

SUMMARY
Androgen-receptor (AR) inhibitors, including enzalutamide, are used for treatment of all metastatic castration-resistant prostate cancers (mCRPCs). However, some patients develop resistance or never respond. We find that the transcription factor CREB5 confers enzalutamide resistance in an open reading frame (ORF) expression screen and in tumor xenografts. CREB5 overexpression is essential for an enzalutamide-resistant patient-derived organoid. In AR-expressing prostate cancer cells, CREB5 interactions enhance AR activity at a subset of promoters and enhancers upon enzalutamide treatment, including MYC and genes involved in the cell cycle. In mCRPC, we found recurrent amplification and overexpression of CREB5. Our observations identify CREB5 as one mechanism that drives resistance to AR antagonists in prostate cancers.
In Brief
Advanced prostate cancers develop resistance to androgen receptor (AR)-targeting therapies. Hwang et al. show that resistant prostate cancers overexpress or amplify CREB5, mediating resistance to AR inhibition. CREB5 suppression reduces the viability of therapy-resistant patient-derived models, suggesting that CREB5 is a target in prostate cancer patients with limited treatment options.
Graphical Abstract
INTRODUCTION
Androgen receptor (AR) regulates the expression of genes that specify the prostate lineage (Watson et al., 2015; Lonergan and Tindall, 2011) and promotes survival and proliferation of prostate cancer cells. Androgen deprivation therapy (ADT), including surgical or chemical castration, remains the mainstay for treating advanced prostate cancers (Watson et al., 2015). New AR-targeted therapies (ARTs), including enzalutamide, a potent competitive inhibitor of AR, or abiraterone, which suppresses androgens to below castration levels (Scher et al., 2012; Attard et al., 2009; Higano et al., 2015), provide additional approaches to inhibit AR signaling after failure of ADT alone (Higano et al., 2015). However, some patients never respond to hormonal therapies, and nearly all patients ultimately develop resistance to all AR-targeted agents (Watson et al., 2015; Bubley and Balk, 2017).
Prior studies have shown that aberrant activation of AR signaling is a common ADT resistance mechanism (Watson et al., 2015). Although rarely seen in primary tumors, amplifications and mutations of AR (Grasso et al., 2012; Robinson et al., 2015) or its enhancer (Takeda et al., 2018; Viswanathan et al., 2018) are frequently observed in metastatic castration-resistant prostate cancer (mCRPC). Re-activated AR signaling also drives resistance to ART, including abiraterone (Romanel et al., 2015) or enzalutamide (Montgomery et al., 2017; Wyatt et al., 2016). In addition, splice variants of AR that no longer require ligand activation drive resistance to ADT in pre-clinical models (Kregel et al., 2016) and are observed in mCRPC (Henzler et al., 2016; Antonarakis, 2016). Upregulation of AR targets via other mechanisms is also associated with ADT resistance. For example, glucocorticoid receptor (GR) signaling regulates the expression of AR targets, leading to enzalutamide resistance (Arora et al., 2013), whereas overexpression of the AR target gene UBE2C independently promotes castration resistance in vitro and in vitro (Wang et al., 2009).
Other signaling pathways are also associated with ADT resistance. The oncogene MYC is amplified in both primary castration-resistant prostate cancer and mCRPC (Cancer Genome Atlas Research Network, 2015; Grasso et al., 2012; Robinson et al., 2015), and overexpression drives castration resistance in prostate cancer cells (Bernard et al., 2003; Gao et al., 2013b). We recently found that MYC was focally amplified as an acquired genetic alteration in an abiraterone-resistant prostate cancer tumor (Han et al., 2017). Mutations of Wnt signaling regulators, including ones that perturb function of the tumor suppressor gene APC or activate CTNNB1 or the Wnt signal enhancer RSPO2, have been found in mCRPC patients (Grasso et al., 2012; Gundem et al., 2015; Robinson et al., 2015). However, alterations in AR, MYC, and Wnt signaling are not found in all mCRPC tumors, suggesting that other mechanisms also contribute to ADT resistance.
To identify other mechanisms that promote resistance to ADT, we performed a genome-scale open reading frame (ORF) screen in androgen-dependent prostate cancer cells exposed to enzalutamide. Integrating information derived from the genomics and transcriptomes of mCRPC samples, we identified the transcription factor CREB5 as a mediator of enzalutamide resistance.
RESULTS
Identification of Genes that Drive Enzalutamide Resistance
To discover genes that promote ADT resistance, we expressed 17,255 uniquely barcoded ORFs (Yang et al., 2011) in the AR-dependent cell line LNCaP. These cells proliferate in vitro in the presence of androgens but arrest under androgen-depleted conditions. We then cultured these cells in androgen-depleted medium (charcoal-stripped serum [CSS]) or in CSS with the AR inhibitor enzalutamide and identified genes that conferred the capacity to proliferate in each setting (Figure 1A). LNCaP cells cultured in androgen-replete medium with fetal bovine serum (FBS) served as a control. ORFs that conferred a proliferative advantage and exhibited enrichment at the end of the screen were considered candidate resistance genes. To compute the relative effects of ORFs, we determined the average barcode representation under each condition at 25 days and compared this with the average initial barcode representations immediately after puromycin selection. We ranked the relative enrichment of each ORF and defined hits as ORFs with a Z score greater than 3 (99.7th percentile). We found 51 hits in the CSS arm and 107 hits in the CSS + enzalutamide arm (Figure 1B; Table S1). The observed consequences of expressing specific ORFs were consistent in both the CSS and CSS + enzalutamide treatment arms (Pearson correlation [R2] = 0.962; Figure 1C), indicating that pathways that promote castration or enzalutamide resistance scored under both of these conditions.
Figure 1. An ORF Screen Identifies Genes that Promote Castration and Enzalutamide Resistance of LNCaP Cells.

(A) Schematic of the positive selection screen in LNCaP cells using an ORF library.
(B) Identification of hits in the CSS and CSS + enzalutamide experimental screening arms (Z score > 3, red dots). The average of three replicates is shown.
(C) Of the 17,255 ORFs, Z scores are displayed for experiments in the CSS (x axis) and CSS + enzalutamide (y axis) treatment arms. CREB5 and several other hits are highlighted (red dots), and the Pearson correlation (R2) score is shown.
(D) Confirming hits in the CSS + enzalutamide arm in an arrayed format. The average population doubling for LNCaP cells expressing each indicated ORF was determined after 14 days, and candidates that conferred significant ADT resistance (pink bars) relative to negative controls are shown (t test, p < 0.005). Green bars represent negative control ORFs (GFP, luciferase), and blue bars represent positive controls (mutant active CTNNB1 and LNCaP cells with genomic deletion of INPP5A). The 8 hits that ranked in the top 20 in both the pooled and arrayed format are displayed in red. * represents the rank of CREB5-overexpressing cells. Mean ± SEM of 8 replicates is depicted.
To validate these hits, we generated stable cell lines expressing the 107 candidates from the CSS + enzalutamide resistance arm in LNCaP cells and re-evaluated their relative resistance. Unlike the pooled screen, we first suppressed residual AR activity by treatment in CSS for 3 days prior to culturing in CSS + enzalutamide for 14 days. We found that overexpression of 56 of the 107 genes significantly promoted proliferation, as assessed by population doubling in CSS + enzalutamide compared with negative control cell lines (GFP, luciferase) (t test, p < 0.005; Figure 1D; Table S2). When we considered both the pooled screen and the arrayed format validation studies performed in enzalutamide, we identified 8 ORFs (CREB5, PHF23, FGF6, MECP2, CDK6, FGFR2, ALX1, and CDK4) that scored among the top 20 candidates in both experiments.
CREB5, Cell Cycle, and Fibroblast Growth Factor (FGF) Signaling Promote Resistance to Androgen Deprivation and Enzalutamide Treatment
Among the candidate genes, we recognized several regulators of the cell cycle and FGF signaling pathways. Specifically, the 56 ORFs that promoted robust enzalutamide resistance in the arrayed experiments (Figure 1D) included the cell cycle kinases CDK4 (rank 17) and CDK6 (rank 6) as well as the FGF signaling pathway effectors FGF6 (rank 3) and FGFR2 (rank 10) (Figure 1D). Of the pooled screen in CSS + enzalutamide, 3 of 3 CDK4 ORFs and 3 of 3 CDK6 ORFs exhibited Z scores above 3 (Table S1; average Z scores: CDK4 = 4.58, CDK6 = 4.13). These ORFs were also top hits in the CSS screen without enzalutamide (Table S1; average Z scores: CDK4 = 3.84, CDK6 = 3.70). These observations are in consonance with previous studies demonstrating that cell cycle (Comstock et al., 2013) and FGF (Bluemn et al., 2017) signaling are associated with ADT resistance.
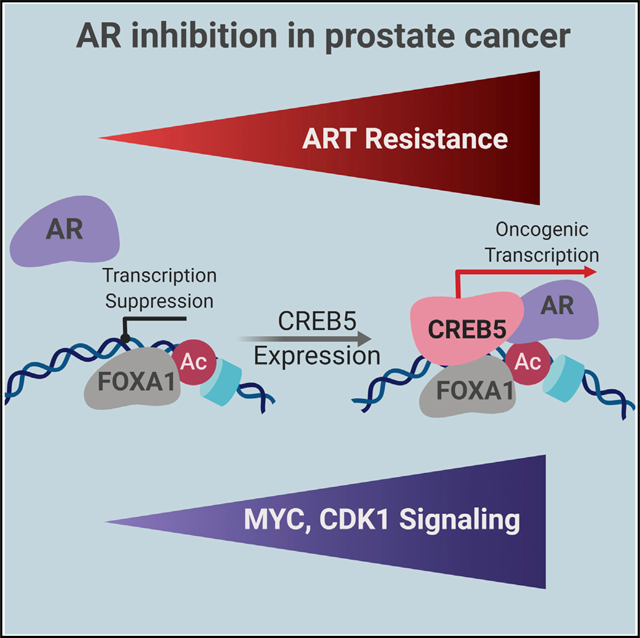
CREB5 was the strongest candidate in the pooled resistance screen in CSS (Z score = 12.7) as well as CSS + enzalutamide (Z score = 14.5) (Figure 1C) and ranked first in the arrayed resistance screen (Figure 1D). CREB5 overexpression did not enhance cell fitness under control culture conditions that included androgens (ZFCS = −1.33). CREB5 is a transcription factor (Nomura et al., 1993) overexpressed in colorectal and ovarian cancers (Qi and Ding, 2014; He et al., 2017) and is required for embryonic development in mice (Smith et al., 2018). Several CREB family members are transcriptional effectors of the protein kinase A (PKA) pathway and have been implicated in castration resistance (Deeble et al., 2007). However, CREB5 does not contain a homologous PKA-regulated activating phosphorylation site. In addition, although CREB5 conferred robust resistance, other ORFs in the PKA pathway failed to confer enzalutamide resistance (Table S1; average Z scores: PRKACA = −2.26, CREB1 = −0.52, CREB3 = −0.36). These findings suggest that CREB5 functions distinctly from other CREB family members in the context of androgen ablation.
CREB5 Enhances the Fitness of Prostate Cancer Cells upon Enzalutamide Treatment
To investigate the mechanism(s) by which CREB5 promotes enzalutamide resistance, we determined the consequences of CREB5 overexpression on other prostate cancer cells cultured in CSS and enzalutamide. We overexpressed CREB5 or luciferase in several prostate cancer cell lines, including LAPC4, 22RV1, PC3, DU145, and C4–2, and cultured the cells in androgen-depleted medium prior to exposure to CSS and enzalutamide. Enzalutamide treatment of the androgen-sensitive lines (LNCaP and LAPC4) led to cell death (Figure 2A). In contrast, CREB5 overexpression rescued cell death and promoted proliferation in both cell lines. We noted that CREB5 expression promoted proliferation in AR-negative DU145 (15%) and PC3 (40%) cells relative to cells expressing a control vector (Figure 2A). These studies showed that CREB5 promoted proliferation of cells under androgen-depleted conditions.
Figure 2. CREB5 Promotes Resistance to Enzalutamide.

(A) Relative proliferation (mean and SEM) of prostate cancer cell lines overexpressing CREB5 (colored bars) or luciferase (LUC) control (black or gray bars) in CSS + enzalutamide (t test, *p < 0.005). Mean ± SD of three replicates is shown. In the immunoblots below, V5 indicates expression of epitope-tagged CREB5 or luciferase. Tubulin was used as a loading control.
(B) The average experimental IC50 is shown for several therapeutic agents for luciferase-overexpressing (LUC, first column) and CREB5-overexpressing (second column) LNCaP cells. The IC50 ratio for these agents was also calculated (CREB5: luciferase cells, third column). Mean ± SD of three replicates is shown.
(C) LNCaP cells were treated with increasing doses of enzalutamide, apalutamide, or darolutamide, and the relative survival (mean and SD) at the indicated doses (left) and relative population doubling (right) under each condition are shown (t test, *p < 0.005). Mean ± SD of three replicates is shown.
(D) LNCaP cells expressing either CREB5 or luciferase (LUC) were implanted into immuno-deficient castrated mice. Tumor formation (volume > 100 mm3) was measured up to 56 days. The final measurement for each xenograft is plotted, as well as the average of all xenografts for luciferase- or CREB5-expressing cells. (Fisher’s exact test, p < 0.005; t test, p < 0.01).
(E) After tumors (250 mm3) were established, mice were castrated and treated with enzalutamide for 4 weeks. Left: the average tumor size is plotted for CREB5 or luciferase (LUC) cells at the start of castration (−7) and throughout the course of treatment (0–28 days) (t test, *p < 0.01). Right: the overall change of individual tumors is plotted, as well as the median change after 28 days of enzalutamide treatment.
(F) The viability in 3D culture was determined for NCI-PC44 prostate cancer organoids cultured in the indicated concentrations of enzalutamide. The mean ± SD of 12 replicates is shown.
(G) De novo infections were performed on NCI-PC44 organoids with RNAi targeting either control (shGFP, black) or CREB5 (3 shCREB5 hairpins, red), and viability was assayed after 14 days. The mean ± SD of 4 replicates is shown. The transcript levels of CREB5 2 days after infection were determined by qRT-PCR and are shown (t test, *p < 0.005).
CREB5 Reduces the Sensitivity of Prostate Cancer Cells to AR Inhibitors
We also examined whether CREB5 mediated resistance to additional AR inhibitors or other antineoplastic agents. We assayed for survival/proliferation of CREB5- or luciferase-expressing control cells after treatment with enzalutamide; other AR inhibitors (apalutamide and darolutamide); chemotherapeutic agents, including docetaxel and mitoxantrone; targeted agents, including the bromodomain inhibitor JQ1, the dual phosphatidylinositol 3-kinase (PI3K), and the mTOR inhibitor LY3023414 or the CDK4/6 inhibitor LEE011. 5-Fold CREB5 overexpression (Figure S2A) promoted enzalutamide resistance by 45-fold, apalutamide resistance by 26-fold, and darolutamide by 13-fold (Figure 2B). In contrast, CREB5 expression failed to affect the sensitivity of cells to treatment with docetaxel, mitoxantrone, JQ1, LY3023414, or LEE011 (Figure 2B; Figure S2B). We noted that treatment with the AR antagonists enzalutamide and apalutamide (10 μM) led to cell death, whereas darolutamide reduced population doubling by 5-fold. In each case, CREB5-overexpressing cells continued to proliferate (Figure 2C, right panels; t test, p < 0.005). We concluded that CREB5 promoted survival in the presence of AR inhibitors but did not promote survival for all therapeutic agents.
CREB5 Promotes Castration and Enzalutamide Resistance In Vivo
We next examined the effects of CREB5 overexpression on tumor growth. We implanted LNCaP cells expressing CREB5 or luciferase in castrated mice. 8 weeks after implantation, we found that CREB5-expressing cells formed larger tumors (Student’s t test, p < 0.01) at higher rates (Fisher’s exact test, p < 0.005) compared with cells expressing luciferase (Figure 2D). We also examined whether CREB5 tumor xenografts were resistant to castration in combination with enzalutamide. Specifically, we allowed LNCaP cells overexpressing CREB5 or luciferase to form tumors, castrated the mice, and then treated with enzalutamide for 4 weeks. CREB5-overexpressing tumors were resistant to castration and continued to grow during the 4 weeks of enzalutamide treatment, whereas the average volume of control tumors decreased during treatment (t test, p < 0.05 on days 14, 21, and 28; Figure 2E). These studies demonstrated that CREB5 overexpression conferred resistance to androgen deprivation and enzalutamide treatment in vivo.
CREB5 Expression Is Necessary for Viability of Patient-Derived Enzalutamide-Resistant Cancer Cells
To assess whether CREB5 mediates enzalutamide resistance in a patient-derived organoid model derived from a metastatic, enzalutamide-resistant prostate adenocarcinoma, we examined CREB5 amplification and expression in an NCI-PC44 organoid (Beshiri et al., 2018) and found that, although CREB5, AR, and MYC were not amplified, the expression of CREB5 was increased 14-fold.
We found that the NCI-PC44 organoid was resistant to enzalutamide (IC50 = 34.01 μM) because 30% of the cells remained viable in the presence of high doses of enzalutamide (100 μM; Figure 2F). To determine whether CREB5 overexpression was necessary for survival, we suppressed endogenous CREB5 expression in the NCI-PC44 organoid using 3 CREB5-targeting shRNAs (Figure S3). CREB5 expression was suppressed by 46%, 29%, and 44% by the three shRNAs, and over 2 weeks of culture, CREB5 suppression led to decreased viability of the NCI-PC44 organoid by 49%, 24%, and 55%, respectively (Figure 2G). We concluded that, in a CREB5-overexpressing patient-derived organoid that proliferates under typically effective doses of enzalutamide, suppression of CREB5 significantly reduced viability.
CREB5 Promotes the Expression of AR Target Genes
Reactivation of AR signaling is a common mechanism of ADT resistance. To determine whether AR signaling is necessary for CREB5-mediated resistance to enzalutamide, we suppressed AR in LNCaP cells that express either CREB5 or luciferase using 3 independent shRNAs. We confirmed that these shRNAs suppressed AR expression by ~75%. We found that the resistance of CREB5-expressing cells in CSS and enzalutamide was reduced by 50% upon AR suppression in the presence of enzalutamide (Figure 3A). This observation indicates that AR is necessary for the survival of CREB5-overexpressing cells.
Figure 3. AR Suppression Attenuates Enzalutamide Resistance, and CREB5 Promotes Transcription of AR Target Genes.

(A) Relative proliferation rates in CSS + enzalutamide without pre-treatment. Mean ± SD of 3 replicates is shown. 3 shRNAs specific for AR and one targeting GFP were introduced into either CREB5-expressing (red) or luciferase-expressing (black) cells. V5 indicates expression of epitope-tagged CREB5 or luciferase, and tubulin is a loading control.
(B) Of RNA obtained from CREB5 (red) or control cells (black), an AR target qPCR array was used with compare relative expression (mean and SEM) of androgen-sensitive genes for cells cultured for 4 days in CSS + enzalutamide. Gene expression is relative to control cells cultured for 4 days in fetal calf serum (FCS) (left). The overall change of the gene set is also shown (right, t test, p = 0.150). Mean ± SD of 3 replicates is shown.
(C) The expression (short exposure) and splicing (long exposure) of AR in CREB5- or luciferase-expressing cells in the presence or absence of ADT are depicted in immunoblots. 22RV1 cells endogenously express an N-terminal AR splice variant, serving as a positive control. Tubulin and HSP90 are used as loading controls.
(D) Assessment of nuclear AR levels by immunoblotting after cell fractionation. Laminin A/C is used as a loading control.
We then examined whether CREB5 regulated known AR transcriptional targets in enzalutamide-treated cells. We found that expression of 16 of the 43 AR target genes was more than 2-fold higher in CREB5-overexpressing cells compared with control cells treated with CSS and enzalutamide (Figure 3B). These transcripts included KLK3 (PSA), FKBP5, ORM1, HERC3, SPDEF, RHOU, SLC45A3, ABHD2, SNAI2, FAM105A, NKX3–1, KLK3, CAMKK2, ELL2, ENDOD1, and SP1. Our observations demonstrate that CREB5 increases the expression of a subset of AR-regulated transcripts upon enzalutamide treatment.
Although CREB5 overexpression increased the expression of several AR targets, CREB5 overexpression failed to lead to significant changes in AR gene expression or splicing (Figure 3C). In addition, although enzalutamide treatment led to a decrease in nuclear localization of AR (Figure 3D), CREB5 expression did not increase AR nuclear localization in the presence of enzalutamide (Figure 3D). These observations show that CREB5 promotes the transcription of a subset of AR target genes without directly altering AR expression or localization.
CREB5 Restores AR Binding Site Interactions Ablated by Enzalutamide
We hypothesized that CREB5 enhanced the ability of the low residual levels of nuclear AR to bind AR target sequences and promote transcription in the presence of enzalutamide. To determine whether CREB5 promoted AR binding to transcription regulatory sites, we performed AR and CREB5 chromatin immunoprecipitation sequencing (ChIP-seq) experiments using chromatin isolated from LNCaP cells expressing CREB5 or luciferase that were cultured in enzalutamide or vehicle control. We found that AR bound to 78,329 loci in luciferase-expressing cells based on a false discovery rate (FDR) q value threshold of 0.01 (Qin et al., 2016; Figure 4A, column 1). Upon enzalutamide treatment, AR binding at all 78,329 sites was reduced to below our threshold for statistical significance (Figure 4A, column 1 versus column 3). In untreated cells, CREB5 overexpression did not globally alter AR binding sites (Figure 4A, column 1 versus column 2). However, in enzalutamide-treated CREB5-overexpressing cells, we found significant AR binding at 32.5% (25,496) of the loci we examined (Figure 4A, column 4). We considered AR binding to be “rescued” by CREB5 at these sites. Furthermore, CREB5 overexpression promoted De novo AR binding at 4,508 sites that were not significant in luciferase-expressing cells even in the absence of enzalutamide treatment (Figure 4A, column 4). We considered AR binding at these sites to be “enhanced” by CREB5.
Figure 4. CREB5 Promotes Site-Specific AR Binding to Chromatin upon Enzalutamide Treatment to Regulate MYC and Cell Cycle Pathways.

(A) Heatmap for AR binding sites identified by ChIP-seq. All AR binding sites are shown for luciferase control (column 1) and CREB5 (column 2) cells without and with enzalutamide (columns 3 and 4). AR binding sites in all columns were sorted based on column 4 into not rescued, rescued, or enhanced.
(B) Heatmap for shared AR and V5-CREB5 binding sites identified by ChIP-seq. AR binding sites (maroon) are depicted for luciferase control (column 1) and CREB5 (column 2) cells without and with enzalutamide (columns 3 and 4). CREB5 binding sites (blue) are depicted for enzalutamide-treated luciferase control (column 5) and CREB5 (column 6) cells. Binding sites in all columns were sorted by the AR binding sites in column 4 into not rescued, rescued, or enhanced.
(C) The relative percentages of non-rescued, rescued, and enhanced AR binding sites are shown for all AR binding sites or shared binding sites of AR and V5-CREB5.
(D) Specific target genes regulate RB and cell cycle pathways. MYC is highlighted in green. Genes in red were regulated by a rescued as well as enhanced AR/CREB5 binding site.
(E) AR and CREB5 binding status at genomic loci upstream of CDK1 (enhancer, promoter) and MYC (promoter). H3K4-me2 status at these loci is used as a marker for active regions for transcription factor binding.
(F) GSEA analysis of RNA-seq data from CREB5 or luciferase cells treated with enzalutamide. The top 10 signatures are shown, along with their normalized p values and FDR q values.
(G) Individual enrichment profiles are shown. MYC is highlighted in green, and RB dominant-negative and cell cycle pathways are shown in red.
When we examined the binding of CREB5 under these same conditions, we observed that CREB5-bound genomic loci frequently overlapped with those that were AR bound (Figure 4B). In total, 46.3% (11,488 of 24,834) of the CREB5-bound sequences in enzalutamide-treated cells also contained AR binding sites. In control cells, enzalutamide treatment ablated AR binding at all of these 11,488 CREB5/AR-co-bound sequences (Figure 4B, column 1 versus column 3), whereas, in CREB5-overexpressing cells, AR was bound to 72.6% (8,399) of the CREB5-bound sequences even after enzalutamide treatment (Figure 4B, column 1 versus column 4). Specifically, CREB5 rescued AR binding at 55.7% (6,393) and enhanced AR binding at 16.9% (1,946) of the 11,488 CREB5/AR-co-bound loci after enzalutamide treatment (Figure 4C; Table S3). These observations demonstrate that overexpressed CREB5 itself bound and robustly promoted AR binding at a subset of AR target sites even after enzalutamide treatment.
CREB5 Promotes an AR Transcriptional Program Associated with Castration Resistance
To identify direct transcriptional targets of CREB5-mediated enzalutamide resistance, we identified transcripts that were differentially regulated and downstream of rescued or enhanced binding sites shared by CREB5 and AR in enzalutamide-treated cells (Figure 4B). In parallel to ChIP-seq experiments (Figures 4A and 4B), we also collected mRNA from luciferase- or CREB5-overexpressing cells treated with vehicle control or enzalutamide. Upon identifying differentially expressed transcripts in CREB5-overexpressing cells post-treatment, we used binding and transcription analysis (BETA) (Wang et al., 2013) to computationally integrate the ChIP-seq with the RNA-seq data to globally determine CREB5/AR direct target genes and the regulatory enhancer, promoter, or distal promoter binding sites of these target genes. We identified 183 upregulated genes and 210 downregulated target genes when comparing CREB5- with luciferase-overexpressing cells after treatment with enzalutamide (Table S4). This list included upregulation of the proto-oncogene MYC (Figures 4D and 4E), which has previously been associated with ADT resistance (Bernard et al., 2003; Gao et al., 2013b). Of the 393 target genes, 22 enriched transcripts mapped to the RB and cell cycle pathways (Figure 4D). The remainder of the CREB5/AR-regulated genes failed to coalesce into known signaling pathways.
To examine global transcriptional programs activated by CREB5 upon enzalutamide treatment, we used gene set enrichment analysis (GSEA) (Barbie et al., 2009; Subramanian et al., 2005) to identify enriched signatures in CREB5 cells after enzalutamide treatment. Upon unbiased examination of 189 oncogenic signatures, we confirmed that enzalutamide-resistant, CREB5-overexpressing cells were enriched in AR-targeted transcriptional pathways that were persistently active in mCRPC patients (Sharma et al., 2013). Specifically, we identified gene signatures that reflect oncogenic MYC activation (ranked third) as well as an activated cell cycle, including dominant-negative RB (ranked fourth) as well as E2F1 (ranked seventh; Figures 4F and 4G). These observations suggest that, in addition to directly regulating the expression of MYC and cell cycle genes, target genes of MYC and the cell cycle were coordinately regulated in enzalutamide-treated, CREB5-overexpressing cells.
FOXA1 Interacts at CREB/AR Binding Sites and Is Required for CREB5-Mediated Enzalutamide Resistance
To further understand how CREB5/AR mediates enzalutamide resistance, we performed motif enrichment analysis at CREB5/AR-bound enhancer and promoter sites using BETA (Wang et al., 2013). At sequences associated with both CREB5 rescued or enhanced genes and for both up- and downregulated target genes, we found a significant enrichment in forkhead domain protein motifs, including the pioneering factor FOXA1 (Figure 5A).
Figure 5. FOXA1 Is Necessary for CREB5-Mediated Enzalutamide Resistance.

(A) Motif enrichment analysis was performed on AR/CREB5-co-bound sequences.
(B) FOXA1 ChIP-seq demonstrated FOXA1 binding at the 11,484 CREB5/AR binding sites in luciferase control or CREB5-overexpressing cells with or without enzalutamide treatment.
(C) FOXA1 binding sites are categorized based on CREB5/AR not rescued (black), rescued (teal), and enhanced (yellow). The integrated binding signal is shown relative to all FOXA1 binding sites (navy).
(D) FOXA1 interactions at binding sites upstream of CDK1 and MYC.
(E) The relative proliferation of cells in CSS + enzalutamide without pre-treatment. FOXA1 was suppressed using two shRNAs in either CREB5-overexpressing (red) or luciferase-overexpressing (black) LNCaP cells. The mean ± SD of three replicates is shown. Immunoblots depict the levels of FOXA1, V5-luciferase, and V5-CREB5. Tubulin is used as a loading control.
We previously demonstrated that overexpression of FOXA1 in prostate epithelial cells promoted AR interactions at AR binding sites and transcription of AR target genes (Pomerantz et al., 2015). To confirm FOXA1 binding status at the CREB5/AR co-bound sites, we performed FOXA1 ChIP-seq experiments on chromatin isolated from either control luciferase- or CREB5-overexpressing cells. Although AR binding at CREB5/AR sites was ablated by enzalutamide in control cells (Figure 4A), enzalutamide treatment (columns 1 and 2 versus columns 3 and 4) led to modest reductions in FOXA1 interactions at individual sites (Figure 5B) and overall binding (Figure 5C) with chromatin in control cells. At these same sites, CREB5 overexpression (column 1 versus column 2 and column 3 versus column 4) led to moderate increases in FOXA1 binding at individual binding sites (Figure 5B) and overall (Figure 5C) after enzalutamide treatment. Relative to “non-rescued” sites, FOXA1 binding at rescued or enhanced sites was 2-fold greater at individual binding sites (Figure 5B) and overall (Figure 5C) after enzalutamide treatment. We also confirmed that FOXA1 bound to cis-regulatory elements upstream of CDK1 and MYC (Figure 5D), demonstrating that FOXA1 interacted at CREB5/AR-co-bound sites that were associated with increased AR target gene expression.
We speculated that CREB5 may require FOXA1 to promote enzalutamide resistance. To test this possibility, we suppressed FOXA1 by 2 distinct shRNAs in either control or CREB5-overexpressing cells and cultured the cells in CSS + enzalutamide. We found that suppression of FOXA1 by 75% led to a 60% reduction of CREB5-mediated enzalutamide resistance (Figure 5E). These observations confirmed that FOXA1 was also bound to CREB5/AR binding sites and that FOXA1 binding was necessary for CREB5-mediated enzalutamide resistance. However, we also observed that, unlike AR binding, enzalutamide treatment does not ablate FOXA1 binding.
Global Chromatin Structure Is Not Altered by Enzalutamide Treatment
To examine the mechanisms by which CREB5 binding induced transcriptional changes, we determined whether CREB5 binding promoted structural changes that primed these genomic loci for transcriptional activation using ATAC-seq (Buenrostro et al., 2015) paired with ChIP-seq on acetylated H3K27 marks derived from luciferase control- or CREB5-overexpressing cells with or without enzalutamide treatment. At the CREB5/AR-co-bound sites upstream of CDK1 and MYC, we observed that CREB5 binding was coordinately aligned to accessible regions based on ATAC-seq results, and these binding sites were flanked by acetylated H3K27 marks (Figure S4A). However, we found that enzalutamide treatment failed to consistently deplete histone marks or change accessibility (Figure S4A).
We also evaluated cumulative structural changes at the 24,838 CREB5-bound sites by integrating peak signals of accessibility (ATAC-seq) and acetylation marks (H3K27 Ac ChIP-seq). Although enzalutamide treatment globally ablated AR binding at the time points tested (Figure 4A), this treatment failed to affect the overall transcription factor accessibility and landscape of active transcriptional marks (Figure S4B). Thus, we found that CREB5 overexpression only modestly increased transcription factor accessibility and transcriptional activation marks after enzalutamide treatment (Figure S4B). These observations support a mechanism whereby CREB5 mediates resistance by promoting AR binding at accessible binding sites primed for transcriptional activity.
CREB5 Amplification in Primary Prostate Cancer and mCRPC
To determine whether CREB5 was altered in mCRPC, we examined copy number alterations present in prostate cancer samples from 17 reported prostate cancer studies (Gao et al., 2013a). We found that up to 26.3% of metastatic prostate cancers (MPC Project) harbored CREB5 amplifications (Figure S5A). In these studies, 88 of 4,406 samples harbored amplification, whereas only 1 sample harbored a deep deletion of CREB5.
To confirm this analysis, we used a second analytical approach, GISTIC2, to identify the gene targets of recurrent copy number gain (Mermel et al., 2011). TCGA used GISTIC2 analysis to identify focal amplifications and deletions in 492 primary prostate tumors (Harvard, 2016). In this analysis, the putative castration resistance genes AR (Xq12), MYC (8q24.21), and CDK6 (7q22.1) were focally amplified, whereas PTEN (10q23.31) was focally deleted (Figure S5B). We found that CREB5 (7p14.3, q value = 0.02772) was one of the 6 enzalutamide resistance screen hits with significant amplifications in prostate tumors (Figure S5C). The 7p14.3 focal peak contained 169 genes, including HOXA1, a gene that scored in both resistance screens but promoted less resistance relative to CREB5 (Figure 1D; Table S1).
To further define the amplification frequency of CREB5, we used FACETs to account for variable tumor purity and ploidy in each sample and to unify calling of copy number changes across distinct WES studies (Shen and Seshan, 2016). This approach allowed us to identify the relative amount of each allele in each sample and then to detect gene amplification or deletion specific to each tumor allele. We determined allele-level copy number changes in 854 prostate cancer tumor samples (326 mCRPC and 528 primary) (Armenia et al., 2018). In this analysis, we examined the relative rates of mono- and bi-allelic amplification as well as heterozygous and homozygous loss of the 56 enzalutamide resistance candidate genes (Figure 6A). We did not observe any cases in which putative negative regulatory genes (PTEN and RB1) were amplified. The frequency of amplifications and deletions for the 56 candidate genes and putative castration resistance genes such as MYC, CDK6, and AR are shown in Figure 6B. Upon visualizing relative enzalutamide resistance (Z score among hits) as a function of allelic amplification and deletion rates in mCRPC, we noted that CREB5 was the most robust hit, based on both functional and genomic analyses (Figure 6B).
Figure 6. CREB5 Is Amplified in Prostate Cancer.

(A) FACET analysis of candidate amplification and deletion status in primary prostate cancer and mCRPC. The top four rows represent the alteration status of the putative prostate cancer genes RB1, PTEN (blue *), MYC, and AR (red *). Primary prostate cancer (left, n = 528) and mCRPC (right, n = 326) were analyzed. CREB5 alterations (black *) are shown. Dotted lines represent the average amplification or deletion rate for the mapped genome. Note that AR amplifications or any copy number alterations on chromosome X or Y are always mono-allelic.
(B) Net allelic changes in mCRPC of 56 candidate genes (percentage of tumor alleles with amplification of a gene subtracted by percentage of tumor alleles with deletion) are plotted against Z scores in CSS + enzalutamide.
(C) Genomic alteration frequencies of the resistance genes AR, MYC, CDK6, PTEN, and RB1 are shown (labeled above) in mCRPC and primary prostate cancer. CREB5 (red box) and another top hit, ETV5, are shown.
(D and E) The post-tumor purity- and ploidy-adjusted amplification frequencies for hits as well as putative regulatory mechanisms are compared between primary prostate cancer (y axis) and mCRPC (x axis); rates for CREB5 (red) and putative castration resistance-regulatory genes (navy) are shown (D). Results for bi-allelic amplification are also depicted (E).
Although these samples were not patient matched, we found that the cohort of ADT treated tumors harbored an increased frequency of CREB5 amplifications (43%) relative to primary prostate cancer (26%; Figure 6C). We noted that CREB5 and another top candidate gene, ETV5, showed high rates of genomic amplification and low rates of deletion, analogous to what was observed for MYC and CDK6 (Figure 6C), which exhibited similar frequencies of increased amplification in mCRPC relative to primary prostate cancer.
Because mono-allelic changes resulting from genomic instability often occur in mCRPC, we also examined bi-allelic gains and homozygous deletions of CREB5. We observed that 1.7% of primary prostate cancers harbored biallelic amplification and that 3.4% of mCRPC harbored bi-allelic amplifications of CREB5 (Figure 6C). Only 1 of 326 mCRPC samples had homozygous deletions of CREB5. In contrast, PTEN and RB1 homozygous deletions were frequent, but bi-allelic gains were not detected. We also examined the relative overall amplification rates (Figure 6D) and bi-allelic amplification rates (Figure 6E) for genes, including CREB5, MYC, CDK4/6, and the 55 other enzalutamide-resistance candidates identified in our screen (Figure 1D). We found that amplification of candidate genes (55 of 56) (Figure 6D) and bi-allelic amplifications (47 of 56) (Figure 6E) were enriched in mCRPCs. Overall, CREB5 amplifications were enriched by 1.8-fold, and bi-allelic CREB5 amplifications were increased by 2.0-fold. In this analysis, the pattern of AR dysregulation was unique compared with all resistance genes we examined. AR amplifications (mono-allelic on chromosome X) were almost exclusive to mCRPC (37%) relative to primary prostate cancer (0.4%), whereas amplifications of other resistance genes were detected in primary prostate cancers but enriched in mCRPC.
These genomic analyses show that CREB5 amplifications occur in primary prostate cancers and at higher frequency in mCRPC. In addition, relative to other enzalutamide resistance candidates, CREB5 was amplified at high rates and deleted at low rates.
CREB5 Overexpression in mCRPC and Prostate Cancer
We also examined whether CREB5 was overexpressed in mCRPC. We examined the RNA sequencing (RNA-seq) dataset from the Stand Up 2 Cancer/Prostate Cancer Foundation (SU2C/PCF) mCRPC cohort (n = 238) (Armenia et al., 2018) by normalizing transcripts per million (TPM) to the median expression values in each sample. Upon examining the overexpression frequency for 14,876 gene transcripts detected in the 238 mCRPCs, the median gene was overexpressed in only 1.7% (Figure 7A). CREB5 (24.7%) was more frequently overexpressed than 99.5% of all gene transcripts (Z score = 4.15; Figure 7A). Because 169 genes reside within the CREB5-inclusive 7p14.3 focal peak predicted by GISTIC2 (Harvard, 2016), we analyzed the expression of 76 of 169 genes expressed in mCRPC, of which 45 of the 76 genes were overexpressed above median rates (Figure 7B). CREB5 was overexpressed at the second-highest frequency (Figure 7B). We found that CREB5 was overexpressed by 5-fold in 24.7%, 10-fold in 15.1%, and 20-fold in 7.1% of mCRPCs (Figure 7C), more than what we observed for MYC and CDK6.
Figure 7. CREB5 Is Overexpressed in Prostate Cancer.

(A) Frequencies by which 14,876 genes were overexpressed 5-fold were determined in 238 mCRPC. The frequencies of overexpression for the median gene (1.7%, dotted red line), ERG (30.3%), and CREB5 (24.7%) are depicted.
(B) Frequency of 5-fold overexpression of CREB5 and 75 other genes in the 7p14.3 focal peak is depicted as open circles. CREB5 is highlighted (red circle).
(C) Expression levels of MYC, CREB5, and CDK6 in 238 mCRPC (TPM divided by median expression). The dotted lines represent 5-fold, 10-fold, and 20-fold expression. The table summarizes the percentage of tumors that express the genes at these respective levels.
(D) Relative expression of CREB5 and AR plotted (left) in TPM. Two well-correlated AR target genes, KLK2 and KLK3 (PSA), are shown on the right. Axes are labeled with TPM values.
(E) The relapse rate is plotted for prostate cancer tumors with amplified or overexpressed (Z score > 2) CREB5 (red) or AR (blue) relative to all other tumors.
We found that overexpression of MYC, CDK6, or CREB5 (Figure S5D) also occurred in mCRPC samples lacking amplifications of these genes. We note that CREB5 transcript upregulation occurs through promoter hypomethylation in colorectal cancers (Molnár et al., 2018) and loss of a negative regulatory miRNA in triple-negative breast cancers (Bhardwaj et al., 2017). Given the co-regulatory transcription functions of AR and CREB5 (Figure 4), we examined whether CREB5 or AR regulated the expression of each other in mCRPC. We found that AR and CREB5 expression were independent (Figure 7D; R = 0.03, Pearson’s correlation). Overall, these findings indicate that in some mCRPC samples CREB5 is amplified and that in others, CREB5 is overexpressed without amplification.
We examined whether CREB5 overexpression was associated with patient outcome. We analyzed the MSKCC primary prostate cancer cohort (Taylor et al., 2010) and found that patients whose tumors exhibited high CREB5 expression (Z score > 2) showed high relapse rates after treatment of primary tumors (6 of 6 versus 17 of 78 in the control group) (Figure 7E). These observations demonstrate that CREB5 overexpression is associated with high rates of relapse of primary prostate tumors.
DISCUSSION
Resistance to androgen deprivation and AR-targeted therapies is often associated with reactivation of AR signaling; however, it is clear that other mechanisms also drive resistance to therapy in mCRPC. We identified CREB5 overexpression as an enzalutamide resistance mechanism that selectively reactivates targets of AR signaling in prostate cancer cells and mCRPC. CREB5 was also the top candidate in our androgen deprivation screen (Figure 1B, CSS, no enzalutamide), and overexpression of CREB5 enhanced the tumorigenicity of LNCaP cells in castrated mice (Figure 2D), suggesting that CREB5 broadly confers resistance to androgen ablation therapies.
7p amplifications (Robinson et al., 2015) and 7p14.3 focal amplifications (Harvard, 2016) have been reported previously in mCRPC and prostate cancers. Our observations indicate that CREB5 is a target of these recurrent amplification events. However, we also found that CREB5 overexpression frequently occurred in the absence of genomic gains (Figure S5D), suggesting that CREB5 overexpression occurs through both copy number gain and increased gene expression. Further studies of patient-matched post-enzalutamide or ADT-treated patients will be needed to determine whether CREB5 amplifications or overexpression are specifically enriched in post-enzalutamide or ADT-treated samples.
CREB5 overexpression represents a resistance mechanism in prostate adenocarcinomas with low AR levels. In the presence of enzalutamide, CREB5- and AR regulated transcription of putative AR target genes, including MYC and numerous cell cycle genes such as CDK1 and E2F2 (Figure 4D). Although AR signaling targets and AR itself were essential for CREB5-mediated resistance (Figures 3A and 3B), CREB5 did not increase the levels of nuclear AR or expression of N-terminal AR splice variants after enzalutamide (Figures 3C and 3D). In clinical mCRPC samples. CREB5 and AR expression also exhibited a limited correlation in mCRPC (Figure 7D). These observations suggest that CREB5 regulates putative AR target genes without directly altering AR expression.
Although CREB5 re-activated AR signaling to promote enzalutamide resistance, we found that CREB5 failed to rescue AR binding at all AR binding sites (Figure 4A) and only significantly enhanced expression of a subset of known AR target genes (Figure 3B). This observation indicates that a comprehensive rescue of the AR transcriptional program was not necessary for enzalutamide resistance. Similarly, signaling by the GR (Arora et al., 2013) and N-terminal splice variants of AR (Kregel et al., 2016) have also been reported to increase expression of a subset of AR target genes in driving resistance. Given that tumors resistant to AR-targeted therapies demonstrate continued AR transcriptional activity (Montgomery et al., 2017), determining how aberrant expression of these “core” resistance target genes downstream of AR altogether mechanistically regulate resistance to enzalutamide is important.
Although less pronounced, CREB5 also enhanced proliferation of AR-negative PC3 and DU145 prostate cancer cells (Figure 2A) and is amplified in some neuroendocrine tumors that do not rely on AR signaling (Beltran et al., 2016). CREB5 signaling may also promote the fitness of AR-negative prostate adenocarcinomas as well as neuroendocrine-like prostate cancers. CREB5 expression has been observed in normal prostate tissue and is observed at much higher levels in the gallbladder, brain, and adipose tissue (Fagerberg et al., 2014). CREB5 has also been reported to be upregulated in colorectal and ovarian cancers (Qi and Ding, 2014; He et al., 2017), and examination of 32 pan cancer datasets (TCGA) identified recurrent CREB5 amplification and overexpression in more than 10% of kidney cancers, sarcomas, lymphomas, lung adenocarcinomas, as well as glioblastomas and gliomas (Figure S6). In prostate cancers, other CREB family members previously implicated in castration resistance (Deeble et al., 2007) and neuroendocrine differentiation (Zhang et al., 2018; Sang et al., 2016) failed to score in our screens (Table S1). CREB5 has limited homology with these family members and heterodimerizes with other CREB proteins or c-Jun in vitro (Nomura et al., 1993). These CREB5 interactions have not been studied in mCRPC cells. Our studies indicate that amplification or overexpression of CREB5 in mCRPC may subvert aspects of normal CREB5 function to enhance AR function under conditions where AR is inhibited.
The pioneering factor FOXA1 regulates AR binding (Jin et al., 2014) and, along with HOXB13, reprograms the AR cistrome in normal prostate tissue, similar to what is observed in prostate cancer (Pomerantz et al., 2015). Our observations implicate FOXA1 in enzalutamide resistance (Figure 5). Prior work has demonstrated that FOXA1 promotes AR binding at low-affinity sites (Jin et al., 2014), and we demonstrated previously that FOXA1 overexpression with HOXB13 regulates expression of AR target genes. Here we found that CREB5 required FOXA1 to promote resistance to enzalutamide. In addition, AR binding in enzalutamide was greater at CREB5 binding sites with relatively higher levels of FOXA1 binding. Outside of CREB5 and FOXA1, characterization of additional chromatin or transcription regulators that mediate AR binding would further elucidate mechanisms of enzalutamide resistance. The positive and negative co-regulators at these sites will inform insights to AR signaling and could act as key targets in prostate cancer resistance to androgen therapies.
Genes involved in the cell cycle (CDK4, CDK6, and CCND3) were among the top hits in the ORF screen (Figure 1D), and cell cycle deregulation is seen in enzalutamide-resistant patients (Han et al., 2017). We also found that CREB5 promoted differential expression of 22 cell cycle regulators, including CDK1, E2F2, BUB1, and BUB1B (Figure 4D). Clinical trials combining CDK4/6 inhibitors with ADT in metastatic prostate cancers are ongoing (NCT numbers and ). Our observations support the rationale of targeting G1/S kinases (CDK4/6) in advanced or metastatic prostate cancer and also suggest that resistance will be associated with G2/M cell cycle regulators (CDK1/2).
In summary, these studies identify CREB5 as a regulator of AR signaling in prostate cancer cells that promotes enzalutamide resistance in vitro and in vivo. These observations underscore the key role of AR signaling in prostate cancer and that mechanisms that upregulate AR-driven transcription dominate clinical resistance. Modulating CREB5 or other castration resistance transcription factors by degrader or epigenetic strategies is an attractive therapeutic approach.
STAR⋆METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by William Hahn at William_Hahn@dfci.harvard.edu. This study did not generate new unique reagents and all reagents are publicly available.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
We utilize xenograft mouse models to study the response of prostate tumors toward castration or inhibition of AR-signaling. LNCaP cells were tested for several strains of potential mycoplasma or mouse virus contamination using “Mouse/Rat Comprehensive CLEAR Panel w/ C.bovis” from Charles River Laboratories.
Mouse model selection
All procedures were performed under the IACUC protocol 03–013 at Dana-Farber Cancer Institute. Male BALB/C immune-deficient mice that were homozygous for Foxn1nu mutation were used. At the time of cell implantation, the mice were between 6 to 8 weeks of age and between 14~20 g. We grafted cells subcutaneously at 20 sites to interrogate the effect of gene expression or treatments on tumor growth when factoring the 30~50% rate of successful tumor formation.
Human subjects
Patient data from previous studies were used in this study. For genes of interest, cancer patient whole exomes and transcripts levels were analyzed based on datasets in cBioPortal (Gao et al., 2013a) or our previous study of prostate cancer patients (Armenia et al., 2018).
Cell lines
Prostate cancer cell lines used in this study were purchased directly from ATCC, who maintains authenticated cell lines by sequencing and comparing Short Tandem Repeats (STR) to parental LNCaP cells in their database. Identity of all other prostate cancer cell lines (LAPC4, PC3, DU145, 22RV1, C4–2) were also confirmed in this manner. Each of these cell lines was cultured in phenol red free RPMI1640, 10% Fetal Calf Serum (FCS) prior to experimental treatments.
Primary cell cultures
NCI-PC44 organoids is an AR-positive adenocarcinoma derived from an enzalutamide-resistant prostate cancer patient as previous described (Beshiri et al., 2018). These were cultured in modified prostate-specific culture medium with advanced DMEM/F12 that includes EGF (50 μg/ml), Noggin (100 μg/ml) and R-Spondin (500 μg/ml).
METHOD DETAILS
Gene overexpression and suppression and experimental confirmation
To perform gene perturbation techniques with reproducibility, we adapted tools and protocols that are publicly available. To assay for direct and indirect effects upon gene induction or suppression, we utilized traditional biochemical approaches. All results were observed 3 or more times and the average or representative experiments are presented.
Lentiviral transduction of cells
Broad Institute Genetic Perturbation Platform protocols (https://portals.broadinstitute.org/gpp/public/resources/protocols) were followed to produce lentivirus particles containing open reading frames (ORFs) of screen hits or control(s) (ORF identification numbers are summarized in Table S1). The number of viral particles were optimized to infect cells at a multiplicity of infection (MOI) of less than 1. Viral particles were next used to infect LNCaP or other prostate cancer cell lines, and cells were continuously cultured in 1 μg/ml puromycin 3 days post-infection. For each resulting cell line, media were refreshed every 2 to 3 days, and cells were split into new flasks after reaching 80% confluency.
RNAi experiments
Several pLKO vectors expressing shRNAs targeting GFP were screened for off-target effects through infection of LNCaP cells. TRCN0000231753 did not induce changes in cell proliferation post infection and selection compared to non-infected cells and was used as a control infection for experiments. Nine CREB5-targeting shRNAs were screened (Figure S3), and shCREB5 #6 was utilized to suppress CREB5 expression in NCI-PC44 cells at viral titers that did not impact baseline viability when comparing shGFP infected cells to none-infected controls (NIC). Lentiviruses expressing shRNAs for AR or FOXA1 were used to infect LNCaP or NCI-PC44 cells. Between 48 and 72 hours post infection protein lysates or RNA were collected to determine extent of suppression. After confirming suppression, respective cells were counted and directly seeded for proliferation experiments using control media or media supplemented with CSS and enzalutamide. All shRNA constructs were acquired from the Broad Institute Genetic Perturbation Platform (https://portals.broadinstitute.org/gpp/public/).
qPCR, qPCR array and RNA-seq experiments
For transcript expression analysis, RNA was collected from the cells using RNeasy plus kit (74104, QIAGEN). 1 μg of RNA from respective samples was used to obtain cDNA through reverse transcription using iSCRIPT (1708841, BioRad). After a 1:1000 dilution, cDNA was mixed with 3 μL of primer at 100nM and 6 μL of Power SYBR Green PCR master mix (4368708, Life Technologies). RT profiler kits were used for the AR qPCR array (330231, QIAGEN), and 1:1000 fold diluted cDNA was mixed with RT SYBR green mix (330503, QIAGEN) at a 1:1 ratio. For qPCR, experimental triplicates were performed 3 times using a CFX384 C1000Touch Thermo cycler (BioRad), 45 cycles at 95°C for 1 minute, 50°C for 1 minute and 72°C for 1 minute. Relative threshold cycles (Ct) of each sample were compared to the average expression of internal control genes B2M and RPLPO for the qPCR array and RPLPO expression for other qPCR experiments. These values were then compared to vehicle control treatments. Relative CREB5 overexpression in LNCaP was 4~6-fold, which was determined utilizing two sets of CREB5 targeting primers (Figure S2A; Table S5). For the qPCR array, only the 43 genes that were downregulated by enzalutamide by 2-fold or more in LNCaP cells were considered AR sensitive and further analyzed (Figure 4B). For RNA-seq experiments, LNCaP cells expressing either luciferase or CREB5 were cultured for 4 days in enzalutamide or DMSO control. For RNA-seq experiments, library preparations, quality control and sequencing on a HiSeq2500 (Illumina) were performed by the Dana-Farber Molecular Biology core facility. Reads were aligned with Tophat 2.0.2 (Trapnell et al., 2010) using the human (hg19) transcriptome and genome annotation from the UCSC genome browser (Kent et al., 2002). Transcript abundance and differential expression between samples were computed using Cufflinks 2.0.2 module (Trapnell et al., 2010) with default setting on GenePattern (Reich et al., 2006).
Immunoblotting
To assay for direct or indirect effects of CREB5 on proteins of interest, cell pellets were collected in 4°C PBS, and cell lysates were collected at 4°C in RIPA lysis buffer (Cell Signaling Technology, 9806S) supplemented with protease and phosphatase inhibitors (Sigma Aldrich, 11836170001, 04906837001). Protein concentration was assayed (Thermo Fisher Scientific PI23225). Up to 10 μg of lysate from each sample was loaded onto NuPAGE Bis-Tris gels (Thermo Fisher Scientific), and gel electrophoresis was subsequently performed in MOPS running buffer (Thermo Fisher Scientific, NP0001). The proteins were subsequently transferred onto nitrocellulose membranes using iBlot apparatus (IB23001). After blocking with Li-Cor blocking buffer (Fisher, NC9877369), primary antibodies were incubated overnight after dilution in the blocking buffer, and anti-mouse (Li-Cor, 926–32210) or anti-rabbit (926–68021) secondary antibodies were subsequently used to image protein expression. Primary antibodies utilized include V5 (1:2000 dilution, Cell Signaling 13202S), Tubulin (1:10,000 dilution, Sigma Aldrich, T9026), AR (1:3000 dilution, Cell Signaling, R96025), HSP90 (1:3000 dilution, Cell Signaling, 4875S) and Lamin A/C (1:1000 dilution, Cell Signaling, 2032S).
Functional screens
In collaboration with the Broad Institute Genetic Perturbation Platform, we obtained lentiviral libraries to examine, at large-scale, genes that regulate prostate cancer cell viability.
Genome-Scale ORF screen
LNCaP cells were used in the screen, as they exhibited the highest AR-dependency based on Project Achilles analysis (Figure S1A). We also determined that 2.5 μM of enzalutamide (Selleckchem, S1250) optimally suppressed expression of AR target genes, including in LNCaP cells stimulated by the androgen analog R1881 (Figure S1B). After infection of the cells with a pooled ORF library (Yang et al., 2011), the cells were subsequently selected with puromycin, and at this point cells were saved to determine initial bar code representation. After 6 days of puromycin selection, 20 million cells were seeded in triplicates and cultured for 25 days in 3 conditions: control (FCS), low-androgen media (CSS) or CSS and enzalutamide. To prevent contact inhibition, we divided the 20 million cells into three separate T175 culture flasks, and 20 million cells were re-plated into fresh flasks as soon as they reached 80% confluency. These counts were used to estimate population doublings in each experimental arm. Otherwise the culture media were refreshed every 3 days. In the screen, proliferation of ORF-infected cell lines were compared to those infected with a GFP control lentivirus (Figure S1C). At the end point, cells were collected for each experimental arm, and genomic DNA from all samples was extracted using the QiaAMP blood Maxi kit (51192, QIAGEN) and barcodes were sequenced via massively parallel sequencing at the Broad Institute core facilities. Barcode representations were determined and de-convoluted and used to calculate individual ORF representation. We evaluated Pearson correlation coefficients (R) from the replicates of each arm. These were tightly correlated and ranged between 0.968 to 0.989 (Figure S1D).
Arrayed Validation Screen of Hits
Of the 107 candidates identified in the CSS and enzalutamide treated arm (Figure 1B), LNCaP cells were individually infected with each ORF at an MOI of less than 1. The cells were subsequently selected with puromycin. Of remaining ORF-expressing cell lines that were viable (101 of 107), cells were first pre-treated in 3 days of media supplemented with CSS, and 200,000 cells of each cell line were seeded into CSS and enzalutamide in quadruplicates. Culture media were refreshed every 3 days. After 14 days, the final cell number was determined by automated counts using Vi-Cell. This experiment was repeated 2 more times, and the average of all replicates was used to determine relative population doubling counts (LOG2 of final divided by initial cell counts).
Viability and tumorigenic assays
Standard cell counting, tumor measurement and viability assays were used in this study to examine the effects of CREB5 overexpression or suppression in prostate cancer cells.
IC50 determination and population doubling
50~200,000 cells were seeded in a 12-well plate and at 7 days, Vi-Cell was used to determine the overall cell count as well as the relative viability of cells. This approach was used to examine response of luciferase control or CREB5- overexpressing LNCaP cells to respective inhibitors (Figures 2B and 2C) at indicated concentrations. Media with drugs were refreshed at day 4. The overall numbers were used to determine IC50 and relative population doubling. The mean and SD of 3 or more experimental replicates are presented.
Tumorigenicity experiments in mice
We implanted cell xenografts subcutaneously (Choudhury et al., 2017) in the two flanks of immuno-deficient male BALB/C (BALBNU, Taconic) mice under Dana-Farber Cancer Institute IACUC protocol 03–013. Approximately 2 million cells were implanted with matrigel at a 1:1 ratio. Post implantation, growth at each site was assessed through measurements by a caliper 2 times a week, and tumor volume was calculated by using the ellipsoid formula. Time lines for additional treatments including castration or enzalutamide treatment (3 doses a week at 25mg/kg via oral gavage) are shown (Figures 2D and 2E). For castration and enzalutamide-treatment experiments, tumors between the size of 100~200mm3 were included in experimental treatments. Weight measurements were also performed after each treatment to ensure health of individual mice. All data points are shown (Figures 2D and 2E).
Organoid transduction
NCI-PC44 organoids were cultured as previously described (Beshiri et al., 2018). To infect, organoids were dissociated into a single cell suspension, counted and plated overnight in 2D, on 6-well plates pre-coated with 2% Matrigel (growth factor-reduced/phenol red-free), in organoid growth media and in the presence of 8 μg/ml polybrene and lentivirus. Following the overnight incubation, the cells were detached from the plate with TrypLE, collected and re-plated in 3D for 48 hours. After 48 hours the organoids were treated with 1mg/ml dispase for 2 hours to free them from the Matrigel. The organoids were then dissociated with TrypLE, counted and re-plated in quadruplicate in 48-well plates at 2,000 cells/20 μL Matrigel drops/well and cultured for 1 or 2 weeks. Relative cell viability was quantified by CellTiter-Glo 3D. The mean and SD from 4 experimental replicates are presented.
ChIP-seq and ATAC-seq experiments
These experiments were used to examine the global interactions of CREB5 and AR with chromatin and the potential impact of overall chromatin structure in experimental conditions. ChIP was performed as previously described (Pomerantz et al., 2015). 10 million cells were fixed with 1% formaldehyde at room temperature for 10 minutes. Cells were quenched with 0.25M glycine, rinsed with ice-cold PBS twice and then collected in lysis buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor (#11873580001, Roche) in PBS). Chromatin was sonicated to 300–800 bp and centrifuged at 13,000 rpm for 10 minutes at 4°C. Antibodies (AR; sc-816x, Santa Cruz, V5; #13202, Cell signaling, tri-acetylated H3K27; C15410196, Diagenode) were incubated with 30 μL of Dynabeads protein A/G (Invitrogen) for at least 3 hours before immunoprecipitation with the sonicated chromatin overnight. Chromatin was washed with RIPA, then with LiCl wash buffer (100 mM Tris pH 7.5, 500 mM LiCl, 1% NP-40, 1% sodium deoxycholate) 4 times for 10 minutes sequentially. After rinsing with TE buffer twice, immunoprecipitated chromatin in elution buffer (1% SDS, 0.1 M NaHCO3) was treated with proteinase K for 6–12 hours at 65C with gentle rocking after RNase A treatment at 37°C for 30 minutes. Sample DNA as well as its input were extracted using QIAGEN Qiaquick columns and were prepared as the sequencing libraries using the ThruPLEX-FD Prep Kit (Rubicon Genomics, Ann Arbor, MI). Libraries were sequenced using 75-bp single reads on the Illumina platform (Illumina, San Diego, CA) at the Dana-Farber Cancer Institute core facility. All samples were processed through the computational pipeline ChiLin (Qin et al., 2016) and binding sites were called with a FDR q-value threshold of 0.01 with consideration of baseline noise of the sequencing data and signal of expected interactions. In luciferase control cells, enzalutamide treatment regulated all analyzed AR binding sites in duplicate experiments. Heatmaps were generated using plotHEATmap. We utilized heatmaps to align all key binding sites and IGV viewer (Robinson et al., 2011) to examine interactions at critical binding sites (TMPRSS2, KLK3, STK39) for all ChIP-seq experiments compared to previous studies (Pomerantz et al., 2015). We also validated AR, and V5-CREB5 interactions at 3 sites with the greatest signal strength using qRT-PCR experiments. Primer sequences are provided (Table S5).
For ATAC-seq samples, nuclei were prepared from 50,000 cells after fixation with 1% formaldehyde for 10 min at RT and incubated with 2.5 μL of transposase (Illumina) in a 50 μL reaction for 30 min at 37°C with mixing. After purification of transposase-fragmented DNA, the library was amplified by PCR and subjected to paired-end 50 bp high-throughput sequencing on an Illumina NextSeq 500 Next Gen Sequencing platform. Data analysis similar to ChIP-seq experiments was subsequently performed to identify regions of accessible chromatin.
CREB5 dysregulation in clinical prostate tumor samples
We performed analysis of clinical genomic and expression data in public portals and previously published datasets to illustrate the dysregulation of CREB5 in prostate cancer.
cBioPortal analysis
17 available prostate cancer studies were queried for CREB5 amplification and deletion rates (Gao et al., 2013a). The 2010 MSKCC study was used to query rates of relapse in patients with CREB5 overexpression.
GISTIC2 analysis
The CREB5-containing focal amplification previously identified at 7p14.3 is considered for further analysis in our study (Harvard, 2016).
Meta-analysis of prostate cancer WES data
Prostate cancer samples were analyzed using whole exome sequencing and subsequently analyzed using standard analytical pipelines (Armenia et al., 2018). Specifically, purity and ploidy were called using FACETs (Shen and Seshan, 2016) and allelic copy number, called by Allelic CapSeg, was adjusted based on the purity and ploidy. The resulting copy number indicated whether each allele was amplified, deleted, or neutral. We aggregated the number of amplifications and deletions in each gene across the metastatic and primary prostate samples. The samples which passed quality control were further analyzed in this study (326 mCRPC and 528 primary).
Expression analysis of CREB5
From an updated combined cohort of men with mCRPC from multiple institutions comprising the SU2C/PCF Prostate Cancer Dream Team (Armenia et al., 2018; Robinson et al., 2015) RNA-seq data, normalized in units of transcripts per million (TPM) was available from 238 patients. Expression data was examined and adjusted for batch effects using ComBat (Johnson et al., 2007) via the R Bioconductor package “sva” (Leek et al., 2012), version V3.22.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined using two tailed Student’s t tests and Fisher’s exact tests and shown in the figure legends whenever applicable. For cell line experiments in culture, the significance is set to 0.005. Since broader range of distributions is generally observed in tumor experiments and clinical samples, the significance threshold level is set to 0.05.
DATA AND CODE AVAILABILITY
The datasets generated during this study are available at NCBI GEO repository under GEO accession number GSE137775. Post analyzed results from the ORF screen, ChIP-seq, ATAC-seq, RNA-seq are in supplementary figures and tables.
No codes were generated in this study.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-mouse secondary | Li-Cor | RRID:AB_621842 |
| Goat anti-rabbit secondary | Li-Cor | RRID:AB_10706309 |
| V5 | Cell signaling | 13202S |
| Tubulin | Sigma Aldrich | RRID:AB_477593 |
| AR for immunoblots | Cell signaling | 5153S |
| HSP90 | Cell signaling | 4875S |
| Lamin A/C | Cell signaling | 2032S |
| AR for ChIP-seq | Santa Cruz | sc-816x |
| H3K27 Ac | Diagenode | RRID:AB_2637079 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Enzalutamide | Selleckchem | S1250 |
| Apalutamide | Selleckchem | S2840 |
| Darolutamide | Selleckchem | S7559 |
| Critical Commercial Assays | ||
| RNeasy plus | QIAGEN | 74104 |
| RT2 profiler PCR array for AR signaling | QIAGEN | 330231 |
| iSCRIPT reverse transcriptase | BioRad | 1708841 |
| Power SYBR Green PCR master mix | Life Technologies | 4368708 |
| Deposited Data | ||
| Raw and processed RNA-seq, ChIP-seq, ATAC-seq | This study | GEO accession number: GSE137775 |
| ORF screen data | This study | Table S1 |
| Experimental Models: Cell Lines | ||
| LNCaP | ATCC | RRID:CVCL_4783 |
| LAPC4 | ATCC | RRID:CVCL_4744 |
| C4–2 | ATCC | RRID:CVCL_4782 |
| PC3 | ATCC | RRID:CVCL_0035 |
| 22RV1 | ATCC | RRID:CVCL_1045 |
| DU145 | ATCC | RRID:CVCL_0105 |
| Experimental Models: Organisms/Strains | ||
| BALB/C nude mice; male | Taconic | RRID:IMSR_TAC:balbnu |
| NCI-PC44, human organoid model | NCI (Beshiri et al., 2018) | N/A |
| Oligonucleotides | ||
| CREB5-F: ATTGACTCACCACCCTGCTG | IDT | Table S5 |
| CREB5-R: GCATGAAGGTGGGAATGGGA | IDT | Table S5 |
| CREB5-F-2: CATTGACTCACCACCCTGCT | IDT | Table S5 |
| CREB5-R-2: GAAGGTGGGAATGGGAGTGG | IDT | Table S5 |
| RLOPO-F: TGGCAGCATCTACAACCCTGAAGT | IDT | Table S5 |
| RLOPO-R: ACACTGGCAACATTGCGGACA | IDT | Table S5 |
| Recombinant DNA | ||
| shRNA-GFP | Broad Institute | TRCN0000231753 |
| shRNA-CREB5 1 | Broad Institute | TRCN0000013486 |
| shRNA-CREB5 2 | Broad Institute | TRCN0000271308 |
| shRNA-CREB5 3 | Broad Institute | TRCN0000271310 |
| shRNA-CREB5 4 | Broad Institute | TRCN0000271247 |
| shRNA-CREB5 5 | Broad Institute | TRCN0000271307 |
| shRNA-CREB5 6 | Broad Institute | TRCN0000013485 |
| shRNA-CREB5 7 | Broad Institute | TRCN0000013483 |
| shRNA-CREB5 8 | Broad Institute | TRCN0000271249 |
| shRNA-CREB5 9 | Broad Institute | TRCN0000013487 |
| shRNA-AR 1 | Broad Institute | TRCN0000350462 |
| shRNA-AR 2 | Broad Institute | TRCN0000314730 |
| shRNA-AR 3 | Broad Institute | TRCN0000003717 |
| shRNA-FOXA1 1 | Broad Institute | TRCN0000358367 |
| shRNA-FOXA1 2 | Broad Institute | TRCN0000014879 |
| ORF-CREB5 | Broad Institute | TRCN0000469202 |
| Software and Algorithms | ||
| Tuxedo Suite RNA-seq analysis package on GenePattern | Previous study (Trapnell et al., 2010) | https://software.broadinstitute.org/cancer/software/genepattern/ |
| ChiLin | Previous Study (Qin et al., 2016) | http://cistrome.org/chilin/ |
| FACETs | Previous Study (Shen and Seshan, 2016) | https://sites.google.com/site/mskfacets/ |
| ComBat | Previous Study (Johnson et al., 2007) | https://rdrr.io/bioc/sva/man/ComBat.html |
| R Bioconductor package | Previous Study (Leek et al., 2012) | https://bioconductor.org/packages/release/bioc/html/sva.html |
| Other | ||
| Analysis of gene dysregulation status in prostate cancer | Previous study (Armenia et al., 2018; Robinson et al., 2015) |
https://doi.org/10.1038/s41588-018-0078-z, 10.1016/j.cell.2015.06.053 |
| Analysis of gene dysregulation status in cancer | cBioPortal (Gao et al., 2013a) | https://www.cbioportal.org/ |
| Analysis of focal copy number change in prostate cancer | Previous study | https://doi.org/10.7908/C14Q7TFZ |
Highlights.
CREB5 promotes resistance to AR inhibitors and androgen therapies in prostate cancer
CREB5 selectively enhances interaction of AR with target genes critical for survival
CREB5 is amplified or overexpressed in therapy-resistant metastatic prostate cancers
Targeting CREB5 is effective in patient-derived models that are therapy resistant
ACKNOWLEDGMENTS
This work was supported by the PCF/SU2C Prostate Cancer Dream Team (to E.M.V.A.), a PCF – V Foundation Challenge Award (to E.M.V.A.), a PCF Young Investigator Award (to E.M.V.A., A.D.C., and A.G.S.), NIH K08 CA188615 (to E.M.V.A.), American Cancer Society – AstraZeneca postdoctoral fellowship PF-16–142-01-TBE (to J.H.), a Susan G. Komen postdoctoral fellowship and a Terri Brodeur postdoctoral fellowship (to J.L.), DoD Prostate Cancer Research Program postdoctoral training award W81XWH-15–1-0659 (to G.J.S.), DoD PCRP Physician Research Training Award W81XWH-12–1-0062 (to A.D.C.), NCI U01 CA176058 (to W.C.H.), and the H.L. Snyder Foundation (to W.C.H. and M.L.F.). We thank Catherine Sypher and Zach Herbert for assistance.
Footnotes
DECLARATION OF INTERESTS
E.M.V.A. is a consultant of Tango Therapeutics, Invitae, Genome Medical, and Novartis. W.C.H. is a consultant for Thermo Fisher, AjuIB, MPM Capital, Frontier Medicines, and Parexel. W.C.H. is a founder and serves on the scientific advisory board of KSQ Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.068.
REFERENCES
- Antonarakis ES (2016). Current understanding of resistance to abiraterone and enzalutamide in advanced prostate cancer. Clin. Adv. Hematol. Oncol 14, 316–319. [PubMed] [Google Scholar]
- Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, et al. ; PCF/SU2C International Prostate Cancer Dream Team (2018). The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 50, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. (2013). Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155, 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, et al. (2009). Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J. Clin. Oncol 27, 3742–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. (2016). Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard D, Pourtier-Manzanedo A, Gil J, and Beach DH (2003). Myc confers androgen-independent prostate cancer cell growth. J. Clin. Invest 112, 1724–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshiri ML, Tice CM, Tran C, Nguyen HM, Sowalsky AG, Agarwal S, Jansson KH, Yang Q, McGowen KM, Yin J, et al. (2018). A PDX/Organoid Biobank of Advanced Prostate Cancers Captures Genomic and Phenotypic Heterogeneity for Disease Modeling and Therapeutic Screening. Clin. Cancer Res 24, 4332–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj A, Singh H, Rajapakshe K, Tachibana K, Ganesan N, Pan Y, Gunaratne PH, Coarfa C, and Bedrosian I (2017). Regulation of miRNA-29c and its downstream pathways in preneoplastic progression of triple-negative breast cancer. Oncotarget 8, 19645–19660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF, Kaipainen A, Corella AN, et al. (2017). Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 32, 474–489.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubley GJ, and Balk SP (2017). Association Between Androgen Receptor Splice Variants and Prostate Cancer Resistance to Abiraterone and Enzalutamide. J. Clin. Oncol 35, 2103–2105. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol, 109, 21.29.1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury AD, Schinzel AC, Cotter MB, Lis RT, Labella K, Lock YJ, Izzo F, Guney I, Bowden M, Li YY, et al. (2017). Castration Resistance in Prostate Cancer Is Mediated by the Kinase NEK6. Cancer Res. 77, 753–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock CE, Augello MA, Goodwin JF, de Leeuw R, Schiewer MJ, Ostrander WF Jr., Burkhart RA, McClendon AK, McCue PA, Trabulsi EJ, et al. (2013). Targeting cell cycle and hormone receptor pathways in cancer. Oncogene 32, 5481–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeble PD, Cox ME, Frierson HF Jr., Sikes RA, Palmer JB, Davidson RJ, Casarez EV, Amorino GP, and Parsons SJ (2007). Androgen-independent growth and tumorigenesis of prostate cancer cells are enhanced by the presence of PKA-differentiated neuroendocrine cells. Cancer Res. 67, 3663–3672. [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013a). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Schwartzman J, Gibbs A, Lisac R, Kleinschmidt R, Wilmot B, Bottomly D, Coleman I, Nelson P, McWeeney S, and Alumkal J (2013b). Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS ONE 8, e63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. (2012). The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, Brewer DS, Kallio HML, Högnäs G, Annala M, et al. ; ICGC Prostate Group (2015). The evolutionary history of lethal metastatic prostate cancer. Nature 520, 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GC, Hwang J, Wankowicz SAM, Zhang Z, Liu D, Cibulskis C, Gaviola GC, Ghazikhanian V, Mckay RR, Bubley GJ, et al. (2017). Genomic Resistance Patterns to Second-Generation Androgen Blockade in Paired Tumor Biopsies of Metastatic Castration-Resistant Prostate Cancer. JCO Precision Oncology, Published online December 11, 2017 10.1200/PO.17.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvard, B. I. O. M. A. 2016. Broad Institute TCGA Genome Data Analysis Center (2016): SNP6 Copy number analysis (GISTIC2). 10.7908/C14Q7TFZ. [DOI]
- He S, Deng Y, Liao Y, Li X, Liu J, and Yao S (2017). CREB5 promotes tumor cell invasion and correlates with poor prognosis in epithelial ovarian cancer. Oncol. Lett 14, 8156–8161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, Liu G, Coleman I, Lakely B, Li R, et al. (2016). Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun 7, 13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higano CS, Beer TM, Taplin ME, Efstathiou E, Hirmand M, Forer D, and Scher HI (2015). Long-term Safety and Antitumor Activity in the Phase 1–2 Study of Enzalutamide in Pre- and Post-docetaxel Castration-Resistant Prostate Cancer. Eur. Urol 68, 795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HJ, Zhao JC, Wu L, Kim J, and Yu J (2014). Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun 5, 3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kregel S, Chen JL, Tom W, Krishnan V, Kach J, Brechka H, Fessenden TB, Isikbay M, Paner GP, Szmulewitz RZ, and Vander Griend DJ (2016). Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget 7, 26259–26274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan PE, and Tindall DJ (2011). Androgen receptor signaling in prostate cancer development and progression. J. Carcinog 10, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár B, Galamb O, Péterfia B, Wichmann B, Csabai I, Bodor A, Kalmár A, Szigeti KA, Barták BK, Nagy ZB, et al. (2018). Gene promoter and exon DNA methylation changes in colon cancer development - mRNA expression and tumor mutation alterations. BMC Cancer 18, 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery B, Tretiakova MS, Joshua AM, Gleave ME, Fleshner N, Bubley GJ, Mostaghel EA, Chi KN, Lin DW, Sanda M, et al. (2017). Neoadjuvant Enzalutamide Prior to Prostatectomy. Clin. Cancer Res. 23, 2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura N, Zu YL, Maekawa T, Tabata S, Akiyama T, and Ishii S (1993). Isolation and characterization of a novel member of the gene family encoding the cAMP response element-binding protein CRE-BP1. J. Biol. Chem 268, 4259–4266. [PubMed] [Google Scholar]
- Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, et al. (2015). The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet 47, 1346–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, and Ding Y (2014). Involvement of the CREB5 regulatory network in colorectal cancer metastasis. Yi Chuan 36, 679–684. [DOI] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, et al. (2016). ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, and Mesirov JP (2006). GenePattern 2.0. Nat. Genet 38, 500–501. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 162, 454. [DOI] [PubMed] [Google Scholar]
- Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, Salvi S, Amadori D, Zafeiriou Z, Rescigno P, et al. (2015). Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med 7, 312re10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang M, Hulsurkar M, Zhang X, Song H, Zheng D, Zhang Y, Li M, Xu J, Zhang S, Ittmann M, and Li W (2016). GRK3 is a direct target of CREB activation and regulates neuroendocrine differentiation of prostate cancer cells. Oncotarget 7, 45171–45185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. ; AFFIRM Investigators (2012). Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med 367, 1187–1197. [DOI] [PubMed] [Google Scholar]
- Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG, and Neal DE (2013). The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 23, 35–47. [DOI] [PubMed] [Google Scholar]
- Shen R, and Seshan VE (2016). FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Blake JA, Kadin JA, Richardson JE, and Bult CJ; Mouse Genome Database Group (2018). Mouse Genome Database (MGD)-2018: knowledgebase for the laboratory mouse. Nucleic Acids Res. 46 (D1), D836–D842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda DY, Spisak S, Seo JH, Bell C, O’connor E, Korthauer K, Ribli D, Csabai I, Solymosi N, Szallasi Z, et al. (2018). A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 174, 422–432.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC, Rhoades J, et al. (2018). Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 174, 433–447.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al. (2009). Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, and Liu XS (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc 8, 2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, and Sawyers CL (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, Haegert A, Warner EW, Mo F, Brahmbhatt S, et al. (2016). Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2, 1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N, Liu Y, Wang Z, Shao L, Ittmann M, et al. (2018). Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun 9, 4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at NCBI GEO repository under GEO accession number GSE137775. Post analyzed results from the ORF screen, ChIP-seq, ATAC-seq, RNA-seq are in supplementary figures and tables.
No codes were generated in this study.