

SUMMARY
Resistance to checkpoint-blockade treatments is a challenge in the clinic. We found that although treatment with combined anti-CTLA-4 and anti-PD-1 improved control of established tumors, this combination compromised anti-tumor immunity in the low tumor burden (LTB) state in pre-clinical models as well as in melanoma patients. Activated tumor-specific T cells expressed higher amounts of interferon-γ (IFN-γ) receptor and were more susceptible to apoptosis than naive T cells. Combination treatment induced deletion of tumor-specific T cells and altered the T cell repertoire landscape, skewing the distribution of T cells toward lower-frequency clonotypes. Additionally, combination therapy induced higher IFN-γ production in the LTB state than in the high tumor burden (HTB) state on a per-cell basis, reflecting a less exhausted immune status in the LTB state. Thus, elevated IFN-γ secretion in the LTB state contributes to the development of an immune-intrinsic mechanism of resistance to combination checkpoint blockade, highlighting the importance of achieving the optimal magnitude of immune stimulation for successful combination immunotherapy strategies.
Graphical Abstract
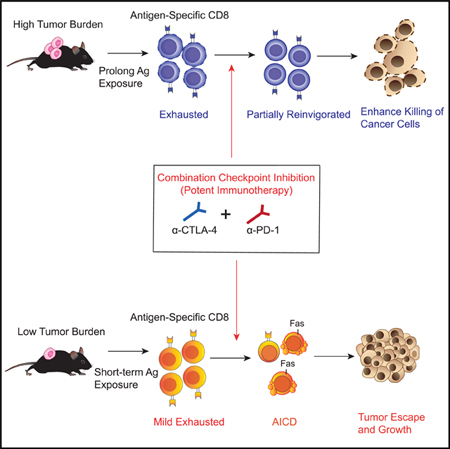
In Brief
Although immune checkpoint blockades are being combined to enhance anti-tumor efficacy, Pai et al. find that this approach can lead to therapy resistance in the low tumor burden setting. Potent immunotherapy in this setting overdrives tumor-reactive T cells, leading to their death. Optimal immunotherapy could therefore be disease-context dependent.
INTRODUCTION
In recent years, immune checkpoint inhibitors have been rapidly approved for the management of advanced malignancies, including melanoma, non-small-cell lung cancer (NSCLC), renal cell carcinoma (RCC), urothelial carcinoma, and head and neck cancer (Callahan et al., 2016). However, only a small subset (10%–30%) of patients respond to single-agent immune checkpoint therapy (Robert et al., 2015), and a myriad of combination strategies are currently being actively investigated in clinical trials with the goal of enhancing anti-tumor immunity and clinical efficacy.
Co-targeting of cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) and programmed death-1 (PD-1) immune checkpoint pathways is one strategy that demonstrates significantly improved clinical outcomes in advanced melanoma (Larkin et al., 2015). Despite these advances, a significant proportion of patients still do not achieve objective responses to checkpoint inhibitors. Recent clinical observations suggest that treatments with checkpoint inhibitors do not always lead to better outcomes in patients. In multiple large randomized trials (Bellmunt et al., 2017; Borghaei et al., 2015; Kwon et al., 2014), patients receiving immune checkpoint inhibitors had worse survival outcomes than did control arms during the initial months of treatment, at a time before immune-related toxicities fully manifest. Indeed, some cancer patients (9%) exhibit accelerated tumor growth upon treatment with immune checkpoint inhibitors, a phenomenon termed “tumor hyper-progression” (Champiat et al., 2017). Therefore, an improved understanding of the mechanisms underlying differential responses to checkpoint inhibition is needed to inform the future development of combinatorial therapeutic strategies.
Several studies have contributed to the understanding of mechanisms underlying differential responses and mechanisms of resistance to immune checkpoint strategies (Sharma et al., 2017). These include adaptive resistance mediated by interferon-dependent expression of inhibitory ligands on cancer cells (Benci et al., 2016; Tumeh et al., 2014), exclusion of CD8+ T cell infiltration by transforming growth factor-β (TGF-β) signaling within the tumor microenvironment (Mariathasan et al., 2018), and the acquisition of resistance by loss-of-function mutations in Janus kinases 1 and 2 (JAK1/2) or truncating mutations in histocompatibility leukocyte antigen (HLA) class I molecules (Zaretsky et al., 2016). Recently, tumor burden has emerged as a key factor determining clinical responses of immune checkpoint blockade (Huang et al., 2017). Early administration of PD-1 blockade in a relatively low disease burden can reinvigorate the dysfunctional T cells, whereas prolonged exposure of tumor antigens can ultimately develop into fixed T cell exhaustion status, resulting in poor response to anti-PD-1 (Schietinger et al., 2016). Similarly, favorable clinical outcomes in patients with low disease burdens treated with PD-1 blockade are associated with a higher ratio of reinvigorated CD8+ T cells to tumor burden (Huang et al., 2017). However, different immune-checkpoint blockade treatments contribute to distinct immune landscapes (Wei et al., 2017), and whether the combining of checkpoint blockades favors the clinical response in low disease burden is questionable. Sub-group analyses of treatment responses of two recent clinical trials significantly favor high, as opposed to low, disease burdens in patients who received anti-CTLA-4 plus anti-PD-1 (42%–29% in RCC and 51%–13% in NSCLC; Hellmann et al., 2018; Motzer et al., 2018), further indicating the complexity of disease burden and therapeutic responses to combination checkpoint blockade. Here, we sought to further understand the effects of different types of immune checkpoint blockade—as monotherapy or combination therapy—in the context of different tumor burdens to investigate potential immune regulatory mechanisms underlying the treatment response.
RESULTS
Combination Checkpoint Inhibition in the Setting of High Tumor Burden
Ipilimumab is a humanized IgG1 antibody targeting CTLA-4, and one of its immunomodulatory mechanisms is engagement with FcγRIIIA to potentially antagonize or deplete regulatory T (Treg) cells (Simpson et al., 2013). However, recent studies have also demonstrated limited capabilities of current CTLA-4 blockades in Treg cell depletion in the clinic (Sharma et al., 2018). In contrast, anti-PD-1 antibodies (such as nivolumab) have been engineered to avoid FcγR binding to prevent depletion of activated T cells through antibody-dependent cellular cytotoxicity (ADCC) (Dahan et al., 2015). To mimic the anti-PD1 antibodies used clinically, we generated an anti-PD-1 antibody without ADCC (anti-PD-1 DANA) for use in our preclinical experiments (Figures S1A–S1C). We also observed that, compared with PD-1 blockade without ADCC, IgG2a PD-1 blockade with intact ADCC depleted activated CD4+ and CD8+ T cells in tumor-bearing mice (Figures S1D–S1G).
We first investigated the anti-tumor activity of single-agent and combined immune checkpoint blockade in the setting of high tumor burden (HTB). To help delineate the difference between low tumor burden (LTB) and HTB states, we utilized TRAMP-C2 as our tumor model because it possesses a relatively slow tumor growth rate and tumors do not become palpable until 30 days after implantation (Figure 1A). Mice were inoculated with TRAMP-C2 cell lines on day 0 and treated with three doses of anti-CTLA-4 alone, anti-PD-1 DANA alone, anti-CTLA-4 plus anti-PD-1 DANA (combo), or IgG2a isotype control on days 43, 46, and 49. We found that anti-CTLA-4 monotherapy demonstrated potent anti-tumor activity compared with that of anti-PD-1 DANA or isotype (p < 0.0001; Figure 1B). Compared with anti-CTLA-4 alone, combination treatment marginally improved tumor control, but this was not statistically significant (Figure 1B). To further evaluate the treatment-induced changes in the immune landscape, we assessed mice for modulated immune responses 3 days after the last dose of treatment. Both CD4+ and CD8+ T cells were expanded in the spleen of the combination group and anti-CTLA-4 treatment group compared with the isotype group (Figures S1H–S1J). Our previous research has described the immunodominant CD8+ T cell epitope (Spas-1) that mediates tumor rejection in the TRAMP-C2 model (Fassò et al., 2008). Using major histocompatibility complex (MHC)-peptide multimers, we tracked the changes in the frequency and number of antigen specific T cells after treatments. Consistent with the anti-tumor activity observed, combined checkpoint blockade induced the highest numbers of Spas-1-specific CD8+ T cells in tumor-draining lymph nodes (Figures S1K and S1L). Overall, these results show that immune checkpoint blockade in mice with established tumors improves anti-tumor activity, at least in part because of the generation of a greater number of activated antigen-specific T cells.
Figure 1. Combination Checkpoint Blockade Enhances Anti-tumor Responses against Established Tumors.

C57BL/6j mice were implanted with TRAMP-C2 tumors and treated with checkpoint inhibitors as indicated.
(A) Schema of mice injected with checkpoint blockade in the HTB state.
(B) Tumor growth curve of the TRAMP-C2 model.
(C) Flow staining of CD45+CD3+CD4+Foxp3+ T cells.
(D) Percentage of CD4+Foxp3+ cells among CD45+ cells.
(E) Flow gating strategy of CD8+ T cell subsets in tumors.
(F) Total numbers of CD8+T cells per tumor weight.
(G) Ratio of CD8+ to CD4+ Treg cells.
(H and I) Tetramer staining among tumor-infiltrating CD8+ T cell populations. Cells were pre-gated on CD45+CD3+CD8+.
Data were collected from at least eight mice per group with two independent experiments. Statistical differences were calculated by one-way or two-way ANOVA with a post hoc Tukey test. *p < 0.05, **p < 0.01, ****p < 0.0001. Data are presented as mean ± SE. See also Figure S1.
To investigate immune infiltration within the tumor microenvironment after treatments, we first evaluated pathologic changes in tumor samples. In the hematoxylin and eosin (H&E) staining, there were peri-tumoral lymphocytic aggregates with prominent perivascular localization and intra-tumoral lymphocytic penetration in the combination-and monotherapy-treated groups as opposed to the isotype control group (Figure S1M). By assessing the regulatory T cells within the tumors with flow cytometry, we found the frequency of CD4+Foxp3+ Treg cells in both anti-CTLA-4 and combination groups to be lower than that of the isotype group (Figures 1C and 1D; p < 0.01 and p < 0.05). Furthermore, the total numbers of infiltrating CD8+ T cells per tumor weight and the ratio of CD8+ to CD4+ Treg cells were significantly increased in the combination group compared with the isotype or monotherapy group (Figures 1E–1G; p < 0.05). To further define the specificity of tumor-infiltrating CD8+ T cell clones, we gated on CD8+ T cells that recognize either the dominant Spas-1 epitope or the subdominant epitope (Spas-2) (Figure 1H), the latter of which can induce IFN-γ secretion from T cells but cannot mediate tumor rejection. Higher frequencies of dominant epitope CD8+ clones were present in the combination-treatment group than in the anti-PD-1 DANA-or isotype-treated group (Figure 1I; 43.08 ± 3.57 versus 32.23 ± 3.72 [p < 0.05] or 24.93 ± 2.30 [p < 0.01]). Conversely, the frequency of minor epitope CD8+ Spas-2-reactive T cells was higher in the isotype group than in the group treated with anti-CTLA-4 alone (p < 0.05). Altogether, anti-CTLA-4 demonstrated potent tumor control in established tumor models by decreasing Treg cells and increasing CD8+ T cells within the tumors. The addition of anti-PD-1 DANA to anti-CTLA-4 also led to increased intra-tumoral CD8+ T cells, particularly those T cell clones that are reactive to the immunodominant epitope Spas-1.
Anti-PD-1DANA Compromises the Anti-tumor Effects of Anti-CTLA-4 in the Setting of LTB
During tumor development, the dynamics of immune microenvironments are altered in parallel with tumor progression, a process termed immunoediting (Dunn et al., 2002). Within different tumor burdens, there exist distinct patterns of immune landscapes. We sought to evaluate whether the combination treatment could also enhance anti-tumor responses in the setting of LTB, a stage when naive T cells are actively becoming effector T cells. For these experiments, mice were treated on days 3, 6, and 9 in the setting of LTB (Figure 2A). Whereas anti-CTLA-4 strongly inhibited tumor growth in comparison with the isotype (Figure 2B; p < 0.0001), anti-PD-1 DANA alone delayed tumor growth more transiently. Surprisingly, the addition of PD-1 blockade attenuated the anti-tumor effects of anti-CTLA-4: mice that received potent combination treatment had significantly larger tumor growth than mice treated with anti-CTLA-4 alone (Figure 2B; p < 0.01). To further investigate the negative role of anti-PD-1 DANA antibody in combination treatment, we titrated the dose of anti-PD-1 DANA antibody. When combined with anti-CTLA-4, a higher dose of PD-1 blockade treatment led to a greater loss of anti-tumor efficacy than did a lower dose of anti-PD-1 DANA antibody treatment (Figures S2A and S2B; p < 0.05). Similar findings were observed when we repeated the experiment with another clone of anti-PD-1 antibody (RMP1–14 clone) (Figure S2C; p < 0.05) or anti-PD-L1 antibody (10F.9G2 clone) (Figure S2D; p < 0.01). In order to evaluate whether other immunotherapies could also compromise the antitumor effects from anti-CTLA-4 treatment, we treated mice with anti-CTLA-4 antibody combined with GM-CSF-secreting cell-based cancer vaccine (GVAX). Compared with anti-CTLA-4 alone, the combination of GVAX and anti-CTLA-4 showed similar anti-tumor control (Figure S2E). Initiation of checkpoint inhibition at an intermediate tumor burden (days 15, 18, and 21 after tumor injection) resulted in similar efficacy between the combination and monotherapy arms (Figures S2F and S2G). We conducted the same experiment with the MOC-1 model, another syngeneic tumor characterized by slow growth. Consistent with findings in TRAMP-C2, compared with anti-CTLA-4 alone in the LTB setting, the combination of CTLA-4 and PD-1 blockade also compromised anti-tumor effects (Figure 2C). In summary, the addition of PD-1 and PD-L1 blockade compromises the anti-tumor efficacy of CTLA-4 blockade in the setting of LTB.
Figure 2. The Effect of Combination Checkpoint Blockade in the LTB State.

(A) Schema of early intervention with dual checkpoint blockade prior to the development of palpable tumors.
(B) TRAMP-C2 tumor growth with early intervention in different treatment groups. The dose for each checkpoint inhibitor was 10 mg/kg.
(C) Tumor growth in the MOC-1 tumor model. The dose for each checkpoint inhibitor was 10 mg/kg. Animal studies were from two independent experiments with eight mice per group. Statistical analyses were calculated by two-way ANOVA with a post hoc test. *p < 0.05, **p < 0.01, ****p < 0.0001.
(D) Comparison of LDH levels between patients treated with anti-PD-1 (monotherapy) and patients treated with anti-PD-1+anti-CTLA-4 (combination therapy).
(E) 153 metastatic melanoma patients treated with checkpoint inhibitors were stratified into three groups with different ranges of baseline tumor size (BTS) as measured by radiographic imaging. The best overall response rate (RECIST v.1.1) in patients treated with monotherapy or combination therapy is presented for each stratum.
Error bars represent SEM. Significance was calculated by the two-sided Mann-Whitney test. *p < 0.05. See also Figure S2.
Clinical Response to Single or Combination Checkpoint Blockade in Advanced Melanoma Patients with Differing Baseline Tumor Burdens
To explore whether clinical responses to single or combined immune checkpoint inhibition might be differentially influenced by baseline tumor burden, we investigated a retrospective cohort of 153 advanced melanoma patients who received either monotherapy (anti-PD-1) or combination therapy (anti-CTLA-4 and anti-PD-1). CTLA-4 blockade shows greater activity in preclinical models, whereas PD-1 blockade demonstrates greater clinical activity (Wolchok et al., 2017). Therefore, the patient cohort receiving anti-PD-1 was studied. The patient demographics were reasonably matched between patients receiving the monotherapy and combination therapy (Table S1). Patients were categorized as responders (complete or partial response) or non-responders (stable or progressive disease) by RECIST v.1.1 (Eisenhauer et al., 2009). Patients were also categorized by normal or elevated serum levels of lactate dehydrogenase (LDH). Serum LDH has been shown to be an important prognostic factor in patients with metastatic melanoma and further defines metastatic categories and delineates tumor burdens (Petrelli et al., 2015). In line with previous reports (Huang et al., 2017), patients who responded to anti-PD-1 treatments demonstrated significantly lower LDH levels (p < 0.01) than did non-responders, indicating a favorable therapeutic response of anti-PD-1 monotherapy in the LTB state (Figure 2D). In contrast, LDH failed to identify clinical responders in the patient cohort that received combination checkpoint inhibitors, suggesting a different pattern of response depending on tumor burden between monotherapy-and combination-therapy-treated patients (Figure 2D). To further evaluate the impact of baseline tumor size (BTS) on clinical response, we assessed patients treated with anti-PD-1 (n = 100) or the combination of anti-CTLA-4 and anti-PD-1 (n = 53) for the best objective response and stratified them by a BTS of ≤6 cm (low), >6 to ≤11 cm (medium), or >11 cm (high). We selected these BTS cutoffs to evenly distribute the patients into three cohorts. As expected, patients in the medium and high BTS cohorts had a higher frequency of visceral metastasis and/or elevated LDH (i.e., M1c disease) than did patients in the low BTS cohort (Table 1). Consistent with our preclinical findings, patients treated with dual checkpoint blockade demonstrated significantly lower response rates than those treated with monotherapy in the low disease settings, but not in higher disease settings (Figure 2E; p < 0.05). In multi-variate risk analysis, there was no statistical difference of prior treatment or BRAF mutation status between monotherapy-and combination-therapy-treated patients, particularly in the low BTS cohort (Table 1). Overall, our pre-clinical and clinical data indicate that combination checkpoint blockade treatment might result in attenuated anti-tumor efficacies in the low disease state.
Table 1.
Patient Demographics and Disease Characteristics in Advanced Melanoma Patients by Baseline Tumor Size
| High BTS (BTS > 11) | Medium BTS (BTS = 6–11) | Low BTS (BTS ≤ 6) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mono | Combo | Mono | Combo | Mono | Combo | |||||||||||
| Variable | Level | n | % | n | % | p | n | % | n | % | p | n | % | n | % | p |
| Best clinical response | PD or SD | 22 | 66.7 | 9 | 64.3 | 1 | 19 | 65.5 | 6 | 40 | 0.194 | 18 | 47.4 | 19 | 79.2 | 0.026a |
| PR or CR | 11 | 33.3 | 5 | 35.7 | 10 | 34.5 | 9 | 60 | 20 | 52.6 | 5 | 20.8 | ||||
| Gender | female | 6 | 18.2 | 3 | 21.4 | 1 | 14 | 48.3 | 3 | 20 | 0.104 | 15 | 39.5 | 13 | 54.2 | 0.384 |
| male | 27 | 81.8 | 11 | 78.6 | 15 | 51.7 | 12 | 80 | 23 | 60.5 | 11 | 45.8 | ||||
| Metastatic status | IIIC | 0 | 0 | 1 | 7.1 | 0.051 | 0 | 0 | 2 | 13.3 | 0.13 | 1 | 2.6 | 4 | 16.7 | 0.041a |
| M1a | 3 | 9.1 | 0 | 0 | 9 | 31 | 2 | 13.3 | 7 | 18.4 | 4 | 16.7 | ||||
| M1b | 7 | 21.2 | 0 | 0 | 7 | 24.1 | 2 | 13.3 | 15 | 39.5 | 3 | 12.5 | ||||
| M1c | 23 | 69.7 | 13 | 92.9 | 13 | 44.8 | 9 | 60 | 15 | 39.5 | 13 | 54.2 | ||||
| LDH | normal | 12 | 37.5 | 1 | 7.1 | 0.072 | 18 | 62.1 | 10 | 66.7 | 1 | 31 | 81.6 | 18 | 85.7 | 1 |
| elevated | 20 | 62.5 | 13 | 92.9 | 11 | 37.9 | 5 | 33.3 | 7 | 18.4 | 3 | 14.3 | ||||
| not available | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 12.5 | ||||
| ECOG | 0 | 18 | 54.5 | 10 | 71.4 | 0.094 | 19 | 65.5 | 13 | 86.7 | 0.171 | 28 | 73.7 | 20 | 83.3 | 1 |
| 1 | 15 | 45.5 | 3 | 21.4 | 10 | 34.5 | 2 | 13.3 | 10 | 26.3 | 3 | 12.5 | ||||
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4.2 | ||||
| 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| 4 | 0 | 0 | 1 | 7.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| Liver metastasis | absent | 18 | 54.5 | 4 | 28.6 | 0.123 | 20 | 69 | 11 | 73.3 | 1 | 28 | 73.7 | 16 | 66.7 | 0.76 |
| present | 15 | 45.5 | 10 | 71.4 | 9 | 31 | 4 | 26.7 | 10 | 26.3 | 8 | 33.3 | ||||
| Lung metastasis | absent | 17 | 51.5 | 7 | 50 | 1 | 14 | 48.3 | 7 | 46.7 | 1 | 20 | 52.6 | 16 | 66.7 | 0.408 |
| present | 16 | 48.5 | 7 | 50 | 15 | 51.7 | 8 | 53.3 | 18 | 47.4 | 8 | 33.3 | ||||
| Brain metastasis | absent | 26 | 78.8 | 7 | 50 | 0.104 | 22 | 75.9 | 11 | 73.3 | 1 | 34 | 89.5 | 19 | 79.2 | 0.29 |
| present | 7 | 21.2 | 7 | 50 | 7 | 24.1 | 4 | 26.7 | 4 | 10.5 | 5 | 20.8 | ||||
| BRAF status | BRAF K601E | 0 | 0 | 0 | 0 | 0.182 | 1 | 3.4 | 0 | 0 | 0.15 | 0 | 0 | 0 | 0 | 0.101 |
| L597 | 0 | 0 | 0 | 0 | 1 | 3.4 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| L597R | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| V600E | 7 | 21.2 | 3 | 21.4 | 5 | 17.2 | 3 | 21.4 | 6 | 15.8 | 9 | 37.5 | ||||
| V600K | 0 | 0 | 2 | 14.3 | 0 | 0 | 2 | 14.3 | 0 | 0 | 0 | 0 | ||||
| V600R | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 7.1 | 0 | 0 | 0 | 0 | ||||
| WT | 25 | 75.8 | 9 | 64.3 | 22 | 75.9 | 8 | 57.1 | 32 | 84.2 | 15 | 62.5 | ||||
| BRAF mutation | mutation | 8 | 24.2 | 5 | 35.7 | 0.654 | 7 | 24.1 | 6 | 42.9 | 0.369 | 6 | 15.8 | 9 | 37.5 | 0.101 |
| WT | 25 | 75.8 | 9 | 64.3 | 22 | 75.9 | 8 | 57.1 | 32 | 84.2 | 15 | 62.5 | ||||
| not available | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 6.7 | 0 | 0 | 0 | 0 | ||||
| Previous chemotherapies | no | 23 | 69.7 | 14 | 100 | 0.022a | 20 | 69 | 15 | 100 | 0.018a | 31 | 81.6 | 23 | 95.8 | 0.136 |
| yes | 10 | 30.3 | 0 | 0 | 9 | 31 | 0 | 0 | 7 | 18.4 | 1 | 4.2 | ||||
| Prior immunotherapies |
no | 23 | 69.7 | 10 | 71.4 | 1 | 15 | 51.7 | 11 | 73.3 | 0.208 | 22 | 57.9 | 16 | 66.7 | 0.672 |
| yes | 10 | 30.3 | 4 | 28.6 | 14 | 48.3 | 4 | 26.7 | 16 | 42.1 | 8 | 33.3 | ||||
| Prior radiation therapy | no | 27 | 81.8 | 11 | 78.6 | 1 | 23 | 79.3 | 11 | 73.3 | 0.714 | 33 | 86.8 | 18 | 75 | 0.397 |
| yes | 6 | 18.2 | 3 | 21.4 | 6 | 20.7 | 4 | 26.7 | 5 | 13.2 | 6 | 25 | ||||
| Prior targeted therapy | no | 24 | 72.7 | 13 | 92.9 | 0.242 | 21 | 72.4 | 13 | 86.7 | 0.452 | 33 | 86.8 | 18 | 75 | 0.397 |
| yes | 9 | 27.3 | 1 | 7.1 | 8 | 27.6 | 2 | 13.3 | 5 | 13.2 | 6 | 25 | ||||
| Primary site | acral or mucosal | 0 | 0 | 1 | 8.3 | 0.279 | 5 | 17.9 | 0 | 0 | 0.502 | 3 | 8.6 | 6 | 30 | 0.136 |
| cutaneous | 28 | 87.5 | 9 | 75 | 20 | 71.4 | 10 | 83.3 | 28 | 80 | 13 | 65 | ||||
| uveal | 4 | 12.5 | 2 | 16.7 | 3 | 10.7 | 2 | 16.7 | 4 | 11.4 | 1 | 5 | ||||
| not available | 1 | 3 | 2 | 14.3 | 1 | 3.4 | 3 | 20 | 3 | 7.9 | 4 | 16.7 | ||||
Abbreviations are as follows: mono, monotherapy (anti-PD-1); combo, combination therapy (anti-CTLA-4 and anti-PD-1).
p < 0.05.
Loss of Antigen-Specific T Cells with Combination Checkpoint Blockade in the LTB
Tumor-specific T cells are a major immune subset that respond to checkpoint blockade and provide anti-tumor immunity (Im et al., 2016). To examine the mechanism underlying the therapeutic resistance of combination therapy in the setting of LTB, we utilized tetramers to investigate the dynamic changes of antigen-specific T cells at different time points (Figure 3A). Two days after the last dose of treatment, Spas-1-specific CD8+ T cells were expanded in the combination-treatment group compared with the groups treated with either anti-PD-1 DANA or isotype (Figures 3B and 3C; p < 0.05). However, 28 days after tumor implantation, Spas-1 CD8+ T cells were found at significant numbers only in the mice treated with anti-CTLA-4 (Figures 3D and 3E; p < 0.01). In contrast, the Spas-2-specific CD8+ T cells did not show significant differences on day 11 (Figure 3F). On day 28, there was an increase in total numbers of Spas-2 CD8+ T cells in the anti-PD-1 DANA group compared with the anti-CTLA-4 or isotype group (Figures 3G and 3H). These results indicate that early treatment with anti-CTLA-4 alone allowed for sustained expansion of CD8+ T cells specific to the dominant Spas-1 antigen. In contrast, anti-PD-1 DANA alone supported the expansion of Spas-2-specific T cells. Combination treatment led to transient induction and then loss of Spas-1-specific CD8+ T cells. We also observed similar kinetics within the antigen-specific T cell population in an LTB metastatic melanoma patient who was treated with combination therapy. Using MHC-peptide multimers, we detected a transient increase in MART-1-specific CD8+ T cells after two cycles of combination treatment, although it was subsequently lost at later time points (Figures S3A–S3F). This paralleled the patient’s clinical course, where the patient had a subsequent tumor progression (Figure S3G). We confirmed that the melanoma tissue sample from this patient expressed MART-1 (Figure S3H). Although these results are consistent with our mouse models, a future prospective clinical trial is needed to further validate the correlation of antigen-specific T cell loss in low BTS patient cohorts who receive combination checkpoint blockade.
Figure 3. Dynamics of Tumor-Specific T Cells after Checkpoint Blockade.

Mice implanted with TRAMP-C2 tumors were treated with checkpoint inhibitors on days 3, 6, and 9. Spleens were harvested on days 11 and 28.
(A) Schema of animal studies.
(B) Flow gating of antigen-specific CD45+CD3+CD8+ T cells against the immunodominant Spas-1 epitope and minor Spas-2 epitope.
(C) Total CD8+ Spas-1 T cells isolated at day 11.
(D) Total CD8+ Spas-1 T cells isolated at day 28.
(E) Dynamic changes of CD8+Spas-1 T cells over time.
(F) Total CD8+ Spas-2 T cells isolated at day 11.
(G) Total CD8+ Spas-2 T cells isolated at day 28.
(H) Dynamic changes of CD8+ Spas-2 T cells over time.
Data were from two or three independent experiments with 9–12 mice per group. Statistical analyses were calculated by one-way ANOVA with a post hoc Tukey test. *p < 0.05, **p < 0.01. Data are presented as mean ± SE. See also Figure S3.
To further assess global changes in the T cell repertoire, we performed T cell receptor (TCR) sequencing on T cells from tumor-draining lymph nodes. We found that although there was no difference in TCR diversity within the anti-CTLA-4-treated group (Figure S3I), the overall TCR diversity increased in the combination-treated mice at day 28 (Figure S3J). When we examined the frequency distribution of each T cell clone, ranking them in descending order, we found that antiCTLA-4-treated groups maintained the same distribution of clonal frequencies between day 11 and day 28. However, the combination-treated mice experienced skewing of their repertoire toward lower-frequency clonotypes (Figure S3K). These results are consistent with a shift from high-abundance T cell clones to many low-frequency clones.
To investigate the alterations induced in these antigen-specific T cells, we sorted Spas-1-specific CD8+ T cells from tumor-draining lymph nodes at day 28 (Figure 4A). The transcriptional profiles of these isolated cells were assessed by RNA sequencing (RNA-seq). As previously reported (Wei et al., 2017), anti-CTLA-4, anti-PD-1 DANA, and combination therapy induced distinct underlying transcriptional programs (Figure 4B). Interestingly, 879 genes were induced in the combination-treated group and were distinct to either monotherapy-treated group (Figure 4C). When we examined the pathways associated with these genes, we found that the combination-treated group had increased gene expression in multiple pathways that are associated with T cell apoptosis and cell death (Figure 4D and Figures S4A–S4E). Consistent with these findings, Spas-1-specific CD8+ T cells isolated from the combination group also had higher expression of the Fas/Fas ligand, caspase, and pro-apoptotic genes than the anti-CTLA-4-treated group (Figure 4E and Figure S4F). In contrast, anti-CTLA-4 treatment alone was associated with higher expression of anti-apoptotic genes than was the combination-treated group (Figure S4G). Flow-cytometry analysis confirmed that the combination treatment induced higher expression of active cleaved-caspase-3 in Spas-1-specific CD8+ T cells than did anti-CTLA-4 alone (Figure 4F; p < 0.01). Spas-1-reactive CD8+ T cells were more susceptible to treatment-induced apoptosis than minor Spas-2-epitope-reactive T cells (Figures S4H and S4I). Overall, these results indicate that the combination of anti-CTLA-4 and anti-PD-1 DANA treatment during early LTB stages of tumor growth can promote apoptosis of antigen-specific T cells, particularly those recognizing the immunodominant tumor epitope.
Figure 4. Tumor-Specific T Cell Loss after Combination Checkpoint Inhibition.

Mice were implanted with TRAMP-C2 tumors and treated with different checkpoint inhibitors.
(A) CD8+ Spas-1 T cells were sorted from draining lymph nodes on day 28 after checkpoint-inhibitor treatment.
(B) RNA-seq was performed on sorted cells, and expression data are presented for each treatment group.
(C) The overlap in overexpressed genes was assessed for different treated groups.
(D) Ingenuity Pathway Analysis was performed from RNA-seq data.
(E) Gene expression for caspase family members was assessed by RT-PCR.
(F) Active caspase-3 expression among CD8+ Spas-1 T cells was determined by flow cytometry. Data represent two independent experiments with ten mice per group.
Statistical analyses were calculated by one-way ANOVA with post hoc Tukey test. *p < 0.05, **p < 0.01. Data are presented as mean ± SE. See also Figure S4.
IFN-γ-Mediated Apoptosis of Activated Antigen-Specific T Cells
PD-1 blockade has been shown to prevent terminal exhaustion of antigen-specific T cells rather than promote apoptosis. We hypothesized that the contraction of antigen-specific T cells observed could result from cytokines mediating T cell contraction (Yajima et al., 2006) and homeostasis (Surh and Sprent, 2008). We first investigated changes in cytokine amounts after treatment during early tumor development. Analysis of serum samples showed a trend toward increased IFN-γ with the combination treatment (Figure S5A). The chemokine ligands CXCL9, LIF, and CCL5 were also increased after combination treatment (Figure S5B). In addition, we found that CD4+ T cells from combination-treated mice secreted significantly higher amounts of IFN-γ than did monotherapy-or isotype-treated mice (Figure 5A). A similar pattern was seen for IFN-γ secretion by CD8+ T cells (Figure 5B). Previously, IFN-γ has been shown to be the most dominant signature in patients who receive dual-checkpoint-blockade treatments (Das et al., 2015). In order to further investigate the immune subsets that are responsible for IFN-γ secretion, we collected peripheral-blood mononuclear cells (PBMCs) from nine patients after dual-checkpoint-blockade treatments and utilized mass cytometry (CyTOF) to analyze IFN-γ amounts across immune cells (Spitzer et al., 2017) (Figures 5C and S5D). We found that CD8+ T cells, natural killer cells, and γδ T subsets were the main source of IFN-γ secretion after dual-blockade treatments in cancer patients (Figure S5E). However, more clinical samples are needed to further validate the results.
Figure 5. IFN-γ Induced Cell Death of T Cells at Effector Stage.

(A and B) Mice implanted with TRAMP-C2 tumors were treated with checkpoint inhibitors on days 3, 6, and 9. Spleens were harvested on day 11, and IFN-γ expression was analyzed in CD4+ and CD8+ T cell subsets by flow cytometry.
(C) PBMCs were isolated from a total of nine cancer patients treated with anti-CTLA-4 plus anti-PD-1 therapies. Data were collected 1 month after the fourth dose of dual-checkpoint-blockade treatments. The data presented here were collected from patient 1, and the other eight patients are shown in Figure S5. Patient 1 is the same patient as reported in Figure S4. The IFN-γ secretion among different immune subsets was analyzed by CyTOF and is presented with SCAFFOLD. The node size represents the abundance of the cell population, and the red color represents the intensity of IFN-γ expression (ASINH ratio of the raw value).
(D) T cells were purified from TRAMP-C2-tumor-bearing mice and subsequently cultured in vitro with the indicated concentrations of IFN-γ.
(E) Cells were harvested 72 h after IFN-γ stimulation and analyzed for active caspase-3 expression among different CD8+ subsets.
(F) IFN-γ receptor expression in different CD8+ T cell subsets.
(G) Four stages of human chimeric antigen receptor (CAR) CD8+ T cell differentiation in vitro are shown.
(H) CD8+ T cells at these different stages of activation were cultured in vitro with human recombinant IFN-γ (3 × 105 IU), and cells were harvested 48 h later. Annexin V expression was determined by flow cytometry.
Statistical analyses were calculated by one-way ANOVA with a post hoc Tukey test. **p < 0.01, ****p < 0.0001. Data are presented as mean ± SE. See also Figure S5.
IFN-γ has been shown to mediate the contraction of antigen-specific CD8+ T cells (Badovinac et al., 2000), induce T cell apoptosis (Refaeli et al., 2002), and increase the expression of CXCL9, LIF, and CCL5 (Guirnalda et al., 2013; Wen et al., 2010). We hypothesized that the loss of Spas-1-specific CD8+ T cells might be related to increased IFN-γ signaling. To evaluate the effects of IFN-γ on antigen-specific T cells, we purified T cells from the spleens of tumor-bearing mice and cultured them with different concentrations of recombinant IFN-γ in vitro (Figure 5D). After 72 h, active caspase-3 expression was examined in different T cell subsets, including naive T cells (CD45+CD8+ CD44−CD62L+), antigen-specific T cells (CD45+CD8+Spas-1), and effector T cells (CD45+CD8+CD44+CD62L−). Activated effector T cells and antigen-specific T cells were more susceptible to IFN-γ-induced active caspase-3 expression than were naive T cells (Figure 5E; p < 0.001). In addition, effector T cells and CD8+Spas-1 T cells showed higher IFN-γ receptor expression than did naive T cells (Figure 5F). It has been shown that T cell homeostasis can be achieved by a decrease in antigen-specific TCR signaling (Gallegos et al., 2016). To investigate whether combination treatment eliminates different T cell clones depending on the strength of TCR binding to the cognate cancer epitope, we gated on CD8+ subsets and investigated Spas-1hi versus Spas-1lo CD8+ T cell clones (Figure S6A). Spas-1hi T cells were more susceptible to caspase-3 induction than Spas-1lo T cells upon IFN-γ co-culture (Figure S6B; p < 0.0001). Finally, PBMCs from four melanoma patients who received anti-PD-1 immunotherapy were isolated and cultured in vitro with IFN-γ stimulation. We also observed an increase in apoptosis corresponding to IFN-γ concentration in CD8+ T cells (Figures S6C and S6D).
A rapidly emerging cellular immunotherapy involves transferring chimeric antigen receptor (CAR) T cells into patients to treat cancer (June et al., 2018). Because CAR T cells are also antigen-specific T cells, we sought to investigate whether CAR T cells might also be susceptible to IFN-γ-mediated apoptosis. To achieve this, we utilized an in vitro CAR-19 model with 4–1BB co-stimulatory molecules (Figure 5G) (Roybal et al., 2016). Four stages of CAR T cells, from naive to effector states, were studied (Figure 5G). As expected, both stage II and stage IV T cells had high expression of the activation marker CD69 (Figure S6E); however, only T cells at stage IV, the effector stage, showed a significant increase in apoptosis when cultured with IFN-γ (Figure 5H). Stage IV T cells also demonstrated high degrees of baseline apoptosis (Figures S6F–S6H), and adding IFN-γ blockade could only partially rescue the effect, indicating that other factors might play significant roles in triggering the apoptosis. Future studies are needed to further investigate the phenomena. Altogether, these data show that elevated IFN-γ can induce the apoptosis of antigen-specific T cells.
Compromised Anti-tumor Memory Responses with Combination Treatment
The generation of long-term T cell memory responses is important for an effective and durable anti-tumor response. Given that we observed the loss of effector T cells in mice treated with combination checkpoint blockades, we sought to evaluate the effect of combination therapy on the formation of memory responses. Mice challenged with TRAMP-C2 were treated and observed for 3 months. At 90 days, 20%–30% of mice treated with combination treatment and 80%–90% of mice treated with anti-CTLA-4 alone were tumor-free. These protected mice were re-challenged with either TRAMP-C2 or MC-38 (control) in the contralateral flank (Figure 6A). We used aged wild-type (WT) mice without prior tumor challenge as controls to evaluate primary responses to these tumors. Mice that received prior combination treatment had compromised protection from TRAMP-C2 tumor re-challenge in comparison with mice previously treated with anti-CTLA-4 alone (Figure 6B; p < 0.05). No differences in tumor outgrowth were observed in MC-38-challenged mice (Figure 6C), indicating that the observed TRAMPC2 tumor control was mediated by tumor-specific memory responses. To investigate the compromised memory responses, we challenged mice with TRAMP-C2 cell lines, treated them with anti-CTLA-4 alone or combination treatment, and harvested tumor-draining lymph nodes on day 28 (Figure S7A). The combination group showed fewer Spas-1-specific CD8+ effector memory T cells (CD45+CD3+CD8+CD44+CD62L−Spas-1) than did the group treated with anti-CTLA-4 alone (Figures S7B–S7D; p < 0.05). The reductions in antigen-specific effector memory cells after dual-blockade treatments corresponded to the tumor outgrowth in mice.
Figure 6. Effective Anti-tumor Response with Dual Checkpoint Blockade Is Restored when T Cells Are Made Unresponsive to IFN-γ.

(A) Eight-week-old C57BL/6j mice were challenged with TRAMP-C2 tumor on day 0 and treated with checkpoint inhibitors on days 3, 6, and 9. Ninety days after tumor implantation, tumor-free mice from CTLA-4 blockade or combination-treatment groups were subsequently re-challenged with either the TRAMP-C2 or MC-38 tumor model. Aged sibling mice without prior tumor challenge were used as control mice.
(B) Mice were re-challenged with TRAMP-C2 tumors.
(C) Mice were re-challenged with MC-38 tumors. To allow a sufficient number of tumor-free mice, each treatment group consisted of 30–45 mice. The numbers of tumor-free mice for re-challenge are labeled correspondingly.
(D) Age-and gender-matched WT or Ifngr1−/− C57BL/6j mice were implanted with tumors at day 0 and treated with checkpoint inhibitors on days 3, 6, and 9.
(E) Comparison of tumor growth curves in the CTLA-4-blockade treatment.
(F) Comparison of tumor growth curves in the isotype-control treatment.
(G) Comparison of tumor growth curves with the combination-blockade treatment.
(H) WT or Ifngr1−/− mice were treated with checkpoint blockade and harvested at day 28.
(I) Total numbers of CD8+ Spas-1 T cells.
(J) WT mice were myeloablated (10.5 Gy) and given an adoptive transfer of bone marrow cells from CD45.2 Ifngr1−/− and CD45.1 Pepc congenic WT mice at a 1:1 ratio. Chimera mice were subsequently implanted with TRAMP-C2 tumors and treated with checkpoint inhibitors on days 33, 36, and 39 after bone marrow transplant.
(K) Mice underwent tail bleeding and were checked for chimerism 30 days after bone marrow transplantation.
(L) Tumor-draining lymph nodes were harvested on day 58. Different CD8+ subsets were pre-gated on flow cytometry and investigated for chimerism.
Experiments included eight (D–G) or five (H and I) mice per group. For (J) and (L), each treatment consisted of five chimera mice per group for a total of 20 chimera mice in these experiments. Data were analyzed by two-way ANOVA with a post hoc test (B–G) or by one-way ANOVA with a post hoc Tukey test (I and L). *p < 0.05, ***p < 0.001, ****p < 0.0001. Data are presented as mean ± SE. See also Figure S6.
Deficiency of the IFN-γ Receptor in Immune Cells Rescues Anti-tumor Activity after Combination Therapy
IFN-γ is essential in triggering potent anti-tumor responses by inducing MHC I expression and enhancing antigen-presenting capabilities (Ikeda et al., 2002). Although neutralization of IFN-γ can potentially prevent antigen-specific T cell loss, neutralization might also abrogate anti-tumor responses. To evaluate whether IFN-γ signaling is important for antigen-specific T cells, we used Ifngr1−/− mice (Figure 6D). T cells from Ifngr1−/− mice can secrete IFN-γ but cannot respond to the cytokine because they lack corresponding IFN-γ receptors. We challenged mice with WT TRAMP-C2 to avoid potential effects from tumor-mediated adaptive resistance (Spranger et al., 2013). After tumor challenge, there was no difference in tumor growth between WT and Ifngr1−/− mice treated with antiCTLA-4 (Figure 6E) or isotype (Figure 6F). In contrast, combination treatment led to significantly improved anti-tumor efficacy in Ifngr1−/− mice compared with WT mice (Figure 6G). To investigate the number of Spas-1-specific CD8+ T cells between Ifngr1−/− and WT mice, we harvested spleens from mice on day 28 (Figure 6H). In WT mice, the total numbers of Spas-1-specific CD8+ T cells were significantly reduced after combination treatment compared with anti-CTLA-4 alone (Figure 6I; p < 0.001). In contrast, in Ifngr1−/− mice, there was no difference in the total numbers of Spas-1-specific CD8+ T cells (Figure 6I).
To determine whether IFN-γ signaling in immune cells or nonimmune cells (e.g., stromal cells) was responsible for these observations, we performed experiments in bone marrow chimera mice. WT mice underwent myeloablative conditioning and were adoptively reconstituted with bone marrow cells from CD45.2 Ifngr1−/− mice and CD45.1 congenic mice in a 1:1 ratio (Figure 6J). We checked hematopoietic recovery and chimerism 30 days after bone marrow transplant to ensure that CD45.2+ and CD45.1+ populations were close to 1:1 in ratio (Figure 6K). Chimera mice were subsequently implanted with TRAMP-C2 cells on day 30 and treated with three doses of checkpoint inhibitors. Tumor-draining lymph nodes were harvested 58 days after the initial bone marrow transplant, and the ratio of CD45.2+ to CD45.1+ cells in different antigen-specific T cell subsets was examined. We found a higher ratio of CD45.2+ to CD45.1+ cells in the combination-treatment group (Figure 6L), indicating a competitive advantage of Spas-1-specific CD8+ T cells from Ifngr1−/−, but not WT, mice after treatment. IFN-γ has been shown to induce ischemia during early tumorigenesis stages that hinder cancer development (Kammertoens et al., 2017). However, we did not observe a statistical difference in angiogenesis between the treatment groups (Figures S7E–S7G). Further studies might be needed to further verify the impacts of IFN-γ on angiogenesis through checkpoint inhibitors. In summary, these data indicate that the dampening of tumor control observed with combination treatment during early tumor growth is at least partially reversed by Ifngr1−/− antigen-specific T cells.
Finally, we sought to investigate the differential effects of checkpoint inhibition on IFN-γ induction in the setting of LTB versus HTB. During tumor development, antigen-specific T cells undergo a dynamic exhaustion process that is correlated with tumor burden (Schietinger et al., 2016). In HTB, persistent antigen exposure with low-grade inflammation can potentially re-program T cells toward exhausted molecular signatures (Wherry and Kurachi, 2015). We first sought to understand whether T cells in the HTB demonstrate bona fide exhausted phenotypes. To investigate this, we isolated T cells from TRAMP-C2-tumor-bearing mice on either day 11 (LTB setting) or day 50 (HTB setting) after tumor injection (Figure S7H). CD8+ T cells isolated from the HTB setting exhibited more exhausted phenotypes (PD-1hiTim-3hi KLRG1lo) than those from the LTB setting (PD-1loTim-3lo KLRG1hi) (Figures S7I–S7K), indicating that the T cell immunological niche varies greatly between the LTB and HTB settings potentially because of the compelling difference in tumor antigen loads and durations of antigen stimulation. In addition, we also found that in both CD4+ and CD8+ T cells, there was significantly less IFN-γ secretion on a per-cell basis from the HTB setting than from the LTB setting (p < 0.01 versus p < 0.05, respectively; Figure S7L). The blunted IFN-γ secretion resulted in preservation of antigen-specific T cells in the setting of HTB, whereas CD8+ Spas-1-reactive T cells were maintained or expanded in both tumors and lymphoid organs (Figures S7M–S7O). Overall, we demonstrated that the T cell immunological niche differs greatly between the LTB and HTB settings, and administration of combination checkpoint blockade might result in different kinetics of antigen-specific T cells.
DISCUSSION
Current cancer immunotherapy strategies aim to counteract the suppressive tumor environment by enhancing antigen recognition of T cell receptors (Torikai et al., 2012), increasing anti-tumor cytotoxicity capabilities via cytokines (Rosenberg, 2014), or unleashing the brakes and preventing terminal T cell exhaustion by blocking different immune checkpoint inhibitors (Sharma and Allison, 2015). The clinical activity of CTLA-4 and PD-1 co-inhibition in melanoma (Larkin et al., 2015) demonstrates the potential of combination immunotherapy to be a viable strategy in improving anti-tumor response. T cells isolated from patients treated with dual checkpoint blockade exhibit significantly higher IFN-γ amounts than do pre-treatment samples (Das et al., 2015), and various combination therapies aimed at enhancing IFN-γ production are the subject of ongoing clinical investigation. We found, however, that potent combination therapy with CTLA-4 and PD-1 blockade in the context of LTB could potentially induce excess amounts of IFN-γ and result in therapeutic resistance.
Whereas combination therapy in mice with established tumors achieved improved tumor control, combination treatment in the context of LTB compromised anti-tumor effects in mice, which was supported by retrospective clinical data from metastatic melanoma patients. Mechanistically, we found that combination treatment during early tumor development led to heightened IFN-γ production, which in turn resulted in apoptosis of the dominant tumor-specific T cells via activation-induced cell death (AICD). IFN-γ has been conventionally demonstrated to have immune stimulatory roles. The secretion of IFN-γ from tumor infiltrating lymphocytes can activate both dendritic cells and macrophages to enhance antigen presentation (Minn, 2015). IFN-γ signaling on cancer cells can also upregulate expression of MHC-I-and STAT-1-associated cyclin-dependent kinase, resulting in immune recognition and apoptosis of tumor cells. (Gao et al., 2016; Ikeda et al., 2002). However, there is also evidence showing the paradoxical role of IFN-γ in cancer immunotherapies, in particular its association with acquired resistance (Zaidi and Merlino, 2011). IFN-γ promotes therapeutic resistance to immune checkpoint blockade by increasing the expression of IDO and PD-L1 (Spranger et al., 2013) and other co-inhibitory receptors (Benci et al., 2016; Koyama et al., 2016). Here, we demonstrated that IFN-γ signaling can be immunosuppressive to mediate therapeutic resistance through a PD-L1-independent pathway. Induction of IFN-γ secretion after dual-blockade treatments can promote apoptosis of tumor-reactive CD8+ T cells while limiting the formation of effector memory anti-tumor responses. In a recent phase II trial study with 298 clinical samples, cancer patients with progressive disease exhibited significantly higher expression of IFN-γR than patients with partial or complete response (p < 0.001), further highlighting how important it is that type II IFN not only account for cytotoxic effects against cancer cells but can also mediate therapeutic resistance of checkpoint-inhibitor treatments.
Tumor burden has emerged as a key prognosis factor associated with cancer patients treated with checkpoint blockades (Huang et al., 2017). In the HTB setting, the duration of chronic antigen exposure and the abundance of antigen loads can alter the exhaustion status and epigenetic profile of T cells. Those factors are, however, intertwined during cancer development. On the other hand, in the setting of LTB, the tumor microenvironment is easily influenced by the micro-inflammation triggered by the tumor injections. Although we were not able to separate out those factors in the current study, our results nonetheless demonstrate a differential effect of checkpoint blockades in two different tumor settings mediated by AICD. AICD is a highly regulated process during the early CD4+ and CD8+ T cell priming stage to prevent hyperactivation that leads to apoptosis (Badovinac et al., 2000; Berner et al., 2007). IFN-γ signaling has been elucidated as the key factor contributing to this process (Refaeli et al., 2002). Lack of PD-1 inhibitory signaling at this stage can also adversely result in terminally exhausted T cells (Odorizzi et al., 2015). In our study, the events of AICD in antigen-specific T cells occurred mostly in the early tumor setting. During priming, T cells upon antigen presentation are actively becoming effector T cells against tumors. Strong activation signals through T cell receptors against dominant antigens in combination with dual immune checkpoint blockades can more easily result in hyperactivation and promote AICD events. In contrast, T cells exhibiting exhausted phenotypes in the HTB are more prone to be reinvigorated rather than hyperactivated after the administration of checkpoint blockades. Tumor-reactive T cells are characterized into two phases on the basis of chromatin states (Philip et al., 2017), and each stage represents a distinct epigenetic mechanism of T cell reprogramming that might lead to a differential response to checkpoint blockades (Schietinger et al., 2016). Therefore, the paradoxical effect might come from the differential exhaustion status of T cells that respond to checkpoint inhibitors. In a recent clinical trial, patients who received combination checkpoint blockades demonstrated remarkable clinical outcomes in the HTB as opposed to the LTB state (complete response [CR] rate: 51% versus 13%) (Hellmann et al., 2018). Additionally, other reports also demonstrate potential hazards toward treatment responses when immunotherapies are combined more frequently. Concurrent dosing of combination therapy is associated with worse outcomes and greater T cell apoptosis than is sequential monotherapy (Messenheimer et al., 2017), and more frequent dosing of dual blockade is associated with a lower overall response rate in lung cancer patients (Hellmann et al., 2017). Clinical trials of combination checkpoint blockade in the adjuvant setting are ongoing and warrant further investigation in future studies.
Altogether, our results indicate that there exists a potential window within which the immune system can optimally respond to cancer. In the setting of LTB, optimal immunotherapy, such as CTLA-4 or PD-1 blockade alone, can induce invigoration of T cells and provide substantial benefits to cancer patients. However, in terms of combining different immunotherapies, there is a need to be cautious because exceeding this window could potentially trigger regulatory mechanisms that hinder anti-tumor effects. As clinical trials are starting to focus on administrating combinational checkpoint blockades in earlier disease states, such as the adjuvant setting, our findings reveal that treatment-induced AICD represents an immune-intrinsic mechanism of resistance that limits maximal anti-tumor activity. Thus, achieving the optimal magnitude of immune stimulation for each disease context and/or concomitant exhaustion status might be critical for successful immunotherapy strategies that provide the best tumor-control rates as well as long-term outcomes for cancer patients.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lawrence Fong (Lawrence.fong@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
8–10 week-old aged control male C57BL/6j, Ifngr KO and CD45.1 congenic (C57BL/6j background) mice were obtained from Jackson Laboratory and used in the experiments. Mice were implanted subcutaneously (S.C.) with either TRAMP-C2 (ATCC Cat# CRL-2731, RRID:CVCL_3615) or MC-38 (RRID: CVCL_B288) cell line at a dosage of 1 × 10^6 cells per mouse at the right flank on day 0, and were treated with different antibodies intraperitoneally (I.P.) on day 3, 6, and 9. In the late intervention TRAMP-C2 group, 1 × 106 TRAMP-C2 cells were similarly implanted S.C. at the right flank on day 0, but allowed to grow for 30–45 days prior to treatment. Mice with tumor volumes within 50–200 mm3 were randomized into different treatment groups before treatment. The average tumor sizes among different treatment groups were checked and ensured to be similar before treatment. Mice were injected with different antibodies I.P. on day 3, 6, and 9. In memory re-challenge experiments, mice were implanted with TRAMP-C2 tumors at a dose of 1 × 106 cells per mouse at the right flank on day 0. Mice were then treated with different immune checkpoint antibodies on day 3, 6, and 9. Tumors were measured twice a week, every 3–4 days. Ninety days after the initial tumor implantations (day 90), tumor-free mice from either anti-CTLA-4 or anti-CTLA-4 and anti–PD-1 DANA combination treatment groups were rechallenged with TRAMP-C2 tumors at the left flank at a dosage of 1 × 106 cells. There were no tumor-free mice treated with anti-PD-1 DANA antibody alone or isotype control. Sibling WT mice without prior tumor challenge or treatment were aged together in the same vivarium and used later as controls for rechallenge experiments. Tumor measurement = L (length) × W (width) × W /2 (mm3); whereas the longer diameter was defined as length and the shorter diameter was defined as width. All mice were maintained at UCSF vivarium in accordance with Institutional Animal Care and Use Committee (IACUC) standards.
Generation of Chimera mice
8–10 week-old C57BL/6j mice (H2b) were used as recipient mice and underwent lethal total body irradiation (1050 cGy; 137Cs source) followed by transplantation from donor CD45.2 Ifngr KO (From where?) mice and CD45.1 congenic mice. T cell-replete bone marrows were mixed in a 1:1 ratio (5 × 105 cells total) and injected intravenously (I.V.) through the tail vein per recipient mouse. Chimera mice were reconstituted and checked for chimerism by tail bleeding. Chimera mice were implanted subcutaneously with 1 × 106 TRAMP-C2 cells into the right flank on day 30. Mice were subsequently injected with different checkpoint inhibitors (10 mg/kg/injection/mouse) on day 33, 36, 39. On day 58, mice were sacrificed, and cells were harvested from tumor draining lymph nodes. The ratio of CD45.2+/CD45.1+ cells in CD8+Spas1 cells was calculated by dividing the total number of CD45.2+CD8+Spas-1 cells by the number of CD45.1+CD8+Spas-1 cells. The proportion of CD8+Spas-2 and CD8+ double negative subsets were similarly derived. All mice were maintained at the UCSF vivarium in accordance with IACUC standards.
METHOD DETAILS
Antibodies generation
Antibodies against mouse PD-1 were generated by immunizing HSD rats with recombinant mouse PD-1 protein (R&D Cat: 1021-PD). Hybridomas were generated by fusing IgG producing cells from immunized mice with myeloma cells (NS0-Mouse Myeloma, PTA-4796), and screened for binding to PD-1. The UC10–4F10–11 hybridoma expressing mouse anti-CTLA-4 antibody was purchased from ATCC (HB-304). The antibody variable domains were cloned from the hybridomas and expressed as murine IgG2a WT or with mutations to inactivate FcR binding (D265A; N297A; DANA). PD-1 and CTLA-4 antibodies were additionally screened for neutralization of the PD-1—PD-L1/L2 or CTLA-4—CD86 interactions, respectively.
Real-time RT-PCR gene cytokine arrays
CD45+CD3+CD8+Spas-1 T cells were sorted from draining lymph nodes from TRAMP-C2 bearing mice on day 28 after treatment. RNA was extracted from sorted CD8+Spas-1 T cells using an Ambion micro RNA isolation kit (AM1931) according to the manufacturer’s protocol, and genomic DNA was eliminated using a DNase kit purchased from QIAGEN. RNA quality was checked by the A260/A280 ratio using NanoDrop Lite (Thermal Scientific). 10ng RNA from each sample was used for subsequent cDNA synthesis. cDNA was synthesized, and cDNA templates were pre-amplified were using a RT2 PreAMP cDNA synthesis kit (QIAGEN Cat 330451) according to the manufacturer’s protocol and a ProFlex PCR machine (Applied Biosystems). Derived cDNA samples were evaluated for apoptotic gene expression arrays using RT2 Profiler™ PCR Array kits purchased from QIAGEN (PAMM-012Zc-12, Cat 330231) with the SYBR Green qPCR Mastermix (QIAGEN Cat 330522). Quantitative real-time RT-PCR arrays were performed using Applied Biosystems Cycler (AB Step-ONE Plus). RT-PCR arrays were analyzed, and gene expression heatmaps were generated using software provided on QIAGEN website under Data Analysis Center. All samples passed quality control (QC). Expression levels for each gene is presented as fold change in comparison to the internal control of housekeeping genes (beta-actin, Gus and Hsp90ab1) in each group. Gene array data are available in GEO database under accession number: GSE95433
RNA sequencing and data analysis
CD45+CD3+CD8+Spas-1 T cells were sorted from draining lymph nodes from TRAMP-C2 bearing mice on day 28 after treatment. Cells from each treatment types were pooled. RNA was extracted from sorted CD8+Spas-1 T cells using an Ambion micro RNA isolation kit (AM1931) according to the manufacturer’s protocol. Bulk RNA sequencing was carried out on 0.25ng of RNA in triplicates from each treatment type using the protocol as previously described (Picelli et al., 2014) with the following modifications. The TSO, Oligo-dT30VN, and ISPCR primers were biotinylated on the 5′ end. 21 cycles of pre-amplification were found to be optimal. Nextera adapters were added and the libraries were pooled and sequenced on an Illumina Hiseq 4000. Approximately 48,710 reads were obtained per library. The Fastq sequences were aligned and quantitated using the CLC genomics workbench RNA-Seq analysis tools (QIAGEN Bioinformatics). The reference library that was used was ensemble_v86 and reseq_GRCm38.p5.r106. Differential expression for RNA-Seq using negative binomial GLM in CLC genomics workbench was carried out for each treatment type versus isotype control resulting in three datasets: Combo versus Isotype, anti-CTLA-4 versus Isotype, and anti-PD-1 DANA versus Isotype. A differential RNA expression for each gene with an FDR p value less than 0.05 was considered significant. Each dataset was run through the Ingenuity Pathway Analysis (IPA) software using the default filter settings and a log2 fold change cutoff of 1 and a comparison pathway analysis for “Diseases & Functions” and “Canonical Pathways” functions was performed. The “Canonical Pathways” function was performed by comparing the p values between each group to generate data while the “Diseases and Functions” function utilized z-scores as a comparison between each group. After filtering out unrelated pathways from both functions, heatmaps, gene networks, and raw datasets were generated and exported. Raw RNA sequence data can be found at NCBI (GSE repository #121694)
T cell receptor sequence
Mice were implanted with TRAMP-C2 subcutaneously at day 0 and treated with different checkpoint inhibitors at day 3, 6 and 9 as described in the manuscript. Draining lymph nodes were harvested at Day 11 and Day 28 post tumor injections and samples were preserved in RNAlater (QIAGEN, Cat NO./ ID: 76106) for subsequent analysis of T cell receptor diversities. Using poly-T-beads, mRNA was isolated and fragmented briefly to generate 600–800bp fragments. Each of these RNA fragments were then converted to double stranded cDNA using random primers, end repaired and an A base was added to each 3′ end of the fragment. Two universal DNA sequences, adapters A and B, were ligated to each end of the fragment respectively. To enrich the library for T cell receptor transcripts, two sets of PCR reactions were performed. The first PCR reaction was done using a Constant region primer and universal sequence A. The second PCR was done using another constant region primer and universal sequence A. In the end Illumina compatible amplicons were generated with greater than 90% specificity to the T cell receptor transcripts. The UMID in the sequenced reads were used to remove PCR duplicates and the reads were mapped to annotated V and J segments. All sequences that had good V, J segment mapping were then translated to amino-acid sequences in all three frames, and sequences with good terminal motifs identifying the 5′ and 3′ ends of the CDR3 were used to identify the CDR3 segments. TCR data analysis was performed using a TCR3D R package as previously described (Zhang et al., 2017). TCR diversity was assessed by Geometric Coefficient Variation (GCV).
Clinical outcomes with immune checkpoint inhibition
Patients were treated with either PD-1 monotherapy with pembrolizumab 2 mg/kg or 10 mg/kg (n = 100), or PD-1/CTLA-4 combination therapy with ipilimumab 3 mg/kg plus nivolumab 1 mg/kg (n = 53). Best objective response rate was determined by RECIST v1.1. Baseline tumor size was calculated by summing the largest diameter of the target lesions per RECIST v1.1. Patients were stratified according to the baseline tumor size into ≤ 6 cm, > 6 and ≤ 11 cm, or > 11 cm. Patients with a complete or partial response were categorized as responders, and those with stable disease or progressive disease as their best response were categorized as nonresponders. The responder fraction was calculated by dividing responders/all patients. The error bars represent SEM. Demographic and clinical characteristics were summarized by descriptive statistics. Please see Statistics section for details.
Antibodies and GVAX
Anti-CTLA-4, anti-PD-1, anti-PD-1 DANA and IgG2a antibodies were obtained from Abbvie or Bioxcell (Table S2). All antibodies were stored in −80°C in small working aliquots to avoid repeated freeze-thaw cycles before use. Antibodies were dissolved in phosphate buffer saline (PBS) and injected I.P. at indicated time points. For combination treatments with anti-CTLA-4 and GVAX, 106 irradiated (10,000 rads) GVAX cells were injected S.C. into the skin over the neck on day 3, 6 and 9, the same time as I.P. antibody injection.
In vitro T cell culture and stimulation with recombinant IFN-γ
Splenocytes were harvested from TRAMP-C2 bearing C57BL/6j mice on day 50 and T cells were purified by magnetic beads according to the manufacturer’s protocol (Miltenyi Biotec Cat 130–095-130). Cells were checked for over 90% purity. Purified T cells were suspended in DMEM (UCSF cell culture core) + 10% fetal bovine serum (Lonza Cat 14–501F) + 1% penicillin/streptomycin (UCSF cell culture core) + murine 20 IU IL-2 (Peprotech Cat 212–12), and seeded in 96 wells at 2 × 105 cells per well. Recombinant murine IFN-γ (Peprotech Cat 315–05) was added into the wells at indicated concentrations. Cells were cultured for 12–72 hours and analyzed by flow cytometry. For the patient sample, peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll (Sigma Cat F4375) and PBMCs were seeded in 96 wells at 2 × 105 cells per well supplemented with 20 IU human IL-2 (Peprotech Cat 200–02). Recombinant human IFN-γ (Peprotech Cat 300–02) was added to the wells. Cells were harvested and analyzed by flow cytometry 72 hours after incubation.
In vitro CAR-19 T cell system
CAR-19 construct with 4–1BB domain was developed as previously described (Roybal et al., 2016). Primary human CD8+ T cells were isolated from an anonymous healthy donor blood apheresis by negative selection (STEMCELL Technologies Cat NO. #15062 and #15063). Blood was obtained from Blood Centers of the Pacific, as approved by the University Institutional Review Board. T cells were cryopreserved in RPMI-1640 (UCSF cell culture core) with 20% human AB serum (Valley Biomedical, Cat NO. HP1022) and 10% DMSO. After thawing, T cells were cultured in human T cell medium consisting of X-VIVO 15 (Lonza Cat NO. 04–418Q), 5% Human AB serum, and 10 mM neutralized N-acetyl L-Cysteine (Sigma-Aldrich Cat NO. A9165) supplemented with 30 units/mL IL-2 (NCI BRB Preclinical Repository) for all experiments. Primary CD8+ T cells were thawed the same day and, after 24 hr in culture, T cells were stimulated with Human T-Activator CD3/CD28 Dynabeads (Life Technologies Cat NO. 11131D) at a 1:3 cell:bead ratio. At 48 hr, viral supernatant was harvested and the primary T cells were exposed to the virus for 24 hr for CAR-19 transfection. At day 4 after T cell stimulation, the Dynabeads were removed, and T cells expanded until day 10 when they were rested and could be used in assays. At stage I, naive T cells (gate on CD45+CD8+CCR7-CD45RO+) prior to beads activation were used for subsequent in vitro experiments. For stage II, T cells stimulated with Dynabeads were used in vitro experiments. Beads were removed after 4 days, and T cells were allowed to expand until day 10 when they were rested to become stage III. For stage IV, CAR-19 T cells were co-cultured 1:1 with the K-562 cancer cell line (ATCC Cat NO CCL-243) or K-562 that was lentivirally transduced to stably express human CD19 at equivalent levels as Daudi tumors. T cells from the different stages as described previously were cultured in vitro under different concentrations of human recombinant IFN-γ (Peprotech Cat 212–12) (2 × 105 cells per well). After in vitro culture, cells were harvested and analyzed for Annexin V expression (BD Bioscience, Cat NO 556547) by flow cytometry.
Cell Culture
Tumor cell lines, TRAMP-C2 and MC-38, were cultured for cell injection into C57BL/6j male mice. TRAMP-C2 cell medium was composed of DMEM (UCSF cell culture core), 5% fetal bovine serum (FBS; Lonza Cat. 14–501F), 5% Nu-serum IV (Corning Cat. 355504), 0.005 mg/mL bovine insulin (Sigma Cat. I0516), 10nM dehydroisoandrosterone (Sigma Cat. D5297), and 1% penicillin/streptomycin (UCSF cell culture core). MC-38 medium was composed of DMEM (Corning Cat 10–013 CM), 10% fetal bovine serum (Lonza Cat. 14–501F), and 1% penicillin/streptomycin (UCSF cell culture core). Frozen cell lines were thawed in the water bath at 37°C before transfer into corresponding pre-warmed media. After wash, cells were then pelleted and resuspended in fresh media before being passaged. Once every two days, culture flasks were checked for confluence with a light microscope. Before cells overcrowd the culture flask (> 90% of confluency), old media from the flask was decanted into waste and 10mL of PBS added as a rinse. After cells were rinsed with PBS, the solution was removed and 5mL of 0.05% trypsin with EDTA (UCSF cell culture core) was introduced into the flask. Subsequently the flask was placed in a CO2 incubator at 37°C for 5 min for trypsinization of adherent cells. After incubation, the trypsin was neutralized with plentiful media, pelleted, and then resuspended in new media before a fractional transfer into new culture flasks. For cell injection into mice, instead of the fractional transfer step, cells were washed and pelleted with PBS twice to remove the presence of FBS. Prior to injection, cells were adjusted with PBS to a concentration of 107 cells per mL with each needle containing 1 × 106 cells in 100 uL.
Mouse Serum Cytokines
Two days after the final checkpoint inhibitor treatment, mouse sera were collected and sent to Eve Technologies (Calgary, Alberta, Canada) for analysis with mouse cytokine 31-plex discovery assay (Cat No: MD31). For the therapeutic model, serum samples were also collected two days after the final treatments and cytokine levels were analyzed by Cytometric Beads Array (BD, San Jose, CA) with cytokine-specific bead sets and standards according to the manufacturer’s protocol. Flow cytometry was performed on BD FACSymphony (BD, San Jose, CA) and data were analyzed by FlowJo software (TreeStar, Ashland, OR). Serum cytokine levels from treatment groups were each divided by the level of the IgG2a control group to be calculated as fold changes. These fold changes were graphed with the Prism 7 software (GraphPad, La Jolla, CA).
Tissue preparation
Spleens were surgically removed with sterilized surgical equipment and crushed with the blunt end of a 10 mL syringe on Petri dishes containing 5mL of PBS. The spleen mixtures were separately filtered through a 70 μM filter into a 50 mL conical tube, centrifuged at 1500rpm for 5 min at 4°C. After wash, cell pellets were resuspended in 5mL of red blood cell lysis solution (Santa Cruz Biotechnology; Cat sc-296258) on ice for 5 min and stopped with the addition of 30mL of PBS. After wash, cells were reconstituted for counting by Vi-Cell (Beckman Coulter, U.S.A.). Draining lymph nodes were extracted with sterilized surgical equipment and crushed between the frosted surfaces of super-frosted microscope slides into wells containing PBS. Cell mixtures were then filtered through a 70 μM filter into 15mL conical tubes. Cells were then washed and counted. Tumors were removed from mice with sterile surgical instruments followed by sectioning for paraformaldehyde fixation or flow cytometry analysis. Tumor tissues for flow analysis were kept moist with 1 mL collagenase IV digest media (DMEM+10%FCS+1% penicillin/streptomycin+Collagenase IV+DNase) and minced with scalpel blades. Tumor cell mixtures were then transferred into 15mL conical tubes and filled with an additional 9 mL of collagenase digest media. Tumor samples were subsequently placed on a 37°C shaker for 1 hour. Samples were filtered through a 100 μM filter into a 50 mL conical tube and washed with PBS before centrifugation. Finally, tumor cell pellets were resuspended and counted before subsequent flow staining.
Antibodies and flow staining
Single cell suspensions (1 million cells) were first incubated with Fc Block (BD PharMingen. San Diego, CA) for 10 min, then co-incubated with antibodies for 20 min at 4°C followed by washing with staining buffer (PBS + 1% FBS). Foxp3 and intracellular staining were performed using an eBioscience intracellular kit (Cat#00–5523-00) according to the manufacturer’s protocol. Active caspase-3 staining was performed by using BD Caspase-3 apoptosis kit (BD Cat 550480), and Annexin V staining was performed using a BD Annexin V apoptosis detection kit (BD Cat 556547) according to the manufacturer’s protocol. Flow cytometry was performed on Fortessa X20 Dual (BD, San Jose, CA), and data analyzed by FlowJo software (TreeStar). Details on flow cytometry antibodies used in this study can be found in Table S3.
Cy-TOF
Experimental protocols were followed as previous described. A summary of all mass cytometry antibodies and clones used for analysis can be found in Table S4. Primary conjugates of mass cytometry antibodies were prepared using the MaxPAR antibody conjugation kit (Fluidigm) according to the manufacturer’s recommended protocol. Following labeling, antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH, Wangen, Germany) containing with 0.5% sodium azide at concentrations of 0.1 and 0.3 mg/mL and stored long-term at 4°C. Each antibody clone and lot were titrated to optimal staining concentrations using primary murine cells. Cryopreserved, cells were live/dead stained with 5 μM cisplatin for 5 min at RT and quenched with Miltenyi Biotec autoMACS Running Buffer (Miltenyi Biotec, Cat 130–091-221, U.S.A.). Cells were then fixed with 1.6% PFA in Miltenyi Biotec autoMACS Running Buffer for 10 min at RT, then washed twice with Miltenyi Biotec autoMACS Running Buffer. Cells were resuspended in Maxpar Barcode Perm Buffer (Fluidigm, Cat 201066) and incubated with specific combinations of 3 of the 6 palladium (Pd) isotopes for 20 min shaking at RT. The excess Pd was removed by washing cells in Miltenyi Biotec autoMACS Running Buffer and combined into one tube for staining, before being resuspended in 100 uL Miltenyi Biotec autoMACS Running Buffer. Antibody against CD16/32 at 20 μg/mL was added to block the Fc receptors for 5 min at RT. An antibody cocktail of cell surface markers were added to a final staining volume of 500 ml, incubated for 30 min at RT while shaking. Following staining, cells were washed twice with Miltenyi Biotec autoMACS Running Buffer, then permeabilized with Maxpar Perm-S Buffer for 30 min at RT. After permeabilization, cells were then washed twice in Miltenyi Biotec autoMACS Running Buffer to remove remaining saponin (Perm-S Buffer), and then stained with intracellular antibodies in 500 μL for 30 min at RT on a shaker. After staining, cells were washed twice with Miltenyi Biotec autoMACS Running Buffer and then stained with 1 mL of 0.25 μM 191/193Ir DNA intercalator (Fluidigm) diluted in saponin-PBS containing 3.2% PFA overnight. Immediately prior to running the samples by Mass Cytometry (Cy-TOF), cells were washed once each in Miltenyi Biotec autoMACS Running Buffer, PBS and MilliQ H20. Mass cytometry samples were resuspended in MilliQ water containing 4 EQ bead standards (Fluidigm) to approximately 106 cells per mL and then run on a Helios equilibrated. Data were analyzed on Cytobank (https://www.cytobank.org/) platform with SCAFFoLD presentation. A detailed gating strategy can be found in Figure S5.
Histology and immunohistochemistry
Tissues harvested from mice were placed in 4% formalin, followed by 70% alcohol and PBS before embedding. Tissues were then embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Tissue sections were evaluated by a board-certified pathologist (M.C.). Images were visualized using an Olympus Vanox AHBS3 microscope with an Olympus SPlan Apo × 20/ 0.70 NA objective (Olympus, Woodbury, NY). A diagnostic instrument spot RT color digital camera utilizing Spot software version 4.0.2 was used to acquire the images (Diagnostic Instruments, Sterling Heights, MI). Murine immunohistochemistry was performed as previously described (DuPage et al., 2011). Tumor tissues were fixed in 4% paraformaldehyde, processed, embedded in paraffin, then cut into 5 mm sections. Paraffin sections were blocked with 3% hydrogen peroxide solution (Sigma Cat H1009), vector streptavidin/biotin (Vector Laboratories cat. SP-2002), and CAS-Block protein block (ThermoFisher Cat. 008120), then stained with CD8 antibody (Biorbyt Cat orb10325). For human immunohistochemistry, blocks were obtained from clinical trials and cut into different sections. Sections were then duo-stained with CD8 antibody (Dako Cat. M7103) and MART-1 antibody (Novus Biotechne Cat. NBP1–30151) using the Envision G2 Doublestain Kit (Dako Cat. K5361).
Angiogenesis studies
8–10 week old B57BL6/j mice were subcutaneously injected with 106 TRAMP-C2 cells and allowed 28 days to grow. Throughout the 28 days, marks were consistently made on the injection site to indicate the area to be harvested. After 28 days, skin containing the injection site was harvested and placed into 4% paraformaldehyde for 48 hours at 4°C and then moved to 70% ethanol at 4°C. Tissues were processed, embedded in paraffin, then cut into 5 um-thick sections. For immunohistochemistry, paraffin sections were blocked with 3% hydrogen peroxide solution (Sigma Cat H1009), vector streptavidin/biotin (Vector Laboratories Cat. SP-2002), and CAS-Block protein block (ThermoFisher Cat. 008120), then stained with CD31 antibody (Invitrogen, Cat. MA1–40074). The slides were scanned using a ScanScope XT Aperio slide scanner at 40X and then analyzed for positive cells using the Strataquest Software (TissueGnostics). A region of interest (ROI) is first drawn around each target section and a smaller region, the test ROI was selected to optimize the parameters needed for analysis. The different marker colors were first established and entered into the software, followed by the creation of a mask that differentiated between tissue and non-tissue. The total tissue area was then measured, and another mask was created to differentiate the different cells and their nuclei. A scattergram was then created to gate in the positive signals based off of average and maximum intensity. The parameters established for the test ROI were applied to all the other ROI’s and analysis was run. After analysis was finished, the data was exported into an excel sheet and the total positive events were divided by the total area and plotted onto Prism 6. Statistics were calculated using an unpaired t test.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Data shown in this manuscript were presented as mean ± SE. Tumor growth curves at different time points were plotted by using Prism 7 and analyzed by two-way ANOVA with a Tukey post hoc test comparison among groups. Flow cytometry data were analyzed by one-way ANOVA with a Tukey post hoc test. Other data were analyzed by 2-tailed Student’s t test as indicated in figure legends. P-values less than 0.05 were considered statistically significant. Gene expression arrays were analyzed by software provided by QIAGEN website under Data Analysis Center. For clinical data, Demographic and clinical characteristics were summarized by descriptive statistics. In general, frequency distribution and percentage were used to summarize categorical variables, and median with interquartile range (IQR) was used to describe continuous variables. Comparison of the continuous variables among groups were assessed using the two-sample t test and analysis of variance (ANOVA) for two groups and more than two groups, respectively. Logarithm transformation with base 10 (log10) was applied to LDH to avoid extreme skewness. Chi-square test was applied to test if there is statistical association between two categorical variables and Fisher exact test was used if the cell count was smaller than 5.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CTLA-4 (In vivo injection) | AbbVie | UC-10 |
| Anti-PD-1 (In vivo injection) | AbbVie | 17D2 |
| Anti-PD-1 (In vivo injection) | BioXCell | RMP1–14 |
| Anti-PD-1 DANA (In vivo injection) | AbbVie | 17D2 |
| Anti-PD-L1 (In vivo injection) | BioXCell | 10F.9G2 |
| CD3 | Biolegend | Cat#100232; RRID: AB_2562554 |
| CD4 | Biolegend | Cat#100447; RRID: AB_2564586 |
| CD8 | BD Bioscience | Cat#552877; RRID: AB_394506 |
| CD16/32 | TONBO | Cat#70–0161-U500; RRID: AB_2621487 |
| CD44 | Biolegend | Cat#103049; RRID: AB_2562600 |
| CD45 | Biolegend | Cat#103126; RRID: AB_493535 |
| CD45.1 | Biolegend | Cat#110737; RRID: AB_11204076 |
| CD45.2 | Biolegend | Cat#109847; RRID: AB_2616859 |
| CD62L | Biolegend | Cat#104433; RRID: AB_10900262 |
| Foxp3 | eBioscience | Cat#11–5773-82; RRID: AB_465243 |
| rIgG2a/k | eBioscience | Cat#11–4321; RRID: AB_470011 |
| PD-1 | Biolegend | Cat#135213; RRID: AB_10689633 |
| rIgG2a/k | Biolegend | Cat#400505; RRID: AB_2736919 |
| Tim-3 | Biolegend | Cat#134003; RRID: AB_1626181 |
| rIgG1/k | Biolegend | Cat#400407; RRID: AB_326513 |
| KLRG1 | Biolegend | Cat#138412; RRID: AB_10641560 |
| Hamster IgG | Biolegend | Cat#402012 |
| IFN-γ | BD Bioscience | Cat#554411; RRID: AB_395375 |
| Rat IgG1 | BD Bioscience | Cat#553924; RRID: AB_479706 |
| CD119 | BD Bioscience | Cat#740897; RRID: AB_2740545 |
| Rat IgG2a | BD Bioscience | Cat#563335 |
| Caspase-3 | BD Bioscience | Cat#51–68654X; RRID: AB_393697 |
| Spas-1 | NIH tetramer Core | Sequence: STHVNHLHCH-2D(b) |
| Spas-2 | NIH tetramer Core | Sequence: IIITFNDLH-2K(b) |
| CD45 (Cy-TOF) | Fluidigm | FLDM#3089003B; RRID: AB_2661851 |
| CD11c (Cy-TOF) | Fluidigm | FLDM#BL 337202 |
| CD235ab/CD61 (Cy-TOF) | Fluidigm | FLDM#BL 306602/BL 336402 |
| CD16 (Cy-TOF) | Fluidigm | FLDM#BL 302002 |
| CD45RA (Cy-TOF) | Fluidigm | FLDM#3143006B; RRID: AB_2651156 |
| CD4 (Cy-TOF) | Fluidigm | FLDM#3145001B; RRID: AB_2661789 |
| CD8a (Cy-TOF) | Fluidigm | FLDM#3146001B; RRID: AB_2687641 |
| BDCA-2 (Cy-TOF) | Fluidigm | FLDM#3147009B; RRID: AB_2714153 |
| CD14 (Cy-TOF) | Fluidigm | FLDM#3151009B |
| FoxP3 (Cy-TOF) | Fluidigm | FLDM#3162011A; RRID: AB_2687650 |
| CD25 (Cy-TOF) | Fluidigm | FLDM#3169003B; RRID: AB_2661806 |
| IFNg (Cy-TOF) | Fluidigm | FLDM#3165002B |
| CD127 (Cy-TOF) | Fluidigm | FLDM#3168017B |
| CD3 (Cy-TOF) | Fluidigm | FLDM#3170001B; RRID: AB_2661807 |
| HLA-DR (Cy-TOF) | Fluidigm | FLDM#3174001B; RRID: AB_2665397 |
| CD56 (Cy-TOF) | Fluidigm | FLDM#3176008B; RRID: AB_2661813 |
| CD27 (Cy-TOF) | Fluidigm | FLDM#3167006B |
| gdTCR (Cy-TOF) | Fluidigm | FLDM#BL331202 |
| TCRVd2 (Cy-TOF) | Fluidigm | FLDM#BL331402 |
| Human: MART-1 (IHC) | Novus Biotechne | Cat#NBP1–30151 |
| Human: CD8 (IHC) | Dako | Cat#M7103; RRID: AB_2075537 |
| Mouse: CD31 (IHC) | Invitrogen | Cat#MA1–40074 |
| Biological Samples | ||
| Blood samples from patients | Clinic | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DNase I | Sigma-Aldrich | Cat#D4263 |
| Xylenes | Fisher | Cat#X3P-1GAL |
| Citric acid monohydrate | Sigma-Aldrich | Cat#C1909–25G |
| TWEEN | Sigma-Aldrich | Cat#P7949–500ML |
| 30% Hydrogen Peroxide | Sigma-Aldrich | Cat#H1009–5ML |
| Gill’s Hematoxylin | Sigma-Aldrich | Cat#GHS216–500ML |
| Permount | Fisher | Cat#SP15–500 |
| DAB+ Substrate Chromagen System | Dako | Cat#K3468 |
| Streptavidin/Biotin Blocking Kit | Vector Laboratories | Cat#SP-2002 |
| Vectastain ABC Kit | Vector Laboratories | Cat#PK-4004 |
| Cas-Block | Life Technologies | Cat#008120 |
| 10X Tris-bufff Saline | Boston BioProducts | Cat#BM-300 |
| Antibody Diluent | Dako | Cat#S0809 |
| Recombinant Mouse PD-1 | R&D Systems | Cat#1021-PD |
| RNAlater | QIAGEN | Cat#76106 |
| Murine IL-2 | Peprotech | Cat#212–12 |
| Human IL-2 | Peprotech | Cat#200–02 |
| Recombinant Murine IFN-γ | Peprotech | Cat#315–05 |
| Recombinant Human IFN-γ | Peprotech | Cat#300–02 |
| 10 mM neutralized N-acetyl L-Cysteine | Sigma-Aldrich | Cat#A9165 |
| RBC Lysis Solution | Santa Cruz Biotechnology | Cat#SC-296258 |
| Collagenase IV | Sigma-Aldrich | Cat#C5138–5G |
| Critical Commercial Assays | ||
| Ambion micro RNA isolation kit | Ambion | Cat#AM1931 |
| Envision G2 Doublestain Kit | Dako | Cat#K5361 |
| RT2 PreAMP cDNA Synthesis Kit | QIAGEN | Cat#330451 |
| RT2 Profiler PCR Array Kit | QIAGEN | Cat#330522 |
| Splenocyte Isolation Kit | Miltenyi Biotec | Cat#130–095-130 |
| Human CD4 Isolation kit | STEMCELL Technologies | Cat#15062 |
| Human CD8 Isolation kit | STEMCELL Technologies | Cat#15063 |
| Mouse Cytokine 31-plex Discovery Assay | Eve Technologies | Cat#MD31 |
| Intracellular Staining Kit | eBioscience | Cat#00–5523-00 |
| Annexin V Apoptosis Detection Kit | BD Biosciences | Cat#556547 |
| Deposited Data | ||
| RNA sequence data | NCBI | GSE repository # 121694 |
| RT-PCR array data | NCBI | GSE repository # 95433 |
| Experimental Models: Cell Lines | ||
| Tramp-C2 | ATCC | Cat#CRL-2731; RRID: CVCL_3615 |
| MC-38 | Kerafast | Cat#ENH204; RRID: CVCL_B288 |
| MOC-1 | Gift from Dr. Uppaluri | MTA transfer from Dana-Farber |
| K562 | ATCC | Cat#CCL-243; RRID: CVCL_0004 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ifngr KO | Jackson Laboratory | 003288 |
| Mouse: B6 Cd45.1 | Jackson Laboratory | 002014 |
| Mouse: C57BL/6j | Jackson Laboratory | 000664 |
| Oligonucleotides | ||
| Biotin-TSOLNA | Exiqon | N/A |
| Biotin-Oligo-dT30VN | Exiqon | N/A |
| Biotin-ISPCR oligo | Exiqon | N/A |
| read1_bc_1_GAGGTAG | Integrated DNA Technologies | N/A |
| read1_bc_2_GCTTAAC | Integrated DNA Technologies | N/A |
| read1_bc_3_GCAATTC | Integrated DNA Technologies | N/A |
| read1_bc_13_GATCCTA | Integrated DNA Technologies | N/A |
| read1_bc_14_TGGTCTC | Integrated DNA Technologies | N/A |
| read1_bc_15_TCCAGTC | Integrated DNA Technologies | N/A |
| read1_bc_25_ACACTTG | Integrated DNA Technologies | N/A |
| read1_bc_26_TACGGCA | Integrated DNA Technologies | N/A |
| read1_bc_27_TCTCGTG | Integrated DNA Technologies | N/A |
| read1_bc_37_GTCATCC | Integrated DNA Technologies | N/A |
| read1_bc_38_ACCGTAC | Integrated DNA Technologies | N/A |
| read1_bc_39_GTACTAC | Integrated DNA Technologies | N/A |
| read2_bc_1_GACGTAC | Integrated DNA Technologies | N/A |
| read2_bc_2_AACTGCA | Integrated DNA Technologies | N/A |
| read2_bc_3_CCACAGT | Integrated DNA Technologies | N/A |
| read2_bc_13_GGTCCTA | Integrated DNA Technologies | N/A |
| read2_bc_14_CCAACGA | Integrated DNA Technologies | N/A |
| read2_bc_15_AGGACAC | Integrated DNA Technologies | N/A |
| read2_bc_25_GTGTGTC | Integrated DNA Technologies | N/A |
| read2_bc_26_GCTACAA | Integrated DNA Technologies | N/A |
| read2_bc_27_CGTAGAC | Integrated DNA Technologies | N/A |
| read2_bc_37_CATGGAT | Integrated DNA Technologies | N/A |
| read2_bc_38_CTGCGAA | Integrated DNA Technologies | N/A |
| read2_bc_39_GAGACGA | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | N/A |
| FlowJo | TreeStar | https://www.flowjo.com/ |
| R | The R Foundation | https://www.r-project.org |
| Ingenuity Pathway Analysis | QIAGEN Bioinformatics | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| StrataQuest | TissueGnostics | http://www.tissuegnostics.com/en/products/analysing-software/strataquest |
| Scaffold | Spitzer et al., 2017 | https://github.com/SpitzerLab/statisticalScaffold |
| CLC Genomics Workbench | QIAGEN Bioinformatics | https://www.qiagenbioinformatics.com/products/clc-genomics-workbench/ |
| Cytobank | Cytobank | https://www.cytobank.org |
| Other | ||
| Human T-Activator CD3/CD28 Dynabeads | Life Technologies | Cat#11131D |
| Recombinant human IFN-g protein | Peprotech | Cat#300–02 |
| Recombinant murine IFN-g protein | Peprotech | Cat#315–05 |
Highlights.
Combination checkpoint blockade leads to impaired efficacy with low tumor burden
This impairment results from IFN-γ-mediated deletion of tumor-reactive T cells
AICD is an immune-intrinsic mechanism of therapeutic resistance to checkpoint blockade
ACKNOWLEDGMENTS
We thank the UCSF Flow Cytometry Core for technical help and the NIH Tetramer Core for the generation of Spas-1 and Spas-2 tetramers. This work was supported by National Cancer Institute grants R01CA163012, R01CA194511, and U01CA233100 and a Prostate Cancer Foundation Challenge Award. This study was also supported by research funding from AbbVie Inc. and Girihlet Inc.
L.F. received research funding from AbbVie, Merck, Roche-Genentech, Janssen, Bavarian Nordic, and Bristol-Myer Squibb Inc. X.L. and J.S. are research scientists at AbbVie Bioresearch Center. D.M.S. and G.A.K. are former research scientists at AbbVie. R.S. and A.J. are research scientists at Girihlet.
Footnotes
DATA AND SOFTWARE AVAILABILITY
RNA sequence data are available at the NCBI GEO repository under accession number GEO: GSE121694. RT-PCR gene array data are available in NCBI GEO repository under accession number GEO: GSE95433.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and five tables and can be found with the article online at https://doi.org/10.1016/j.immuni.2019.01.006.
DECLARATION OF INTERESTS
The other authors declare no conflicts of interest.
REFERENCES
- Badovinac VP, Tvinnereim AR, and Harty JT (2000). Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science 290, 1354–1358. [DOI] [PubMed] [Google Scholar]
- Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK, et al. ; KEYNOTE-045 Investigators (2017). Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 376, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner V, Liu H, Zhou Q, Alderson KL, Sun K, Weiss JM, Back TC, Longo DL, Blazar BR, Wiltrout RH, et al. (2007). IFN-gamma mediates CD4+ T-cell loss and impairs secondary antitumor responses after successful initial immunotherapy. Nat. Med. 13, 354–360. [DOI] [PubMed] [Google Scholar]
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. (2015). Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Postow MA, and Wolchok JD (2016). Targeting T cell coreceptors for cancer therapy. Immunity 44, 1069–1078. [DOI] [PubMed] [Google Scholar]
- Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, Chaput N, Eggermont A, Marabelle A, Soria JC, and Ferté C (2017). Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 23, 1920–1928. [DOI] [PubMed] [Google Scholar]
- Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, and Ravetch JV (2015). FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell 28, 285–295. [DOI] [PubMed] [Google Scholar]
- Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, and Dhodapkar KM (2015). Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J. Immunol. 194, 950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, and Schreiber RD (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998. [DOI] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, and Jacks T (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. (2009). New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247. [DOI] [PubMed] [Google Scholar]
- Fassò M, Waitz R, Hou Y, Rim T, Greenberg NM, Shastri N, Fong L, and Allison JP (2008). SPAS-1 (stimulator of prostatic adenocarcinoma-specific T cells)/SH3GLB2: A prostate tumor antigen identified by CTLA-4 blockade. Proc. Natl. Acad. Sci. USA 105, 3509–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos AM, Xiong H, Leiner IM, Sušac B, Glickman MS, Pamer EG, and van Heijst JW (2016). Control of T cell antigen reactivity via programmed TCR downregulation. Nat. Immunol. 17, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167, 397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirnalda P, Wood L, Goenka R, Crespo J, and Paterson Y (2013). Interferon γ-induced intratumoral expression of CXCL9 alters the local distribution of T cells following immunotherapy with Listeria monocytogenes. OncoImmunology 2, e25752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, Ready NE, Gerber DE, Chow LQ, Juergens RA, et al. (2017). Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 18, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, Ni A, Novik JB, Mangarin LMB, Abu-Akeel M, et al. (2018). Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33, 843–852.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Old LJ, and Schreiber RD (2002). The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 13, 95–109. [DOI] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC (2018). CAR T cell immunotherapy for human cancer. Science 359, 1361–1365. [DOI] [PubMed] [Google Scholar]
- Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, Ivanov A, Szymborska A, Patone G, Kunz S, et al. (2017). Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature 545, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. (2016). Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7, 10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, et al. ; CA184–043 Investigators (2014). Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 15, 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, Gough MJ, Urba WJ, and Fox BA (2017). Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin. Cancer Res. 23, 6165–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ (2015). Interferons and the immunogenic effects of cancer therapy. Trends Immunol. 36, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthélémy P, Porta C, George S, et al. ; CheckMate 214 Investigators (2018). Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 378, 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi PM, Pauken KE, Paley MA, Sharpe A, and Wherry EJ (2015). Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 212, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli F, Cabiddu M, Coinu A, Borgonovo K, Ghilardi M, Lonati V, and Barni S (2015). Prognostic role of lactate dehydrogenase in solid tumors: a systematic review and meta-analysis of 76 studies. Acta Oncol. 54, 961–970. [DOI] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Refaeli Y, Van Parijs L, Alexander SI, and Abbas AK (2002). Interferon gamma is required for activation-induced death of T lymphocytes. J. Exp. Med. 196, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. ; KEYNOTE-006 investigators (2015). Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA (2014). IL-2: the first effective immunotherapy for human cancer. J. Immunol. 192, 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, and Lim WA (2016). Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 167, 419–432.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, et al. (2016). Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, and Sharma P (2018). Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin. Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 210, 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic immunity is required for effective cancer immunotherapy. Cell 168, 487–502.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, and Gajewski TF (2013). Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 5, 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh CD, and Sprent J (2008). Homeostasis of naive and memory T cells. Immunity 29, 848–862. [DOI] [PubMed] [Google Scholar]
- Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovich B, et al. (2012). A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119, 5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, and Allison JP (2017). Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 170, 1120–1133.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Kudo T, Payne L, Wang X, Rodgers L, and Suzuki Y (2010). Predominant interferon-γ-mediated expression of CXCL9, CXCL10, and CCL5 proteins in the brain during chronic infection with Toxoplasma gondii in BALB/c mice resistant to development of toxoplasmic encephalitis. J. Interferon Cytokine Res. 30, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. (2017). Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 377, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima T, Yoshihara K, Nakazato K, Kumabe S, Koyasu S, Sad S, Shen H, Kuwano H, and Yoshikai Y (2006). IL-15 regulates CD8+ T cell contraction during primary infection. J. Immunol. 176, 507–515. [DOI] [PubMed] [Google Scholar]
- Zaidi MR, and Merlino G (2011). The two faces of interferon-γ in cancer. Clin. Cancer Res. 17, 6118–6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Cham J, Paciorek A, Trager J, Sheikh N, and Fong L (2017). 3D: diversity, dynamics, differential testing-a proposed pipeline for analysis of next-generation sequencing T cell repertoire data. BMC Bioinformatics 18, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.