

Abstract
Quality of response to immunosuppressive therapy and long-term outcomes for pediatric severe aplastic anemia remain incompletely characterized. Contemporary evidence to inform treatment of relapsed or refractory severe aplastic anemia for pediatric patients is also limited. The clinical features and outcomes for 314 children treated from 2002 to 2014 with immunosuppressive therapy for acquired severe aplastic anemia were analyzed retrospectively from 25 institutions in the North American Pediatric Aplastic Anemia Consortium. The majority of subjects (n=264) received horse anti-thymocyte globulin (hATG) plus cyclosporine (CyA) with a median 61 months follow up. Following hATG/CyA, 71.2% (95%CI: 65.3,76.6) achieved an objective response. In contrast to adult studies, the quality of response achieved in pediatric patients was high, with 59.8% (95%CI: 53.7,65.8) complete response and 68.2% (95%CI: 62.2,73.8) achieving at least a very good partial response with a platelet count ≥50×109L. At five years post-hATG/CyA, overall survival was 93% (95%CI: 89,96), but event-free survival without subsequent treatment was only 64% (95%CI: 57,69) without a plateau. Twelve of 171 evaluable patients (7%) acquired clonal abnormalities after diagnosis after a median 25.2 months (range: 4.3-71 months) post treatment. Myelodysplastic syndrome or leukemia developed in 6 of 314 (1.9%). For relapsed/refractory disease, treatment with a hematopoietic stem cell transplant had a superior event-free survival compared to second immunosuppressive therapy treatment in a multivariate analysis (HR=0.19, 95%CI: 0.08,0.47; P=0.0003). This study highlights the need for improved therapies to achieve sustained high-quality remission for children with severe aplastic anemia.
Introduction
Acquired severe aplastic anemia (SAA) is a rare disorder characterized by peripheral bi- or pancytopenia and bone marrow (BM) hypoplasia. Initial therapy for younger patients with SAA is a matched sibling hematopoietic stem cell transplantation (HSCT) or immunosuppressive therapy (IST) if a matched sibling donor is not available.1 There is a paucity of data for children with SAA treated during the modern era from the ethnically and geographically diverse population of North America. Pediatric studies from the National Institutes of Health,2 the Japanese Childhood Aplastic Anemia Study Group,3-6 Brazil,7 and the Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplant (SAAWP-EBMT)8,9 have been reported. However, diagnostic evaluation for constitutional disorders has been limited, and duration of follow up has often been variable and short. Many published studies report outcomes with rabbit anti-thymocyte globulin (rATG) due to the withdrawal of horse ATG (hATG) from some European and Asian markets in 2007; however, a large prospective study of upfront rATG versus hATG reported inferior response rates and lower survival with rATG.10 Thus, contemporary data to inform therapeutic decisions in pediatric patients treated with IST in North America are of interest given international differences in treatment regimens, recent advances in diagnosis of genetic marrow failure disorders, improvements in HSCT outcomes, and better supportive care.
To facilitate collaborative clinical studies of pediatric aplastic anemia, a consortium of 25 institutions (now numbering 39) across North America, named the North American Pediatric Aplastic Anemia Consortium (NAPAAC), was formed in 2014.11 The urgent need for evidence-based guidelines for patient management in pediatric SAA was highlighted by a NAPAAC survey of clinical practices of member institutions which revealed considerable variability in diagnostic evaluation and management.11 Recognizing the challenges of performing a prospective trial to address these critical knowledge gaps, NAPAAC conducted a retrospective study of presentation and outcomes of pediatric patients diagnosed from 2002 to 2014 with SAA and treated with IST with a minimum of two years of follow up in member institutions. The objectives of the study were to determine the rates of response and survival following IST, to assess rates of clonal abnormalities, and to explore patient-specific factors contributing to survival, refractory disease, relapse, and clonal progression following IST. This NAPAAC effort represents the first large study of pediatric SAA reflecting the racially diverse population of children afflicted by SAA across multiple centers in North America. These contemporary data from a large number of pediatric patients will inform future studies of additional diagnostic or prognostic testing, and guide evidence-based clinical management.
Methods
Patients
A retrospective chart review was conducted by member institutions of all patients aged 1-20 years treated with IST as the first therapy for pediatric SAA between 1st January 2002 and 30th June 2014 for whom follow up was available for a minimum of 24 months or until death. This study was approved by the Institutional Review Board (IRB) at each participating institution or via a reliance agreement with the central study IRB at Boston Children’s Hospital. Each site abstracted data from the local medical record and entered them into a central Red Cap database. The data included demographics, disease characteristics at diagnosis, treatment, and outcomes. When available, the original anonymized written reports of specific studies including BM aspirate and biopsy, cytogenetics, fluorescence in situ hybridization (FISH), clonal analysis for paroxysmal nocturnal hemoglobinuria (PNH), telomere flow-FISH analysis, immunological analyses, and autopsies were uploaded for central review.
Submitted cases were included only if the diagnostic BM was reported as hypocellular and the patient had at least two peripheral cytopenias: 1) absolute neutrophil count (ANC) < 0.5 ×109/L; 2) platelet count (Plts) < 20×109/L; 3) hemoglobin (Hb) < 8 g/dL. Central review of diagnostic marrow slides was not feasible but marrow pathology reports were reviewed to confirm that the marrow cellularity was <25% or hypocellularity was stated to be consistent with the diagnosis of SAA without a specified percentage cellularity. Patients with a local diagnosis of an inherited BM failure syndrome were excluded from this study, as were subjects with an HLA-matched sibling who went to transplant upfront. Date of diagnosis was considered to be the date of the BM biopsy. Date of treatment was considered the first day of IST, and all outcomes were timed from the first day of treatment. Structured reporting of status and blood counts was required at initiation of IST, as well as at 3, 6, 12, 24, 36, 48, and 60 months, and at last follow up.
Definitions
Overall survival (OS) was measured from the time from first day of IST until death or date last known alive. Event-free survival (EFS) was measured from the time from start of IST until an event (death or start of a second therapy for SAA, either HSCT or a second course of IST) or the date last known to be without an event.
Response12 was defined using hemoglobin (Hb), absolute neutrophil count (ANC) and platelets (Plts). Complete response (CR), very good partial response (VGPR) and partial response (PR) required the indicated levels in all three lineages as noted below; no response (NR) was defined as failure in any lineage:
CR: Hb ≥10 g/dL and ANC ≥1×109/L and Plts ≥100×109/L
VGPR: Hb ≥8 g/dL and ANC ≥0.5×109/L and Plts ≥50×109/L
PR: Hb ≥8 g/dL and ANC ≥0.5×109/L and Plts ≥20×109/L
NR: Hb <8 g/dL or ANC <0.5×109/L or Plts <20×109/L
Patients receiving transfusions of packed red blood cells within six weeks or platelets or granulocyte-colony stimulating factor (G-CSF)/granulocyte/macrophage-colony stimulating factor (GM-CSF) within two weeks of evaluation were deemed to have had NR at that time point. An objective response (OR) was defined as at least a PR (PR+VGPR+CR) and a deep response (DR) was defined as at least a VGPR (VGPR+CR). Duration of response (DOR) was defined as time from start of response to an event (death or start of a second therapy for SAA, either HSCT or a second course of IST).
Statistical analysis
Summary statistics included median and range for continuous variables and frequency and proportion for binary variables. Fisher’s exact test and Wilcoxon rank sum test were used to compare proportions and medians, respectively. The proportion of subjects with an OR, DR, and CR were reported along with the exact binomial 95% confidence interval (95%CI). OS, EFS and DOR were estimated using the Kaplan-Meier method (log-log transformation for Confidence Interval) and compared using the log-rank test. A Cox proportional hazards model was used to compare EFS by treatment adjusting for covariates of interest [age, gender, time from initial IST treatment to 2nd treatment, and lymphocytopenia (lymphocyte count <1×109/L)]. Reference groups in this model were: IST treatment, age at second treatment ≥10 years, male gender, lymphocyte count at diagnosis ≥1×109/L. An indicator variable was included in the model for missing lymphocyte data as these data were not provided for all subjects. The median follow up among all subjects was 62 months. There was 80% power to detect differences of at least 16% difference in proportions for 314 and 264 subjects, respectively (two-sided Fishers exact test, alpha=0.05). In terms of precision, the maximum Confidence Interval width of the exact binomial 95%CI for an observed proportion was 0.12 and 0.11, respectively, with n=314 and n=264. R language was used for analysis (R Core Team, 2016, Vienna, Austria; https://www.R-project.org).
Results
Patients’ characteristics at diagnosis
A total of 314 pediatric patients treated with IST for SAA were identified by systematic retrospective chart review across 25 NAPAAC institutions. Table 1 summarizes baseline demographics of study subjects. Nine patients (0.03%) were 1-2 years of age. Gender was evenly distributed. The population was racially diverse, reflecting the ethnic diversity of North America. A family history of aplastic anemia, none of whom were first-degree relatives, was noted in four patients.
Table 1.
Baseline demographics and characteristics.

Hepatitis was noted prior to diagnosis in 43 (13.7%) patients, of whom 33 (12.5%) were treated with hATG/cyclosporine (CyA). Laboratory features at diagnosis are summarized in Table 2. PNH test results were available at diagnosis for 140 patients and a PNH clone was noted in 55 (39.3%). In most cases, the clone size was small [interquartile range (IQR): 0-0.12%]. Only 5 patients had PNH clones larger than 10% (range: 12.56-28.4%) in the granulocyte lineage and none had clinically evident hemolysis or thrombosis at diagnosis.
Table 2.
Laboratory features at diagnosis.

The most commonly investigated inherited BM failure syndromes at diagnosis were Fanconi anemia, Shwachman-Diamond syndrome, and dyskeratosis congenita. Fanconi anemia screening was negative for 292 patients (93%), 18 patients were not tested, and results were not available for four subjects. Genetic testing for Shwachman-Diamond syndrome was available for 46 patients (14.6%); all of these were negative. Perhaps reflecting the evolution of understanding the role of dyskeratosis congenita in BM failure over the past five years, telomere length was assessed at the time of diagnosis in only 115 patients (36.6%) and a report of a clinical telomere flow-FISH test was available for 93 patients (29.6%). No patient exhibited a pattern suspicious for a primary telomere disorder (telomeres <1st percentile in at least 3 different lymphocyte subsets).13 However, telomere lengths <1st percentile were found in total lymphocytes in six patients, of whom only two had a complete 6-panel analysis.
The current diagnostic criteria for SAA includes an ARC (absolute reticulocyte count) of <20-60×109/L.12,14,15 Many institutions relied on hemoglobin (Hb) rather than the ARC as a more clinically relevant indicator of erythroid hypoplasia to inform diagnosis and treatment decisions. Comparison of ARC versus Hb for the 231 subjects with ARC <100 revealed a lack of concordance [(estimated Pearson correlation coefficient of 0.15 (95%CI: 0.02, 0.27)] between the commonly utilized diagnostic criteria of ARC of <60 ×109/L and significant anemia defined as Hb <8 g/dL (Figure 1). A subset of patients meeting ARC criteria of ARC <60 ×109/L had Hb >8 g/dL (12.7%, 28 of 220), and conversely, some patients who did not meet diagnostic criteria for ARC had Hb levels <8 g/dL (74%, 26 of 35).
Figure 1.

Correlation between absolute reticulocyte count and hemoglobin at diagnosis. Absolute reticulocyte counts <100×109/L (n=231) were plotted against the hemoglobin at diagnosis. CI: Confidence Interval.
Treatment
Treatment groups are outlined in Table 3. The majority of patients (n=264) received hATG plus CyA. Of these subjects, one patient received hATG but switched to rabbit ATG (rATG) due to an anaphylactic reaction. Overall, the demographics of the hATG/CyA population was similar to that of the entire group (Tables 1-3). The small number of subjects in groups treated with alternative IST regimens limited intergroup comparisons, and so only outcomes of the entire population and that of the hATG/CyA group were analyzed.
Table 3.
Treatment.

Of the 282 subjects who were treated with hATG,187 (71%) received 40 mg/kg/day for four days, 77 were dosed with a different regimen containing about the same total dose of hATG, and in 18 the dose was not available. CyA target trough levels were variable (100-400), with the majority 200-400. Following treatment with hATG/CyA, data regarding the cyclosporine taper were available for 194 patients. CyA was discontinued by six months for 13 subjects, between 6-12 months for 40 subjects, between 12-18 months for 34 subjects, between 18-24 months for 37 subjects, and after two years for 34 subjects. CyA was not discontinued at last follow up for 36 subjects.
Median time from diagnosis to treatment for all subjects was 24 days with an IQR of 12-40 days for the entire cohort. Similar time to treatment was noted for the hATG/CyA group, with a median time from diagnosis of 23 days and an IQ range of 12-40 days.
Response
For the cohort of 314 patients and for the subset treated with hATG/CyA, median time to initial response was six months (range: 3-48 months; IQR: 3-12 months). Best responses for all patients and for the hATG/CyA group are summarized in Tables 4 and 5.
Table 4.
Best response to immunosuppressive therapy.

Table 5.
Response outcome in each treatment group.

For the 264 subjects treated with hATG/CyA, OR was 71.2% (95%CI: 65.3,76.6), consistent with results from prior adult and pediatric studies.8,9,16-20 The quality of response to hATG/CyA was good, with 59.8% (95%CI: 53.7,65.8) achieving a CR. Further, since the magnitude of a partial platelet response carries clinical implications for quality of life, the group attaining a deep response (Plts ≥50×109/L) was separately analyzed. A deep response was achieved by 68.2% (95%CI: 62.2,73.8). No response (NR) was noted in 66 subjects (25%) and 10 subjects (3.8%) were not evaluable (NE, Tables 4 and 5). Responses at six months post hATG/CyA were as follows: CR 21.6%, VGPR 19.7%, PR 8%, NR 47%, NE 3.7%.
The duration of response among subjects who had any initial response (CR, VGPR, or PR) is shown for all subjects (Figure 2A) and for those treated with hATG/CyA (Figure 2B). The estimated probability of sustained response for all subjects was 94% (95%CI: 89,96) at 24 months and 83% (95%CI: 76,88) at 60 months. For the subset of subjects treated with hATG/CyA, the estimated probability of sustained response was 94% (95%CI: 90,97) at 24 months and 84% (95%CI: 76,90) at 60 months. However, there was no plateau for loss of response observed over time even after five years post treatment.
Figure 2.

Duration of response. Kaplan-Meier analysis of duration of response for (A) all subjects or (B) subjects treated with horse anti-thymocyte globulin (rATG)/cyclosporine (CyA) who achieved at least a partial response.
Factors potentially affecting OR or DR for subjects treated with hATG/CyA were evaluated. No correlation with response was detected for telomere lengths <1st or <10th percentiles by flow-FISH, presence of a PNH clone, or red cell macrocytosis (mean corpuscular volume ≥100 fL) (P ≥0.17) (Online Supplementary Table S1). There was also no correlation between OR and median lymphocyte count (1.1 vs. 1.3;Wilcoxon rank sum test, P=0.51). There was no significant difference in median time from diagnosis to treatment between responders and non-responders (P≥0.29) (Online Supplementary Table S1).
Survival
The estimated overall survival is summarized in Figure 3. Median follow up amongst all 314 subjects was 62 months (59 months for those still alive) and 61 months among the hATG/CyA group (59 months for those still alive). At the time of this analysis, there were 29 (9.3%) deaths amongst all subjects; 21 (8%) amongst hATG/CyA-treated patients. Estimated OS (95%CI) for the entire cohort at 12, 24 and 60 months was 96% (95%CI: 94,98), 95% (92,97), and 92% (88,95), respectively (Figure 3A). Among the subjects who received hATG/CyA, estimated OS (95% CI) at 12, 24 and 60 months was 97% (94,99), 96% (93,98), and 93% (89,96), respectively (Figure 3B). Six individuals died after one treatment with hATG/CyA without receiving additional therapy.
Figure 3.

Immunosuppressive therapy: survival. Kaplan-Meier analysis of overall survival for (A) all subjects or (B) subjects treated with horse anti-thymocyte globulin (rATG) / cyclosporine (CyA). Kaplan-Meier analysis of event-free survival for (C) all subjects or (D) subjects treated with hATG/CyA.
Of the 314 subjects, 119 (38%) died or required an additional therapy. The estimated EFS (95%CI) for all subjects at 12, 24, and 60 months was 76% (95%CI: 70,80), 71% (95%CI: 66,76), and 62% (95%CI: 56,68), respectively (Figure 3C). The estimated median EFS for all subjects was 133 months. For patients treated with hATG/CyA, 98 of 264 subjects (37%) had an event. Median estimated EFS was 133 months (Figure 3D). Estimated EFS (95%CI) at 12, 24, and 60 months was 76% (95%CI: 70,81), 72% (95%CI: 66,77), and 64% (95%CI: 57,69), respectively, with events continuing to accrue even after five years post treatment.
No differences were detected in OS (P=0.13), EFS (P=0.26), or response (P>0.25) versus age at diagnosis for all subjects. No differences were detected in OS (P=0.09), EFS (P=0.22), or response (P>0.20) versus age at diagnosis for subjects treated with hATG/CyA.
Cytogenetics and clonal progression
Of the 271 patients in the cohort who had BM metaphase cytogenetics (n=254) and/or FISH (n=133) assessments performed at diagnosis, seven (3%) had detectable clonal chromosomal abnormalities (Table 6). Six of these patients had follow-up cytogenetic assessments. Two patients had a del(13q) clone at diagnosis, which remained detectable through the duration of follow up (range: 33-49 months) but was not associated with acquisition of additional chromosomal abnormalities. In contrast, other small clones present at diagnosis, including del(7q) in one patient and del(16q) in one patient, were no longer detectable at follow-up assessment.
Table 6.
Clonal cytogenetic abnormalities at diagnosis and at follow up.

Of the 171 total patients who had follow-up BM metaphase cytogenetics (n=160) and/or FISH (n=109) assessment performed after IST initiation, 12 (7.0%) patients had evidence of new clonal chromosomal abnormalities (Table 7). One additional subject had a clonal abnormality [+der(14;21)(q10;q10)] at 143 months from the time of initial diagnosis; however, the baseline status was unknown. The most common genetic alteration after IST was loss of chromosome 7 [either −7 or del (7q)] occurring in six patients (3.5%), all of whom had normal cytogenetics at baseline. Three patients had a del(13q) clone detected during follow-up BM assessments of which one was acquired after treatment. Among those patients with no clonal abnormalities at presentation, but who subsequently developed abnormalities at follow up, the median time to observe an abnormality was 25.2 months (range: 4.3-71.0 months; IQ range: 5.3-66.6 months). Interestingly, only four patients had BM pathology reports confirming the diagnosis of myelodysplastic syndrome of whom two proceeded to HSCT prior to further clonal evolution. One patient developed acute lymphoblastic leukemia and three developed acute myeloid leukemia.
Table 7.
Comparison of baseline cytogenetic clones with subsequent clonal abnormalities.

Complications
Complications following initiation of IST were common with bleeding (gastrointestinal, intracranial, hematuria or other significant hemorrhage) being most frequent and reported in 74 (23.6%) of all subjects (23.5% of hATG/CyA subjects). Infections, including bacteremia, fungal infections, cellulitis, meningitis, or pneumonia, were reported in 177 (56.4%) of all subjects (53.4% of hATG/CyA subjects). Renal failure requiring dialysis was reported in five patients (1.6%), all in the hATG/CyA group (1.9%). Causes of death classified by type of therapy and survival interval are detailed in Online Supplementary Table S2.
Outcomes after second-line therapy
Subsequent treatments following upfront hATG/CyA are summarized in Table 8.
Table 8.
Subsequent treatments.
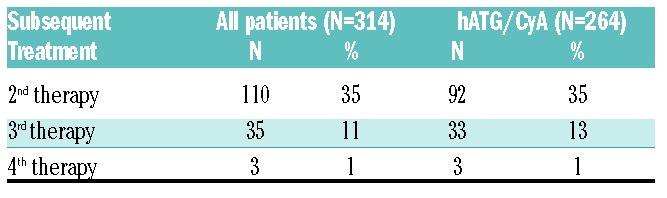
Overall, 110 of 314 (35%) subjects received a second treatment, 35 of the 110 a third treatment, and three a fourth therapy.
Of the 38 patients undergoing HSCT for second-line therapy, response to initial treatment with hATG/CyA was as follows: 31 had refractory disease and seven had relapsed disease. Of the seven patients undergoing HSCT for relapsed disease, responses to initial hATG/CyA at three months and six months were as follows: one CR and six NR at three months; three CR, one VGPR, two NR, and one NE at six months. Of the 52 patients receiving a second IST treatment, response to initial treatment with hATG/CyA was as follows: 36 had refractory disease, 15 had relapsed disease, and one patient lacked available response data. Of the 15 patients undergoing second IST for relapsed disease, responses to initial hATG/CyA at three months and six months were as follows: one CR, one VGPR, one PR, and 12 NR at three months; one CR, four VGPR, five PR, and five NR at six months. Two additional patients received tacrolimus for relapsed disease. Additional information is provided in the Online Supplementary Table S1.
For the 80 subjects who underwent second-line HSCT therapy, donors included 12 matched sibling donors, 61 matched unrelated donors (MUD), and 7 haplo-identical donors. Stem cell sources within the MUD cohort consisted of 42 from BM, five from peripheral blood, 12 from cord blood, and two without available data. Transplant preparative regimens varied widely both within and between institutions.
Overall survival for patients receiving a second treatment is shown in Figure 4A. Among all subjects (n=110) and the hATG/CyA group (n=92) who received second-line treatment, 20 (18.2%) and 15 (16.3%) died, respectively. In a Cox proportional hazards model, there was no significant effect of time from IST to second treatment on OS for either the entire cohort (HR=1.0, 95%CI: 0.98,1.02; P=0.76) or the hATG/CyA group (HR=0.99, 95%CI: 0.98,1.03; P=0.69). EFS is shown in Figure 4B. Among all subjects receiving second-line treatment (n=110), 49 (44.6%) failed with a median time to failure of 88.5 months (95%CI: 45.6,131.5). Among the 92 subjects from the hATG/CyA group receiving second-line treatment, 43 (46.7%) failed with a median time to failure of 64.4 months (95%CI: 44.2,131.5)
Figure 4.

Outcomes after second-line therapy for relapsed/refractory disease. Kaplan-Meier analysis of (A) overall survival and (B) event-free survival for all subjects or subjects treated with horse anti-thymocyte globulin (rATG)/cyclosporine (CyA). (C) Log-rank test was used to compare event-free survival after second-line treatment with hematopoietic stem cell transplantation (HSCT) versus immunosuppressive therapy (IST) for all subjects or subjects treated with hATG/CyA.
Outcomes of second-line treatment with HSCT versus IST were compared for all subjects and for the hATG/CyA treatment group. Among all subjects receiving subsequent treatment (n=110), 45 received HSCT and 65 received IST. Among the hATG/CyA group receiving second-line treatment (n=92), 38 received HSCT and 54 received a second course of IST. Due to the significantly longer follow up with second-line IST as compared to bone marrow transplantation for both the entire cohort (69 vs. 36 months; log rank, P=0.05) and the hATG/CyA group (74 vs. 36 months; log rank, P=0.026), the data were censored at 36 months in the analysis to minimize the impact of differential follow up. The analysis shows that EFS is significantly longer with the second-line treatment of HSCT compared with IST (log rank, P≤0.011) (Figure 4C); this effect remains after adjusting for other variables (Online Supplementary Figure S1).
Discussion
We report a multi-institutional study of the presentation and outcomes of 314 North American pediatric SAA patients treated with IST. Although retrospective studies are limited by potential confounding factors and data availability, this multicenter study provides a contemporary analysis of the diagnostic evaluation and treatment outcomes for pediatric SAA. Since the natural history, risks, benefits, and outcomes of treatments are not identical between children versus adults with SAA, the study of rare diseases such as pediatric SAA requires collaborative effort through large consortia with the goal of improving diagnosis and treatment. As with any rare disease, national registries would greatly advance the prospective study of pediatric aplastic anemia.
Although traditionally reticulocytopenia has been used as a diagnostic criterion for SAA, in this large study, reticulocytopenia with an ARC <60×109/L did not correlate with a Hb <8 g/dL in a subset of patients. We found that the majority of clinicians were using clinically significant anemia and need for transfusion, rather than the ARC, to inform diagnosis and initiation of therapy. Indeed, in the setting of severe anemia without BM failure, the reticulocyte count would be expected to be markedly higher, so even an ARC within the normal range may be a sign of impaired erythropoiesis.
Red cell macrocytosis may be indicative of the time frame from evolution of reduced hematopoietic stem cell numbers and clinically significant cytopenias. In addition, red cell macrocytosis may be associated with dysplastic processes. However, no association between macrocytosis and likelihood of hematologic response or survival was observed. Lymphopenia in pediatric patients with other cytopenias may be seen with primary immunological disorders; however, no association with response was observed. Development of cytogenetically abnormal clones or overt hematologic clonal disease was rare, although post-treatment marrow surveillance was not uniformly conducted or captured due to the retrospective nature of this analysis.
A correlation between short leukocyte telomere length at diagnosis, measured by quantitative polymerase chain reaction, with increased risk of relapse, clonal evolution, and reduced survival has been reported.21 Since current clinically available telomere length testing utilizes flow-FISH,13 we explored whether the results of this clinical telomere length assay correlated with outcomes. We did not detect a correlation between telomere lengths less than either the 1st or 10th percentile for age with response to IST.
We observed some notable differences from reported outcomes for adult SAA patients treated with hATG/CyA. Since the magnitude of blood count recovery is especially important to support normal growth and activity in children, we examined the quality of response to IST. Of those pediatric patients who responded to hATG/CyA, the quality of response was high, with 59.8% achieving a CR and 68.2% achieving a DR. In contrast, typical rates of CR cited for adults is 10%.12 In addition, a lower rate of clonal progression was noted for children in comparison to the 10-15% clonal progression reported in adults, although this observation is potentially limited by the retrospective nature of this study and short follow-up time.22,23 However, EFS was low, with events continuing to accrue well past five years post IST without an apparent plateau. EFS is of particular concern for pediatric patients given their long future lifespan. Accordingly, both clinical decisions and evaluation of new therapies should be based on pediatric data whenever possible, rather than on extrapolation of data from adult cohorts. For patients with refractory or relapsed disease, EFS was superior for patients receiving second-line treatment with HSCT compared with IST even after adjusting for age, gender, time from initial IST to second treatment, and lymphocytopenia. A prospective study of 21 pediatric SAA patients receiving a second course of IST for refractory disease reported anaphylaxis in three patients and a trilineage response in only two (11%) of the remaining 18 patients, with a 5-year failure-free survival of only 9% at five years post second-line therapy.6 Together with the excellent contemporary outcomes of MUD transplantation,24-26 these data strongly suggest that allogeneic transplantation with a MUD is a superior second-line therapy for relapsed or refractory SAA after IST for pediatric patients. In addition, these data suggest a potential role for MUD HSCT as upfront therapy in young patients. A randomized pilot and feasibility trial comparing hATG/CyA versus MUD HSCT in newly diagnosed SAA patients lacking an HLA-matched family donor is currently underway.
Supplementary Material
Acknowledgments
The authors are grateful to Susan Kornetsky for her guidance and expertise. We also thank Tim Colby, Brian Sheehan, and Melissa Rose for their assistance with this project.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/10/1974
Funding
This project was supported by the Copeman Fund, Julian’s Dinosaur Guild, and an NIH R24 DK 099808 to AS and DAW, the Rauch Family Foundation to TDC, the Campini Foundation to JNH and U54 DK106857 to GMK.
References
- 1.Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16(1 Suppl):S119-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheinberg P, Wu CO, Nunez O, Young NS. Long-term outcome of pediatric patients with severe aplastic anemia treated with antithymocyte globulin and cyclosporine. J Pediatr. 2008;153(6):814-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kojima S, Horibe K, Inaba J, et al. Long-term outcome of acquired aplastic anaemia in children: comparison between immunosuppressive therapy and bone marrow transplantation. Br J Haematol. 2000; 111(1):321-328. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida N, Kobayashi R, Yabe H, et al. First-line treatment for severe aplastic anemia in children: bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica. 2014;99(12):1784-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamio T, Ito E, Ohara A, et al. Relapse of aplastic anemia in children after immunosuppressive therapy: a report from the Japan Childhood Aplastic Anemia Study Group. Haematologica. 2011;96(6):814-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosaka Y, Yagasaki H, Sano K, et al. Prospective multicenter trial comparing repeated immunosuppressive therapy with stem-cell transplantation from an alternative donor as second-line treatment for children with severe and very severe aplastic anemia. Blood. 2008;111(3):1054-1059. [DOI] [PubMed] [Google Scholar]
- 7.Garanito MP, Carneiro JD, Odone Filho V, Scheinberg P. Outcome of children with severe acquired aplastic anemia treated with rabbit antithymocyte globulin and cyclosporine A. J Pediatr (Rio J). 2014; 90(5):523-527. [DOI] [PubMed] [Google Scholar]
- 8.Dufour C, Pillon M, Passweg J, et al. Outcome of aplastic anemia in adolescence: a survey of the Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation. Haematologica. 2014;99(10):1574-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dufour C, Pillon M, Socie G, et al. Outcome of aplastic anaemia in children. A study by the severe aplastic anaemia and paediatric disease working parties of the European group blood and bone marrow transplant. Br J Haematol. 2015;169(4):565-573. [DOI] [PubMed] [Google Scholar]
- 10.Scheinberg P, Nunez O, Weinstein B, Biancotto A, Wu CO, Young NS. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365(5):430-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams DA, Bennett C, Bertuch A, et al. Diagnosis and treatment of pediatric acquired aplastic anemia (AAA): An initial survey of the North American Pediatric Aplastic Anemia Consortium (NAPAAC). Pediatr Blood Cancer. 2014;61(5):869-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. N Engl J Med. 2017;376(16):1540-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alter BP, Baerlocher GM, Savage SA, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110(5):1439-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camitta BM, Storb R, Thomas ED. Aplastic anemia (first of two parts): pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306(11):645-652. [DOI] [PubMed] [Google Scholar]
- 15.Camitta BM, Storb R, Thomas ED. Aplastic anemia (second of two parts): pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306(12):712-718. [DOI] [PubMed] [Google Scholar]
- 16.Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. The German Aplastic Anemia Study Group. N Engl J Med. 1991;324(19):1297-1304. [DOI] [PubMed] [Google Scholar]
- 17.Rosenfeld SJ, Kimball J, Vining D, Young NS. Intensive immunosuppression with antithymocyte globulin and cyclosporine as treatment for severe acquired aplastic anemia. Blood. 1995;85(11):3058-3065. [PubMed] [Google Scholar]
- 18.Fuhrer M, Burdach S, Ebell W, et al. Relapse and clonal disease in children with aplastic anemia (AA) after immunosuppressive therapy (IST): the SAA 94 experience. German/Austrian Pediatric Aplastic Anemia Working Group. Klin Padiatr. 1998; 210(4):173-179. [DOI] [PubMed] [Google Scholar]
- 19.Kojima S, Hibi S, Kosaka Y, et al. Immunosuppressive therapy using antithymocyte globulin, cyclosporine, and danazol with or without human granulocyte colony-stimulating factor in children with acquired aplastic anemia. Blood. 2000; 96(6):2049-2054. [PubMed] [Google Scholar]
- 20.Bacigalupo A, Bruno B, Saracco P, et al. Antilymphocyte globulin, cyclosporine, prednisolone, and granulocyte colony-stimulating factor for severe aplastic anemia: an update of the GITMO/EBMT study on 100 patients. European Group for Blood and Marrow Transplantation (EBMT) Working Party on Severe Aplastic Anemia and the Gruppo Italiano Trapianti di Midolio Osseo (GITMO). Blood. 2000;95(6):1931-1934. [PubMed] [Google Scholar]
- 21.Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA. 2010;304(12):1358-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Socie G, Rosenfeld S, Frickhofen N, Gluckman E, Tichelli A. Late clonal diseases of treated aplastic anemia. Semin Hematol. 2000;37(1):91-101. [PubMed] [Google Scholar]
- 23.Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289(9):1130-1135. [DOI] [PubMed] [Google Scholar]
- 24.Samarasinghe S, Steward C, Hiwarkar P, et al. Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol. 2012;157(3):339-346. [DOI] [PubMed] [Google Scholar]
- 25.Anderlini P, Wu J, Gersten I, et al. Cyclophosphamide conditioning in patients with severe aplastic anaemia given unrelated marrow transplantation: a phase 1-2 dose de-escalation study. Lancet Haematol. 2015;2(9):e367-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bacigalupo A, Giammarco S, Sica S. Bone marrow transplantation versus immunosuppressive therapy in patients with acquired severe aplastic anemia. Int J Hematol. 2016;104(2):168-174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.