

Abstract
Nucleic acid based adjuvants recognized by Toll-like receptors (TLR) are potent immune system stimulants that can augment the antitumor immune responses in an antigen-specific manner. However, their clinical uses as vaccine adjuvants are limited primarily due to lack of accumulation in the lymph nodes, the anatomic sites where the immune responses are initiated. Here, we showed that chemical conjugation of type B CpG DNA, a TLR9 agonist to dextran polymer dramatically enhanced CpG’s lymph node accumulation in mice. Dextran conjugation did not alter CpG ODN’s uptake, internalization, and bioactivity in vitro. Delivery of Dextran-CpG conjugate markedly increased the uptake by antigen presenting cells in the lymph nodes and enhanced CD8+ T cell responses primed by protein vaccines, leading to improved therapeutic antitumor immunity. Furthermore, immunization with Dextran-CpG mixed with necrotic whole tumor cells induced a protective antitumor response in a murine model, suggesting that this approach was not limited to molecularly defined antigens. This simple method might also be applicable for the delivery of many other nucleic acid based adjuvants in cancer vaccines.
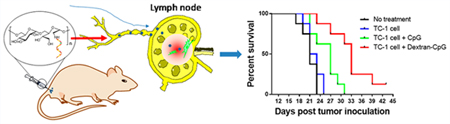
Graphical Abstract
INTRODUCTION
Vaccination represents the single most effective medical intervention in modern medicine, saving millions of lives each year by providing protection against various disease epidemics.1–3 However, vaccines have not succeeded in therapeutic settings such as cancer.3–6 This is in part due to the difficulties associated with priming cytotoxic T cells that can overcome the tumor-related immune suppression and can specifically attack and destroy cancer cells.3–5 To overcome this limitation, adjuvants are routinely added to vaccine formulations to promote the generation of antigen-specific cytotoxic CD8+ T cells and to break the immune suppression.6–9 Today, a multitude of adjuvants are available. For example, ligands binding to toll-like receptors activate the innate immunity and support the subsequent development of adaptive immunity, and have been shown to potently prime CD8+ T cells and exhibit anticancer effects against established tumors.10–12 However, a method to rationally design adjuvants that specifically activate immune cells, leading to potent, tailored immune responses, is lacking.13–15 A major obstacle is the poor physicochemical and pharmacokinetic properties of adjuvants in vivo, which lead to suboptimal lymph node (LN) drainage and retention after injection.16–24 As a result, no vaccine with molecularly defined adjuvants is currently available for cancer patients.
The lymphatic system is designed to filter the lymph fluid and remove foreign materials such as bacteria and virus entering the body, and mounts immune reactions toward these foreign materials. Lymph nodes could be strategic targets for vaccine delivery because of their important roles in initiating adaptive immunity.19–24 The molecular sizes greatly affect the uptake of vaccine molecules in the LN following parenteral injection.14,15,24,25 While small molecules (<5 nm) quickly diffuse into blood by crossing the small gaps of blood capillaries, larger molecules (5–200 nm) are primarily drained to the lymphatic system through the gaps in the wall of the lymph vessels and accumulate in the draining LNs.14,15,24,25 Therefore, controlling the hydrodynamic sizes of vaccine components is critical to achieving LN targeting of vaccines in vivo. Antigens and molecular adjuvants conjugated to or encapsulated in size-optimized nanoparticles (NP) have frequently been used to promote LN targeting.13–15,19–25 NP conjugation dramatically increases the hydrodynamic sizes of molecular adjuvants, preventing them from diffusing into the blood circulation and retargeting them to the lymphatics. Apart from NPs, linear polymers have high molecular weight, tunable biological functions, and are widely used for drug delivery purposes.26,27 Despite considerable efforts to develop polymeric imaging agents for sentinel LN mapping, few reports have been conducted using polymer as an adjuvant carrier to target LN.28–30
Here we report a simple strategy to fulfill the size requirements for LN targeting. We hypothesize that covalently linking an oligonucleotide agonist (CpG DNA) to dextran polymer could markedly increase the hydrodynamic size of CpG DNA and subsequently transport it to the lymph node. Enhanced LN targeting of CpG adjuvant would enhance the activation of LN-resident antigen presenting cells (APCs), leading to augmentation of T-cell priming. Dextran was chosen as the LN targeting carrier because (1) fluorescently labeled dextran has been widely used to stain lymphatic capillaries due to its specificity and high affinity toward cell surface proteins such as mannose receptor and DEC-205, which are widely expressed on antigen presenting cell surface;31–33 (2) dextran is nontoxic and has been used as an FDA-approved polymer for lymph node imaging;34,35 and (3) versatile chemistries can be used in functionalizing dextran polymers for bioconjugation. Our results demonstrate that Dextran-CpG conjugation is a simple and effective approach to enhance the vaccine-elicited CD8+ T cell responses by targeting CpG oligonucleotide adjuvant to LN at low doses. Importantly, this simple LN-targeting adjuvant approach is not limited to molecularly defined antigens, as protective antitumor immunity and prolonged survival were also observed when Dextran-CpG was mixed with whole tumor cell vaccines. Thus, LN targeted delivery of oligonucleotide adjuvant by dextran polymer conjugation might serve as a plug and play adjuvant applicable to many current vaccines.
RESULTS
Synthesis and Characterization of Dextran-CpG DNA Conjugate.
Due to its small molecular weight, CpG DNA injected subcutaneously primarily diffuses into blood circulation and only a small fraction enters the lymphatic system.16 We hypothesize that a carbohydrate polymer carrier would dramatically increase the hydrodynamic size of CpG and block its diffusion into blood circulation. To test this idea, we chose linear dextran polymer with average molecular weight of 70K. Linear hyaluronan polymer with similar molecular weight has been shown to have the largest LN area-under-the-curve following s.c. injection.29 The class B CpG ODN (CpG 1826) specific for murine TLR9 was conjugated to periodate oxidized dextran via reductive amination of aldehydes (Figure 1a). The conjugate efficiency was characterized by agarose gel electrophoresis. As shown in Figure 1b, unmodified CpG ODN showed a sharp band with fast mobility, while the crude conjugate product exhibited a long, smeared band with retarded mobility, indicating an increased molecular weight and heterogeneous nature of the dextran and CpG conjugate. Notably, simply mix CpG and dextran without reaction resulted in a single sharp band that was identical to unmodified CpG, demonstrating that dextran polymer alone did not interact with CpG in the gel (Figure 1b, lane 2). Dynamic light scattering characterization of Dextran-CpG gave an average effective diameter of 6.5 nm, a size that was significantly bigger than unmodified CpG. This conjugation method yields a quantitative Dextran-CpG conjugate that can be easily purified by dialysis. The final conjugate contains approximately 1.33 nmol CpG per mg of dextran after purification.
Figure 1.

Synthesis and in vitro characterization of Dextran-CpG conjugate. (a) 5′-Terminal amine modified CpG ODN (CpG 1826) was conjugated to oxidized dextran by reductive amination. (b) Characterization of Dextran-CpG conjugate by agarose gel electrophoresis. (c) Laser scanning confocal microscopy images showing similar intracellular distribution of CpG or Dextran-CpG (100 nmol) in DC2.4 cells after 2 h incubation at 37 °C. Scale bar: 10 μm. (d) Quantification of cellular uptake by flow cytometry. (e) Dextran-CpG conjugate retains the immune stimulatory activity in mouse TLR9 NF-κB/SEAP transfected HEK cells. (f) Immunostimulatory activities of dextran, Dextran-CpG, and free CpG in RawBlue cells. Data show the mean values ± SEM: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s., not significant.
Dextran-CpG Conjugate Does Not Enhance the Cellular Uptake in Vitro.
Polysaccharide-based natural molecules play important roles in the recognition processes that occur at the cell surface.36 In fact, polysaccharide conjugates are widely used to improve the poor cell- or tissue-specific delivery of various molecules through cell-receptor-mediated endocytosis.37 A number of receptors specific for polysaccharide are expressed at the surface of antigen presenting cells.31,33,38 We, therefore, test whether dextran conjugate enhances the binding and subsequent uptake of CpG DNA in cell culture. DC2.4 cells (a mouse dendritic cell line) were incubated with fluorescently labeled CpG, or Dextran-CpG (100 nmol), and the cells were subsequently washed and imaged. After 2 h incubation, both CpG and Dextran-CpG showed extensive uptake in DC2.4 cells. Interestingly, in comparison to unmodified CpG, Dextran-CpG exhibited reduced cellular uptake, as demonstrated by both fluorescent image (Figure 1c) and flow cytometry analysis (Figure 1d). To improve in vivo stability, type B CpG is typically modified with phosphorothioate backbones, which are known to nonspecifically bind to cell surface components, with no consensus about the cellular entry, docking, and intracellular distribution.39,40 We believe that this “sticky” feature of CpG DNA might explain the observed reduction of uptake after dextran conjugation. However, other possibilities, such as a surface CpG receptor on DC, cannot be ruled out.41 Live cell imaging demonstrated that CpG and Dextran-CpG had indistinguishable intracellular distributions in DC2.4 cells (Figure 1c).
Dextran-CpG Conjugate Does Not Compromise the Adjuvant Activities of CpG in Vitro.
Covalent chemical modifications on certain types of CpG ODN has been shown to affect their immunostimulatory activity, especially at the 5′-end.42 We thus first determined whether Dextran-CpG conjugate retains its bioactivity in vitro. TLR-9 transfected HEK cells secrete embryonic alkaline phosphatase (SEAP) upon activation of the NF-κB pathway, allowing quantitative measurement of cell stimulation. Thus, TLR-9 cells were incubated with Dextran-CpG or soluble CpG, and activations of NF-κB were measured by quantifying SEAP levels in the supernatant. As shown in Figure 1e, Dextran-CpG conjugate induced high levels of NF-κB, comparable to unmodified CpG, indicating that dextran conjugation did not affect the CpG’s immune stimulatory activity. This result was consistent with previous observations for type B CpG, where covalent modification at the 5′ terminal had minimal effect on its bioactivity.16,43 To test whether dextran polymer alone can be an immune adjuvant, we used a reporter macrophage cell line. RawBlue cells derived from murine Raw 264.7 macrophages express a wide variety of pattern recognition receptors, and activation of these receptors induces the production of SEAP. Dextran alone did not activate RAW-Blue cells, indicating a low level of endotoxin in dextran polymer. Further, dextran modification did not enhance the stimulatory potency of CpG in these cells (Figure. 1f), suggesting dextran polymer alone was nonstimulatory.
Dextran-CpG Conjugate Enhances Lymph Node Uptake.
To test whether conjugation between CpG and dextran enhances the lymph node accumulation of CpG, we injected C57BL/6 mice with fluorescent CpG-dextran conjugate at the tail base, using dextran and free CpG as a control. Twenty-four hours after injection, draining LNs were excised and CpG uptakes were analyzed using flow cytometry. Both the inguinal nodes and the axillary nodes increased in size following s.c. injection of CpG or Dextran-CpG (Figure 2a,b), as compared with PBS or dextran controls. However, Dextran-CpG had the highest local activities, showing a 2- to 3-fold increase in LN size. LN-accumulating CpGs were mainly associated with F4/80+ macrophages and CD11c+ dendritic cells, key antigen presenting cells in the lymph nodes. However, Dextran-CpG conjugate showed significantly better accumulation and cellular uptake as compared to free CpG (Figure 2c,d). In mice injected with Dextran-CpG, nearly 23% of inguinal lymph node DCs and 22% of macrophages stained positive for CpG. In contrast, CpG positive DCs were less than of 10% of the cells in LN in mice injected with free CpG. These results demonstrated that Dextran-CpG conjugate efficiently accumulated in key antigen presenting cells in the draining lymph nodes after injection.
Figure 2.

Dextran-CpG conjugate enhances the lymph node accumulation after subcutaneous injection. (a,b) C57BL/6 mice (4 lymph nodes/group) received PBS, dextran, 3.3 nmol of fluorescein labeled CpG, Dextran + CpG, or Dextran-CpG; 24 h later, inguinal nodes and axillary nodes were isolated and sizes were determined two-dimensionally by digital analysis of scaled photographs. (c,d), The above lymph nodes were digested and lymph node cells were stained with antibodies against CD11c and F4/80. Shown are representative flow cytometry plots of CD11c and F4/80 staining (c) versus CpG fluorescence in viable cells. (d) Percentages of CpG+ cells in the LNs determined by flow cytometry at 24 h. Data show the mean values ± SEM: *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Dextran-CpG Conjugate Enhances CD8+ T-cell Immune Responses When Combined with Soluble Protein Antigen.
To access the immunostimulatory potential of Dextran-CpG conjugate in vivo, mice were immunized in combination with ovalbumin (OVA), a model antigen that is widely used in adjuvant studies. OVA antigen was simply mixed with Dextran-CpG or CpG and administered subcutaneously at the tail base. C57BL/6 mice were immunized twice at 2 weeks apart and 6 days after the final injection, peripheral blood samples were collected and OVA-specific T cells were analyzed by tetramer staining. In mice immunized with Dextran-CpG, nearly 12% of the blood CD8+ T cells were specific for the MHC-I-restricted peptide OVA257–264, compared with 2.1% in mice treated with free CpG (Figure 3a,b). Interestingly, conjugating OVA antigen to dextran completely suppressed the immune activity of OVA, even when adjuvanted with LN targeting Dextran-CpG (Figure 3a,b). Carbohydrate-conjugated antigens have been previously used to elicit an immune response against pathogens including bacteria and yeasts.44–46 It was not clear why and how Dextran-OVA conjugation suppressed the OVA-specific CD8+ T-cell reactivity in vivo. As demonstrated in numerous previous studies, it is highly unlikely that the conjugation chemistry completely abrogates the immunogenicity of OVA.45,46 However, several recent studies demonstrated the immunosuppressive activities of saccharide when conjugated to antigens.47–49 Further studies are needed to better understand the immunosuppressive mechanism on Dextran-OVA conjugate.
Figure 3.

Dextran-CpG conjugate, when combined with a soluble protein antigen, elicits robust expansion of antigen-specific CD8+ T-cells with therapeutic benefits, as compared to soluble formulations. (a,b) C57BL/6 mice were primed on day 0 and boosted on day 14 with Dextran-CpG and Dextran-OVA (10 μg OVA protein) or equivalent soluble protein/CpG vaccines. Six days post boost, mice were bled and analyzed for tetramer positive CD8+ T-cells in peripheral blood. (a) Representative flow cytometry dot plots of H2Kb/SIINFEKL tetramer staining of CD8+ cells. (b) Mean percentages of OVA-specific CD8+ T cells. (c,d) C57BL/6 mice (n = 8/group) were inoculated with 3 × 105 EG7 tumor cells s.c. in the flank and immunized with soluble or dextran-conjugated vaccines on days 6 (10 μg OVA, 1.24 nmol CpG) and 13 (20 μg OVA, 1.24 nmol CpG). Kaplan−Meier survival curves of eight mice per group are shown in (c). (d) Frequencies of OVA-specific CD8+ T cells in tumor bearing mice were determined 6 days post the final treatment. (e) On day 70 post tumor inoculation, tumor-free mice were bled and OVA-specific T cells in the blood were measured by tetramer staining. Data show the mean values ± SEM: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s., not significant.
Therapeutic Benefit of Dextran-CpG Conjugate in a Mouse EG7 Model.
Antitumor immunity requires the development of a strong antigen-specific CD8+ T cell response. To test whether the Dextran-CpG conjugate provides therapeutic protection from the development of the tumor, C57BL/6 mice were inoculated with EG7 cells (an OVA-expressing murine T-cell lymphoma cell line).48 Mice bearing 6-day established EG7 tumors were treated with 2 injections (prime/boost) of vaccines at day 6 and day 13. Tumor growth and immune responses were monitored at various time points. Tumor grew vigorously when mice were treated with Dextran + OVA, Dextran-OVA + CpG, or Dextran-OVA + Dextran-CpG (Figure 3c). However, tumor growth rate was effectively controlled in mice immunized with Dextran-CpG + OVA or free CpG + OVA. Dextran alone had minimal effect on tumor growth, as demonstrated in the Dextran + OVA group and Dextran + OVA + CpG group, showing similar results in mice treated with PBS or OVA + CpG, respectively. Notably, 4 out of 8 of the mice treated with Dextran-CpG and 3 out of 8 of those receiving CpG completely rejected the tumor. The reduction in tumor growth was correlated with an increase in survival (Figure 3c). Measuring the immune responses elicited by these vaccines in tumor-bearing mice on day 18 post tumor inoculation indicated that Dextran-CpG induced high frequencies of OVA-specific CD8+ T cells. In mice immunized with free CpG, 8.1% of the T cells in the blood are OVA-specific (Figure 3d). In contrast, 15% of the blood CD8+ T cells are OVA-specific in mice treated with Dextran-CpG. Consistent with the survival data, mice treated with Dextran-OVA showed low frequencies of OVA-reactive CD8+ T cells (Figure 3c,d).
Long-Term Memory T-cells of Dextran-CpG Conjugate That Prevents Tumor Recurrence.
To assess whether Dextran-CpG treatment can induce memory T cells, all the survival mice were again challenged by injecting a lethal dose of tumor cell on day 70 post tumor inoculation. No tumor growth can be detected (data not shown). Strikingly, the OVA-specific CD8+ T cells in the blood were still detectable (Figure 3e) in both treatment groups 70 days post tumor inoculation, suggesting a development of long-lived memory CD8+ T cells. Mice treated with Dextran-CpG exhibited significantly stronger OVA-specific CD8+ T cells compared with mice treated with soluble CpG (Figure 3e).
Therapeutic Benefit of Dextran-CpG Conjugate Combined with Whole Tumor Cells.
The therapeutic benefits of Dextran-CpG conjugate in the mouse EG7 model promote us to test its efficacy in vaccines with whole tumor cells. The advantage of whole tumor cells used as vaccine rather than a specific protein or peptide tumor antigen is that tumor cells contain all the potential antigens. Previous studies have demonstrated the importance of coadministration of an immunostimulating adjuvant in whole-cell vaccines.50,51 Freeze-and-thaw-disrupted tumor cells were prepared from the murine TC-1 tumor cell line and were combined with Dextran-CpG as therapeutic vaccines. TC-1 cell is a human papillomavirus E7-expressing murine cell line.52 Cells underwent three freeze−thaw cycles using a 37 °C water bath and liquid nitrogen to prevent in vivo replication. C57BL/6 mice were inoculated subcutaneously in the flank with 3 × 105 live TC-1 cells, which were allowed to establish into a palpable solid tumor for 6 days. Mice were then treated by two s.c. injections of vaccines composed of killed TC-1 cells, or TC-1 cells adjuvanted with Dextran-CpG, or TC-1 cells + free CpG. As shown in Figure 4a, compared to PBS control, mice immunized with killed TC-1 cells and TC-1 cells + CpG barely slowed tumor growth, while mice treated with tumor cells + Dextran-CpG significantly inhibited tumor growth. The reduction of tumor growth was also correlated with an increase in survival (Figure 4b). To access the mechanism of this therapeutic effect, we measured the frequencies of E7 peptide (an immune dominant epitope) specific CD8+ T cell in blood 11 days post tumor induction. Although the antigen-specific T cells have been diluted because whole tumor cell antigens were used, Dextran-CpG elicited a significantly high frequency of anti-E7 CD8+ T cells compared to CpG, even at day 11 post tumor cell inoculation.
Figure 4.

Dextran-CpG inhibits tumor growth when combined with whole tumor cell vaccine. C57BL/6 mice (n = 8/group) were inoculated with 3 × 105 TC-1 tumor cells s.c. in the flank and treated with PBS or immunized with soluble or Dextran-CpG + TC-1 tumor cells on days 5 and day 12 (1.24 nmol CpG, 1 × 106 freeze−thawed TC-1 cells). Tumor growth (a) and Kaplan−Meier survival (b) of mice were monitored over time. (c), on day 11 post tumor inoculation, mice were bled and antigen-specific CD8+ T cells were analyzed by H-2Db restricted E7 peptide (an immune dominant epitope) tetramer staining. Data show the mean values ± SEM: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s., not significant.
DISCUSSION
The development of vaccines that can overcome tumor-related immune suppression has been greatly hampered by the lack of an effective method to elicit antigen-specific cytotoxic CD8+ T cells. Adjuvants that skew the vaccine toward Th1 responses are thus highly desired in cancer immunotherapy. However, many traditional adjuvants such as alum and mineral oil are able to induce good antibody (Th2) responses; they have little capacity to stimulate cellular (Th1) response.53 Recent advances in our understanding of immunology have enabled the identification of pattern-recognition receptors (PRRs) of innate systems, particularly Toll-like receptors (TLRs) in amplifying adaptive immunity.11–15 Molecularly defined ligands that can engage TLRs have the potential to elicit cytotoxic T-cell responses, resulting in the cell-mediated attack and elimination of malignant host cells, while at the same time reducing the toxicity.16 However, most TLR ligands injected parenterally do not reach lymph nodes, the anatomic sites where the immune responses are orchestrated.15,16 Adjuvants failing to reach LN are largely ignored by the immune system, leading to unresponsiveness. In contrast, adjuvant formulations targeting TLR agonists to LN have been shown to dramatically augment the efficacy of molecular adjuvants.15,16,20–23 An effective strategy to enhance the LN accumulation is to formulate the TLR adjuvants with the nanosized particulate carriers.15,20,23 Structurally optimized nanoparticle carriers, such as polymer particles, inorganic particles, or liposomes, can promote adjuvant transport to draining LNs through lymphatics, enabling activation of antigen-presenting cells in the LN, and promoting cellular immunity.20–23 This approach has been demonstrated in a number of TLR agonists using different nanoparticle formulations.20,23
Motivated by these previous findings, here we reported an alternative approach to target molecular adjuvants to lymphoid tissues through the use of a linear polymer carrier. We demonstrated that dextran polymer is an efficient carrier for concentrating CpG DNA in the LNs. Conjugation of CpG DNA to dextran polymer dramatically enhanced the hydro-dynamic sizes of CpG adjuvant, leading to increased LN accumulation. Importantly, Dextran-CpG conjugate effectively adjuvanted the cellular immune response when simply admixed with protein antigens without requiring the co-conjugation of antigen and adjuvant on the same polymer. This new adjuvant formulation was also effective in whole tumor cell vaccines, where a vast amount of T cell epitopes were available for activating CD4+ T helper and CD8+ cytotoxic lymphocytes simultaneously.
We choose dextran with a molecular weight of 70K as the polymeric carrier. Class B CpG DNA, a TLR9 agonist with the ability to prime potent CD8+ T cell responses, was covalently conjugated to dextran. Carbohydrate polymer with similar molecular weight has been optimized previously for sentinel LN imaging.29,30 Remarkably, the conjugate greatly increased the LN accumulation of CpG, leading to potent expansion of vaccine-elicited cytotoxic T lymphocyte. These massive cellular responses to Dextran-CpG conjugate also correlated with tumor protection in a murine model.
Many previous studies showed the avid in vitro uptake of proteins/drugs after carbohydrate conjugation. Our finding that Dextran-CpG conjugate did not promote the cellular binding and uptake in an in vitro setting, suggesting the enhanced immune response was mainly due to LN targeting. We believe that the reduced cellular uptake reflects the sticky nature of CpG DNA’s phosphorothioate (PS) backbone.39,40 However, other adjuvants lacking the PS modification might still benefit from the enhanced uptake via carbohydrate carrier. Surprisingly, contrary to many previous findings, where antigen–carbohydrate conjugates promote the development of immune responses, OVA conjugated to dextran completely abrogated OVA-specific CD8+ T cell responses, even when adjuvanted with LN targeting dextran-CpG, suggesting conjugation strategy could markedly impact the antigen immunogenicity.
Many tumor antigens are not fully characterized and thus well-defined epitopes are usually not available. The use of Dextran-CpG with whole tumor cell vaccine was very effective in halting tumor growth and resulted in a measurable enhancement in response against one of the well-characterized antigens. Dextran-CpG was able to induce detectable (E7 peptide) tetramer positive CD8+ T cells and long-term memory T cells.
The breadth of therapeutic opportunities of dextran polymer conjugate to CpG strongly suggests that dextran might also serve as an effective carrier for other types of TLR agonists, including RNA oligonucleotides and non-oligonucleotide ligands. For example, small molecule immune response modifiers that specifically activate immune cells via TLR-7/8 might be conjugated to dextran to ensure the delivery to APCs in the LN, avoiding a nonspecific and generalized immune activation.
CONCLUSION
In conclusion, we show that dextran is an efficient polymeric carrier to target CpG adjuvant to the antigen presenting cells in the draining lymph node. Dextran-CpG conjugate dramatically enhances the hydrodynamic size of CpG ODN, prevent it from rapidly diffusing into the systemic blood circulation, and retargeting it to the lymphatic capillaries. Targeting CpG to lymph nodes via dextran conjugate not only enhances the CD8+ T cell responses when combined with a protein antigen, but also improves the antitumor immunotherapy when simply mixed with a whole tumor cell vaccine. Our results suggest dextran conjugation might be a simple and effective strategy to improve many current vaccines.
EXPERIMENTAL PROCEDURES
Mice.
Animals were cared for in the USDA-inspected WSU Animal Facility under federal, state, local and NIH guidelines for animal care. Female C57BL/6 mice were purchased from Jackson Laboratory. Mice were 5 to 8 weeks of age at the onset of experiments.
Reagents and Cell Lines.
All DNA synthesis reagents including the MMT-Hexylaminolinkerphosphoramidite were purchased from Glen Research or Chemgenes and used according to the manufacturer’s instructions. Ovalbumin protein was purchased from Worthington Biochemical Corporation; dextran polymers were purchased from Sigma-Aldrich. Murine MHC class I tetramers were obtained from Beckman Coulter (Beckman Coulter, Inc., San Diego, CA). All other reagents were from Sigma-Aldrich and used as received except where otherwise noted. DNA was synthesized using an ABI 394 synthesizer on a 1.0 μmol scale. DNA were purified by a reverse phase HPLC using a C4 column (BioBasic-4, 200 mm × 4.6 mm, Thermo Scientific), 100 mM triethylamine−acetic acid buffer (TEAA, pH 7.5), methanol (0–30 min, 10–100%) as an eluent. HPLC was achieved using an Agilent 1100 chromatography system (Agilent Technologies, Santa Clara, CA, USA) with a variable-wavelength UV detector. TC-1 tumor cells were obtained from Dr. T.C. Wu of Johns Hopkins University. DC2.4 cell line was a gift from Dr. Z.W. Wei of Wayne State University. EG7 tumor cells were purchased from ATCC. All cells were cultured in RPMI media supplied with supplemented 5% FBS, and antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2.
Synthesis of Dextran-CpG Conjugate.
Dextran (from Leuconostoc app. Mr: 70k) was purchased from Sigma and was dialyzed against water before use. Dextran was deemed free of endotoxin using an assay with RawBlue cells. Dextran was oxidized into activated polysaccharides containing aldehyde groups in the presence of periodate in aqueous media. Briefly, 500 mg of dextran was dissolved in aqueous solution of sodium periodate (0.1 M, 25 mL). The reaction mixture was stirred in the dark at 4 °C for 6 h. The resulted solution was dialyzed (Spectra dialysis tubing MWCO 10K) against deionized H2O at 4 °C in the dark for 24 h. Finally, the aqueous solutions of oxidized dextran were lyophilized. 1 mg of 5′-amine-modified CpG (full PS backbone CpG 1826:5′-amine-tccatgacgttcctga-cgtt-3′) was then mixed with 100 mg of oxidized dextran, the reaction was agitated for 6 h before 0.05 M solution of sodium borohydride in 0.05 M borate buffer of pH 9.5 was added for 24 h at room temperature. The final conjugates were dialyzed against water and lyophilized. For fluorescent labeling, 3′-fluorescein CpG was used for the conjugation. The degree of conjugation was estimated by moles of CpG added/final product weight.
Agarose Gel Electrophoresis.
Each ODN sample (1 μg) was analyzed by electrophoresis for about 90 min, under constant voltage 75 V, through a 1% agarose gel in 1×TBE (tris(hydroxymethyl)aminomethane (Tris, 89 mM), ethylenediaminetetraacetic acid (EDTA, 2 mM), and boric acid (89 mM), pH 8.0) buffer. The DNA bands were visualized by UV illumination (312 nm) after ethidium bromide staining and photographed by a digital camera.
In Vitro Cellular Uptake and Distribution.
DC2.4 cells were pulsed with 100 nM fluorescein labeled CpG or Dextran-CpG for 2 h at 37 °C. Cells were washed and subsequently imaged and were also subjected to flow cytometry analysis to quantify the CpG uptake.
In Vitro TLR Reporter Assay.
Dextran, Dextran + CpG, Dextran-CpG, or free CpG (500 nM) was incubated for 24 h with 1 × 105 InvivoGen HEK-Blue murine TLR9 reporter cells, or with RAW-Blue cells which secreted embryonic alkaline phosphatase (SEAP) upon activation. SEAP levels were quantified by incubating the supernatant with Quanti-Blue substrate for 1 h and reading the absorption at 620 nm, following manufacturer’s instructions.
Lymph Node Imaging and Antigen Presenting Cell Uptake.
After injection of fluorescein-labeled probes, animals were sacrificed and inguinal and axillary LNs were excised and imaged using a digital camera. Lymph nodes were digested with 0.8 mg/mL Dispase and 0.2 mg/mL collagenase P (both from Roche) and 0.1 mg/mL DNase I (invitrogen). Cells were stained with antibodies against F4/80, CD11c, and analyzed by flow cytometry. Unless stated otherwise, data were presented as mean ± s.e.m.
Immunizations.
C57Bl/6 mice (5–8 weeks, 3–4 mice/ group) were vaccinated by a homologous prime-boost regimen; animals were primed on day 0 and boosted on day 14 (unless stated otherwise) with 10 μg OVA and 1.24 nmol CpG (in soluble or conjugation forms) suspended in PBS. Mice were typically injected subcutaneously at the tail base. The volume of all vaccine injections was 100 μL. In tumor cell vaccine, C57BL/6 mice were injected with 1 × 106 tumor cells and were treated with 2 injections of 1.24 nmol CpG + 10 μg of OVA. For whole tumor vaccine studies mice were treated with 1 × 106 freeze–thawed TC-1 tumor cells mixed with 1.24 nmol CpG formulations.
Flow Cytometry.
All antibodies were purchased from BD Pharmingen or ebioscience. The following primary antibodies were used: anti-CD16/CD32 (BD bioscience, Cat#: 553142, clone: 2.4G2), anti-CD8-APC (ebioscience, Cat#: 17–0081-83, clone: 53–6.7), anti-CD11c-PE (ebioscience, Cat#: 12–01184-02, clone: N418), anti-F4/80-APC (ebioscience, Cat#: 17–4801-82, clone: BM8). Flow data were acquired on an Attune focus flow cytometer (Life Technologies) and analyzed using Attune Cytometric Software.
Tetramer Staining.
Blood was collected and red blood cells were depleted by ACK lysing buffer. Cells were then blocked with Fc-blocker (anti-mouse CD16/CD32 monoclonal anti-body) and stained with phycoerythrin-labeled tetramers (Beckman Coulter) and anti-CD8-APC (ebioscience, Cat#: 17–0081-83, clone: RMUL.2) for 30 min at room temperature. Cells were washed twice, resuspended in FACS buffer (5 μg/ mL DAPI), and analyzed on an Attune Focus flow cytometer (Life Technology). Analysis typically gated on live, CD8+, tetramer positive live cells.
Tumor Inoculation and Tumor Therapy Experiments.
C57BL/6 mice (6–8 weeks) were anaesthetized and inoculated subcutaneously on the right hind flank with 3 × 105 TC-1 cells (a tumor cell line derived from primary lung epithelial cells of C57BL/6 mice and transformed with human papillomavirus 16 (HPV-16) E6/E7) or EG7 Cells (an OVA expressing murine T-cell lymphoma cell line). Tumors were allowed to establish for 6 days before the treatment. TC-1 tumor bearing mice were randomized into groups and were vaccinated on day 6 (1 × 106 killed TC-1 cells, 1.24 nmol CpG), day 13 (1 × 106 TC-1 cells, 1.24 nmol CpG). OVA expressed EG7 tumor bearing mice were treated on day 6 (10 μg OVA, 1.24 nmol CpG) and day 13 (10 μg OVA, 1.24 nmol CpG). Tumor sizes were measured every 1–2 days by electronic calipers and calculated as the product of 2 orthogonal diameters (D1 × D2).
Statistical Analysis.
Based on pilot immunization and tumor treatment studies, we used group sizes of 4 animals/group for immunogenicity measurements and at least 8 animals/group for tumor therapy experiments to obtain 80% power at the 95% confidence level to detect 30% differences in T-cell expansion or functionality. All plots show mean values and error bars represent the SEM. Comparisons of mean values of two groups were performed using unpaired Student’s t tests. One-way analysis of variance (ANOVA), followed by a Bonferroni post-test was used to compare >2 groups. *, p < 0.05; **, p < 0.01; ***, p < 0.001 unless otherwise indicated. Log-Rank test was performed on the Kaplan−Meier survival curves. Statistical analysis was performed using GraphPad Prism software (San Diego, CA).
ACKNOWLEDGMENTS
This work is supported in part by NIH (R56DK103651), American Cancer Society (11–053-01-IRG), and Wayne State University President’s Research Enhancement Program.
ABBREVIATIONS
- ODN
oligonucleotide
- LN
lymph node
- APC
antigen presenting cells
- DC
dendritic cells
- OVA
ovalbumin
- TLRs
Toll-like receptors
- PRRs
pattern-recognition receptors
- DNA
DNA
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Pulendran B, and Ahmed R (2011) Immunological mechanisms of vaccination. Nat. Immunol. 131, 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rappuoli R, Mandl CW, Black S, and De Gregorio E (2011) Vaccines for the twenty-first century society. Nat. Rev. Immunol 11, 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Scully T (2014) The Age of Vaccines. Nature 507, S2–S3. [DOI] [PubMed] [Google Scholar]
- (4).Berzofsky JA, Ahlers JD, and Belyakov IM (2001) Strategies for designing and optimizing new generation vaccines. Nat. Rev. Immunol 1, 209–219. [DOI] [PubMed] [Google Scholar]
- (5).Pardoll DM (2002) Spinning molecular immunology into successful immunotherapy. Nat. Rev. Immunol. 2, 227–238. [DOI] [PubMed] [Google Scholar]
- (6).Melief CJ, van Hall T, Arens R, Ossendorp F, and van der Burg SH (2015) Therapeutic cancer vaccines. J. Clin. Invest. 125, 3401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, and Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907. [DOI] [PubMed] [Google Scholar]
- (8).Dunn GP, Old LJ, and Schreiber RD (2004) The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360. [DOI] [PubMed] [Google Scholar]
- (9).van der Burg SH, Arens R, Ossendorp F, van Hall T, and Melief CJ (2016) Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 16, 219–33. [DOI] [PubMed] [Google Scholar]
- (10).Adams S (2009) Toll-like receptor agonists in cancer therapy. Immunotherapy 1, 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dowling JK, and Mansell A (2016) Toll-like receptors: the swiss army knife of immunity and vaccine development. Clin. Transl. Immunol 5 (5), e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bhardwaj N, Gnjatic S, and Sawhney NB (2010) TLR AGONISTS: Are They Good Adjuvants? Cancer J. 16, 382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).O’Hagan DT, and Fox CB (2015) New generation adjuvants–from empiricism to rational design. Vaccine 33, B14–B20. [DOI] [PubMed] [Google Scholar]
- (14).Liu H, and Irvine DJ (2015) Guiding principles in the design of molecular bioconjugates for vaccine applications. Bioconjugate Chem. 26, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Irvine DJ, Hanson MC, Rakhra K, and Tokatlian T (2015) Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev. 115, 11109–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liu H, Moynihan KD, Zheng YR, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, and Irvine DJ (2014) Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Engel AL, Holt GE, and Lu H (2011) The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Expert Rev. Clin. Pharmacol. 4, 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wu TY, Singh M, Miller AT, De Gregorio E, Doro F, D’Oro U, Skibinski DA, Mbow ML, Bufali S, Herman AE, et al. (2014) Rational design of small molecules as vaccine adjuvants. Sci. Transl. Med. 6, 263ra160. [DOI] [PubMed] [Google Scholar]
- (19).Hubbell JA, Thomas SN, and Swartz MA (2009) Materials engineering for immunomodulation. Nature 462, 449–460. [DOI] [PubMed] [Google Scholar]
- (20).Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, and Hubbell JA (2007) Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol 25, 1159–1164. [DOI] [PubMed] [Google Scholar]
- (21).Swartz MA, and Lund AW (2012) OPINION Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat. Rev. Cancer 12, 210–219. [DOI] [PubMed] [Google Scholar]
- (22).Johansen P., Mohana D., Martínez-Gómez JM., Kündig TM., and Gander B. (2010) Lympho-geographical concepts in vaccine delivery. J. Controlled Release 148, 56–62. [DOI] [PubMed] [Google Scholar]
- (23).Moon JJ, Huang B, and Irvine DJ (2012) Engineering nano- and microparticles to tune immunity. Adv. Mater. 24, 3724–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bachmann MF, and Jennings GT (2010) Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 10, 787–796. [DOI] [PubMed] [Google Scholar]
- (25).Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, and Bachmann MF (2008) Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 38, 1404–1413. [DOI] [PubMed] [Google Scholar]
- (26).Duncan R (2006) Polymer conjugates as anticancer nano-medicines. Nat. Rev. Cancer 6, 688–701. [DOI] [PubMed] [Google Scholar]
- (27).Knop K, Hoogenboom R, Fischer D, and Schubert US (2010) Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem., Int. Ed. 49, 6288–308. [DOI] [PubMed] [Google Scholar]
- (28).Noh YW, Kong SH, Choi DY, Park HS, Yang HK, Lee HJ, Kim HC, Kang KW, Sung MH, and Lim YT (2012) Near-infrared emitting polymer nanogels for efficient sentinel lymph node mapping. ACS Nano 6, 7820–7831. [DOI] [PubMed] [Google Scholar]
- (29).Bagby TR, Cai S, Duan S, Thati S, Aires DJ, and Forrest L (2012) Impact of Molecular Weight on Lymphatic Drainage of a Biopolymer-Based Imaging Agent. Pharmaceutics 4, 276–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nune SK, Gunda P, Majeti BK, Thallapally PK, and Forrest ML (2011) Advances in Lymphatic Imaging and Drug Delivery. Adv. Drug Delivery Rev. 63, 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kato M, Neil TK, Fearnley DB, McLellan AD, Vuckovic S, and Hart DNJ (2000) Expression of multilectin receptors and comparative FITC–dextran uptake by human dendritic cells. Int. Immunol. 12, 1511–1519. [DOI] [PubMed] [Google Scholar]
- (32).Apostolopoulos V, Thalhammer T, Tzakos AG, and Stojanovska L (2013) Targeting Antigens to Dendritic Cell Receptors for Vaccine Development. J. Drug Delivery 2013, 869718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tacken PJ, de Vries IJ, Torensma R, and Figdor CG (2007) Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 7, 790–802. [DOI] [PubMed] [Google Scholar]
- (34).Fernandez E, Faintuch B, Teodoro R, Wiecek D, Pirmettis I, Duatti A, and Pasqualini R (2009) Radiolabeling optimization of a cysteine-dextran lymph node seeking molecule using 99mTc-tricarbonyl core. J. Nucl. Med. 50, 1923–1923. [Google Scholar]
- (35).Marcinow AM, Hall N, Byrum E, Teknos TN, Old MO, and Agrawal A (2013) Use of a novel receptor-targeted (CD206) radiotracer, 99mTc-tilmanocept, and SPECT/CT for sentinel lymph node detection in oral cavity squamous cell carcinoma: Initial institutional report in an ongoing phase 3 study. JAMA otolaryngology-head & neck surgery 139, 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Brandley BK, and Schnaar RL (1986) Cell-surface carbohydrates in cell recognition and response. J. Leukoc Biol. 40, 97–111. [DOI] [PubMed] [Google Scholar]
- (37).Doh KO, and Yeo Y (2012) Application of polysaccharides for surface modification of nanomedicines. Ther. Delivery 3, 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ezekowitz RA, Williams DJ, Koziel H, Armstrong MY, Warner A, Richards FF, and Rose RM (1991) Uptake of Pneumocystis carinii mediated by the macrophage mannose receptor. Nature 351, 155–8. [DOI] [PubMed] [Google Scholar]
- (39).Haas T, Metzger J, Schmitz F, Heit A, Müller T, Latz E, and Wagner H (2008) The DNA sugar backbone 2′ deoxyribose determines toll-like receptor 9 activation. Immunity 28, 315–23. [DOI] [PubMed] [Google Scholar]
- (40).Heeg K, Dalpke A, Peter M, and Zimmermann S (2008) Structural requirements for uptake and recognition of CpG oligonucleotides. Int. J. Med. Microbiol. 298, 33–8. [DOI] [PubMed] [Google Scholar]
- (41).Lahoud MH, Ahmet F, Zhang JG, Meuter S, Policheni AN, Kitsoulis S, Lee CN, O’Keeffe M, Sullivan LC, Brooks AG, et al. (2012) DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 109, 16270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kandimalla ER, Bhagat L, Yu D, Cong Y, Tang J, and Agrawal S (2002) Conjugation of ligands at the 5′-end of CpG DNA affects immunostimulatory activity. Bioconjugate Chem. 13, 966–74. [DOI] [PubMed] [Google Scholar]
- (43).Tom JK, Mancini RJ, and Esser-Kahn AP (2013) Covalent Modification of Cell Surfaces with TLR Agonists Improves & Directs Immune Stimulation. Chem. Commun. 49, 9618–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Adams EW, Ratner DM, Seeberger PH, and Hacohen N (2008) Carbohydrate-mediated targeting of antigen to dendritic cells leads to enhanced presentation of antigen to T cells. ChemBioChem 9, 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lybaert L, Vanparijs N, Fierens K, Schuijs M, Nuhn L, Lambrecht BN, and De Geest BG (2016) A Generic Polymer-Protein Ligation Strategy for Vaccine Delivery. Biomacromolecules 17, 874–81. [DOI] [PubMed] [Google Scholar]
- (46).Slütter B, Soema PC, Ding Z, Verheul R, Hennink W, and Jiskoot W (2010) Conjugation of ovalbumin to trimethyl chitosan improves immunogenicity of the antigen. J. Controlled Release 143, 207–14. [DOI] [PubMed] [Google Scholar]
- (47).Tzianabos AO (2000) Polysaccharide Immunomodulators as Therapeutic Agents: Structural Aspects and Biologic Function. Clin. Microbiol. Rev. 13, 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Perdicchio M, Ilarregui JM, Verstege MI, Cornelissen LA, Schetters ST, Engels S, Ambrosini M, Kalay H, Veninga H, den Haan JM, et al. (2016) Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc. Natl. Acad. Sci. U. S. A. 113, 3329–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Carbone FR, and Bevan MJ (1990) Class I-restricted processing and presentation of exogenous cell-associated antigen in vivo. J. Exp. Med. 171, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Goldstein MJ, Varghese B, Brody JD, Rajapaksa R, Kohrt H, Czerwinski DK, Levy S, and Levy R (2011) A CpG-loaded tumor cell vaccine induces antitumor CD4+ T cells that are effective in adoptive therapy for large and established tumors. Blood 117, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chiang CL, Coukos G, and Kandalaft LE (2015) Whole Tumor Antigen Vaccines: Where Are We? Vaccines 3, 344–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August T, Pardoll DM, and Wu T-C (1996) Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 56, 21–26. [PubMed] [Google Scholar]
- (53).HogenEsch H (2013) Mechanism of Immunopotentiation and Safety of Aluminum Adjuvants. Front. Immunol. 3, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]