

Abstract
Internal tandem duplication in Fms-like tyrosine kinase 3 (FLT3-ITD) is the most frequent mutation observed in acute myeloid leukemia (AML) and correlates with poor prognosis. FLT3 tyrosine kinase inhibitors are promising for targeted therapy. Here, we investigated mechanisms dampening the response to the FLT3 inhibitor quizartinib, which is specific to the hematopoietic niche. Using AML primary samples and cell lines, we demonstrate that convergent signals from the hematopoietic microenvironment drive FLT3-ITD cell resistance to quizartinib through the expression and activation of the tyrosine kinase receptor AXL. Indeed, cytokines sustained phosphorylation of the transcription factor STAT5 in quizartinib-treated cells, which enhanced AXL expression by direct binding of a conserved motif in its genomic sequence. Likewise, hypoxia, another well-known hematopoietic niche hallmark, also enhanced AXL expression. Finally, in a xenograft mouse model, inhibition of AXL significantly increased the response of FLT3-ITD cells to quizartinib exclusively within a bone marrow environment. These data highlight a new bypass mechanism specific to the hematopoietic niche that hampers the response to quizartinib through combined upregulation of AXL activity. Targeting this signaling offers the prospect of a new therapy to eradicate resistant FLT3-ITD leukemic cells hidden within their specific microenvironment, thereby preventing relapses from FLT3-ITD clones.
Introduction
The Fms-like tyrosine kinase 3 (FLT3) gene encodes a class III receptor tyrosine-kinase (RTK) that is well expressed in hematopoietic stem progenitor cells (HSPC) and strongly activates PI3K/AKT and MAPK pathways upon ligand binding.1 Internal tandem duplication (ITD) in FLT3 is one of the most frequent mutations found in acute myeloid leukemia (AML).2 Although the FLT3-ITD mutation is a late event in leukemogenesis,3 it is an important target for the disease.4 Indeed, FLT3-ITD mutation is associated with a poor prognosis,5-7 and its frequent occurrence at relapse suggests that FLT3-ITD AML-initiating cells are key targets for long-lasting remission.
The FLT3-ITD mutation induces constitutive activity of the receptor and a distinct pattern of activated signaling pathways, the principal change being the activation of the transcription factor STAT5.8 FLT3 tyrosine kinase inhibitors (FLT3-TKI), which were developed as ATP-competitive inhibitors, were initially tested in clinical trials and produced variable benefits according to the disease heterogeneity. Among these treatments, quizartinib (AC220), a FLT3-TKI specifically designed for FLT3, induces a hematologic improvement in monotherapy associated with approximately 50% of response.9 However, bone marrow (BM) blasts show little noticeable cell apoptosis, but are associated with cell-cycle arrest and terminal differentiation.10 Remissions are of short duration, with the emergence of resistance related to several mechanisms. Intrinsic mechanisms include the activation of bypass signaling pathways11 and activation loop or gatekeeper mutations.4 Extrinsic mechanisms include cell-to-cell interactions and secretion of cytoprotective factors.12
AXL belongs to the TAM receptor family, which also includes TYRO3 and MER.13 This RTK is activated by homodimerization upon binding of its major ligand growth arrest-specific 6 (GAS6).14 The GAS6/AXL pathway contributes to cell growth, survival, invasiveness, chemotaxis, apoptotic body clearance and immunity.15 AXL is ectopically-or over-expressed in a wide variety of cancers and has always been associated with a poor prognosis.16 We have reported resistance mechanisms involving AXL in chronic myeloid leukemia.17 In AML, AXL and GAS6 levels of expression have been related to poor outcomes.18,19 Paracrine AXL activation has been shown to induce AML resistance to conventional chemotherapies but also to FLT3-targeted therapy.20-23 However, no information is available concerning the regulation of AXL expression in the context of the AML-supportive hematopoietic niche, which sustains AML resistance in vivo.
Indeed, AXL expression and function have been shown to be modulated by stress, nutrient deprivation and low oxygen (O2) concentration in various solid tumors.14,24 These characteristics are quite similar to those within the hematopoietic microenvironment of AML-initiating cells. We show here that the hematopoietic microenvironment educates AML cells to over-express AXL through a cytokine-dependent STAT5 activation and low O2 concentration.
Methods
Cell culture, apoptosis and AXL, GAS6 and STAT5 gene expression knock-down using shRNA
All cell lines (MV4-11, MOLM-13, MOLM-14, UT7-mpl, K562, MS5, OP9, HS27a) were cultured in RPMI1640 or αMEM medium, supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin. Hypoxia was induced by incubating cells in a specific O2 chamber (BioSpherix). The UT7-mpl cell medium was supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF, 2.5ng/mL, Diaclone, France), as previously described.25 UT7-mpl cells were pre-incubated for 18 hours (h) in the absence of GM-CSF, before being activated by cytokines. Where indicated, cells were cultured with vehicle or thrombopoietin peptide (Sigma), interleukin (IL)-3 (Miltenyi Biotec), AXL-Fc chimeric proteins (R&D Systems), AC220 (quizartinib) (LC laboratories), R428 (Selleckchem), Ly294002, pimozide, JAK inhibitor-I (Calbiochem/Merck). AC-4-130, a selective STAT5 inhibitor, was provided by JANPIX Ltd. (UK) under the license from Prof. Patrick Gunning’s group (University of Toronto, Canada).26 Cell apoptosis was assessed using an APC-conjugated Annexin V labeling detection kit coupled to flow cytometry and BDFACSDIVATM software (BD Bioscience). For shRNA, viral supernatants were titrated by serial dilutions and a FACS analysis of GFP co-expressed marker in transduced K562 cells 72 h later (Accuri C6, Beckton Dickinson). Lentiviruses were added once, at a multiplicity of infection of 5-20 according to the cells. Transduced cells were sorted 2-3 days after transduction using GFP. Two different shRNA were used for all targets (Online Supplementary Table S1). To confirm silencing, AXL, GAS6 or STAT5 protein expressions were analyzed by immunoblotting.
Acute myeloid leukemia patient and cord blood donor biological samples
Acute myeloid leukemia samples were obtained from patients at the University Hospital of Bordeaux and University Hospital Paris Centre who gave written informed consent for the use of biological samples for research, in accordance with the Declaration of Helsinki. This allowed the collection of clinical and biological data in an anonymized database, registered at the “Commission Nationale de l’Informatique et des Libertés” (authorization n. 915285) and “Comité de Protection des Personnes” (authorization n. 2015-08-11D). For FLT3-ITD AML patient samples, selection criteria were high leukemic burden (blast cells level > 70%) and high FLT3-ITD/WT ratio (ITD/WT > 0.5) (Online Supplementary Table S2). Bone marrow mononu-clear cells were co-cultured with MS5 stromal cells in H5100 medium (Stem Cell Technologies), unless otherwise stated. When indicated, AML cells were preincubated for 18 h in serum-free IMDM medium in the absence of MS5 before being incubated for 7 h in the presence of a cytokine cocktail that included IL-3 (20 ng/mL), GM-CSF (10 ng/mL) and TPO (20 nM). Human cord blood (CB) units were collected according to institutional guidelines. Cord blood CD34+ were purified and cultured in a four-cytokine-supplemented serum-free IMDM medium, as previously described.27
Further details about the methods used are available in the Online Supplementary Appendix.
Results
Stromal protection of FLT3-ITD AML cells to quizartinib correlates with STAT5 activation
We first confirmed the protective effect of three stromal cell lines on MV4-11, MOLM-13 and MOLM-14 FLT3-ITD AML cell lines exposed to quizartinib (AC220), an ATP-competitive FLT3-inhibitor. Co-culture of AML cells with one human (HS27a) and two murine (OP9 or MS5) stromal cells decreased apoptosis triggered by FLT3-TKI treatment compared to AML cells cultured without stroma (Figure 1A and Online Supplementary Figure S1A and B). This decrease persisted at high AC220 concentra tions (10-20 nM) (Online Supplementary Figure S1C) and was not due to a decrease in active AC220 concentration by the metabolism of stromal cells (Online Supplementary Figure S1D). To investigate further the mechanism of such protection, stromal/AML cell co-cultures were performed with a transwell separating the two kinds of cells, thereby avoiding cell-to-cell contact. Transwell-separated stromal cells still protected FLT3-ITD AML cells from quizartinib-induced apoptosis, but the protective effect was weaker (Figure 1B), suggesting the involvement of both cell-to-cell contact and diffusible factors. We then analyzed signaling pathways in cells treated with AC220 in the presence or absence of stroma. Whereas AC220 treatment of FLT3-ITD AML cell lines (MV4-11, MOLM-14) decreased Y591FLT3, T202/Y204ERK, Ser473AKT and Y694/Y699 STAT5 phosphorylation in the absence of stroma, the presence of co-cultured stromal cells (MS5, OP9, HS27A), along with AC220 treatment, specifically sustained Y694STAT5A/Y699STAT5B phosphorylation (hereafter referred to as pYSTAT5) (Figure 1C and Online Supplementary Figure S1E). STAT5 Tyr phosphorylation was still conserved at higher AC220 concentrations (Online Supplementary Figure S1F).
Figure 1.

Stromal cells protect MV4-11 cells from AC220-induced apoptosis and correlate with enhanced STAT5 activation. (A) MV4-11 FLT3-ITD acute myeloid leukemia (AML) cell line was incubated in the absence (Vehicle) or presence of AC220 (3 nM) for 48 hours, without (None) or with the indicated murine (OP9 or MS5) or human (HS27a) stromal cell lines. Cell apoptosis was determined by Annexin V/DAPI labeling followed by flow cytometry analysis. (B) MV4-11 FLT3-ITD AML cell line was incubated in the absence (Vehicle) or presence of AC220 (3 nM) for 48 h, without (None) or with OP9 cells separated (Stroma TW) or not (Stroma) by transwells. Apoptosis induction was determined as in (A). (C) MV4-11 cells were incubated in the absence (Vehicle) or presence of AC220 (1 nM), without (none) or with OP9 stromal cells (Stroma) for 48 h. Upon cell lysis, immunoblot analysis of the indicated protein with β actin as a loading control, each of protein-dedicated immunoblot without (none) or with (stroma) OP9 co-culture was performed on the same membrane. Results shown are representative of three experiments. Graphs show the mean±Standard Error of Mean of results of at least three independent experiments. *P<0.05; **P<0.01; ***P<0.001.
Therefore, stromal protection of FLT3-ITD AML cells is associated with an FLT3-ITD-independent conservation of STAT5 tyrosine phosphorylation.
AXL contributes to stromal cell-dependent FLT3-ITD AML resistance to FLT3-TKI
To determine how STAT5 contributes to TKI-resistance of AML cells by stromal cells, we looked for STAT5 target genes in myeloid cells. We searched for STAT5 regulated genes by conducting a comparative large-scale genome expression analysis of STAT5 knocked-down (KD) and control cells using normal human primitive CD34+ CD38− HSPC. Among the top-ranked down-regulated transcripts upon STAT5 KD, we looked for mediators that might be involved in stromal protection. AXL was the only RTK down-regulated in these STAT5 KD cells (Online Supplementary Table S3) and was the only one reported in resistance to treatment in AML.21,23 To examine the role of AXL in cancer resistance, we investigated whether there was a link between STAT5-dependent AXL regulation, stroma-dependent STAT5 tyrosine phosphorylation maintenance and stromal protection for AC220. We found that AXL mRNA and protein were both decreased in STAT5 KD HSPC (Online Supplementary Figure S2A). Like HSPC, STAT5 KD induced AXL loss in MV4-11 and MOLM-14 FLT3-ITD or UT7 FLT3 wt AML cell lines (Figure 2A and Online Supplementary Figure S2B). STAT5 is encoded by two highly similar genes, STAT5A and STAT5B, whose combined KD led to cell death. However, STAT5A-restricted KD did not affect AML cell survival; it allowed us to perform rescue experiments. Silencing of STAT5A followed by expression of either murine STAT5A or murine STAT5B enhanced AXL expression, thus providing evidence that both STAT5 regulated AXL expression (Online Supplementary Figure S2B). Finally, we found that stromal cells enhanced both STAT5 phosphorylation and AXL expression in MV4-11 and MOLM-14 cells (Figure 2B). These activities were both prevented when cells were incubated in the presence of the STAT5 inhibitor AC-4-130 (Online Supplementary Figure S2C). Overall, these data indicated that stroma supports STAT5 activation of AML cells, which enhances AXL expression of AML cells.
Figure 2.

STAT5 up-regulates AXL which contributes to FLT3-ITD acute myeloid leukemia (AML) cell survival. (A) MOLM-14 and MV4-11 cells were transduced with the indicated shRNA encoding lentiviral vectors and lysed three days later. Immunoblot analysis of the indicated proteins was performed with β actin as a loading control. (B) MOLM-14 and MV4-11 cells were co-cultured in the absence (−) or presence (+) of OP9 stromal cells for 48 hours (h) before being isolated and lysed. The indicated proteins were analyzed by immunoblotting. (C) MV4-11, MOLM-13 and MOLM-14 cells were treated in the absence (Vehicle) or presence of minimal effective (MV4-11, MOLM-14) or suboptimal (MOLM-13) dose of AC220 (1 nM) and R428 (0.3 μM) or both (Combo) for 48 h. Apoptosis induction was determined as in Figure 1. (D) Primary FLT3-ITD AML blasts (n=2, AML#1-2) were cultured without (−) or with (+) human stromal cells (HS27a) for two days and lysed. Immunoblot analysis of the indicated proteins was performed with β actin as a loading control. (E) Primary FLT3-ITD AML blasts (n=7, AML#3-9) were co-cultured on MS5 stromal cells for 24 h, then co-incubated in the presence of AC220 (1 nM), R428 (0.3 μM) or both for 48 h. Apoptosis induction was assessed by Annexin V/DAPI labeling and flow cytometry analysis. Results are from seven AML samples treated in independent experiments. (F) MV4-11 cells were incubated with AC220 (1 nM), without (none) or with stromal cells (OP9) added with Ctrl-Fc or AXL-Fc (1 μg/mL). Cell apoptosis was determined as indicated in Figure 1. (G) MV4-11 cells were incubated in the absence (Vehicle) or presence of AC220 (3 nM), without (None) or with OP9 stromal cells that express shCtrl or shGAS6. Apoptosis induction was determined as described above. Graphs show the mean±Standard Error of Mean of results of at least three independent experiments. *P<0.05; **P<0.01; ***P<0.001.
We thus analyzed the role of AXL in AML cell survival with a well-characterized and clinically investigated AXL-TKI compound, R428.28 To determine whether AXL and FLT3-ITD pro-survival activity were connected, we first determined the minimal effective AC220 and R428 dose to induce apoptosis after 48 h of incubation (Online Supplementary Figure S2D). The minimal effective AC220 dose was 1-3 nM, depending on the FLT3-ITD AML cell line, whereas for R428 it was 0.3 μM for all. Using these doses, we observed that AXL and FLT3 co-inhibition triggered additive apoptosis (MOLM-13) and even a weak synergistic activity (MV4-11 and MOLM-14) as compared to single drug treatment (Figure 2C).
Next, we investigated whether such effects were observed in primary AML blasts. In all primary AML samples tested, HS27a stroma induced upregulation of AXL expression (AML#1, #2 in Figure 2D and Online Supplementary Table S2). However, this increase did not provide extra AML cell survival when AXL activity was inhibited. Instead, AXL and FLT3 co-inhibition induced a significantly stronger apoptosis than single drug treatments on primary AML blasts cultivated ex vivo on stromal cells (AML #3 to #9 in Figure 2E and Online Supplementary Table S2).
AXL is activated through the binding of its ligand GAS6, for which autocrine and paracrine activities have been reported in various contexts, including AML. Although both stromal and leukemic cells are known to secrete GAS6, stromal secretion was 10-to 20-fold more abundant than leukemia ones, as detected by species-specific ELISA (Online Supplementary Figure S2E).29 We analyzed the contribution of GAS6 to the protective activity of stromal cells toward AC220-induced AML cell apoptosis. Protection of FLT3-ITD AML cells by stromal cells was significantly reduced yet was not suppressed in the presence of AXL-Fc, a well-known GAS6-neutralizing molecule22 (Figure 2F). Moreover, GAS6-KD OP9 stromal cells provided weaker protection than parental OP9 (Figure 2G and Online Supplementary Figure S2F), confirming the results observed with AXL-Fc.
Taken together, using an AXL inhibitor (R428) or a GAS6-neutralizing molecule (AXL-Fc) and genetically engineered stromal cells (OP9 shGAS6), these results confirm that AXL activity contributes to the stromal protection of FLT3-ITD AML.
STAT5-activating cytokines up-regulate AXL expression and activity
Since AXL expression is associated with STAT5 activation, we wondered whether and how stroma activates STAT5. We first analyzed GAS6 activity but did not detect any STAT5 tyrosine phosphorylation upon GAS6 treatment in AML cells. We therefore wondered whether STAT5-activating cytokines, secreted by stromal cells, could trigger AXL overexpression. Therefore, we first used the UT7-mpl AML cell line (FLT3 wild type) which is highly sensitive to GM-CSF and TPO for its growth and survival.25 Both GM-CSF and TPO activated STAT5 and enhanced AXL expression within a few hours (Figure 3A and B). These cytokines are known to activate STAT5 by binding to their receptors through subsequent activation of JAK2 kinase. In the presence of the STAT5 inhibitor pimozide (pi) or the JAK2 inhibitor-I (ji), GM-CSF and TPO no longer up-regulated AXL expression, whereas inhibition of the PI3K/AKT pathway by LY294002 (ly) had no effect (Figure 3B). We further observed that AXL activation was also induced by GM-CSF and TPO in AML cells, as assessed by Tyr779 AXL phosphorylation detection and by global Tyr phosphorylation in AXL immunoprecipitates (Figure 3B and C). Similar results were obtained using primary CB CD34+ HSPC in the presence of interleukin (IL)-3 or TPO. However, this was not the case with the FLT3 ligand, which did not activate STAT5 (Online Supplementary Figure S3A). These results indicated that STAT5-activating cytokines can up-regulate both AXL expression and activation in normal HSPC and FLT3 wild-type AML.
Figure 3.

Activation of STAT5 up-regulates AXL gene expression and phosphorylation. (A) UT7-mpl acute myeloid leukemia (AML) cells were treated with GM-CSF (2.5 ng/mL) or TPO (20 nM) for the indicated time and lysed. Immunoblot analysis of the indicated proteins; HSP60 was used as a loading control. (B) UT7-mpl cells were treated with GM-CSF (2.5 ng/mL) or TPO (20 nM) for the indicated time in the absence or presence of pimozide (pi, 2 μM), JAK inhibitor-I (ji, 1 μM) or Ly294002 (ly, 20 μM). Total cell lysates were analyzed by western blotting with the indicated antibodies. HSP60 was used as a loading control. (C) UT7-mpl cells were incubated with (+) or without (−) TPO (20 nM) for one hour (h) before being lysed. AXL immunoprecipitates were prepared and analyzed by immunoblotting with the indicated antibodies. (D and E) MOLM-14 or MV4-11 cells were pre-incubated with (+) or without (−) AC220 (3 nM) for 3 h before adding IL-3 (ng/mL, MOLM-14) or TPO (nM, MV4-11) at the indicated concentration. Cells were incubated for 4 h before being lysed. (F and G) MS5 co-cultured AML primary blasts were maintained in serum-free medium without stroma for 18 h before being incubated in the presence of a IL-3 (20 ng/mL)/GM-CSF (10 ng/mL)/TPO (20 nM) cytokine cocktail for 7 h and then lysed for protein (F, n=6, AML#10-12 and 15-16) and mRNA (G, n=5, AML#10-14) purification. Immunoblot analysis of the indicated proteins using β actin or HSP60 as loading controls; real-time quantitative polymerase chain reaction (RT-qPCR) quantification of AXL mRNA levels in cytokine-treated and untreated primary AML cells, normalized to GAPDH expression, and expressed relative to untreated cells. (H and I) UT7-mpl cells were incubated with (+) or without (−) TPO (20 nM) for 30 minutes. Chromatin-immunoprecipitation (IP) assays were performed using control immunoglobulins (IP IgG), STAT5 (IP STAT5) or RNA polymerase II (IP PolII) antibodies. Immunoprecipitated DNA were analyzed by qPCR using primers spanning the conserved STAT5-responsive element of AXL gene sequence and expressed relative to total lysates (input). Graphs show the mean±Standard Error of Mean of at least three independent experiments. *P<0.05; **P<0.01; ***P<0.001; NS: not significant.
FLT3-ITD AML cells are known to co-express cytokine receptors such as IL-3 and TPO receptors, whose activity remains ill-defined.29,30 We wondered whether cytokines could trigger AXL upregulation, specifically in FLT3-ITD AML cells. Despite the presence of FLT3-ITD inhibitor AC220, IL-3 sustained STAT5 phosphorylation (Figure 3D). This activation was correlated with an increase in AXL protein expression (Figure 3E). Similarly, in primary AML samples, an IL-3/GM-CSF/TPO cytokine cocktail increased AXL RNA expression (AML#10-12 and #15-16 in Figure 3F and Online Supplementary Table S2) associated with STAT5 activation (AML#10-14 in Figure 3G and Online Supplementary Table S2).
To unravel how STAT5 up-regulates AXL transcript levels, we focused on the AXL genomic sequence. STAT5 transcription factors bind to TTCN3GAA STAT-Response Element (SRE) on their target genes. One SRE that is conserved among species including rat, mouse and human was identified within intron 16 and located at Chr19(+) [41,725,104-41,767,672] (www.genome.ucsf.edu). Oligonucleotide pull-down assays showed that this identified SRE was functional, since it bound STAT5 as efficiently as the control canonical SRE (ONc) from the IRF1 promoter sequence (Online Supplementary Figure S3B). Chromatin immunoprecipitation (ChIP) assays with STAT5 antibodies showed that STAT5-activation triggered binding to the AXL genomic sequence (Figure 3H). Together with STAT5 binding, recruitment of RNAPolII to the AXL gene was enhanced, as assessed by anti-RNAPolII ChIP (Figure 3I). These data indicated that STAT5 binds to the AXL gene and increases AXL promoter activity.
Taken together, these data indicated that STAT5 mediates stroma-dependent AXL upregulation.
Low O2 concentration enhances AXL expression
In addition to stromal cells and cytokines, the hematopoietic niche is also characterized by low O2 concentrations, which have been linked to AXL expression in other cancer cells.24,31 We thus assessed the contribution of low O2 levels (1% O2) in this model. Interestingly, hypoxia enhanced AXL expression in both FLT3-ITD AML cell lines and primary FLT3-ITD AML blasts (Figure 4A and AML#17 in Figure 4B and Online Supplementary Table S2). Several Hypoxia-Response Elements (HRE) have been described within a 2.4 kb fragment of the AXL promoter.24 AXL transcript levels were thus assessed in hypoxic culture for both AML cell lines and primary AML blasts. In all cells, hypoxia enhanced AXL RNA expression, thereby indicating that O2 levels affect AXL expression (Figure 4C and AML#18-20 in Figure 4D and Online Supplementary Table S2).
Figure 4.
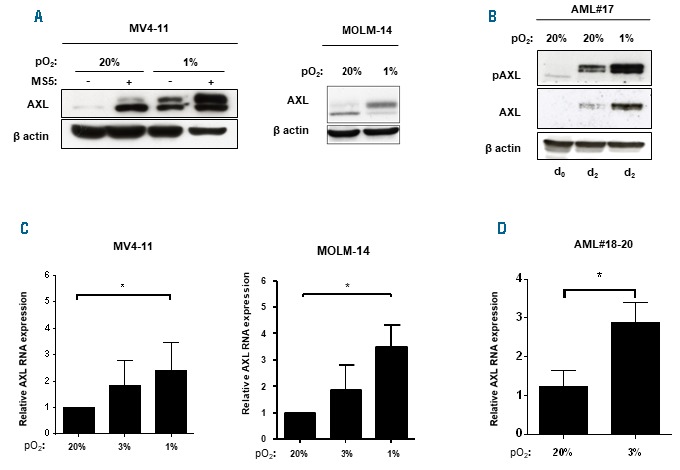
Low O2 concentration up-regulates AXL expression of FLT3-ITD acute myeloid leukemia (AML) cells. (A) MV4-11 cells were incubated for 48 hours (h) at 20% or 1% O2 with or without MS5 stromal cells as indicated; MOLM-14 cells were incubated for 48 h at 20% or 1% O2 without stroma. Immunoblot analysis of the indicated proteins was performed with βactin as a loading control. (B) Primary FLT3-ITD AML blasts (AML#17) were cultured with HS27a stromal cells for two days in normoxia (20%) and hypoxia (1%). Samples were lysed at day zero and two days after growth on stromal cells. Immunoblot analysis of the indicated proteins was performed with βactin as a loading control. (C) MV4-11 and MOLM-14 cells were incubated for 48 h at 20%, 3%, 1% O2. Real-time quantitative polymerase chain reaction (RT-qPCR) analysis of AXL mRNA was performed. Results are normalized to GUSB expression and expressed relative to 20% O2 cultured cells. (D) Primary AML blasts (n=3, AML#18-20) were incubated for 48 h at 20% and 3% O2. RT-qPCR analysis of AXL mRNA was performed as in (B). Graphs show the mean±Standard Error of Mean of results of at least three independent experiments. *P<0.05; **P<0.01; ***P<0.001.
As AXL contributed to FLT3-ITD AML cell survival in normoxia (see Figure 2C), we then analyzed its involve ment in AML cell survival in hypoxia by using the AXL-TKI R428. In the absence of stroma, R428-induced apoptosis was similar in hypoxia and normoxia. Conversely, in the presence of stroma, R428 triggered much stronger apoptosis under hypoxia (Online Supplementary Figure S4). Overall, these data indicate that both low O2 concentrations and STAT5-activating stromal cytokines, in addition to GAS6, regulate AXL activity, thus mediating stronger AXL-dependent protection of AML cells.
AXL mediates microenvironment-dependent resistance of FLT3-ITD acute myeloid leukemia cells to AC220 treatment in vivo
Having shown that AXL mediates microenvironment-dependent protection against quizartinib in vitro, we analyzed the contribution of AXL in vivo. To better investigate the role of AXL on a long timescale, we generated stable AXL KD FLT3-ITD AML cells through lentivirus-mediated RNA interference (Online Supplementary Figure S5A). MV4-11 shCtrl and MV4-11 shAXL cells were engineered to stably express the firefly luciferase gene. They were xenografted by vein injection and leukemic cell engraftment was analyzed by bioluminescence imaging (BLI). BLI analysis did not evidence any significant difference in engraftment (timing, intensity) between MV4-11 shCtrl and MV4-11 shAXL cells after seven days (Online Supplementary Figure S5B). Half of the mouse cohort was then treated with AC220 (5 mg/kg/day) by daily oral gavage for seven days, and response to treatment was assessed by BLI at day 14. The tolerance to treatment was checked by body weight control twice a week. A similar bioluminescence indicative of tumor progression was observed for MV4-11 shCtrl and MV4-11 shAXL cells in the vehicle-treated cohort (Figure 5A and B). In contrast, a significant decrease in total bioluminescent signal was observed in response to AC220 in MV4-11 shAXL-engrafted mice as compared to MV4-11 shCtrl-engrafted animals (P<0.01) (Figure 5B). To strengthen these data in the specific setting of the microenvironment, another set of experiments was performed with the same sequence of conditioning, engraftment and treatment, but BLI was performed upon treatment on femurs and tibias ex vivo at sacrifice. Again, a significant decrease in bioluminescence was observed in bones from AC220-treated MV4-11 shAXL-engrafted mice compared to bones from MV4-11 shCtrl-engrafted animals, whereas no significant difference was observed between these two cohorts in the absence of treatment (Figure 5C). Immunohistochemistry analysis of BM was then performed to trace human cells in the murine bone marrow tissue. Similar amounts of human cells were detected in MV4-11 shCtrl-and shAXL-injected mice in the absence of treatment (Figure 5D). In the AC220-treated cohort, as expected, lower amounts of human cells were detected in MV4-11 shCtrl-engrafted BM than in the untreated cohort. Interestingly, almost no human cells were detectable in MV4-11 shAXL-treated animal samples, so BLI detection was under the threshold in MV4-11 shAXL-treated animals (Figure 5D). Altogether, these results show that AXL plays a key role in the AC220 response of FLT3-ITD AML cells in the specific context of the hematopoietic niche, thus confirming our in vitro and ex vivo data.
Figure 5.

AXL sustains FLT3-ITD acute myeloid leukemia (AML) cell resistance to AC220 in the bone marrow hematopoietic niche. (A) MV4-11 shCtrl-Luc and MV4-11 shAXL-Luc were injected at day 0 (106 cells/mouse) in the retro-orbital sinus vein of busulfan-pretreated NSG female mice. Mice were then treated or not with AC220 (5 mg/kg/day body weight) from day 8 to day 14. At day 14, animals (B) or bones (femurs, tibias), collected from the indicated animals (C) were subjected to bioluminescent imaging (BLI). The dotted line shows the median background signal indicating the threshold for BLI sensitivity. Ph: photon; s: second; sr: steradian. (D) Immunohistochemistry analysis of Human Leukocyte Antigen expression in bone marrow biopsies collected from the indicated AML cell-injected mice in the absence or presence of AC220 at day 14. *P<0.05; **P<0.01; ***P<0.001; NS: not significant.
Next, we wondered whether the bone marrow niche protection of FLT3-ITD AML cells is specific to the hematopoietic niche conditions or if it would be similar in other microenvironments. We thus evaluated the impact of quizartinib on subcutaneous xenograft tumor growth using control (MV4-11 shCtrl) and AXL KD (MV4-11 shAXL) AML cells. Subcutaneous implantation of these cells into immunodeficient NSG mice resulted in solid tumors without any difference between MV4-11 shCtrl and MV4-11 shAXL cells (Online Supplementary Figure S5C). After 21 days, when tumors reached 300-500 mm3, half of the mouse cohort received AC220 by daily oral gavage (5 mg/kg/day). This resulted in complete regression of the tumors after 14 days. We did not observe any difference between MV4-11 shCtrl and MV4-11 shAXL in terms of response to AC220 treatment regarding both the kinetic and intensity of tumor regression (Online Supplementary Figure S5C). The daily administration of AC220 was ceased after full tumor regression, allowing tumor regrowth 12 days after AC220 withdrawal. Again, a similar progression was observed post-relapse between MV4-11 shCtrl and MV4-11 shAXL-injected cohorts both in time to relapse and in tumor volumes, thus confirming the specific role of the hematopoietic niche (Online Supplementary Figure S5C). Similar tumor weights at sacrifice confirmed these observations (Online Supplementary Figure S5D).
The absence of any difference in subcutaneous tumor growth between MV4-11 shCtrl and MV4-11 shAXL-injected animals shows that the instrumental role of AXL in protecting AML cells against FLT3-targeted therapy in vivo is specifically mediated by the hematopoietic niche.
Discussion
These findings show that the bone marrow hematopoietic niche provides specific protection for FLT3-ITD AML cells against quizartinib by multiple signaling pathways converging to AXL upregulation and activation. In addition to the established role of the AXL canonical ligand GAS6, the bone marrow niche enhances AXL expression and the activity of AML cells through both STAT5-activating soluble factors and local hypoxic environment.
The microenvironment was already known to promote AML cell resistance via several mechanisms such as CXCL12-CXCR4 signaling,32 adhesion molecules such as CD44 and selectins,33 vasculature by VEGF,34 and angiopoi-etins/TIE2 signaling35 and well-reviewed by Brenner et al.36 More recently, FLT3-ligand and FGF1/2 were also identified as protective molecules against FLT3-TKI in vitro.37 Hypoxia, a key factor of the microenvironment, was also reported to drive pro-tumoral signaling in AML via a HIF1α/MIF/IL8 pathway that is also thought to play a role in chronic lymphocytic leukemia survival.38 Stroma has also been shown to sustain AML cell resistance to cytarabine via activation of the AXL receptor upon AML cell-dependent education of stroma to secrete GAS6 in vitro.21 We now provide evidence that several microenvironment messages converge to enhance AXL expression and activation, which sustain pro-survival signals to selectively protect FLT3-ITD AML cells from quizartinib treatment in situ. Beyond demonstrating the value of AXL as a therapeutic target for AML burden, our data suggest a more appealing approach in which prevention of AXL expression will be key to alleviating protection against treatment of the leukemic initiating cells provided by their hematopoietic niche.
The role of cytokines and growth factors inside the hematopoietic niche with regard to AML cell survival has long been debated. TPO receptor expression level has been shown in approximately 50% of AML patients.30 In addition, primary human AML engrafts with higher efficacy in mice in which human versions of CSF1, CSF2, IL3 and TPO genes are knocked-in into murine loci, suggesting the central role of these cytokines.39 The IL-3 receptor (CD123), whose expression carries a poor prognosis,40 is thought to be a marker for AML-initiating cells41 and to be closely related with FLT3-ITD mutation.42 Our data now demonstrate a new survival mechanism provided by STAT5/hypoxia in the upregulation of AXL.
AXL expression is often up-regulated in solid tumors. Its activation has been mostly observed under stress conditions associated with metastatic disease or in situations where tumor cells are under severe nutrient deprivation. Knowledge about the extracellular messages that regulate AXL expression is limited. However, interferon-α has been reported to enhance AXL expression in monocytes through a STAT1-dependent pathway, but the mechanism by which this is achieved has not yet been investigated.43 Our results show for the first time that the AXL gene exhibits a conserved SRE that binds cytokine-activated STAT5 which up-regulates AXL gene expression through enhanced recruitment of RNAPolII. All the cytokines studied (IL-3, GM-CSF and TPO) activated both STAT5A and STAT5B, but we observed that STAT5A-selective knockdown triggered massive AXL downregulation, whatever the levels of STAT5B expression and activation (Dumas et al., 2019, personal communications). These observations suggest that STAT5A plays a dominant role in AXL regulation, an observation that fits with major regulation by STAT5A of other genes like HIF2α or PHD3 that promote HSPC and leukemia cell maintenance, as we and others have observed.44 Whether other STAT5-activating factors also enhance AXL expression and contribute to tumorigenesis and resistance of other myeloid leukemia, non-myeloid leukemia and/or solid tumors awaits further investigation.
The current findings show that STAT5-activating factors not only enhance AXL expression but also trigger AXL activation. Indeed, TK receptors such as EGFR or VEGFR, which activate Src kinases,45 and FLT3, were shown to activate AXL.22 AXL also functions as a docking site for non-receptor kinases such as Syk and Lyn,46 which could be activated by hematopoietic niche signals. Whether other kinases mediate AXL activation remains to be studied.
AXL expression is known to be modulated by O2 concentration in various solid tumors.24,31 Our present data extend these observations to AML. Low O2 concentrations are a well-known BM niche hallmark.47 Hypoxia was shown to down-regulate FLT3 and FLT3-ITD signaling in AML cells.48 Under such conditions, the PI3K/AKT pathway was shown to sustain AML cell survival, rescuing FLT3 activity,49 while AXL is known to activate PI3K/AKT signaling in AML cells. We and others have previously shown that cytokine-activated STAT5 enhances HIF2αexpression directly44 but also indirectly by inhibiting one of its destabilizers, the prolyl 4-hydroxylase domain protein 3 (PHD3) in both normal hematopoietic progenitors and leukemic cells.27 Such activities might reinforce the hypoxia-induced message coming directly from the microenvironment and strengthen AXL overexpression.
Finally, our data show that the hematopoietic niche provides selective signals regulating AXL expression. This plays a pivotal role in the selective response of FLT3-ITD AML cells to quizartinib within the specific location of the bone marrow, where leukemic stem cells are well maintained. Our study suggests that, in addition to the dual inhibition of AXL and FLT3 through TKI combination or a dual TK inhibitor such as gilteritinib, targeting AXL upstream signaling steps could be investigated in targeting FLT3-ITD AML-initiating cells in the hematopoietic niche.9,50
Supplementary Material
Acknowledgments
We thank Mrs. Bernadette De Buhan and her family for their generous support. We thank the Cytometry and Genomic Facilities of Institut Cochin/Inserm U1016, the Cytometry Facility, Vectorology Facility, Animals Facility and Histology Facility of FR TransBioMed, Bordeaux University, for performing cell sorting, microarray screening and handling animals. We thank the Obstetric Unit at Orsay Hospital and the Cell Therapy Center at St-Louis hospital (Paris, France) for providing CB samples. P-YD was a recipient of the SIRIC Brio for research fellowship for one year, then the MD-PhD program from the University Hospital of Bordeaux for two years. CN and SM-L were fellows of the “Association pour la recherche contre le Cancer”, “Ligue Nationale contre le Cancer” and Région Ile de France DIM-Biothérapies.We thank David Galeazzi for accurate data collection concerning patients at Bordeaux University Hospital. We also acknowledge the Centre de Ressources Biologiques Cancer, Bordeaux Biothèques Santé (BB-0033-00036) at Bordeaux University Hospital as well as the CRBP at the Hematology unit at Cochin Hospital (CPP: 2015-08-11 DC) for providing biological material.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/10/2017
Funding
AG was funded by the SIRIC Brio (grant INCa-DGOS-Inserm 6046) for his work at the UMS 005 TBM Core histopathology platform. This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), Paris-Descartes University, Agence Nationale de Recherche (ANR-2011-RPIB-009-04), Ligue Nationale contre le Cancer (RS13/75-6; EL2014-3), Ligue Régionale contre le Cancer (Comité Pyrénées Atlantique et comité Ile de Paris) and Cancéropôle Grand Sud-Ouest (2015-0389). We thank the data management unit at Toulouse University Hospital and the CAPTOR (Cancer Pharmacology of Toulouse Oncopole and Region) project (ANR-11-PHUC-001) for its financial support enabling e-CRF for the AML database. The funders had no role in study design, data collection or analysis, decision to publish or preparation of the manuscript. We thank Prof. D. Bouscary, Dr P. Auberger and Dr E. Lauret for their critical reading of the manuscript.
References
- 1.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532-1542. [DOI] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirsch P, Zhang Y, Tang R, et al. Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat Commun. 2016;7:12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith CC, Wang Q, Chin C-S, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stirewalt DL, Kopecky KJ, Meshinchi S, et al. Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood. 2006; 107(9):3724-3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlenk RF, Dohner K, Krauter J, et al. Mutations and Treatment Outcome in Cytogenetically Normal Acute Myeloid Leukemia. N Engl J Med. 2008; 358(18):1909-1918. [DOI] [PubMed] [Google Scholar]
- 7.Fischer M, Schnetzke U, Spies-Weisshart Br, et al. Impact of FLT3-ITD diversity on response to induction chemotherapy in patients with acute myeloid leukemia. Haematologica. 2017;102(4):e129-e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choudhary C, Brandts C, Schwable J, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110(1):370-374. [DOI] [PubMed] [Google Scholar]
- 9.Cortes JE, Tallman MS, Schiller GJ, et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD mutated, relapsed or refractory AML. Blood. 2018;132(6):598-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sexauer A, Perl A, Yang X, et al. Terminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3/ITD AML. Blood. 2012;120(20):4205-4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green AS, Maciel TT, Hospital MA, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci Adv. 2015;1(8):e1500221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato T, Yang X, Knapper S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011; 117(12):3286-3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linger RMA, Keating AK, Earp HS, Graham DK. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer. Advances in Cancer Research. In: Klein GFVWaG , ed. Volume 100 ed: Academic Press, 2008:35-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoumacher M, Burbridge M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr Oncol Rep. 2017;19(3):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769-785. [DOI] [PubMed] [Google Scholar]
- 16.Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting axl and mer kinases in cancer. Mol Cancer Ther. 2011; 10(10):1763-1773. [DOI] [PubMed] [Google Scholar]
- 17.Gioia R, Leroy C, Drullion C, et al. Quantitative phosphoproteomics revealed interplay between Syk and Lyn in the resistance to nilotinib in chronic myeloid leukemia cells. Blood. 2011;118(8):2211-2221. [DOI] [PubMed] [Google Scholar]
- 18.Rochlitz C, Lohri A, Bacchi M, et al. Axl expression is associated with adverse prognosis and with expression of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): results from a multicenter trial of the Swiss Group for Clinical Cancer Research (SAKK). Leukemia. 1999; 13(9):1352-1358. [DOI] [PubMed] [Google Scholar]
- 19.Whitman SP, Kohlschmidt J, Maharry K, et al. GAS6 expression identifies high-risk adult AML patients: potential implications for therapy. Leukemia. 2014;28(6):1252-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong CC, Lay JD, Huang JS, et al. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008; 268(2):314-324. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Batalla I, Schultze A, Wroblewski M, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine cross-talk of leukemia cells with bone marrow stroma. Blood. 2013; 122(14):2443-2452. [DOI] [PubMed] [Google Scholar]
- 22.Park I-K, Mishra A, Chandler J, Whitman SP, Marcucci G, Caligiuri MA. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: implications for Axl as a potential therapeutic target. Blood. 2013; 121(11):2064-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park IK, Mundy-Bosse B, Whitman SP, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382-2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rankin EB, Fuh KC, Castellini L, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014;111(37):13373-13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porteu F, Rouyez MC, Cocault L, et al. Functional regions of the mouse thrombopoietin receptor cytoplasmic domain: evidence for a critical region which is involved in differentiation and can be complemented by erythropoietin. Mol Cell Biol. 1996;16(5):2473-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wingelhofer B, Maurer B, Heyes EC, et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia. 2018;32(5): 1135-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casetti L, Martin-Lanneree S, Najjar I, et al. Differential contributions of STAT5A and STAT5B to stress protection and tyrosine kinase inhibitor resistance of chronic myeloid leukemia stem/progenitor cells. Cancer Res. 2013;73(7):2052-2058. [DOI] [PubMed] [Google Scholar]
- 28.Holland SJ, Pan A, Franci C, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70(4):1544-1554. [DOI] [PubMed] [Google Scholar]
- 29.Angelini DF, Ottone T, Guerrera G, et al. A Leukemia-Associated CD34/CD123/CD25/CD99+ immunophenotype identifies FLT3-mutated clones in acute myeloid leukemia. Clin Cancer Res. 2015;21(17): 3977-3985. [DOI] [PubMed] [Google Scholar]
- 30.Rauch PJ, Ellegast JM, Widmer CC, et al. MPL expression on AML blasts predicts peripheral blood neutropenia and thrombocytopenia. Blood. 2016;128(18):2253-2257. [DOI] [PubMed] [Google Scholar]
- 31.Mishra A, Wang J, Shiozawa Y, et al. Hypoxia Stabilizes GAS6/Axl Signaling in Metastatic Prostate Cancer. Mol Can Res. 2012;10(6):703-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braun M, Qorraj M, Buttner M, et al. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia. 2016;30(8):1788-1792. [DOI] [PubMed] [Google Scholar]
- 33.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167-1174. [DOI] [PubMed] [Google Scholar]
- 34.Dias S, Hattori K, Zhu Z, et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106(4):511-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schliemann C, Bieker R, Padro T, et al. Expression of angiopoietins and their receptor Tie2 in the bone marrow of patients with acute myeloid leukemia. Haematologica. 2006;91(9):1203-1211. [PubMed] [Google Scholar]
- 36.Brenner AK, Nepstad I, Bruserud O. Mesenchymal Stem Cells Support Survival and Proliferation of Primary Human Acute Myeloid Leukemia Cells through Heterogeneous Molecular Mechanisms. Front Immunol. 2017;8:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Traer E, Martinez J, Javidi-Sharifi N, et al. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016;76(22):6471-6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdul-Aziz AM, Shafat MS, Sun Y, et al. HIF1alpha drives chemokine factor protumoral signaling pathways in acute myeloid leukemia. Oncogene. 2018;37(20): 2676-2686. [DOI] [PubMed] [Google Scholar]
- 39.Ellegast JM, Rauch PJ, Kovtonyuk LV, et al. inv(16) and NPM1mut AMLs engraft human cytokine knock-in mice. Blood. 2016;128(17):2130-2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vergez F, Green AS, Tamburini J, et al. High levels of CD34+/CD38low/CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucémies Aiguës et Maladies du Sang (GOELAMS) study. Haematologica. 2011;96(12):1792-1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myeloge-nous leukemia stem cells. Leukemia. 2000;14(10):1777-1784. [DOI] [PubMed] [Google Scholar]
- 42.Riccioni R, Diverio D, Riti V, et al. Interleukin (IL)-3/granulocyte macrophage-colony stimulating factor/IL-5 receptor alpha and beta chains are preferentially expressed in acute myeloid leukaemias with mutated FMS-related tyrosine kinase 3 receptor. Br J Haematol. 2009;144(3):376-387. [DOI] [PubMed] [Google Scholar]
- 43.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124-1136. [DOI] [PubMed] [Google Scholar]
- 44.Fatrai S, Wierenga AT, Daenen SM, Vellenga E, Schuringa JJ. Identification of HIF2alpha as an important STAT5 target gene in human hematopoietic stem cells. Blood. 2011;117(12):3320-3330. [DOI] [PubMed] [Google Scholar]
- 45.Ruan G-X, Kazlauskas A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012;31(7):1692-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gioia R, Tregoat C, Dumas PY, et al. CBL controls a tyrosine kinase network involving AXL, SYK and LYN in nilotinib-resistant chronic myeloid leukaemia. J Pathol. 2015; 237(1):14-24. [DOI] [PubMed] [Google Scholar]
- 47.Spencer JA, Ferraro F, Roussakis E, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sironi S, Wagner M, Kuett A, et al. Microenvironmental hypoxia regulates FLT3 expression and biology in AML. Sci Rep. 2015;5:17550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin L, Tabe Y, Lu H, et al. Mechanisms of apoptosis induction by simultaneous inhibition of PI3K and FLT3-ITD in AML cells in the hypoxic bone marrow microenvironment. Cancer Lett. 2013;329(1):45-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017; 18(8):1061-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.