang B, Zheng X, et al. IL-17 disrupts corneal barrier following desiccating stress. Mucosal Immunol. 2009;2(3):243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. International immunology. 1993;5(11):1461–1471. [DOI] [PubMed] [Google Scholar]
- 12.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–271. [DOI] [PubMed] [Google Scholar]
- 13.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. [DOI] [PubMed] [Google Scholar]
- 14.Leon F, Contractor N, Fuss I, Marth T, Lahey E, Iwaki S, et al. Antibodies to complement receptor 3 treat established inflammation in murine models of colitis and a novel model of psoriasiform dermatitis. J Immunol. 2006;177(10):6974–6982. [DOI] [PubMed] [Google Scholar]
- 15.Valatas V, He J, Rivollier A, Kolios G, Kitamura K, Kelsall BL. Host-dependent control of early regulatory and effector T-cell differentiation underlies the genetic susceptibility of RAG2-deficient mouse strains to transfer colitis. Mucosal Immunol. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouma G, Kaushiva A, Strober W. Experimental murine colitis is regulated by two genetic loci, including one on chromosome 11 that regulates IL-12 responses. Gastroenterology. 2002;123(2):554–565. [DOI] [PubMed] [Google Scholar]
- 17.Stern ME, Beuerman RW, Fox RI, Gao J, Mircheff AK, Pflugfelder SC. The pathology of dry eye: the interaction between the ocular surface and lacrimal glands. Cornea. 1998;17(6):584–589. [DOI] [PubMed] [Google Scholar]
- 18.Labbe A, Alalwani H, Van Went C, Brasnu E, Georgescu D, Baudouin C. The relationship between subbasal nerve morphology and corneal sensation in ocular surface disease. Invest Ophthalmol Vis Sci. 2012;53(8):4926–4931. [DOI] [PubMed] [Google Scholar]
- 19.Kim JC, Cheong TB, Park GS, Park MH, Kwon NS, Yoon HY. The role of nitric oxide in ocular surface diseases. Advances in experimental medicine and biology. 2002;506(Pt A):687–695. [DOI] [PubMed] [Google Scholar]
- 20.Bergmanson JPG, Doughty MJ. Anatomy, Morphology and Electron Microscopy of the Cornea, and Conjunctiva In: Bennett ES, Weissman BA, editors Clinical Contact Lens Practice. Philadelphia: Lippencott Williams & Wilkins; 2005. p. 11–39. [Google Scholar]
- 21.De Paiva CS, Villarreal AL, Corrales RM, Rahman HT, Chang VY, Farley WJ, et al. Dry eye-induced conjunctival epithelial squamous metaplasia is modulated by interferon-gamma. Invest Ophthalmol Vis Sci. 2007;48(6):2553–2560. [DOI] [PubMed] [Google Scholar]
- 22.Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(31):11008–11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunological reviews. 2014;259(1):88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Chauhan SK, Saban DR, Sadrai Z, Okanobo A, Dana R. Interferon-gamma-secreting NK cells promote induction of dry eye disease. J Leukoc Biol. 2011;89(6):965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Volpe EA, Gandhi NB, Schaumburg CS, Siemasko KF, Pangelinan SB, et al. NK cells promote Th-17 mediated corneal barrier disruption in dry eye. PLoS One. 2012;7(5):e36822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller LJ, Pels L, Vrensen GF. Ultrastructural organization of human corneal nerves. Invest Ophthalmol Vis Sci. 1996;37(4):476–488. [PubMed] [Google Scholar]
- 27.Liu Y, You C, Zhang Z, Zhang J, Yan H. Roles of Treg/Th17 Cell Imbalance and Neuronal Damage in the Visual Dysfunction Observed in Experimental Autoimmune Optic Neuritis Chronologically. Neuromolecular Med. 2015;17(4):391–403. [DOI] [PubMed] [Google Scholar]
- 28.Moynes DM, Vanner SJ, Lomax AE. Participation of interleukin 17A in neuroimmune interactions. Brain Behav Immun. 2014;41:1–9.24642072 [Google Scholar]
- 29.Noma N, Khan J, Chen IF, Markman S, Benoliel R, Hadlaq E, et al. Interleukin-17 levels in rat models of nerve damage and neuropathic pain. Neurosci Lett. 2011;493(3):86–91. [DOI] [PubMed] [Google Scholar]
- 30.St. Leger A, Desai JV, Drummond RA, Kugadas A, Almaghrabi F, Silver P, et al. An Ocular Commensal Protects against Corneal Infection by Driving an Interleukin-17 Response from Mucosal γδ T Cells. Immunity 2017;47(1):148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Adoptive transfer of CD4+CD45RBhigh naïve T cells to RAG2−/− mice. (A) CD4+CD45RBhigh T cells from spleen of C57BL/10 donor mice were sorted by FACS sorting after negative selection by MACS. One to three thousand cells were adoptively transferred to C57BL/10 RAG2−/− mice by i.p. injection. (B) CD4+CD45RBhigh T cells and CD4+CD45RBlow T cells from spleen of C57BL/10 donor mice were isolated by FACS after negative selection by MACS. One to three thousand mixed (1:1) cells were adoptively transferred to C57BL/10 RAG2−/− mice by i.p. injection.
Supplemental Figure 2. No evidence of microbial infection. Section of the corneal tissue were evaluated by Gram (A, B), Giemsa (C, D), and Silver nitrate (E, F) staining for excluding the infection by bacteria or fungi in corneal lesions. Original magnification X100 (A, C, E), X600 (B, D, F).