

Abstract
Observing structure and conformational dynamics of membrane proteins at high-resolution in their native environments is challenging because of the lack of suitable techniques. We have developed an approach for high-precision distance measurements in the nanometer range for outer membrane proteins (OMPs) in intact E. coli and native membranes. OMPs in Gram-negative bacteria rarely have reactive cysteines. This enables in-situ labeling of engineered cysteines with a methanethiosulfonate functionalized nitroxide spin label (MTSL) with minimal background signals. Following overexpression of the target protein, spin labeling is performed with E. coli or isolated outer membranes (OM) under selective conditions. The interspin distances are measured in-situ using pulsed electron-electron double resonance spectroscopy (PELDOR or DEER). The residual background signals, which are problematic for in-situ structural biology, contributes specifically to the intermolecular part of the signal and can be selectively removed to extract the desired interspin distance distribution. The initial cloning stage can take 5–7 d and the subsequent protein expression, OM isolation, spin labeling, PELDOR experiment, and the data analysis typically take 4–5 d. The described protocol provides a general strategy for observing protein-ligand/substrate interactions, oligomerization, and conformational dynamics of OMPs in the native OM and intact E. coli.
Keywords: membrane protein, outer membrane, gram-negative, E. coli, DEER, PELDOR, double electron-electron resonance, Pulsed Electron-Electron Double Resonance, electron paramagnetic resonance, EPR, methanethiosulfonate, MTSL, spin label, cellular structural biology, protein conformation, conformational dynamics, ligand binding, substrate binding, interspin distance, pulsed dipolar EPR spectroscopy, PDS
EDITORIAL SUMMARY
This protocol describes how to label bacterial outer membrane proteins with spin labels to study conformational changes and their interaction with ligands and substrates in native membranes and cells using Pulsed Electron-Electron Double Resonance (PELDOR or DEER) spectroscopy.
TWEET
A new protocol for studying conformational changes and ligand/substrate interactions of bacterial outer membrane proteins in-situ.
COVER TEASER
Studying membrane protein conformations in-situ
INTRODUCTION
Membrane proteins often sample a broad conformational landscape and the activity of channels, transporters, or receptors often involves large-scale domain movements1. Thus, a mechanistic description of their function necessitates an understanding of conformational changes and equilibrium dynamics. Biomolecular structures of membrane proteins have primarily been determined with cryoEM, solution NMR, and X-ray crystallography. All these techniques require the isolation of the target molecules from the native environment, which masks the effects of cellular conditions such as the lipid environment, interaction with other molecules/ions, molecular crowding, pH or ionic gradients, and the specific localization. All these factors may critically influence protein structure, function, and dynamics. For example, there is increasing evidence for the vital role of the native lipid environment on membrane protein folding, structure, and activity2,3. In this protocol we describe how to use Pulsed Electron-Electron Double Resonance (PELDOR or DEER) to observe outer membrane protein (OMP) structure and dynamics in their native environment, either in E. coli or in purified outer membranes.
EPR spectroscopy of intrinsically diamagnetic biomolecules
Continuous wave (CW) electron paramagnetic resonance (EPR) spectroscopy is a powerful technique for conformational studies on membrane protein as it can provide information on water accessibility, polarity of the surrounding environment, dynamics, and intra- or intermolecular distances between sites in the range of 1–2 nm4. However, most biomolecules are not paramagnetic, and for EPR spectroscopy they must be modified with an appropriate spin label. For proteins, labels are normally attached by covalently linking a functionalized spin label to cysteines engineered using site-directed mutagenesis (SDM)5. This requires the removal of native reactive cysteines in the target protein, and this usually does not impair protein function. Alternatively, spin probes may be incorporated using genetic encoding in response to a nonsense codon6–8. The development of these site-directed spin labeling approaches led to a rapid growth of EPR spectroscopy as a powerful tool for structural biology. The nitroxide-based methane thiosulfonate spin label (MTSL, which forms the side chain denoted as R1) is the most preferred spin label for proteins (see Table 1). There exist numerous studies with MTSL and rotamer libraries have been created to describe the internal motion of R14. It carries an unpaired electron localized along the N-O bond. The small size, high specificity, and reactivity make MTSL an ideal spin label for structural studies. It is also used in paramagnetic relaxation enhancement (PRE) NMR experiments to determine long-range distance constraints (up to ~35 Å).
Table 1 |.
Spin labels for in-situ PELDOR experiment of proteins/peptides
| Label | Linkage | Advantages/disadvantages | Ref. |
|---|---|---|---|
| (A) Nitroxide labels | |||
MTSL (R1)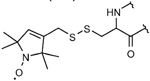
|
–S–S– (disulfide) bond via cysteines | small size, high specificity and reactivity, and very well-studied label | 35,36,46,47 |
| reduction of nitroxide label and the –S–S– bond under in-situ conditions |
|||
| 3-maleimido-PROXYL | |||
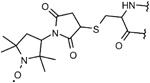 |
–C–S– (thioether) bond via cysteine | stable covalent attachment to the protein less specific and irreversible linkage, may react with Arg or Lys, bulkier than MTSL, and limited literature data |
12 |
| M-TETPO | |||
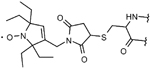 |
–C–S– (thioether) bond via cysteine | very stable under reducing conditions and covalent attachment to the protein | 38 |
| disadvantages similar to 3-maleimido-PROXYL | |||
| (B) Gadolinium(Gd3+) labels | |||
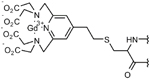
Gd(III)-DOTA-M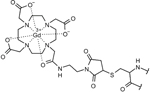
|
–C–S– (thioether) bond via cysteine | stable under reducing conditions and higher sensitivity bulky and less specific, endogenous Mn2+ may interfere with in-situ experiments, requires higher frequency (94 GHz) for optimal sensitivity, thiol exchange with glutathione or hydrolysis of the succinimide ring may take place in-situ |
14,40 |
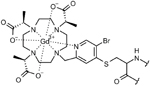
| Gd-PyMTA | |||
 |
–C–S– (thioether) bond via cysteine | smaller linker and higher cysteine specificity, lower affinity than DOTA limited literature data |
39 |
| GdL | |||
 |
–C–S– (thioether) bond via cysteine | smaller linker and higher affinity (for Gd3+) and reactivity to cysteines limited literature data |
41 |
| (C) Trityl labels | |||
| TAM1 | |||
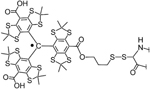 |
–S–S– (disulfide) bond via cysteines | stable under reducing conditions and higher sensitivity bulky, low water solubility, and tendency for aggregation |
44,45 (for the latter, the linker is shorter by one bond) |
Interspin distances beyond 2 nm can however not be determined using CW-EPR because the line broadening due to dipolar interaction becomes too small to be extracted within the linewidth of CW-EPR spectrum. In order to resolve the week dipolar couplings resulting from longer distances, several pulsed dipolar EPR spectroscopy (PDS) techniques have been developed. Pulsed Electron-Electron Double Resonance (PELDOR or DEER) is presently the most widely utilized tool among a growing number of PDS techniques9,10. It is a powerful technique for structural investigation of biomolecules in solution, membrane, or cellular environments11–15. PELDOR can resolve distance distributions between spin pairs in the range of 1.5–16.0 nm16. In addition, interspin distances derived using PELDOR have been used to explore conformational changes, equilibrium dynamics, and structural heterogeneity of several membrane protein complexes17–19. Combined with simulations and modeling, such constraints can validate existing structures, provide novel structural information, and visualize alternate conformations that have not been yet observed in the crystal structures17,19,20.
As in any probe-based technique, PELDOR provides sparse distance restraints. The distances are determined between the unpaired electrons, which are connected to the protein backbone through flexible linkers. The degrees of freedom for the rotation of the dihedral angles in the linker lead to a rotameric distribution of the spin label ( in the absence of protein backbone motion)1. Changes in the interspin distance provide direct information on the extent and the nature of the conformational changes. However, to extrapolate those changes to the backbone or to use the distance distribution for structural modelling, the rotameric states of the spin label need to be described. This has been rather well established for MTSL21, which permits the comparison of an available structure with experimental data or allows modeling a new functional- or an oligomeric state from an existing structure19.
Theoretical rationale behind DEER/PELDOR spectroscopy
PELDOR employs a refocused Hahn echo with the pulse sequence π/2–τ1–π–τ1–echo1 –τ2–π– τ2–echo2 applied at the observer frequency νA on the A spin (Fig. 2, in grey). A second inversion pulse at the pump frequency νB is applied on the B spin at a variable time t with respect to the observer echo. The observer sequence refocuses the inhomogeneous broadening of the A spin arising from g value dispersion, hyperfine interaction, and coupling with other electron spins. Inversion of the B spin by the pump pulse changes the resonant frequency of the A spin by the dipolar coupling frequency ωdip. Thus, varying the timing of the pump pulse leads to a phase gain of the A spin by and oscillation of the echo amplitude . The resulting PELDOR is a product of two contributions9:
| (1) |
Figure 2 |. Pulse sequences for electron-electron double resonance spectroscopy (DEER/PELDOR).

(a) Echo-detected field sweep spectrum of MTSL at Q-band (34 GHz, 50 K). The positions and a schematic view of the excitation profiles for the observer (in grey) and pump pulses (in red or blue) are shown. The excitation profile for the rectangular pulse is a sinc function, whereas a shaped pulse like the sech/tanh pulse (at the bottom in d) provides a larger and uniform excitation (in dashed blue) of the spins. (b) Pulse sequence for the 4-pulse DEER. The modulation of the intensity of a refocused Hahn echo is monitored as a function of the timing of the pump pulse. (c) Pulse sequence for the 5-pulse DEER. The observer sequence is similar to the 4-pulse DEER, but are applied under a Carr-Purcell (CP) condition to prolong the observation window. The first pump pulse is fixed in time and the modulation of the observer echo intensity is monitored as a function of the timing of the second pump pulse. (d) Pulse sequence for the 7-pulse CP-PELDOR. The observer pulse sequence contains an additional π pulse accompanied with a pump pulse. Shaped sech/tanh pulses are employed to minimize the artefacts due to non-uniform excitation by the successive pump pulses. The second pump pulse is fixed in time and the echo intensity is monitored while the first and the third pump pulses are moved in equal increments in the time domain.
, the intermolecular contribution, commonly known as the background function arises from the interaction between spins in the neighboring biomolecules. , which is the wanted intramolecular interaction arises from the interaction of the spins within the same biomolecular unit being observed. For a single isolated spin pair, can be expressed as8:
| (2) |
For a macroscopically disordered sample as that of a biomolecule, can be calculated from an ensemble average according to the following equation:
| (3) |
The indexing refers to the B spins. The averaging runs for all the distances () over all the orientations () of the interspin vectors with respect to the external magnetic field (). The parameter is the fraction of the coupled B spins that are excited by the pump pulse (also known as the inversion efficiency), which leads to a decay of to . For nitroxide spin labels separated by distances longer than 1 nm, the unpaired electron can be approximated to be localized at the center of the N-O bond with the quantization axis parallel to the direction of . Thus, the dipole-dipole tensor can be described by the point-dipole interaction. The exchange coupling through bonds is negligible for distances larger than 1.5 nm, especially when determined with biomolecules in frozen buffers or membranes. Typically, the difference between the pump and observer frequency (Δν) is much larger than and therefore the pseudosecular term in the Hamiltonian for electron-electron interaction can be neglected22. Now under the approximation of weak coupling (Δν > ) is given by:
| (4) |
Where is the permeability of vacuum, is the reduced Planck’s constant, and is the angle between the interspin vector and . With an isotropic value of approximated for nitroxide spins, has a value of 52.04 MHz nm−3. The intermolecular contribution , which arises from dipolar interaction between randomly distributed spins on neighboring objects can also be expressed according to Eq. 39. Such intermolecular interactions lead to an exponential decay function as below:
| (5) |
In which, is the concentration of the A spins that interacts through intermolecular interaction and is the fraction of the B spins excited by the pump pulse. The spins can have a homogenous distribution in space (with ) or could have a spatial distribution with fractional dimension as in the case of membrane proteins reconstituted into proteoliposomes (with ). Values greater than three indicate contributions from exclusion volume effects due to large physical separation between the spins23. The constant is given as:
| (6) |
The modulation depth of (Fig. 3c) is the product of (inversion efficiency in Eq. 3) and the labelling efficiency (). Thus, depending on the spin labeling efficiency, decays to a final value of (when the labeling efficiency is less than 100%, in Eq. 2 and 3 should be substituted with ). One of the critical steps in PELDOR data analysis is the separation of from the original data. When the dipolar evolution is observed for sufficiently long times, this can be achieved by dividing by (Eq. 1). Reliable fit for requires that the dipolar evolution is observed for much longer time after the dipolar oscillations have fully decayed. Owing to the flexibility of the spin label and the protein backbone, the PELDOR data contains a distribution of frequencies/distances. The corresponding distance distribution is characterized by its mean, width, and shape. The width and the shape of the distance distribution is encoded in the decay rate and the shape of the decay envelope respectively of the dipolar oscillations. The observed put an upper limit for the accurate determination of the mean distance by and of the width by 13. In order to determine the shape of the distance distribution, should be even longer with extremely high signal-to-noise (S/N) ratio.
Figure 3 |. In-situ PELDOR in native OM.

(a) Position 188 in the second extracellular loop and the TEMPO-labeled hydroxycobalamin (TEMPO-HOCbl, 25 μM) are highlighted on the BtuB crystal structure (PDB 1NQH). The TEMPO-HOCbl was synthesized as described in Box 1. (b) Original PELDOR data obtained in native OM as indicated. The data are slightly shifted along the vertical axis for clarity. For the 188R1 mutant, the data perfectly fit into a stretched exponential decay (). (c) The dipolar evolution (in yellow) obtained for 188R1/TEMPO-HOCbl PELDOR after correction for the intermolecular contribution () and the corresponding fit from Tikhonov regularization (TR) is overlaid (in black). The modulation depth (Δ) is indicated. Overall, the data suggests a two-dimensional distribution of the spins over the large cell surface and deviation of the value for (from 2.0) might be for other reasons including the membrane curvature and sample inhomogeneity. (d) The dipolar spectrum obtained with Fourier transformation (in yellow) or TR (in black) of c. Frequencies corresponding to the parallel (θ = 0) and perpendicular (θ = 90) orientations of the interspin vectors to the B0 are indicated. (e) Interspin distance distributions obtained from TR of c.
The molecules in the sample and hence the interspin vectors () are randomly distributed. When all the orientations are excited, Fourier transformation of gives a dipolar spectrum (or Pake pattern) and the interspin distance can be obtained directly from the frequencies at which the singularities () appear (Fig. 3d). Normally, the inherent flexibility of the protein backbone and the spin label cause broadening of the Pake pattern and the singularities are not defined anymore, which leads to inaccuracies in the probability distribution of the interspin distances. Such a scenario leads to an inverse problem in which the need to be computed from or the dipolar spectrum. This is an ill-posed problem as the noise in the time- or frequency-domain data can have an even larger effect on the computed distance distribution. To address this ill-posed problem, current approaches impose a variable smoothness to stabilize the solution against artifacts. Tikhonov regularization (TR) is one of the most common methods employed to solve ill-posed problems. TR has also been used for PELDOR data analysis24,25 and is implemented in the MATLAB-based software DeerAnalysis26. During TR, the time-domain signal for a given distance distribution is simulated and fitted with the experimental form factor with minimum deviation. Straight fitting of to would result in strong noise artifacts and TR employs a regularization parameter , which is related to the smoothness of . Large means a broad distance distribution and for well-defined narrow distances a smaller is required. The optimum is calculated by the L curve criterion. This whole procedure works with , which requires a priori estimation of For data with short dipolar evolution times, a priori estimation of might be difficult or could lead to unrealistically large errors. Also, when consists of a mixture of narrow and broad components, this procedure may result in splitting of the broad component into multiple narrow peaks to fit the single value of the regularization parameter used. In order to quantify the effect of the choice of on the uncertainty in , a Bayesian statistical approach may be used with TR27. Alternatively, the original PELDOR data may be simultaneously fitted for both and in a model-based approach using simple analytical expression of such as Gaussian components28. This does not require a priori estimation of and also permits a rigorous statistical analysis on the fit parameters. However, none of these approaches can circumvent the length of the dipolar evolution time required for reliable determination of and . Even when this condition is achieved, it is worthwhile to compare the shapes of obtained with Tikhonov regularization and model-based analysis.
The observable length of the dipolar evolution time is limited by the loss of phase memory due transverse relaxation, spectral-, spin-, and instantaneous diffusion, which altogether lead to an exponential decay with a time constant called as the phase memory time (). Under the typical conditions of PELDOR measurements on membrane proteins (50–100 μM spin and 50–80 K), is dominated by fluctuating hyperfine interaction with nearby protons, including those from the methyl groups and the higher local spin concentration. For membrane-reconstituted proteins, this reduces the to 1–2 μs (from typical values between 3–4 μs in detergent solution) and thereby limits the upper range of the accessible distances to ~5 nm29. Sample deuteration16 and recently, multiple refocusing pulses at the observer frequency have been implemented to prolong and increase the range of the accessible distances. This leads to the extension of the 4-pulse DEER towards 5-pulse and 7-pulse versions (known as the 5-pulse DEER and the 7-pulse CP-PELDOR respectively)29,30 both of which can prolong the up to two-fold for membrane-reconstituted proteins (Fig. 2). Each of these observer π pulses, which are applied under a Carr-Purcell condition is accompanied by an inversion π pulse at the pump frequency. The non-ideal behaviour of the successive pump pulses leads to uneven excitation of the B spins and dipolar signals. Therefore, additional data processing is required to remove the artefacts and extract the wanted dipolar pathway (in which the B spins are excited by all the successive pump pulses). While PELDOR is a double frequency experiment, there exist other single frequency techniques for distance measurements such as double quantum coherence (DQC)31, single frequency technique for refocusing dipolar couplings (SIFTER)32, and RIDME33. DQC and SIFTER are used less frequently, mainly due to the requirement for non-selective broadband pulses, which can excite the whole spectrum (or both the coupled spins). RIDME has been shown to be a very sensitive technique for distance measurements involving fast-relaxing metal centers33,34. However, a stronger background decay than in the PELDOR experiment makes the separation of for longer distances more complicated.
Spin labels for in-situ PELDOR
Routinely, spin labeling and PELDOR are performed following the extraction and purification of the protein from the native environment. With recent developments in spin label chemistry, PELDOR experiments have been performed in a few cases under in-situ conditions (Table 1). For soluble proteins, in-situ PELDOR has been demonstrated following exogenous addition of the protein to E. coli, HeLa cells, or oocytes12,14,35,36. However, with MTSL, the disulfide bond that connects the spin label with the cysteine in the protein of interest becomes less stable under the reducing cellular environment. The maleimido-proxyl label (3-Maleimido-2,2,5,5-tetramethyl-1-pyrrolidinyloxy), which is attached to the protein via a C-S bond, is more stable in-situ12, although the nitroxide moiety may still be reduced into a hydroxylamine. In addition, the maleimido-proxyl may produce undesirable side reactions with amines. Sterically shielding the nitroxide moiety by substituting the gem-dimethyl groups with gem-diethyl groups has been shown to provide kinetic stability against reduction37. Such a protected label when functionalized with the maleimide group (called as M-TEMPO) is shown to be more stable in the cellular environment38. The limited stability of the nitroxide spin labels stimulated the application of redox-stable Gd3+ and trityl (triarylmethyl) spin labels for in-situ EPR. Gd3+ is found to be stable inside several cell types and enabled PELDOR on exogenously introduced molecules with high sensitivity14,39–41. With the ongoing efforts to further reduce the size of the chelator and the linker, Gd3+ would become an ideal spin label for in-situ EPR42. Trityl is also stable under reducing conditions and provides higher sensitivity due to its narrow EPR spectrum43. Trityl-nitroxide PELDOR has been shown to be a versatile tool for observing protein-ligand interaction in native E. coli membranes44. Trityl-Fe3+ distances have been determined using relaxation-induced dipolar modulation enhancement (RIDME) experiments on cytochrome P450 inside Xenopus laevis oocytes45. The major drawbacks of trityl labels are their large size and the tendency to aggregate when exposed to membranes or whole cells44. Genetic encoding has been tested for direct incorporation of an unnatural amino acids, which can be spin labelled6 or carrying a nitroxide label7, but reduction of nitroxide in the cellular environment has prevented the in-situ application of this approach. Despite these progresses, in-situ EPR is limited only for those molecules that can be purified and spin labeled in-vitro. For membrane proteins this approach is not feasible as the protein needs to be spin labeled directly in the complex native environment. As a result, the labeling of membrane proteins in native environments for spectroscopic investigation presents a major challenge for structural biologists.
In situ PELDOR on bacterial outer membrane proteins
We have developed a general strategy to make accurate distance measurements within outer membrane proteins in intact E. coli and native outer membranes using PELDOR. The cell envelop of Gram-negative bacteria consists of an inner membrane (IM) surrounding the cytoplasm and an outer membrane (OM) that protects the cells from harsh conditions including antibiotics (Fig. 1). The periplasm separates these two membranes. The OM is an asymmetric bilayer made up of phospholipids (PL) and lipopolysaccharides (LPS) and it harbors numerous β-barrel proteins (OMPs). OMPs range in size between 8 to 26 β-strands and many of them are being explored as targets for novel drugs. These proteins rarely have reactive cysteines, which decreases non-specific signals that might result from in-situ spin labeling46,47. We exploited this natural cysteine-exclusion for selective MTSL labeling and in-situ PELDOR measurements of OMPs44,47–49. In E. coli, spin labeling can be performed at solvent-exposed sites on the extracellular side under ambient conditions. Alternatively, the use of isolated OM preparations provides access to both membrane surfaces for spin labeling and ligand binding. In this protocol, we provide a detailed procedure for the site-directed mutagenesis, protein expression, OM isolation, spin labeling in E. coli and OM, sample preparation, PELDOR set up, and data analysis.
Figure 1 |. Schematic view of the cell envelop of Gram-negative bacteria.

The cell envelope of Gram-negative bacteria consists of an inner membrane (IM) and an outer membrane (OM), which are separated by the periplasm. The IM is a phospholipid (PL) bilayer, whereas the OM is an asymmetric bilayer consisting of PL and lipopolysaccharide (LPS). The IM contains α-helical proteins and the OM harbors numerous β-barrel proteins (or outer membrane proteins, OMPs) including the porins, which are essential for bacterial growth or pathogenicity. The OM also contains peripherally attached lipoproteins (LP). The OMPs rarely have reactive thiols and their cysteine mutants can be labeled with MTSL in E. coli or isolated OM with minimal background labeling.
Applications of the method
OMPs consist of autotransporters, channels for specific substrates, general porins, protein folding machinery, transporters, and proteins involved in adhesion, invasion, or evasion of host cells. They are involved in diverse essential physiological processes such as membrane biogenesis, motility, infection, immune response, transport, resistance to toxic compounds, and signaling etc., and many of them are highly sought-after targets for novel drugs50. Currently, there is a need to understand the structure, function, and dynamics of OMPs in their native environments. Over the past few years, there is increasing evidence that the native environment might significantly influence folding, function, oligomerization, and structure of OMPs51–53. For example, it has been observed that the detergent extraction required for the high-resolution methods can significantly alter OMP conformation54,55. Some of the OMP complexes such as the β-barrel assembly machinery (BAM)51,52,54,55 or the lipopolysaccharide transport system (Lpt)56 themselves are responsible for the OM biogenesis. Unlike the other membrane transport complexes which transport substrates across the membrane, BAM and Lpt insert their substrates directly into the membrane. Thus, the asymmetric OM (which is too complicated to be reconstructed in-vitro) forms an integral part of the overall structure of these complex machines. Our protocol describes an approach to validate OMP structures in the native OM and in intact E. coli. A comparison of in-silico PELDOR simulations with experimental data can reveal the similarity or the differences in the structure between in-vitro and in-situ conditions36,37 (Fig. 4e).
Figure 4 |. In-situ PELDOR in E. coli.

(a) The extracellular loops carrying the positions 188 and 399 are highlighted on the BtuB crystal structure (PDB 1NQH). (b) Original PELDOR data in E. coli as indicated. For WT BtuB (which is naturally Cys-less), the data fit into a stretched exponential decay (), which could not be measured longer due to the weak signal. The data are slightly shifted along the vertical axis for clarity. (c) The dipolar evolution (in yellow) obtained for the 188R1/399R1 PELDOR after correction for the intermolecular contribution () and the corresponding fit from TR (in black). The modulation depth (Δ) value is indicated. Overall, the data suggests a two-dimensional distribution of the spins over the large cell surface and deviation of the value for d (from 2.0) might be for other reasons including the membrane curvature and sample inhomogeneity. (d) The dipolar spectrum obtained with Fourier transformation (in yellow) or TR (in black) of c. (e) Interspin distance distributions obtained from TR of c. The corresponding simulation on the BtuB crystal structure (PDB 1NQH) using the MMM software is overlaid (in violet), which suggests a very good agreement between the conformations observed in the crystal structure and E. coli.
OMPs show variations in the oligomeric structure from monomers to trimers50. It remains unknown whether such oligomerization persists in-situ. Recent biochemical and biophysical data suggest that the membrane environment can induce homo-oligomerization as well as heterologous protein-protein interaction for proteins, which were observed as a monomer in the crystal structures53,57. Such interactions may govern their local and global dynamics as well as spatial clustering into micro-domains called OMP islands53. Our protocol can be used to address such homo- or heteromolecular interactions at high-resolution in the native environments.
OMPs can undergo large conformational changes or sometimes a shift in the overall conformational equilibrium during function48–50,55,56. Such changes might be mediated through interactions with other molecules like the lipids in the OM, substrates, ligands, toxins, transducers, or a subunit of the molecular complex. As we demonstrated earlier, our protocol enables the observation of such long-range conformational changes directly in E. coli and native OM48 (Fig. 4 and Supplementary Fig. 1). In this example, the extracellular ligands or the interacting molecules could be added from outside. Alternatively, such investigations can be performed in the absence of an interacting protein (for e.g., through the knockout of a gene, which encodes an interacting protein located in the OM or the periplasm) to elucidate the role of a particular protein-protein interaction in an oligomeric complex for the overall structure and conformational changes. Our approach can also be used to observe protein-ligand/substrate interactions44,47,49 or to follow conformational changes of a cell surface receptor or its cognate ligand (such as a toxin or viral proteins) following their interaction. The OM preparation offers a versatile solution in which spin labeling can be achieved on either side of the membrane (Fig. 3, Supplementary Figs. 1 and 2). Also, protein-ligand interactions or conformational changes can be monitored from both the extracellular and the periplasmic sides47–49. This permits the in-situ observation of cross membrane allosteric coupling following events on one side of the membrane49. For the heterooligomeric complexes, PELDOR using orthogonal spin labels, as we demonstrated in the native OM44, would be a potential tool to monitor the intersubunit interactions as well as the intrasubunit conformational changes within the same sample.
Comparison with other methods
Our protocol offers several advantages for in-situ structural biology of membrane proteins over existing structural biology tools. Although DNP-enhanced ssNMR58 is emerging as a promising tool for structural investigation of membrane proteins under native conditions, difficulties with selective labeling, lower sensitivity and spectral resolution, insensitivity to longer distances (> 0.2 nm), and maintaining cell integrity during the measurement period are major challenges. Cryo-electron tomography with subtomogram averaging allows structural investigation in the native environment59. However, reconstruction of a membrane protein structure in-situ with sufficient resolution has not been achieved yet. PELDOR spectroscopy and single-molecule Fluorescence Resonance Energy Transfer (smFRET) are unique tools to address conformational changes and equilibrium dynamics of large membrane protein complexes19,60. Both PELDOR and smFRET can be used to determine distances between two labeled sites. In particular, smFRET is useful for observing conformational dynamics with spatio-temporal resolution and superior sensitivity. However, the requirement for site-specific orthogonal labeling on two distinct sites, limited range of the distances for a given FRET pair, and the large size as well as the uncertainty in the mutual orientation of the fluorophores pose major challenges. Fluorescent proteins have been widely used for in-situ fluorescence studies, but their large size limits the scope for smFRET. Current approaches for in-situ smFRET employ exogenous introduction of biomolecules labeled with organic fluorophores61, which is limited to soluble molecules. In contrast, EPR spectroscopy uses identical spin labels at both sites that are relatively small in size and provide structural information on the ensemble with high precision over a wide range of distances (1.5–16 nm)16. Moreover, for OMPs spin labeling and distance measurements can be performed in E. coli or native OM to observe their structure and conformational changes.
Overall, the protocol described here is relatively straightforward. In principle, structural and dynamic information can be obtained for any OMP in E. coli and native OM. For example, with the high-copy-number pAG1 vector, BtuB can be overexpressed up to ~105 copies/cell47,62, which is similar to the normal expression level of many OMPs53. With different approaches currently available for protein expression, such levels can be achieved for any OMP of interest. We have not observed any protein aggregation or any structural distortion following overexpression of the target protein (Figs. 3 and 4). With the single cysteine mutant of BtuB (T188C), interspin distances could be measured only when a spin labeled hydroxycobalamin (TEMPO-HOCbl, which binds to BtuB) is added from outside (see Box 1 and Supplementary Figs. 3–5 for TEMPO-HOCbl synthesis and characterization). Spin labeling of cobalamin did not significantly alter its affinity for BtuB49. However, it is possible that non-specific protein-protein interaction or aggregation may be triggered depending on the expression vector, growth conditions, or the E. coli strain. The expression can be done either with a minimal or a rich medium in a constitutive or induced manner47,63. The labeling procedure is rather easy and as PELDOR uses identical spin labels at both sites, site-specific labeling with orthogonal labels as in FRET is not required. Labeling is achieved by adding MTSL to the cell exterior and unreacted spin labels can be removed by few washing steps. As we previously demonstrated with a trityl label44, this protocol can be adopted to attach other spin labels, which are functionalized to react with cysteines. In practice, any solvent exposed sites can be spin labeled with high efficiency under the optimized labeling conditions described here. The signals arise exclusively from the spin labels; therefore, the background signals are significantly suppressed. Sarkosyl solubilization of the IM64 further reduces the background labeling and thereby enriches the signals for the cysteine mutants in the isolated OM (Supplementary Fig. 2a-b). Compared to E. coli, OM provides several advantages. Overall, sample handling and spin labeling is easier as there are no concerns of cell viability or spin label reduction (see limitations). Also, OM can be concentrated to a higher degree, thereby providing a higher S/N while keeping the protein in the native membrane environment.
Box 1 |. TEMPO-hydroxycobalamin (TEMPO-HOCbl) synthesis, purification, and characterization.
TEMPO-HOCbl synthesis
TEMPO-HOCbl was synthesized according to a protocol we previously described for the synthesis of TEMPO-cyanocobalamin (TEMPO-CNCbl)44. First, the ribose-5’-hydroxyl was activated with 1,1-Carbonyl-di-(1,2,4-triazole) (CDT, Sigma) and then reacted with 4-amino TEMPO (Sigma). The synthesis was performed using dry solvents under argon atmosphere. 0.1 g hydroxycobalamin was dissolved in 50 mL DMSO and 0.036 g CDT was added, and the reaction mixture was stirred for 30 minutes at room temperature. Then 0.13 g 4-amino-TEMPO was added, and the reaction mixture was stirred at room temperature for 12 h. TEMPO-HOCbl was precipitated by adding 200 mL 1:1 mixture of acetone and diethylether. The precipitate was separated by centrifugation at 4 °C for 15 min at 4000xg. The centrifugation was repeated once more with the supernatant. The pooled precipitate (TEMPO-HOCbl) was washed with acetone, centrifuged as before, dried overnight under air, and freeze-dried. The yield of the raw product was 0.091 g (0.059 mol), which corresponds to 80% of the theoretical yield.
TEMPO-HOCbl purification and characterization
The crude product from the above reaction was analyzed using high performance liquid chromatography (HPLC, Agilent 1200 series) on a BDS-C18 column (5 μM, 2×250 mm from Hewlett-Packard) with detection at both 254 and 316 nm (Supplementary Fig. 3). 8 μL of 1 mM TEMPO-HOCbl was injected and eluted at 1 mL/min flow rate with 400 bar pressure at room temperature. The following solvent system was used for elution: solvent A: water; solvent B: methanol and elution was performed at a linear gradient of 15–60% B in 40 min. MALDI-ToF-MS was performed on a Voyager STR Workstation DE pro (Applied Biosystems) with 100 kW laser peak power (337 nm) in a matrix made of 2,5 Dihydroxybenzoic acid and 6-Aza-thiothymine (Supplementary Fig. 4). The LC-ESI-MS measurements were performed with Shimadzu LCMS-2020 system (Supplementary Fig. 5). A water/acetonitrile solvent system (both containing 0.1% formic acid) was used for column elution with a capillary voltage of 3–4 kV, 5 bar N2 pressure, and a mass range of 80–2000 Da with single quad detection.
Limitations
The protocol described here can be used for studying OMPs in E. coli and native OM. In E. coli, labeling is limited to solvent-exposed residues on the extracellular side. We have shown that positions located on the extracellular loops and the barrel lumen can be labeled in E. coli47,48. Spin labeling can however not be achieved in the periplasm under the conditions described here using MTSL or the maleimido proxyl label, which is attached to the protein through a C-S covalent bond (Table 1). Although our optimized protocol provides good labeling efficiency, labeling has a limited time window due to reduction of the MTSL molecules. After addition to the cell suspension, MTSL is rapidly reduced with a half-life of ~10 min (Supplementary Fig. 6) and the maximum labeling happens already within in the first few minutes (Fig. 5c). The mechanism for nitroxide reduction by E. coli remains unknown as of now. Currently, we are working on alternate strategies to introduce more stable spin labels (such as the protected nitroxide, Gd3+, or Cu2+), which would also allow us to extend spin labeling into the periplasm. Notably, spin label reduction is not an issue with the native OM preparation.
Figure 5 |. In-situ MTSL labeling of BtuB in E. coli.

(a) RT CW-EPR spectra of BtuB obtained in live E. coli after labeling with 500 μM MTSL at OD600 = 25 for 10 min at 25 °C. (b-d) MTSL labeling of BtuB 188C-399C in E. coli at 25 °C. Spin concentrations of the E. coli (normalized to unit OD600) are given on the y-axis. (b) Spin concentration after labeling with different MTSL concentrations. Labeling was performed at OD600 = 25 for 10 min. (c) Spin concentration after labeling for different time intervals. MTSL labeling was performed at OD600 = 25 with 500 μM MTSL for different times as indicated. For the zero-time point, cells were pelleted immediately after mixing with MTSL (overall, which took an additional 6–7 min including centrifugation and EPR measurement). (d) Spin concentration after labeling at different OD600 values. Labeling was performed with 500 μM MTSL for 10 min at different OD600 values as indicated. The inset shows a contour plot summarizing the experiments in c and d. The shaded area indicates a small window for the incubation time and the cell density under which maximal labeling can be achieved. Error bars indicate a 15% error, which is typical for spin quantification using RT CW EPR spectroscopy. Similar trends were observed between independent experiments.
Although the OMPs are devoid of reactive cysteines, we always observed background labeling with E. coli and OM (see the wild-type (WT) in Figs. 4b and 5a). It is possible that the labels bind to the membranes through physical adsorption. In agreement, previously we observed that a more hydrophobic trityl label shows a higher binding (and aggregation) in the OM as compared to MTSL44. Thus, adhesion to the cell membrane might be the major contribution, if not alone for the non-specific labeling. In general, background labeling is one of the major hurdles for in-situ structural biology. Here, the signals from background labeling contribute exclusively to the intermolecular part ( in Eq. 1) of the PELDOR signal. In effect this reduces the modulation depth to 8–10% ( in Eq. 2), but most importantly background signals do not interfere with distance measurements on the target proteins (Figs. 3b and 4b). Even though the bulk spin concentration is in the range of 50–100 μM, the background of the PELDOR data shows a rather fast decay in both E. coli and OM. This indicates inhomogeneous distribution of BtuB (spin labels) and possibly spatial clustering into micro domains with other proteins. Despite such a background decay and a lower modulation depth, we are able to measure a high quality 4 μs trace within ~12 h. The decreased modulation depth value together with the background decay somewhat reduce the overall sensitivity, but do not interfere with the outcome or reliability of the data analysis.
Our in-situ protocol requires the target OMP to be overexpressed (~105 copies/cell). The copy number of OMPs vary from few hundreds to hundreds of thousands (102–105) of copies per cell. Thus, some of the OMPs (such as porins) can be studied at their native expression level. E. coli contains ~103 BtuB/cell and in our experiments, it was overexpressed up to ~105 copies/cell47, which did not cause any aggregation or oligomerization. However, in some cases it could happen that overexpression alters the local environment of the protein by modulating interactions with other proteins or lipids, which may lead to structural distortion or aggregation. For OMPs that interact with partners located in periplasm or inner membrane, the fraction of molecules engaged in such interaction would be significantly reduced unless all of them are simultaneously overexpressed. Also, it should be kept in mind that the genetic background of the cells used for protein expression itself may influence the outcome of an experiment or the observed structural properties.
Due to fast relaxation of MTSL at higher temperatures, PELDOR is routinely performed in frozen solution. The sample is frozen in presence of a cryoprotectant, usually 10–30% glycerol, and other than slightly modifying the spin label rotamer distribution, freezing as such might not alter the structural properties existing in solution65. Glycerol has been widely used as a vitrifying agent in biochemistry to eliminate protein aggregation. In order to suppress spin relaxation from nuclear spin diffusion, deuterated glycerol (glycerol-d8) is used. Spin labels attached to BtuB in E. coli or OM displayed a TM value of ~2 μs and addition of 15% d8-glycerol extends the TM up to 3.5 μs47. At this concentration, glycerol did not show any effect on the tested interspin distance distributions (data not shown) and such an enhancement of TM is typical for solvent exposed positions in detergent solubilized membrane proteins. However, placing labels closer to membrane region would further reduce TM (to 1–2 μs)29 and thereby limit the upper range of accessible distance to ~5 nm. Sample deuteration or the application of 5-pulse DEER or the 7-pulse CP PELDOR are approaches that could be taken to determine longer distances in such scenarios.
Experimental design
In this protocol, we describe a detailed protocol for spin labeling and PELDOR measurements on OMPs in E. coli and native OM44,47–49. The protocol consists of the following stages: SDM to introduce cysteines (Steps 1–2), protein expression in E. coli (Steps 3–4), spin labeling in E. coli (Steps 5–7 and 8(A)i-vii) or OM (Steps 5–6 and 8(B)i-xii), PELDOR measurements (Step 9–21), and data analysis (Step 22). Our procedure enables the observation of protein-protein or protein-ligand interactions as well as the characterization of long-range conformational changes of OMPs in the native environments.
Introducing cysteines for spin labeling.
SDM is done using standard protocols to introduce cysteines at the desired positions in the WT protein. OMPs rarely have reactive cysteines or when present they are either buried or cross-linked and often there is no need to create the Cys-less background. When performing PELDOR measurements in E. coli, positions located on the extracellular loops and the plug domain inside the β-barrel can be mutated to cysteines for labeling. In contrast, when performing PELDOR measurements on native OM, mutation and spin labeling can be performed at positions on either side of the membrane. The accessibility of the target site for the spin label is critical for successful labeling and buried sites should therefore be avoided. This also precludes any possible structural distortion of the protein. In addition, residues with functional or structural roles such as Gly, His, Pro, and charged or aromatic amino acids should be avoided when possible. Residues like Ala, Ile, Leu, Met, Ser, Thr, or Val would be more favored for SDM and labeling. Whenever possible, the functional integrity of the mutants should be verified (using in-situ or in-vitro functional assays) after spin labeling. It is recommended that an in-silico spin labelling and PELDOR experiment be performed for the selected sites (when a structure is available), e.g. by using one of the available programs such as MMM21, MtsslWizard66, or PRONOX67 before proceeding with SDM.
OMP expression.
The OMP may be expressed either in a constitutive or induced manner, in a minimal or rich media, and the best conditions should be tested in a case-by-case manner. Although we did not encounter any issues with expression levels in different strains; different vectors, strains, media, and protocols may be tested when the expression level is not sufficient. The RK5016 strain (MC4100, metE70, argH, btuB, recA)68 we used for BtuB expression is derived from the E. coli K-12 / MC4100 strain, which has been extensively used for genetic experiments and also its genome is well characterized69. This strain allowed us to perform in-situ PELDOR in both whole cells and native OM. Another E. coli K-12 strain KDF541 was used for in-situ EPR of the ferric enterobactin transporter FepA in whole cells46. In both cases, the protein was expressed in a constitutive manner (BtuB from the lac promoter and FepA from the native promoter) in minimal medium. E. coli BL21(DE3) cells, one of the most popular and commercially available strains also can be used as we demonstrated for in-situ PELDOR of the ferrichrome transporter FhuA in native outer membranes63. We could achieve similar results for the ferric enterobactin transporter FecA as well. For spin labeling in native outer membranes, an identical protocol works with both K-12 and BL21(DE3) strains, suggesting that our protocol can be used for studying any of the OMPs in the native membranes. In our experience, the K-12 strains appear to be better suited for investigations in whole cells. However, we anticipate that BL21(DE3) or other commercial strains will also be useful for whole cell investigations with further optimization for protein expression and spin labeling.
Obtaining efficient spin labeling.
Calculating the labeling efficiency in-situ is a difficult task. If a spin labeled ligand (or an interacting protein fragment) is available, binding analysis using room temperature (RT) CW–EPR can be used to quantify the expression level and the labeling efficiency of the protein of interest47. Signals for the WT background (under identical conditions) should be subtracted to account for non-specific labeling and ligand binding. For surface-exposed positions, a high labeling efficiency of >80% can usually be achieved. Most often, overexpression of the OMP does not cause protein aggregation in the membrane and it can be checked by measuring interspin distances between singly labeled variants (Fig. 3b). When there is evidence for non-specific interaction or aggregation, the expression vector, E. coli strain, or the growth conditions may be further optimized. Spin labeling in the complex cellular environment requires more selective and stringent conditions as compared to the labeling of purified proteins. Labeling is fraught with background labeling and the fast reduction of MTSL. Thus, the labelling conditions as well as the properties of the spin label itself become crucial for successful labeling. We tested labeling at different cell densities, MTSL concentrations, and incubation times. In our experience, maximal labeling is achieved with 50 μM MTSL, already in the first few minutes of incubation. Higher MTSL concentrations have no effect and prolonging the incubation time reduces the overall signal intensity (Figs. 5b and 5c). Overall, labeling appears to be more efficient at lower OD600 (< 10) values and MTSL is reduced faster at higher cell densities (Fig. 5d). By prolonging the incubation time at higher spin concentrations (for e.g. 1 h with 150 μM MTSL), it is possible to reduce signals from non-specific labeling47, although the labeling of the cysteine as well is decreased. Following incubation, free spin labels are easily removed by 2–3 rounds of washing and resuspension. Free spin label reduces the modulation depth and hence the overall sensitivity; therefore, the number of wash steps may be adjusted as necessary to reduce unbound label below detection.
Isolation of the OM.
Removal of the IM (from the cell envelop), which carries several α-helical proteins is necessary for efficient spin labeling in the native OM (Supplementary Fig. 2a-b). When labeled on the intact envelope, cysteine mutants and the WT sample revealed similar spectra47. Selective solubilization of IM using sarkosyl reduces non-specific labeling and gives a much larger signals for cysteine mutants compared to WT sample. Cysteines located on either the extracellular or the periplasmic sides can be labeled with isolated OM (Fig. 3 and Supplementary Fig. 2c-d) and excess of MTSL is removed after few rounds of washing. Spin labeled ligands or proteins (tested up to 10 kDa) can be added to observe their interaction with the target OMP from either side of the membrane without any extrusion or freeze-thaw cycles47–49, meaning that the native OM are also accessible to larger substrates. Although our current labeling protocol provides samples with sufficient quality for the PELDOR experiment, it may be possible to reduce the background labeling through further optimization of the labeling conditions. Also, alternative methods for OM isolation such as density gradient centrifugation70 may be tested.
Sample handling and PELDOR measurements.
We have seen that the MTSL signals are gradually lost even if the cells are kept on ice. Therefore, the cells should be transferred to the PELDOR tubes and be frozen immediately after spin labeling. Care should be taken to ensure that the cells are not too concentrated as they may lyse during transfer into the EPR tubes. Although significantly smaller than the signals for the double cysteine mutants, the WT cells also give (non-specific) signals under the optimized labeling conditions (Figs. 4b and 5a). The more apolar the spin label, the higher the non-specific labeling becomes47, suggesting a physical adsorption of the labels to the cells. Therefore, it is strongly recommended to always measure the WT sample to rule out the contribution from background labeling. Due to the stochastic nature of the non-specific labeling, the WT sample does not yield any particular distance and only contributes to the intermolecular component () of the PELDOR signal. The PELDOR experiments described in this protocol are performed following standard procedures11,71 (Steps 9–21). Addition of 20% d8-glycerol significantly prolong TM in both E. coli and OM47. This enables the observation of the dipolar evolution for up to 4 μs within ~12 h. By combining the advanced sequences such as the 5-pulse DEER or the 7-pulse CP-PELDOR with complete deuteration (of proteins and lipids by growing in deuterated media), it might be possible to significantly reduce the measurement time and prolong the observation window of the dipolar evolution.
MATERIALS
REAGENTS
E. coli K-12 / RK501668 (MC4100, metE70, argH, btuB, recA) or BL21(DE3) cells. Another K12 strain KDF541 has been used for FepA46. E. coli RK5016 cells are available upon request from the corresponding author.
Expression vectors for the protein of interest (POI); in the example described in this protocol, we use a pUC8-based plasmid (pAG1) carrying btuB under the control of the lac promoter for constitutive expression. Inducible expression from the T7 promoter under the control of the lac operator (pHK763) also worked for FhuA63. These plasmids are available upon request from the corresponding author.
QuikChange Lightning Site Directed Mutagenesis Kit (Agilent Technologies, cat. no. 210519)
Plasmid purification kit (Qiagen, cat. no. 27104)
K2HPO4 (Sigma-Aldrich, cat. no. P8281)
KH2PO4 (Sigma, cat. no. P5655)
(NH4)2SO4 (Sigma, cat. no. A4418)
Sodium citrate (Aldrich, cat.no. W302600)
L-Methionine (Sigma-Aldrich, cat. no. M9625)
L-Arginine (Sigma-Aldrich, cat. no. A5006)
D-(+)-Glucose (Sigma-Aldrich, cat. no. G5767)
MgSO4 (Sigma, cat. no. M2643)
CaCl2 (Sigma, cat. no. C5670)
Thiamine (Sigma-Aldrich, cat. no. T4625)
Ampicillin 100 mg/mL (Sigma, cat. no. A5354)
MOPS (Sigma, cat. no. M9024).
Sodium chloride (Sigma-Aldrich, cat. no. S7653)
PMSF (Sigma, cat. no. 78830) ! CAUTION PMSF has acute toxicity and can cause damage to skin or eyes. Wear appropriate eye and skin protection.
N-Lauroylsarcosine (or Sarkosyl, Sigma, cat. no. 61743) ! CAUTION Sarkosyl is very toxic and can cause serious damage to eyes and skin. Wear appropriate eye and skin protection.
(1-Oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) Methanethiosulfonate spin label (MTSL), (TRC, cat. no. O875000)
TEMPO (Aldrich, cat. no. 214000)
DMSO (Sigma, cat. no. D2650) ! CAUTION DMSO is an irritant for eyes and skin. Wear appropriate eye and skin protection.
Sodium hydroxide (NaOH, Sigma, cat. no. S2770) ! CAUTION NaOH is corrosive to skin and can cause serious eye damage. Wear appropriate eye and skin protection.
d8-glycerol (Aldrich, cat. no. 447498)
EQUIPMENT
Microcentrifuge (up to 20,000xg, Thermo Scientific Heraeus, cat. no. 75002410)
15- and 50-mL Falcon tubes (Fisher Scientific, cat. no. 12–565-269 and 12–565-271 respectively)
Petri Dishes (Fisher Scientific, cat. no. FB0875713)
Thermal cycler (Eppendorf, Catalog No. 6311000010)
ThermoMixer (Eppendorf, Catalog No. 5382000015)
Syringe filters for sterilization (PVDF, 0.22 μM from Millipore, cat. no. SLGV033RS)
Standard equipment for bacterial cell culture (pipettes, autoclave, clean bench, culture flasks, incubator shakers etc.)
Centrifuges for pelleting cells (~5000xg, Sorvall, cat. no. 70900171)
Centrifuges for pelleting membranes (≥200,000g): Beckman Coulter Optima XE 90 ultracentrifuge (cat. no. A94471) and Sorvall MTX 150 Micro-Ultracentrifuge (cat. no. 46962)
French pressure cells and press (SLM Instruments, Inc.,)
Ultrasonic water bath (Branson, cat. no. CPX-952–539R)
Micropipettes (20 μL, BRAND GMBH, cat. no. 708718)
Micropipette controller (BRAND GMBH, cat. no. 25800)
Disposable syringe needles (120 mm length, Ø 0.8 mm, B Braun, cat. no. 466564/3)
Disposable syringes (1 mL, B Braun, cat. no. 9166017V)
Liquefied N2 (Linde) and He (AirLiquide) ! CAUTION Very toxic gases. Inhalation leads to oxygen-deficient symptoms and contact with skin can cause frostbite.
Bruker E500 CW X-band (9.4 GHz) spectrometer equipped with a SHQE cavity.
ELEXSYS E580 pulsed Q-band (34 GHz) EPR spectrometer (Bruker) equipped with a PELDOR unit (E580−400U), continuous-flow helium cryostat (CF935, Oxford Instruments), temperature control system (ITC 502, Oxford Instruments), ELEXSYS SuperQ-FT accessory unit, 150 W TWT amplifier (Applied Systems Engineering Inc.), and a Bruker EN5107D2 cavity.
Suprasil quartz EPR tubes (outer Ø 1.6 mm, Wilmad-LabGlass, cat. no. WG-222T-RB)
Software
LINUX (for EPR data acquisition) and Windows or Mac environments (for data processing and analysis)
Xepr from Bruker for EPR data acquisition
MATLAB from MathWorks: https://www.mathworks.com/products/matlab.html
MATLAB-based DeerAnalysis software: http://www.epr.ethz.ch/software.html
MATLAB-based MMM software: http://www.epr.ethz.ch/software.html (or MtsslWizard: https://pymolwiki.org/index.php/MtsslWizard)
Pymol or similar software for molecular visualization
Any software for nucleotide sequence analysis
ORIGIN or similar software for spectroscopic data analysis
REAGENT SETUP
10x minimal medium Dissolve 105 g K2HPO4, 45 g KH2PO4, 10 g (NH4)2SO4, and 5 g sodium citrate in 1 L Milli-Q water and autoclave. It can be stored at room temperature (RT: 22–26 °C) for several weeks. Alternatively, a rich medium such as LB (Luria-Bertani) or TB (Terrific Broth) also may be used.
2% (wt/vol) Methionine and Arginine Dissolve 2 g each in 100 mL Milli-Q water and filter sterilize.
20% (wt/vol) Glucose + thiamine Dissolve 20 g glucose and 50 mg thiamine in 100 mL Milli-Q water and filter sterilize.
1 M MgSO4 Dissolve 12.04 g in 100 mL Milli-Q water and filter sterilize.
1 M CaCl2 Dissolve 1.11 g in 10 mL Milli-Q water and filter sterilize. These sterile solutions can be stored at RT for several weeks when properly handled to prevent microbial contamination.
1x minimal medium Mix 100 mL of the 10x medium with 5 mL each of 2% Arginine and Methionine, 10 mL of 20% (wt/vol) glucose + thiamine, 3 mL of 1 M MgSO4, and 300 μL of 1 M CaCl2 and make up the final volume to 1 L. CRITICAL Prepare fresh each time and perform mixing on the clean bench to keep the medium sterile.
Plasmids for the expression of BtuB T188C or T188C-G399C mutations were introduced in BtuB (which is cloned into a pAG1 vector) using QuikChange Lightning Site Directed Mutagenesis Kit.
100 mM PMSF Dissolve 174.2 mg in 10 mL isopropanol and store at −20 °C for several months.
Spin labeling buffer Dissolve 5.2 g MOPS and 1.6 g NaCl in Milli-Q water and adjust the pH to 7.5 in a final volume of 500 mL. Prepare the buffer fresh before the experiment.
100 mM MTSL Dissolve 10 mg in 377 μL DMSO and store at −20°C for up to an year.
TEMPO standard (calibration standard for estimating spin concentration) Dissolve 1.57 mg in 10 mL Milli-Q water for a 1 mM stock. Perform a serial dilution to prepare 4–5 samples in the range of 50–300 μM. The solutions can be stored at −20°C for up to an year.
PROCEDURE
Plasmid construction and mutagenesis. TIMING: 5–7 days
1| Clone the POI into a suitable bacterial expression vector. Please see reagents for the expression vectors which worked in our hands. In the example described here, we used the pUC-8-based pAG1 plasmid, which constitutively expresses btuB (see Reagents). The plasmid is available upon request from the corresponding author.
CRITICAL STEP The POI can be expressed either in a constitutive or induced manner and should be tested in a case-by-case manner (see Experimental Design).
PAUSE POINT Once the plasmid has been constructed, it can be stored in −20°C until proceeding with the next step.
2| Introduce the cysteines using SDM following the manufacturer’s instructions at the positions which will be labeled with MTSL.
CRITICAL STEP If the POI has any native cysteines, they should first be mutated to generate a Cys-less protein. If the cysteines are buried with low accessibility for MTSL, they need not be mutated. Native cysteines may be mutated to alanine, valine, serine, or to a combination of those amino acids. With regard to hydrophobicity, alanine or valine is a better match for cysteine; however, difference in the overall size may lead to structural or functional distortions in some cases. Serine is a good match in terms of the size, but its increased polarity may have an adverse effect on the protein. Therefore, it is recommended to test the function of the Cys-less construct with purified protein when feasible.
CRITICAL STEP It may be necessary to generate several cysteine mutants for optimal levels of expression and spin labeling (see Experimental Design). With the double cysteine mutations for PELDOR measurements, it is recommended to measure one or both of the corresponding single cysteine mutants to rule out aggregation or to identify oligomerization, if any. When feasible, it is advised to test spin labeling efficiency and PELDOR with the purified protein as well.
PAUSE POINT The mutated plasmids can be stored in −20°C until proceeding with the next step.
? Troubleshooting
Transformation. TIMING: 1.5 hours
3| Transform the plasmid into E. coli as follows: Mix 1 μL of the plasmid with 50 μL of E. coli cells in a sterile 1.5 mL microcentrifuge tube. Incubate at 42°C for 45 seconds and transfer the tube into ice for 2 min. Add 250 μL of sterile LB medium and incubate at 37°C for 1 h. Spread 100 μL of the culture on a sterile LB Agar plate containing 100 μg/mL ampicillin and incubate at 37°C for overnight.
CRITICAL STEP Alternatively, the BL21(DE) strain can be used for IPTG-induced expression and spin labeling. Both K-12 and BL21(DE3) strains are useful for investigations in native membranes, whereas K-12 strains appear to be better suited for studies in whole cells (see Experimental Design for a detailed discussion).
CRITICAL STEP Perform transformation with the WT (or with the Cys-less background for proteins having native cysteines) plasmid as well to characterize the signals from background labeling.
Cell culture and protein expression. TIMING: 24 hours
4| Next day, transfer a single colony from the agar plates into 25 mL of 1x minimal media (see reagent setup) and incubate at 33°C for 6–8 h until the culture turns cloudy. Transfer 5 mL culture into 1 L sterile minimal medium in a 3 L Erlenmeyer flask and incubate at 33°C for 12–16 h.
CRITICAL STEP The incubation time and temperature should be thoroughly optimized to avoid non-specific protein-protein interaction or aggregation. Overgrowth may lead to cell lysis, which can result in MTSL reduction and poor spin labeling efficiency in E. coli. Expression may be performed in a rich medium such as LB as well63.
CRITICAL STEP Typically, a 1 L culture (on both minimal and rich media) gives cells far more than required for preparing several CW and PELDOR samples. A smaller culture volume could be used if the protein expression level is not negatively affected.
? Troubleshooting
In-situ spin labeling of the protein in E. coli and in the outer membrane. TIMING: 2–3 hours
5| Following protein expression, pellet the cells by centrifugation at 4°C in 500 mL tubes at 5,000xg for 10 min using a Sorval SLA-3000 rotor.
6| Discard the supernatant and resuspend the cell pellet from each 500 mL tube into 30 mL of precooled spin labeling buffer supplemented with 1% (wt/vol) glucose and keep on ice.
CRITICAL STEP The cells should be handled as gently as possible to avoid lysis. Precool the buffer on ice before use. Immediately proceed to the next step.
7| Determine the OD600 value. Dilute the sample to measure within the linear range (OD600 = 0.01–0.4). The OD600 value is usually between 70 and 100 at this point.
8| Spin label the protein in-situ in E. coli or in the outer membrane by following Option A or Option B respectively.
Option A: In-situ spin labeling of the protein in E. coli. TIMING: 2–3 hours
i| Transfer an appropriate amount of the cell suspension into a sterile 1.5 mL Eppendorf tube in duplicate and dilute with spin labeling buffer to 1 mL and a final OD600 value of 10. Store the remaining suspension on ice until used in Step 15.
CRITICAL STEP Higher OD600 values lead to rapid reduction of MTSL and lower signals (see Fig. 5d). Prepare the samples in duplicate for CW-EPR and PELDOR experiments. Multiple mutants may be prepared; however, the samples need to be rapidly processed in the subsequent steps, which may make it difficult to handle large number of samples simultaneously.
ii| Add 1 μL of 100 mM MTSL stock solution (100 μM final concentration) and incubate on a thermal mixer at 25°C for 1–5 min.
CRITICAL STEP Longer incubation leads to rapid reduction of MTSL and lower signals. Our experience shows that shorter incubation times give higher signals (see Fig. 5c). Prolonged incubation (150 μM MTSL for 1 h at RT) can be used to reduce the background signals47, however this will reduce the overall signals for the cysteine mutants as well.
iii| Pellet the cells by centrifugation at 5,000xg for 5 min at 4°C.
iv| Discard the supernatant, suspend the cells in 1 mL of precooled spin labeling buffer and pellet as described in Step iii.
CRITICAL STEP These washing steps should almost completely remove the free MTSL; otherwise the number of washing steps may be increased as required.
v| Discard the supernatant and remove any residual buffer with a micropipette. Suspend the cells into 20 μL precooled spin labeling buffer and keep on ice. immediately proceed with steps vi and vii.
CRITICAL STEP The volume of spin labeling buffer may be adjusted according to the amount of the cell pellet. Larger volumes will dilute the spin concentration and too small volumes would make it difficult to transfer the cells into the EPR tubes. Keeping the cells too long may lead to loss of signals.
vi| Add 20% d8-glycerol (vol/vol) to the sample and mix gently. Transfer 10–15 μL of the sample into a Suprasil quartz EPR tube (Ø 1.6 mm) using a syringe needle and immediately freeze in liquid N2.
PAUSE POINT The EPR tubes with the sample can be stored at −80°C (for several months) until performing the PELDOR measurements.
vii| Transfer the duplicate sample into a 20 μL micropipette. Measure the signal with a Bruker E500 CW X-band spectrometer (or a similar instrument) and estimate the spin concentration from an external calibration curve of TEMPO. Measure the cells expressing WT protein to estimate background labeling.
CRITICAL STEP 1 mL of a OD600 = 10 E. coli suspension contains ~8×109 cells. Depending of the final volume of the E. coli suspension (30–50 μL, from step v), the bulk spin (MTSL) concentrations varies between 30–100 μM, which is in a range obtained for purified membrane proteins. The active volume of the resonator is ~5 μL, which means the DEER/PELDOR experiments are performed with ~109 cells. A part of the signal may arise from background labeling depending on the labeling conditions.
? Troubleshooting
Option B: In-situ spin labeling of the protein in the outer membrane. TIMING: 8 hours
-
i|
Isolation of the outer membrane (Steps i-vi): Add 300 μL of 100 mM PMSF stock solution (1 mM final concentration) to the cell suspension from Step 6.
-
ii|
Lyse the cells with a French Press (press for 2–3 times until the suspension is translucent) at ~10,000 psi using 1/8” nylon balls.
-
iii|
Remove cell debris by centrifugation at 17,000xg for 20 min at 4°C in a Sorval SS-34 rotor.
-
iv|
Collect the supernatant, which contains cell envelope (OM+IM). Solubilize the IM with 0.5% sarkosyl (vol/vol)64 and mix it gently at RT for 2–3 min.
CRITICAL STEP Mix gently, but thoroughly. Incomplete solubilization of the IM can result in larger background signals or even produce additional distances.
-
v|
Dilute the volume to 60 mL with spin labeling buffer and pellet the OM by centrifugation at 220,000xg for 1.5 h at 4°C in a T-647.5 rotor.
-
vi|
Remove the supernatant and suspend the pellet into 7 mL SL buffer in a 15 mL Falcon tube.
PAUSE POINT The pellet after suspending into the spin labeling buffer can be stored at −20°C for several months.
-
vii|
In-situ spin labeling of the protein in the outer membrane (Steps vii-xii): TIMING: 5 hours. Homogenize the OM suspension by gentle mixing in an ultrasonic water bath.
CRITICAL STEP The membranes form small clusters during ultracentrifugation in Step v and complete homogenization is necessary for efficient spin labeling.
-
viii|
Add 7 μL of 100 mM MTSL (100 μM final) and incubate at RT for 1 h.
-
ix|
Remove free MTSL by pelleting the OM with centrifugation at 500,000xg for 45 min at 4°C in a S100-AT4 rotor (lower g may be used with prolonged centrifugation, for e.g., 1.5 h at 200,000xg).
-
x|
Discard the supernatant and collect the OM-containing pellet. Wash the OM by suspending in 7 mL spin labeling buffer and homogenize by repeatedly pipetting up and down or using an ultrasonic water bath. Centrifuge as in step ix.
CRITICAL STEP These washing steps should reduce the free MTSL concentration below detection; otherwise the washing steps may be increased as required.
-
xi|
Remove the supernatant and collect the OM by suspending the pellet into a final volume of 250–500 μL spin labeling buffer. For preparing PELDOR samples, take 10–20 μL of the OM suspension and add 20% (vol/vol) d8-glycerol.
PAUSE POINT The OM suspension can be stored in −80°C (for several months) until performing the PELDOR measurements.
-
xii|
Take 20 μL of the sample and measure the spin concentration as detailed in step 8 Option A.vii. Measure the OM from cells expressing WT protein to estimate background labeling.
CRITICAL STEP OM isolated from 1 L culture when suspended into 300–500 μL buffer typically gives 100–150 μM spin after MTSL labeling. Similar to E. coli, some of those signals arise from background labeling.
? Troubleshooting
DEER/PELDOR measurement in E. coli or outer membranes. TIMING: 12–24 hours
9| DEER/PELDOR experiments were performed according to standard protocols11,71 with some modifications. The procedure is described for a Q-band (34 GHz) Bruker E580 spectrometer having independent Microwave Pulse Forming Units (MPFUs) and equipped with a 150 W Traveling Wave Tube (TWT) amplifier, a continuous-flow helium cryostat (CF935, Oxford Instruments), an Intelligent Temperature Control system (ITC 502, Oxford Instruments), an ELEXYS SuperQ-FT accessory unit (Bruker), and a EN5107D2 resonator. Some modifications may be required depending on the spectrometer configuration (for e.g. for a spectrometer having an inbuilt Arbitrary Wave Form Generator (AWG)).
10| Cool the cryostat with the resonator down to 50 K using liquid helium and wait until the temperature is stable. The cryostat and the transfer line must be evacuated to enable efficient cooling.
11| Turn on the spectrometer and the TWT amplifier. Press the tuning button in the ‘Microwave Bridge Tuning’ panel, switch to tune mode, and change the microwave (mw) power from 60 dB to 20 dB, turn the reference arm off, and insert the frozen sample into the resonator. A frequency shift will be visible in the tune window. Change the frequency to find the resonance dip and adjust the position of the sample holder to overcouple the resonator. Readjust the frequency to the center of the resonator frequency.
? Trouble shooting (see Table 2)
TABLE 2 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2 | The cysteine mutant is expressed at very low levels | The mutated residue(s) is important for protein expression/stability | Choose another position for SDM. Test different vectors, strains, or mode of expression |
| 4 | No or poor cell growth after overnight culture | (1) Only few cells are seeded (2) Some of the minimal media components are missing |
(1) Ensure that the pre-culture has sufficient cell density (OD600 of ~0.3–0.5). (2) Check that all the supplements are added to the minimal media (see reagent setup) |
| (2) Some of the minimal media components are missing | (2) Check that all the supplements are added to the minimal media (see reagent setup) | ||
| 8(A)vii and 8(B)xii | Weak signals for the cysteine mutants after spin labeling | (1) Labeling and sample handling under non-optimal conditions, resulting in cell lysis. | (1) For E. coli, process cells quickly after labeling |
| (2) Unsuccessful SDM | (2) Verify the cysteine mutation(s) or choose other positions for SDM | ||
| (3) Limited accessibility for the target sites | (3) Optimize the labeling conditions or choose other positions for SDM | ||
| Large amount of free MTSL (as evident from the narrow lines in the RT CW EPR spectrum) | Insufficient washing of the cells or the OM | Increase the number of washing steps after labeling in Step 8(A)iv or 8(B)x | |
| No difference in signal intensity between WT and the mutant (under identical conditions) | (1) High background labeling | (1) Optimize the labeling and washing steps to reduce background labeling. | |
| (2) Poor labeling of the target cysteines | (2) Optimise the labeling conditions or change the target sites for labeling. | ||
| (3) For OM preparations, incomplete solubilization of the IM | (3) Use a fresh stock of sarkosyl. | ||
| 11 | When the sample is inserted, there is no shift of the resonator frequency | The sample tube is not inside the cavity | Remove the sample and position it correctly |
| 14 | The protection switches (defense pulses) are not visible | Reference arm is off or too low Bias | Switch the reference arm on and open the bias by completely by sliding it to the right-had side. If the problem persists, increase the number of averages and the Video Amplifier Gain. |
| 21 | The PELDOR data shows no decay or only an exponential decay | (1) Incorrect ELDOR channel settings (no decay) | (1) Set the mw frequency, which is used for pump pulse optimization as the current ELDOR frequency. Check for the correct ELDOR power |
| (2) Low labeling efficiency or very long interspin distances (only an exponential decay) | (2) Check the labeling efficiency and improve it or change the labeling positions as required. | ||
| S/N ratio is not sufficient | (1) Low spin concertation | (1) Try to Increase the amount of sample in the active volume of the resonator | |
| (2) τ2 is too long | (2) Decrease τ2, but at least one full oscillation should be observed. Add d8-glycerol or deuterate the protein | ||
| The data the WT sample cannot be measured for sufficiently long time window | Small background labeling | Observe the dipolar evolution as long as possible or the measurement may be skipped if the signal is too weak. | |
| 22 | The WT or the single cysteine mutant shows an intramolecular contribution | (1) Incorrect fitting of the intermolecular background function | (1) Optimize the fitting of B(t). The WT or the Cys-less data often fit into an exponential decay (Figs. 3b and 4b). |
| (2) The target protein either aggregates or oligomerizes in-situ | (2) In case of non-specific interaction/aggregation, try to optimize the vector, growth conditions, or the expression strain. For natural oligomers, the oligomeric state could be further characterized with additional single cysteine mutants. | ||
| The doubly labeled protein shows some long-distance contributions | Incorrect fitting of the intermolecular background function | Check the dimensionality of the background function using single cysteine mutants. For the whole cell or the native membrane samples, a value of d between 2.0 to 2.5 fits for the background function. | |
12| Turn the CW microwave power off (attenuation of 60 dB), switch the spectrometer to operate mode and turn the reference arm on.
13| With the ‘New Experiment’ tab, create a new experiment by selecting the ‘Pulse’ tab with the option ‘Advanced’ and click on the activate button (parameter to hardware). In the ‘FT Bridge’ panel, change the ‘Bridge Configuration’ from CW to Pulse mode.
14| Safety test (also see the Bruker E580 user manual): make sure that the TWT amplifier is in standby mode and run a two-pulse Hahn echo sequence π/2 – τ – π, in one of the pulse channels (for e.g., in the +x channel with a 16 ns(π/2) – 500(τ) – 32 ns(π) sequence). Under ‘Patterns’ in the ‘FT EPR Parameters’ panel, create an acquisition trigger by entering an acquisition start point and an integration window and ensure that the protection switches (defense pulses) are visible in the ‘SpecJet window.
? Trouble shooting (see Table 2)
15| Switch the TWT amplifier to operate mode and slowly decrease the attenuation to 0 dB (in ‘Receiver Unit’ under the panel ‘FT Bridge’) to increase the mw power. Change the detection video bandwidth to 20 MHz. There should be no cavity ringing visible after the protection switches in the ‘SpecJet window.
16| Hahn echo optimization: optimize the echo intensity observed in the SpecJet window by changing the magnetic field and adjusting the power and phase of the used pulse channel, video gain (from the ‘FT Bridge’ panel), and short repetition time (~ 2–3 ms for nitroxides, from the ‘FT EPR Parameters’ panel). Acquire a field-swept spectrum (sweep width ~150 G) and move the magnetic field to the maximum of this spectrum.
? Trouble shooting (see Table 2)
17| To get a first impression on the maximum possible time window for the 4-pulse PELDOR measurement, it is helpful to measure the phase memory time () by increasing the delay τ in the two-pulse Hahn echo sequence and record a time-dependent decay curve. The acquisition trigger position displacement should be twice as large as the time increment for the π pulse. Fit the curve with a mono- or stretched exponential decay to get the time constant (). As a thumb of rule, the maximum feasible time window for the PELDOR experiment is ~ 2 .
? Trouble shooting (see Table 2)
18| Pump (ELDOR) pulse optimization: Insert a 12 ns long pulse in the ELDOR channel 500 ns before the two-pulse Hahn echo sequence optimized in Step 7 (therefore shift the pulses of the Hahn echo and the acquisition trigger by 500 ns). Enter the spectrometer frequency as the ‘Current ELDOR frequency’ (in ‘Microwave’ under the ‘FT EPR Parameters’ panel). Increase the ELDOR mw power until the maximum inversion of the Hahn echo. Ideally, a nutation experiment should be performed by gradually increasing the ELDOR pulse length at a fixed mw power (of the ELDOR channel). Note down the optimal ELDOR mw attenuation (for a 12 ns π pulse).
CRITICAL STEP With the TWT amplifier, shorter ELDOR pulsed (<12 ns) could be used. However, with the used resonator and rectangular pulses, this can lead to a stronger overlap of the excitation bandwidths between the pump and observer pulses resulting in decreased signal to noise (S/N) ratio and artifacts. A large-bandwidth resonator71 or G aussian pulses72 have been shown to eliminate the above problems.
19| Set the observer pulses: Decrease the mw power to 60 dB and change the frequency by −70 MHz in the ‘Microwave Bridge Tuning’ panel. Carefully optimize the observer pulses to obtain a 16 ns π/2 pulse in the +<x> and -<x> channels with a phase difference of 180° and a 32 ns π pulse in the +<y> channel as described in step 5.
CRITICAL STEP Longer pulses will give smaller echo amplitude and shorter pulses can lead to an overlap of the excitation bandwidths between the pump and observer pulses. A larger frequency offset may be used when stronger pump or observer pulses are employed; however, this will considerably reduce the echo amplitude and the S/N. Note that for spectrometers generating the pulses with an AWG, additional phase cycling steps are required to eliminate the echo crossing artifacts arising from coherence transfer pathways73.
20| Setting up the 4-pulse PELDOR: Press the ‘PulseSpel’ button in the ‘Acquisition’ tab under ‘FT EPR Parameters’ panel and load the PulseSpel Program with the corresponding variable definitions (available from Bruker). Load the observer pulse sequence π/2–τ1–π–τ2–π. Set the τ1 between 100–200 ns depending on the spectrometer configuration. To optimize the acquisition trigger and the echo integration window, set the delay τ2 initially to 1 μs. On the SpecJet phase the echo and change the acquisition trigger to integrate symmetrically (for e.g., for the length of the observer π pulse) over the maximum of the echo.
21| Choose the delay τ2 according to the phase memory time of the sample. It has to be long enough to observe at least one full oscillation of the PELDOR signal (which depends on the expected distance) but short enough to get a reasonable S/N within 12–24 h. Adjust the variables to increase τ1 by 16 ns for 8 steps to average out the deuterium modulation. Also, move the pump pulse (by 8 or 16 ns for e.g.) between the two observer π pulses with sufficient delays to avoid temporal overlap. The signals from the phase cycling of the first π/2 pulses are subtracted to eliminate the receiver offset. Accumulate as many scans as needed to get a reasonable S/N.
? Trouble shooting (see Table 2)
DEER/PELDOR data analysis. TIMING 10–20 min
22| Perform data analysis using the MATLAB-based DeerAnalysis software26, which can be downloaded at http://www.epr.ethz.ch/, (see the manual for details of data analysis and usage).
CRITICAL STEP Analysis of both WT (or the Cys-less background for proteins having native cysteines) and the mutant is critical for reliable results. The signal from WT or single cysteine mutants contributes to an exponential decay devoid of any particular distances (Figs. 3 and 4). The entire dipolar evolution for the WT or the single cysteine mutant fits into a stretched exponential decay (with , which is in agreement with a two-dimensional spatial distribution of the spins over the large membrane surface). The data for the doubly labeled protein gives the dipolar spectrum and the corresponding distance distribution. Fitting data with leads to the appearance of a small population of longer distances accompanied with distortions in the dipolar spectrum (Pake pattern, see Figs. 3–4). We strongly recommend checking for the shape of the Pake pattern after fitting to ensure quality of background correction. When data quality is good (with sufficient S/N and time window), background can automatically be determined with simultaneous fitting for and the time window. The dipolar evolution should be acquired for sufficiently long time window as described earlier in the section - DEER/PELDOR spectroscopy.
? Troubleshooting
TIMING
Steps 1 and 2, Plasmid construction and site directed mutagenesis: 5–7 days. Additional time may be required to introduce the optimal mutations.
Day 1:
Step 3, transformation (for up to three samples): 1.5 h
Day 2:
Step 4, preculture preparation and protein expression overnight: 24 h
Day 3:
Steps 5–7, preparing the cells for in situ spin labeling: 30 min
Step 8 (A)i-vii, In-situ MTSL labeling of E. coli: 2–3 h
Step 8(B)i-vi, isolation of native OM: 3 h
Day 4:
Step 8(B).vii-xii, spin labeling of native OM: 5 h
Day 4–5:
Steps 9–21, PELDOR experiment: 12–24 h (for a 4 μs long time trace)
Step 22, PELDOR data analysis: 10–20 min
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2
ANTICIPATED RESULTS
With the conditions for protein expression and spin labeling described here, a 4 μs long PELDOR trace can be obtained for the double cysteine mutants within 12–24 h of measurement in E. coli or even less with OM. When the background labeling is small, it will be difficult to observe a long time trace for the WT sample. Typical modulation depth () values for the double cysteine mutants are ~8% (the maximum under our experimental setup is ~25–30%). Although the background labeling in E. coli and OM does not give any particular distances, in effect it reduces the modulation depth. Such values despite the background labeling suggest a good labeling efficiency, at least 60–70% or even more. As shown in Fig. 3, the interspin distances derived from the PELDOR experiments can provide structural information for ligand binding. In the absence of the ligand, the PELDOR data fits into an exponential decay, and ligand binding leads to visible oscillations. The high data quality allows reliable determination of the Pake pattern and the interspin distance distribution. When compared with simulations on the corresponding crystal structure, such measurements can reveal the similarity or the difference for ligand binding between in-vitro and in-situ conditions47. Also, the modulation depth () can provide quantitative information for the amount of ligand binding74. By performing such experiments using orthogonal labels (for e.g., MTSL combined with trityl), greater sensitivity and selectivity can be obtained44. A similar experiment performed with a doubly labeled protein enabled the observation of the extracellular loop conformation in BtuB (Fig. 4). A comparison between the experimental distance distribution with the simulation performed on the corresponding crystal structure revealed a rather good agreement. Thus, it is a powerful approach for observing or validating OMP conformation in the native environments. Moreover, we observed that the second extracellular loop undergoes large conformational changes in presence of the substrate (cyanocobalamin) or the ligand (Ca2+)48. Thus, our protocol could be used to elucidate the changes in conformation or conformational equilibrium18 of OMPs during function in their native environments. …
Supplementary Material
Please indicate up to four primary research articles where the protocol has been used and/or developed.
Joseph, B. et al. Distance Measurement on an Endogenous Membrane Transporter in E. coli Cells and Native Membranes Using EPR Spectroscopy. Angew. Chem. Int. Ed. 54, 6196–6199 (2015).
Joseph, B., Sikora, A. & Cafiso, D. S. Ligand Induced Conformational Changes of a Membrane Transporter in E. coli Cells Observed with DEER/PELDOR. J. Am. Chem. Soc. 138, 1844–1847 (2016).
Joseph, B. et al.s Selective High-Resolution Detection of Membrane Protein-Ligand Interaction in Native Membranes Using Trityl-Nitroxide PELDOR. Angew. Chem. Int. Ed. 55, 11538–11542 (2016).
Sikora, A., Joseph, B., Matson, M., Staley, J. R. & Cafiso, D. S. Allosteric Signaling Is Bidirectional in an Outer-Membrane Transport Protein. Biophys. J. 111, 1908–1918 (2016).
ACKNOWLEDGEMENTS
Authors thank Marie-Curie COFUND Postdoctoral program (PCOFUND-GA-2011–291776, GO-IN), Adolf-Messer Foundation (B.J.), Deutsche Forschungsgemeinschaft (SFB807 to B.J. and T.F.P), and National Institutes of Health, NIGMS (GM035215, D.S.C) for financial support.
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare that they have no competing financial interests.
DATA AVAILIBILITY STATEMENT
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1.McHaourab HS, Steed PR & Kazmier K. Toward the fourth dimension of membrane protein structure: insight into dynamics from spin-labeling EPR spectroscopy. Structure 19, 1549–1561 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laganowsky A. et al. Membrane proteins bind lipids selectively to modulate their structure and function. Nature 510, 172–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta K. et al. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeschke G. Conformational dynamics and distribution of nitroxide spin labels. Prog. Nucl. Mag. Res. Sp 72, 42–60 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Hubbell WL & Altenbach C. Investigation of structure and dynamics in membrane proteins using site-directed spin labeling. Curr. Opin. Struct. Biol 4, 566–573 (1994). [Google Scholar]
- 6.Fleissner MR et al. Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc. Natl. Acad. Sci. USA 106, 21637–21642 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt MJ, Borbas J, Drescher M & Summerer D. A genetically encoded spin label for electron paramagnetic resonance distance measurements. J. Am. Chem. Soc 136, 1238–1241 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Abdelkader EH et al. Protein conformation by EPR spectroscopy using gadolinium tags clicked to genetically encoded p-azido-L-phenylalanine. Chem. Commun 51, 15898–15901, (2015). [DOI] [PubMed] [Google Scholar]
- 9.Milov AD, Ponomarev AB & Tsvetkov YD Electron Electron Double-Resonance in Electron-Spin Echo - Model Biradical Systems and the Sensitized Photolysis of Decalin. Chem. Phys. Lett 110, 67–72 (1984). [Google Scholar]
- 10.Martin RE et al. Determination of End-to-End Distances in a Series of TEMPO Diradicals of up to 2.8 nm Length with a New Four-Pulse Double Electron Electron Resonance Experiment. Angew. Chem. Int. Ed 37, 2833–2837 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Schiemann O. et al. Spin labeling of oligonucleotides with the nitroxide TPA and use of PELDOR, a pulse EPR method, to measure intramolecular distances. Nat. Protoc 2, 904–923 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Igarashi R. et al. Distance determination in proteins inside Xenopus laevis oocytes by double electron-electron resonance experiments. J. Am. Chem. Soc 132, 8228–8229 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Jeschke G. DEER distance measurements on proteins. Ann. Rev. Phys. Chem 63, 419–446 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Martorana A. et al. Probing protein conformation in cells by EPR distance measurements using Gd3+ spin labeling. J. Am. Chem. Soc 136, 13458–13465 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Azarkh M. et al. Site-directed spin-labeling of nucleotides and the use of in-cell EPR to determine long-range distances in a biologically relevant environment. Nat. Protoc 8, 131–147 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Schmidt T, Walti MA, Baber JL, Hustedt EJ & Clore GM Long Distance Measurements up to 160 A in the GroEL Tetradecamer Using Q-Band DEER EPR Spectroscopy. Angew. Chem. Int. Ed 55, 15905–15909 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claxton DP, Kazmier K, Mishra S & McHaourab HS Navigating Membrane Protein Structure, Dynamics, and Energy Landscapes Using Spin Labeling and EPR Spectroscopy. Methods Enzymol. 564, 349–387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barth K. et al. Conformational coupling and trans-inhibition in the human antigen transporter ortholog TmrAB resolved with dipolar EPR spectroscopy. J. Am. Chem. Soc (2018). [DOI] [PubMed] [Google Scholar]
- 19.Bordignon E & Bleicken S. New limits of sensitivity of site-directed spin labeling electron paramagnetic resonance for membrane proteins. Biochim. Biophys. Acta 1860, 841–853 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Dastvan R, Fischer AW, Mishra S, Meiler J & McHaourab HS Protonation-dependent conformational dynamics of the multidrug transporter EmrE. Proc. Natl. Acad. Sci. USA 113, 1220–1225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polyhach Y, Bordignon E & Jeschke G. Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys 13, 2356–2366 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Jeschke G & Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys 9, 1895–1910 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Kattnig DR, Reichenwallner J & Hinderberger D. Modeling excluded volume effects for the faithful description of the background signal in double electron-electron resonance. J. Phys. Chem. B 117 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Jeschke G, Panek G, Godt A, Bender A & Paulsen H. Data analysis procedures for pulse ELDOR measurements of broad distance distributions. Appl. Magn. Reson 26, 223–244 (2004). [Google Scholar]
- 25.Chiang YW, Borbat PP & Freed JH The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J. Magn. Reson 172, 279–295 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Jeschke G. et al. DeerAnalysis2006 - A Comprehensive Software Package for Analyzing Pulsed ELDOR Data in Appl. Magn. Reson Vol. 30 473–498 (2006). [Google Scholar]
- 27.Edwards TH & Stoll S. A Bayesian approach to quantifying uncertainty from experimental noise in DEER spectroscopy. J. Magn. Reson 270, 87–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein RA, Beth AH & Hustedt EJ A Straightforward Approach to the Analysis of Double Electron-Electron Resonance Data. Methods Enzymol. 563, 531–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borbat PP, Georgieva ER & Freed JH Improved Sensitivity for Long-Distance Measurements in Biomolecules: Five-Pulse Double Electron-Electron Resonance. J. Phys. Chem. Lett 4, 170–175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spindler PE et al. Carr-Purcell Pulsed Electron Double Resonance with Shaped Inversion Pulses. J. Phys. Chem. Lett 6, 4331–4335 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Saxena S & Freed JH Theory of double quantum two-dimensional electron spin resonance with application to distance measurements. J. Chem. Phys 107, 1317–1340 (1997). [Google Scholar]
- 32.Jeschke G, Pannier M, Godt A & Spiess HW Dipolar spectroscopy and spin alignment in electron paramagnetic resonance. Chem. Phys. Lett 331, 243–252 (2000). [Google Scholar]
- 33.Milikisyants S, Scarpelli F, Finiguerra MG, Ubbink M & Huber M. A pulsed EPR method to determine distances between paramagnetic centers with strong spectral anisotropy and radicals: The dead-time free RIDME sequence. J. Magn. Reson 201, 48–56 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Razzaghi S. et al. RIDME Spectroscopy with Gd(III) Centers. J. Phys. Chem. Lett 5, 3970–3975 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Lawless MJ et al. Analysis of Nitroxide-Based Distance Measurements in Cell Extracts and in Cells by Pulsed ESR Spectroscopy. ChemPhysChem 18, 1653–1660 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Dunkel S, Pulagam LP, Steinhoff HJ & Klare JP In vivo EPR on spin labeled colicin A reveals an oligomeric assembly of the pore-forming domain in E. coli membranes. Phys. Chem. Chem. Phys 17, 4875–4878 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Jagtap AP et al. Sterically shielded spin labels for in-cell EPR spectroscopy: analysis of stability in reducing environment. Free Radic Res. 49, 78–85 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Karthikeyan G. et al. A Bioresistant Nitroxide Spin Label for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. Int. Ed 57, 1366–1370 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Qi M, Gross A, Jeschke G, Godt A & Drescher M. Gd(III)-PyMTA label is suitable for in-cell EPR. J. Am. Chem. Soc 136, 15366–15378 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Theillet FX et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Yang Y. et al. A Reactive, Rigid Gd-III Labeling Tag for In-Cell EPR Distance Measurements in Proteins. Angew. Chem. Int. Ed 56, 2914–2918 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Prokopiou G. et al. Small Gd(III) Tags for Gd(III)-Gd(III) Distance Measurements in Proteins by EPR Spectroscopy. Inorg. Chem 57, 5048–5059 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Reginsson GW, Kunjir NC, Sigurdsson ST & Schiemann O. Trityl radicals: spin labels for nanometer-distance measurements. Chemistry 18, 13580–13584 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Joseph B. et al. Selective High-Resolution Detection of Membrane Protein-Ligand Interaction in Native Membranes Using Trityl-Nitroxide PELDOR. Angew. Chem. Int. Ed 55, 11538–11542 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Jassoy JJ et al. Versatile Trityl Spin Labels for Nanometer Distance Measurements on Biomolecules In Vitro and within Cells. Angew. Chem. Int. Ed 56, 177–181 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Jiang X. et al. Ligand-specific opening of a gated-porin channel in the outer membrane of living bacteria. Science 276, 1261–1264 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Joseph B. et al. Distance Measurement on an Endogenous Membrane Transporter in E. coli Cells and Native Membranes Using EPR Spectroscopy. Angew. Chem. Int. Ed 54, 6196–6199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joseph B, Sikora A & Cafiso DS Ligand Induced Conformational Changes of a Membrane Transporter in E. coli Cells Observed with DEER/PELDOR. J. Am. Chem. Soc 138, 1844–1847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sikora A, Joseph B, Matson M, Staley JR & Cafiso DS Allosteric Signaling Is Bidirectional in an Outer-Membrane Transport Protein. Biophys. J 111, 1908–1918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fairman JW, Noinaj N & Buchanan SK The structural biology of beta-barrel membrane proteins: a summary of recent reports. Curr. Opin. Struct. Biol 21, 523–531 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gessmann D. et al. Outer membrane beta-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proc. Natl. Acad. Sci. USA 111, 5878–5883 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schiffrin B. et al. Effects of Periplasmic Chaperones and Membrane Thickness on BamA-Catalyzed Outer-Membrane Protein Folding. J. Mol. Biol 429, 3776–3792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rassam P. et al. Supramolecular assemblies underpin turnover of outer membrane proteins in bacteria. Nature 523, 333–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iadanza MG et al. Lateral opening in the intact beta-barrel assembly machinery captured by cryo-EM. Nat. Commun 7, 12865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu Y. et al. Structural basis of outer membrane protein insertion by the BAM complex. Nature 531, 64–69 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Okuda S, Sherman DJ, Silhavy TJ, Ruiz N & Kahne D. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat. Rev. Microbiol 14, 337–345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robert V. et al. Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. Plos Biology 4, 1984–1995 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pinto C. et al. Studying assembly of the BAM complex in native membranes by cellular solid-state NMR spectroscopy. J. Struct. Biol (2017). [DOI] [PubMed] [Google Scholar]
- 59.Wagner J, Schaffer M & Fernandez-Busnadiego R. Cryo-electron tomography-the cell biology that came in from the cold. FEBS Lett. 591, 2520–2533 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Dimura M. et al. Quantitative FRET studies and integrative modeling unravel the structure and dynamics of biomolecular systems. Curr. Opin. Struct. Biol 40, 163–185 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Sustarsic M & Kapanidis AN Taking the ruler to the jungle: single-molecule FRET for understanding biomolecular structure and dynamics in live cells. Curr. Opin. Struct. Biol 34, 52–59 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Fuller-Schaefer CA & Kadner RJ Multiple extracellular loops contribute to substrate binding and transport by the Escherichia coli cobalamin transporter BtuB. J. Bacteriol 187, 1732–1739 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarver JL, Zhang M, Liu LS, Nyenhuis D & Cafiso DS A Dynamic Protein-Protein Coupling between the TonB-Dependent Transporter FhuA and TonB. Biochemistry 57, 1045–1053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Filip C, Fletcher G, Wulff JL & Earhart CF Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J. Bacteriol 115, 717–722 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Georgieva ER et al. Effect of freezing conditions on distances and their distributions derived from Double Electron Electron Resonance (DEER): A study of doubly-spin-labeled T4 lysozyme. J. Magn.Reson 216, 69–77 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagelueken G, Ward R, Naismith JH & Schiemann O. MtsslWizard: In Silico Spin-Labeling and Generation of Distance Distributions in PyMOL. Appl. Magn. Reson 42, 377–391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hatmal MM et al. Computer modeling of nitroxide spin labels on proteins. Biopolymers 97, 35–44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cadieux N & Kadner RJ Site-directed disulfide bonding reveals an interaction site between energy-coupling protein TonB and BtuB, the outer membrane cobalamin transporter. Proc. Natl. Acad. Sci. USA 96, 10673–10678 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peters JE, Thate TE & Craig NL Definition of the Escherichia coli MC4100 genome by use of a DNA array. J. Bacteriol 185, 2017–2021 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baker LA, Daniels M, van der Cruijsen EA, Folkers GE & Baldus M. Efficient cellular solid-state NMR of membrane proteins by targeted protein labeling. J Biomol Nmr 62, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Polyhach Y. et al. High sensitivity and versatility of the DEER experiment on nitroxide radical pairs at Q-band frequencies. Phys. Chem. Chem. Phys 14, 10762–10773 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Teucher M & Bordignon E. Improved signal fidelity in 4-pulse DEER with Gaussian pulses. J. Magn. Reson 296, (2018). [DOI] [PubMed] [Google Scholar]
- 73.Tait CE & Stoll S. Coherent pump pulses in Double Electron Electron Resonance spectroscopy. Phys. Chem. Chem. Phys 18, 18470–18485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bode BE et al. Counting the monomers in nanometer-sized oligomers by pulsed electron - Electron double resonance. J. Am. Chem. Soc 129, 6736–6745 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.