

Summary
Diagnoses of behavioral disorders such as autism spectrum disorder and schizophrenia are based on symptomatic descriptions that have been difficult to connect to mechanism. Although psychiatric genetics provide insight into the genetic underpinning of such disorders, with a majority of cases explained by polygenic factors, it remains difficult to design rational treatments. In this review, we highlight the value of understanding neural circuit function both as an intermediate-level of explanatory description that links gene to behavior as well as a pathway for developing rational diagnostics and therapeutics for behavioral disorders. As neural circuits perform hierarchically organized computational functions and give rise to network-level processes (e.g. macroscopic rhythms, goal-directed or homeostatic behaviors), correlated network-level deficits may indicate perturbation of a specific circuit. Therefore, identifying such correlated deficits or a ‘circuit endophenotype’ would provide a mechanistic point of entry, enhancing both diagnosis and treatment of a given behavioral disorder. We focus on a circuit endophenotype of the thalamic reticular nucleus (TRN) and how its impairment in neurodevelopmental disorders gives rise to a correlated set of readouts across sleep and attention. Because TRN neurons express several disorder-relevant genes identified through genome wide association studies, exploring the consequences of different TRN disruptions may be of broad translational significance.
Introduction
Neuropsychiatric and neurodevelopmental disorders are a substantial source of morbidity and collectively contribute to a large economic burden (Kessler et al., 2005; Olesen et al., 2012; Whiteford et al., 2013). According to the World Health Report of 2001 from the WHO, mental and neurological conditions make up for >30% of the global burden of disease with six neuropsychiatric conditions ranking among the top 20 contributing disorders, leading to a formal recognition of mental health importance and 2013–2020 action plan (Saxena et al., 2013). Therefore, there is a substantial unmet public health need for devising effective treatments to target these disorders. However, in comparison to most medical disciplines, modern psychiatry faces unique challenges when attempting to translate basic science into treatments.
Compared to other organ systems, understanding the basic cellular elements of the brain, although extremely important, provides only limited insight into how its collective function is realized as thoughts and behavior (Dehaene and Changeux, 1997; Dehaene et al., 2014; Graziano and Aflalo, 2007; Jazayeri and Afraz, 2017). For example, based on a biophysical understanding of the cardiac action potential (Weidmann, 1951), it is easy to appreciate why hERG1 or Kv11.1 (encoded by KCNH2) loss-of-function mutation is a primary genetic substrate for congenital long QT syndrome (LQTS) and therefore predisposition to cardiac arrhythmia (Curran et al., 1995). In contrast, the link between the polymorphism of this same gene and schizophrenia (SCZ) (Huffaker et al., 2009) cannot directly be explained by its involvement in neural excitability or synaptic transmission. This difficulty stems from the fact that thought and behavior are not a direct result of individual cellular processes, but rather of transformations that take place across hierarchical levels of organization (Kanai et al., 2015).
In addition to a lack of basic mechanistic understanding to inform disorder classification, there is substantial overlap both at the behavioral and genetic levels between disorders (Bulik-Sullivan et al., 2015; Lee et al., 2013; Smoller et al., 2013). Gaining mechanistic access to these somewhat arbitrary entities has proven difficult given their heterogeneous and polygenic nature (Geschwind and Flint, 2015; McCarroll and Hyman, 2013). To address the heterogeneity, one approach has been to identify an ‘endophenotype’; a heritable factor associated with a behavior or set of behaviors within an otherwise heterogeneous diagnostic entity (Gottesman and Gould, 2003; John and Lewis, 1966). This approach has met limited success; on one end, even simple behavioral deficits can arise from multiple different genetic causes as well as have highly polygenic genetic underpinnings (Flint et al., 2014; Pappa et al., 2016) and on the other, a single genetic perturbation can give rise to multiple behavioral deficits (e.g. Mecp2 in Rett’s Syndrome (Amir et al., 1999))(Figure 1A–B). Similar limitations are associated with extended definitions of the endophenotype to include non-behavioral readouts (Flint et al., 2014). For example, neither electroencephalographic (EEG) measures such as reductions in the P50 evoked response (Adler et al., 1985; Cadenhead et al., 2000) or mismatch negativity (MMN) (Javitt et al., 1993; Shelley et al., 1991; Umbricht and Krljes, 2005), nor structural readouts such as changes in white matter structure, grey matter thickness or subcortical volume (van Erp et al., 2016; Kuperberg et al., 2003; Lener et al., 2015) have been successful at identifying genetic associations or mechanisms in SCZ (Greenwood et al., 2013; Harrisberger et al., 2016; Prasad and Keshavan, 2008; Reus et al., 2017; Voineskos et al., 2016).
Figure 1. Circuit endophenotype connects genetic and behavioral disruption at the level of neuronal circuits.
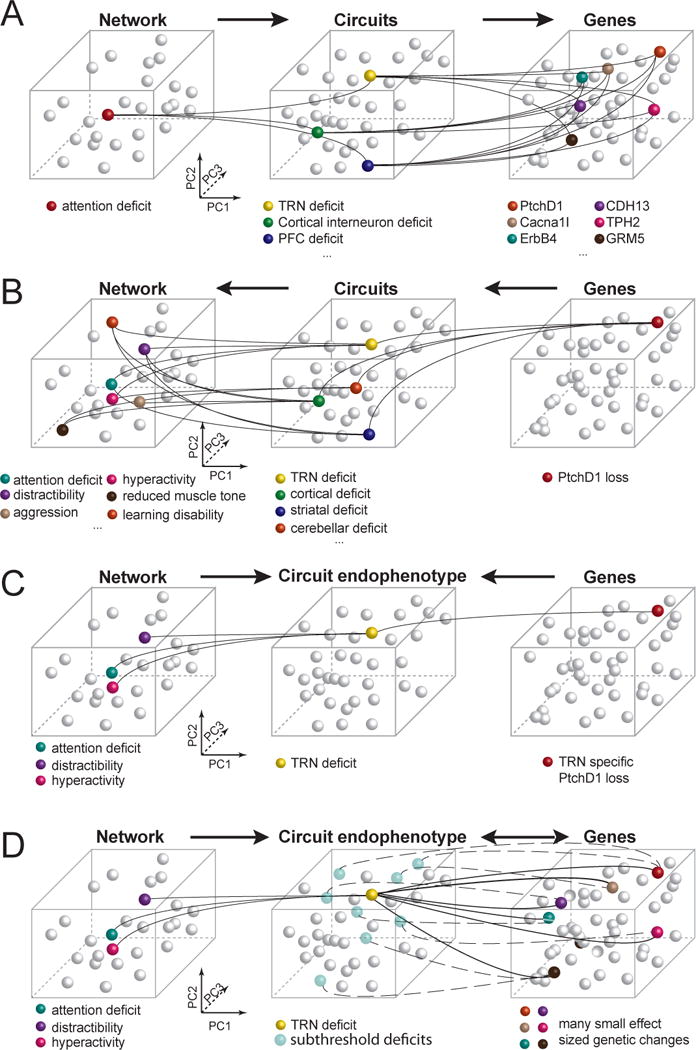
A. The challenge in finding the causes of a clinically identified deficit is that it can result from disruptions in any one of many neural circuits and genes affecting those circuits. B. Disruption in a single gene generally affects multiple neural circuits and thus can lead to a large number of clinically identified symptoms. C. Using animal models, a genetic disruption can be limited to a specific neural circuit, revealing the extent of a single circuit endophenotype and its manifestation at the behavioral level. D. When individual genetic changes are of small effect size, they may not lead to perturbation of a circuit. However together, these genetic changes can be enriched at and expressed as a more restricted circuit endophenotype as can be seen from the network level changes observed.
To address the challenge of mechanistically understanding behavioral brain disorders (neurodevelopmental and psychiatric disorders), a neural circuit description will be important, as has been suggested previously (Airan et al., 2007; Dubin et al., 2012; Hall et al., 2011; Vaghi et al., 2017; Yeh et al., 2017). On one end, neural circuits utilize biophysical mechanisms (e.g. excitability and synaptic transmission) that can be linked to genetic factors (Claes et al., 2001; Saitsu et al., 2008). On the other, their hierarchical organization into networks and systems is what gives rise to network-level processes, including thought and behavior that can be probed through tasks (Dehaene and Changeux, 1997; Dehaene et al., 2014; Graziano and Aflalo, 2007; Jazayeri and Afraz, 2017), as well as physiological readouts that can be probed through electrophysiology or imaging (e.g. oscillatory cross-regional interactions in visuospatial attention (Womelsdorf et al., 2006)). As such, circuits are the level by which networks can be link into mechanism. However, as can be appreciated by inspecting panels A and B of Figure 1, a single network-level dysfunction can have multiple circuit causes, and similarly, a single genetic dysfunction can impair multiple circuits. However, a specific circuit can, in principle, act as a regressor for a set of correlated network-level processes on one end and a set of correlated genetic-level processes on the other, effectively reducing the dimensionality in both and tying them together through an intermediate explanatory level. In a disease context, this is what a circuit endophenotype is, which has the added advantage of identifying a putative mechanism in addition to the set of correlated readouts. While efforts to identify genetic correlations have been successful at identifying potential molecular mechanistic pathways (Glessner et al., 2009; O’Dushlaine et al., 2015; Parikshak et al., 2015), it is not immediately obvious how they can be linked to circuits. A set of correlated network-level readouts, on the other hand, makes circuit endophenotype identification more tractable. In fact, we think that appropriate animal models uniquely enable this link, and ultimately a full mechanistic description of a network-level perturbation, as we explain throughout this review (Figure 1C). Additionally, it is important to consider that the majority of genetic changes have small effect sizes. However, if a subset of such genes has an overlapping expression pattern across one or more circuits, their contribution to a disorder could be additive and one that could be understood by studying where circuit ‘hits’ converge. The predicted circuit endophenotype would further be supported by observation of corresponding network level deficits (Figure 1D).
The concept of circuit-level endophenotype has been previously explored (Airan et al., 2007; Dubin et al., 2012; Hall et al., 2011; Vaghi et al., 2017; Yeh et al., 2017). In this review we particularly stress the importance of using correlated readouts, rather than a single measure, to identify circuit endophenotypes. For example, the limitation of single readouts having multiple underlying circuit underpinnings can be easily appreciated when considering how attention impacts sensory processing; both cortical inhibitory (Buia and Tiesinga, 2008; Cardin et al., 2009; Kim et al., 2016; Vinck et al., 2013) and thalamic reticular nucleus (TRN) neurons are associated with computations relevant for attentional selection (McAlonan et al., 2006; Wells et al., 2016; Wimmer et al., 2015). However, while cortical interneurons are associated with cortical gamma oscillations (Jadi and Sejnowski, 2014; Kim et al., 2016; Sohal et al., 2009; Veit et al., 2017), TRN neurons are linked to spindle oscillations (Halassa et al., 2011, 2014; Steriade et al., 1986). Thus while either TRN or cortical interneurons deficits could, in principle, lead to broadly overlapping symptomatic descriptions of attention deficits, adding EEG recordings within a controlled-task setting (among other readouts) would help in narrowing down the mechanistic perturbation.
For the rest of this review, we will first briefly discuss large-scale human genetic studies and the insight they have given into the heterogeneity of neurodevelopmental disorders (Fromer et al., 2014; Genovese et al., 2016; Krumm et al., 2015; Leppa et al., 2016; Marshall et al., 2017; Purcell et al., 2014; Ripke et al., 2014; De Rubeis et al., 2014). These studies support looking across disorders at genes and symptoms (Bulik-Sullivan et al., 2015; Fromer et al., 2014; Lee et al., 2013; Purcell et al., 2014; Smoller et al., 2013) and the utility of employing a circuit endophenotype framework. To explore what a circuit endophenotype could look like we turn to the specific example of the TRN for two reasons. First, several disease-linked genes are highly expressed in the TRN including neurodevelopmental disorder/autism spectrum disorder (ASD) gene PTCHD1, SCZ GWAS hits Cacna1i and Grm3, and ASD risk gene CHD2 (Figure 2), suggesting this structure may reflect a source of common vulnerability across a subset of neurodevelopmental disorders. Second, recent discoveries have provided new insight into TRN function, allowing predictions to be made about how its perturbations would lead to network-level dysfunction, helping define TRN circuit endophenotypes. To identity TRN circuit-endophenotypes in human neurodevelopmental disorders, we review the clinical literature for appropriate network-level readouts on one end, and the basic science literature of TRN-specific mouse models on another. By bringing together these disparate levels of enquiry through the example of the TRN, we aim to present an approach that through future iteration can bring about a better understanding of the relationship between affected genes, neural circuits and behavioral changes in a subset of psychiatric disorders.
Figure 2. Neurodevelopmental disorder associated genes enriched in TRN.
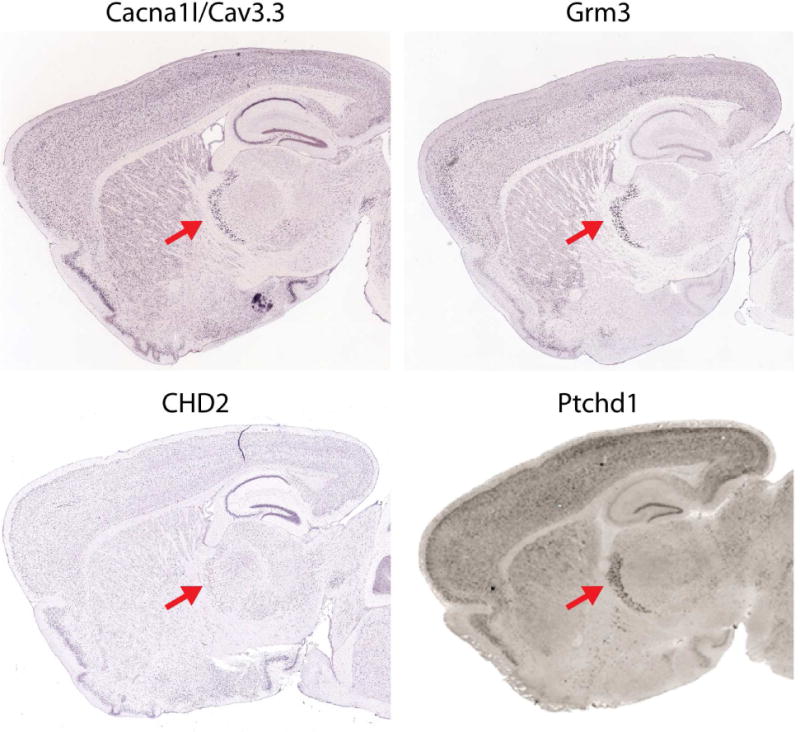
Cacna1I associated with schizophrenia (Ripke et al., 2014). Grm3 associated with schizophrenia (Ripke et al., 2014). CHD2 associated with autism (Iossifov et al., 2012). Ptchd1 associated with autism with intellectual disability (Chaudhry et al., 2015; Pinto et al., 2010). Red arrow indicates TRN. Images are from Allen Brain atlas except that of Ptchd1 (Wells et al., 2016).
Genetics of neurodevelopmental disorders
Heterogeneity and polygenicity within a disorder
Neurodevelopmental disorders have long been held to be heritable with twin and family studies suggesting heritability around 60–80% (Lichtenstein et al., 2009; Posthuma and Polderman, 2013; Sullivan et al., 2003). Recently, large-scale genomic efforts have firmly established the genetic contribution to these disorders by identifying associations between these disorders and common genetic variants (common single nucleotide polymorphisms), rare mutations and copy number variations (CNVs) (Fromer et al., 2014; Genovese et al., 2016; Iossifov et al., 2014; Krumm et al., 2015; Leppa et al., 2016; Marshall et al., 2017; Purcell et al., 2014; Ripke et al., 2014; De Rubeis et al., 2014; Singh et al., 2016). However, these studies have also highlighted the underlying challenge, that these disorders are highly heterogeneous as well as polygenic. This is seen from the very large sample sizes needed to begin to uncover statistically robust associations as well as the complicated genetic architecture with contributions from common variants, CNVs, de novo and inherited mutations (Geschwind and Flint, 2015; Iossifov et al., 2014; Weiner et al., 2017). Although common variants account for around a quarter of the heritability (Gaugler et al., 2014; Lee et al., 2013), they are of small effect size and relatively few have been individually identified (Anney et al., 2012, 2017; Ripke et al., 2014). Similarly, although there is clear enrichment of mutations identified by whole exome sequencing in ASD and SCZ, few individual variants have been identified and those identified are in few patients (Genovese et al., 2016; Kosmicki et al., 2017; Singh et al., 2016). In an ASD study, despite being able to identify likely causal variants with high confidence, only 7% of patients had an identified mutations (Kosmicki et al., 2017). In a SCZ study, despite a very large sample size, no single gene disruption was found statistically significant despite the study being >90% powered to detect presence in 1% of cases (Genovese et al., 2016). These findings point to the heterogeneity and polygenicity of these disorders. Despite these difficulties, there is hope that a finer parsing of behavioral traits may be accessible at the genetic level, supporting an endophenotype approach. For instance, although cognitive function is a very broadly defined behavioral disruption in schizophrenia, a negative genetic correlation between cognitive function and schizophrenia has been found (Bulik-Sullivan et al., 2015; Hill et al., 2016; Trampush et al., 2017) as well as a couple associated common variants (Trampush et al., 2017).
Genetic overlap between disorders
Another facet emerging from human genetic studies is a significant overlap in the genetics underlying different disorders, supporting a blurring of lines between different psychiatric disorders. For instance familial heritability studies suggest that the presence of SCZ in the family increases the risk for ASD (Larsson et al., 2005; Rapoport et al., 2009). Multiple studies show ASD-like social deficits and repetitive behaviors in children diagnosed with ADHD and vice versa (Grzadzinski et al., 2016). Furthermore, siblings of children with ADHD have an increased risk to develop symptoms characteristic for ASD, supporting a familial, genetic relationship between the two sets of symptoms (Ghirardi et al., 2017; Mulligan et al., 2009). Studies taking advantage of whole genome measures of heritability also support genetic overlap in these disorders: Looking at heritability from common genome-wide SNPs shows a modest but significant correlation between disorders of around 0.16 between ASD and SCZ (Bulik-Sullivan et al., 2015; Lee et al., 2013; Smoller et al., 2013). Similar modest overlap is seen between ASD and SCZ in de novo coding variants (Fromer et al., 2014) and in affected gene groups (Purcell et al., 2014). Such an overlap of genetics both from familial studies and at the variant level supports taking an across-disorders approach when trying to link behavioral changes to genetic changes.
In summary, genetic heterogeneity within disorders and overlap between disorders have important consequences for diagnosis and subsequent clinical studies investigating potential treatments. Clinical studies, especially those with detailed electrophysiology or behavioral testing, use sample sizes on the order of 5–30 patients and controls, despite the contribution of mutations on the order of hundreds. Thus, each person in a study likely has a different underlying genetic deficit, while being treated as having the same disorder and set of behaviors. Designing studies around circuit endophenotypes could partly mitigate what is likely to be a major limitation of such underpowered studies, by decreasing the variance within experimental groups due to heterogeneous etiologies.
Circuit endophenotype
Given the genetic and behavior heterogeneity among disorders, is there hope for better diagnosis and treatment? We think yes: neural circuits and computations offer an intermediate description that links the genetics and behavior levels of organization (Figure 1B). Given the association of many psychiatric disorders with perturbed behavioral and thought patterns, it is natural to think about putative dysfunction of cortical circuits (Bush, 2011; Cohen and Servan-Schreiber, 1992; Friston and Frith, 1995; Minshew and Keller, 2010; Yizhar et al., 2011). However, the cortex does not operate in isolation but in partnership with several subcortical structures such as the thalamus and basal ganglia (Graybiel and Grafton, 2015; Nakajima and Halassa, 2017; Saalmann and Kastner, 2011). In fact, recent studies have revealed central roles for thalamic circuits in coordinating and regulating cortical function well beyond their classical description of relaying sensorimotor signals (Bolkan et al., 2017; Ito et al., 2015; Komura et al., 2013; Schmitt et al., 2017; Xu and Sudhof, 2013). Below we discuss these findings and highlight the utility of understanding thalamic circuit organization given its readily observable dynamics and what they could reveal about cognitive and sensorimotor function. These dynamics, such as organized global rhythms in sleep, make thalamic function and dysfunction uniquely accessible to biomarker development and the identification of robust circuit endophenotypes relevant to psychiatric diagnoses.
A brief overview of thalamic function
The thalamus is composed of excitatory neurons traditionally grouped into nuclei with specific connectivity patterns to cortex and plays critical roles in transmitting sensorimotor signals as well as maintaining and switching functional connectivity within and across cortical regions (Figure 3) (Nakajima and Halassa, 2017; Saalmann and Kastner, 2011). The classical view of the thalamus is that of a relay of categorical information to or across cortical regions. For example, extensive study of the lateral geniculate nucleus (LGN), a sensory thalamic nucleus, has resulted in detailed knowledge about the role of the LGN in relaying retinotopic inputs to primary visual cortex (Golden et al., 2016; Sherman, 2005; Usrey and Alitto, 2015; Vukadinovic, 2014). This success in understanding sensory thalamic function has greatly influenced thinking on the function of other less studied thalamic nuclei. Even nuclei that receive their primary driving input from cortical structures, rather than the periphery, have been thought to relay information from one cortical region to another (Guillery and Sherman, 2002; Ketz et al., 2015; Sherman, 2016). However, the exclusive view of thalamic function as a relay has been changing recently for several reasons. First, anatomical evidence suggests that this view is incomplete. Thalamic projections to cortex are anatomically-heterogeneous which may vary continuously their degree of spatial spread and local density (Clasca et al., 2012), suggesting that only a subset of these inputs are well-suited for topographical and faithful information transmission. Similarly, cortical inputs to several thalamic nuclei exhibit anatomical features that suggest a high degree of convergence onto individual thalamic neurons (Rovo et al., 2012), which again does not seem to be compatible with topographical relay. Second, a number of recent studies show that the thalamus can engage in cognitive processes in a manner that may not include relaying categorical information (Bolkan et al., 2017; Halassa and Kastner, 2017; Ito et al., 2015; Komura et al., 2013; Schmitt et al., 2017; Xu and Sudhof, 2013). For example, in two tasks requiring maintenance of representations within the prefrontal cortex (PFC) [spatial working memory, sustained attention], mediodorsal thalamic input was necessary for maintaining these representations. At least in one of these tasks (Schmitt et al., 2017), this thalamic input was devoid of categorical specificity observed in the PFC, clearly indicating that relaying this information was not the actual function. Instead, this input amplified local synaptic recurrence that was required for sustained representations and behavioral output (Schmitt et al., 2017). Enhancing this thalamic input augmented behavioral performance in both tasks (Bolkan et al., 2017; Schmitt et al., 2017). Together, these findings demonstrate that the thalamus plays important roles both in cognitive functions as well as in the relay of sensory information.
Figure 3. TRN in the thalamocortical circuit.
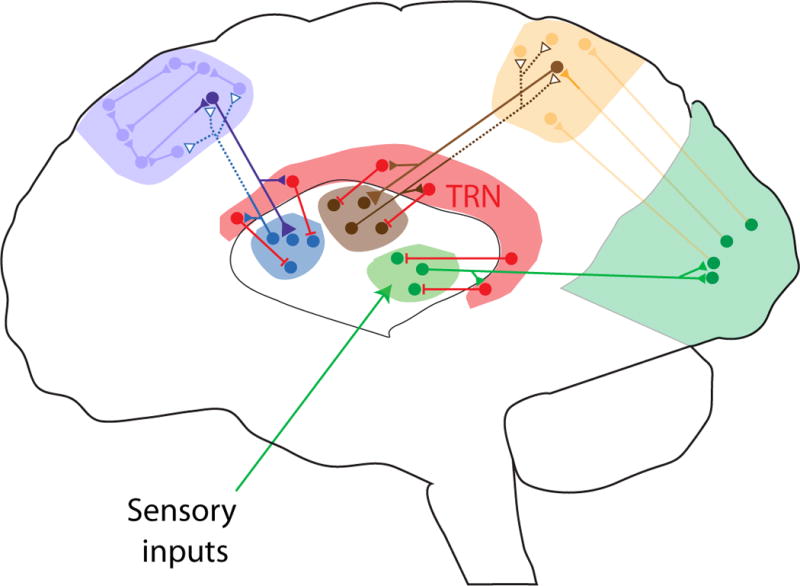
Thalamus plays a critical role in relaying sensorimotor signals (green circuit, e.g. LGN; solid lines indicate thalamic control of spike timing) as well as maintaining and switching functional connectivity (indicated by dashed line) within (blue circuit, e.g. MD) and across cortical regions (brown circuit, e.g. pulvinar). The TRN is a central control unit in the thalamocortical circuit, receiving input from both thalamus and cortex while only inhibiting thalamic targets. It is anatomically segregated into subnetworks that project to spatially discrete thalamic targets allowing the TRN to influence multiple levels of functional thalamic organization. These include global arousal and specific cognitive states such as attention. The exact location of TRN subnetworks is schematized loosely based on anatomy.
The role of TRN in controlling thalamic function
Given the role of the thalamus in sensorimotor and cognitive functions, controlling input-output transformations within and across thalamic circuits is of central importance. Inhibitory input is one powerful mechanism to achieve such control. Compared to cortex, the number and variety of thalamic local interneurons are substantially limited (Halassa and Acsady, 2016). Instead, a major thalamic source of inhibition is the TRN, a thin shell of GABAergic neurons that surround thalamic projection circuits (Pinault, 2004; Steriade et al., 1986). The TRN receives inputs from both cortex and thalamus while its output is limited to thalamic structures (Figure 3). TRN is composed of subnetworks that project to spatially discrete thalamic targets (Halassa et al., 2014; Pinault and Deschênes, 1998; Wimmer et al., 2015; Zikopoulos and Barbas, 2012). Thus neurons that project to the LGN appear to occupy a particular spatial location within the TRN, that is largely non-overlapping with those occupied by other spatially-discrete subnetworks (Halassa et al., 2014; Pinault, 2004; Wimmer et al., 2015). Importantly, such anatomical segregation is associated with multiple levels of functional organization that span global arousal and more specific cognitive states (Halassa et al., 2014; McAlonan et al., 2006, 2008; Wimmer et al., 2015). TRN subnetworks that project to thalamic circuits associated with sensory transmission contain a high proportion of cells that increase spiking during sleep compared to subnetworks that project to limbic-associated thalamic circuits (Chen et al., 2016; Halassa et al., 2014). Such enhanced spiking is expected to translate to augmented inhibitory control of sensory transmission during sleep. However, the exact contribution of this process to decreasing behavioral sensitivity to sensory stimulation during sleep does not appear to be straightforward, as multiple studies have shown qualitatively preserved sensory responses of primary cortical regions during sleep (Issa and Wang, 2011; Nir et al., 2015; Portas et al., 2000).
While little is known about the impact of TRN on non-sensory thalamic circuits, several studies have investigated TRN’s role in sensory processing (Chen et al., 2016; Fisher et al., 2017; Halassa et al., 2014; McAlonan et al., 2006; Wimmer et al., 2015; Yu et al., 2009). For example, in sensory transmission regulating the magnitude of a signal that varies in amplitude and time requires appropriate filtering routines that account for such variance if the behavioral use of this signal demands invariance (Carandini and Heeger, 2011). For a visual signal to be faithfully transmitted regardless of its absolute luminance, but rather in relation to its luminance to its local surround, specific filters need to be applied to match that local spatial structure. Recent experiments on visual responses of LGN neurons suggest that such filtering is likely implemented through inhibitory inputs from the TRN (Fisher et al., 2017; Schmitt and Halassa, 2017). Further, the contribution of modality-specific TRN subnetworks to suppressing distracting sensory inputs as well as that process’ control through executive function has also been recently established (Wimmer et al., 2015). Thus, in tasks requiring volitional control over sensory processing and distractor suppression, TRN plays a critical part in implementing top-down instructions. Taken together, thalamic inhibition through circuit-specific TRN inputs are critical for regulating sensory processing in the context of cognition and behavior (Schmitt and Halassa, 2017).
A TRN circuit endophenotype in human disease?
These TRN studies have defined clear roles for TRN in top-down distractor suppression, partly through modality-specific normalization of sensory input. This mechanistic understanding of TRN function makes clear predictions of the collection of deficits expected in neurodevelopmental disorders that would collectively point to a TRN circuit endophenotype. Observing such a collection of deficits together would point to TRN disruption and might appear to cross traditional diagnostic boundaries as is also observed at the genetic level. On a behavioral level, to ask whether there are any indications in the human clinical literature for disruption of TRN-related functions in neurodevelopmental disorders, we must consider what generally brings people to the psychiatrist. Thus, symptoms relevant to a TRN disruption might include relatively broad complaints about deficits in attention or abnormal sensitivity to sensory stimuli.
Indeed attention deficits are considered a hallmark of ADHD and are also highly prevalent in ASD (Grzadzinski et al., 2016; Keehn et al., 2013) and SCZ patients (McGhie and Chapman, 1961). Many different kinds of observations of attention deficits are reported. For instance in ASD, observation of infants at risk for ASD reveals a decrease in orientation and shift of visual attention to stimuli including people (Keehn et al., 2013) and this deficit is predictive of later diagnoses (Zwaigenbaum et al., 2005). In older individuals, continuous performance tests can assess reaction times and deficits in sustained attention in response to visual or auditory cues and comparable deficits are seen in children with ASD and ADHD (Corbett and Constantine, 2006).
Sensory processing is also disrupted across multiple neurodevelopmental disorders including ASD, ADHD and SCZ. In ASD in particular a diagnostic criterion in DSM-5 includes “hyper- or hyporeactivity to sensory input or unusual interests in sensory aspects of the environment” with both hypersensitivity to auditory, visual and tactile sensation reported as well as occasional occurrences of hyposensitivity (Blakemore et al., 2006; Cascio et al., 2008; Marco et al., 2011; Takahashi et al., 2014). Evaluations are often done using simple caregiver report questionnaires, such as the Short Sensory Profile used to evaluate sensitivity across multiple different senses including vision, sound and touch. This approach is very useful for higher-throughput studies and is able to reveal pervasive problems in sensory processing with a relatively large study of 281 children with ASD and controls (Tomchek and Dunn, 2007). This study found that 95% of children with ASD had some degree of sensory processing dysfunction as evaluated by the Short Sensory Profile. Hypersensitivity was also found in ADHD patients both through parental reports on the Short Sensory Profile as well as through direct skin conductance measurement showing larger physiological responses to various sensory stimuli (Mangeot et al., 2001). The Sensory Gating Inventory, a different self-report questionnaire evaluating metrics including discomfort with loud sounds and effects of background noise, revealed more perceptual abnormalities in ADHD and SCZ patients compared to controls (Micoulaud-Franchi et al., 2015a, 2015b; Sable et al., 2012).
The studies described above illustrate that complaints potentially consistent with TRN-related perturbations (either intrinsic or input/output related) are present across neurodevelopmental disorders. However, a major challenge is that much of the characterization of these deficits is done at a very broad level. Deficits in sensory processing and attention can arise for many, fundamentally different, reasons including those of perception or executive control (Keehn et al., 2013; Marco et al., 2011). Broad measures are therefore insufficient to discern whether TRN may or may not be involved. For example, attention deficits could result from an inability to engage in the task because of a deficit in directed arousal, which is inconsistent with known TRN roles. Alternatively, a deficit in identifying and suppressing distractors would be consistent with TRN involvement.
To help address these challenges, it is necessary to administer more carefully constrained tasks, which are more capable of revealing specific deficits of how distractor suppression arises. In SCZ research a sensory gating malfunction hypothesis has led to the development of several tasks with the goal to separate sensory processing deficits from those resulting from executive function or general task engagement. In one study, Smucny et al. 2013 administered a visual attention task in the presence or absence of an auditory distractor. The authors saw a reduction in accuracy in schizophrenics compared to controls as well as a reduction in reaction time. In a second study, the authors tried to distinguish between bottom up and top down attention mechanisms to enable detection of visual stimuli in the presence of visual distractors (Gold et al., 2007). In one version of the task, the visual distractor shared no features with the stimuli, requiring only bottom up feature detection. In the second version, more features were shared and detection required incorporation of executive control. Patients with SCZ exhibited increased response times with both tasks but were worse at the second compared to controls. These examples illustrate that simple tasks, more specifically implicating a circuit dysfunction, can detect differences between patients and controls (Gold et al., 2007). Although these tasks have not been applied for the most part to other neurodevelopmental disorders, they can be in the future.
While symptoms observed in patients can be consistent with TRN deficits, human studies do not currently allow for a direct test of TRN involvement and even the best behavioral assessments are unlikely to be sufficient. Genetics offer one way to more firmly link changes in neurodevelopmental disorders with dysfunction in specific circuits. First, as additional candidate genes emerge from large-scale human genetic studies, evidence may converge upon certain circuits. Second, creating animal models of human disease-relevant manipulations, especially when the manipulations are limited to specific circuits, can directly test links between neurodevelopmental disorders, genes and behaviors in humans and animals and connect them to changes in circuit activity (Figure 1C). There is probably as yet insufficient genetic data to specifically implicate the TRN above other structures using untargeted genetic approaches. However, several genes implicated by large-scale genetic studies in neurodevelopmental disorders are highly enriched in the TRN including neurodevelopmental disorder/ASD gene PTCHD1 and SCZ GWAS hits Cacna1i, Grm3, and ASD risk gene CHD2 (Figure 2). As we are still in the very early days of identifying genes involved in these disorders, this number is expected to increase over time.
Only one of these risk genes, PTCHD1, has been thoroughly interrogated in terms of circuit mechanism. Mutations in PTCHD1, an X-chromosome gene, have been found in families with ASD and intellectual disability, first in de novo CNVs and subsequently in many additional families. PTCHD1 deletion is associated with ADHD, autistic traits, motor dysfunction, sleep disturbance and intellectual disability (Chaudhry et al., 2015). In mice, Ptchd1 expression is restricted to the TRN during development and continues to be TRN-enriched throughout life. Ptchd1 mutant mouse models were created to examine both the consequences of a full Ptchd1 knockout (Ung et al., 2017; Wells et al., 2016) as well as TRN-specific deletion of Ptchd1 (Wells et al., 2016). The full knockout mice show many phenotypic abnormalities relevant to symptoms found in patients including attention deficits, hyper activity, motor deficits, aggression and sleep disruption (Ung et al., 2017; Wells et al., 2016). In particular, Ptchd1 knockout mice exhibit reduced performance on a visual detection task in the presence of distractors. In the TRN, slice recordings demonstrated reduced bursting of TRN neurons. Furthermore, in vivo, Ptchd1 loss results in diminished TRN output following a visual stimulus as directly measured by chloride photometry in the thalamus. Importantly, deleting Ptchd1 selectively from the TRN replicates the attention deficit, hyperactivity and sleep disruption found in the full knockout mice, but not other disease-related behavioral phenotypes. In addition, pharmacological rescue of TRN biophysical dysfunction, determined by whole cell recordings in slice, also selectively rescues ADHD-related behaviors in the full knockout Ptchd1 (Wells et al., 2016). These manipulations allowed linking a genetic alteration to changes in biophysical properties of TRN neurons and further impairment in behaviors depending on intact TRN function. Of note, this study also demonstrated that it was possible to parse TRN dependent behaviors from others, further strengthening the idea of a TRN-specific circuit endophenotype.
A second gene, CACNA1I, is a top hit in a SCZ GWAS study (Ripke et al., 2014) and also in a study of de novo mutations with 105 probands, where amino acid changes were found in two separate individuals (Gulsuner et al., 2013). Cacna1I encodes the major pore-forming subunit of CAV3.3, a low-voltage activated (T-type) calcium channel with an enriched expression profile in the TRN compared to other T-type calcium channels (Liu et al., 2011; Talley et al., 1999). Deletion of Cacna1I in the mouse alters TRN cell discharge properties as well as distinct EEG characteristics as discussed later in the review (Astori et al., 2011; Lee et al., 2014). Additionally, a point mutation identified in schizophrenia patients (Gulsuner et al., 2013), which leads to an amino acid change in an extracellular loop of CAV3.3, reduced glycosylation and surface expression of the channel. Modeling the expected reduction in current predicts a substantial deficit in TRN rebound bursting (Andrade et al., 2016). Indeed, as predicted, TRN slice recordings from mice with the engineered point mutation demonstrated a reduced number and efficiency of TRN bursting (Ghoshal et al., SfN poster 173.15). These results directly link a defined mutation from patients with a specific change in TRN firing properties. Ongoing studies will likely reveal circuit-level defects related to TRN function.
Additionally, ERBB4, although not yet reaching genetic significance in well powered large scale studies (Leppa et al., 2016; Marshall et al., 2017), has also been studied in the context of the TRN in relevance to various disorders. Rare mutations in the receptor tyrosine kinase ERBB4 have been linked to susceptibility for SCZ (Mei and Nave, 2014) including rare deletions in SCZ (Walsh et al., 2008) and ASD (Pinto et al., 2010; Prasad et al., 2012). Although mutations in ERBB4 have been found in relatively small, poorly-powered cohorts, its prominent role in TRN function may help to push for further genetic studies in defined subpopulation of patients. The potential significance of ERBB4 rare mutations is supported by highly significant under-representation of loss of function mutations in the very well powered EXAC cohort, a 60,000+ sample of exome sequenced control individuals (Lek et al., 2016). Nonetheless, demonstrating how a gene-centric approach can reveal circuit-specific function, Ahrens et al. (2015) showed that TRN-specific ErBb4 deletion in mice is associated with reversal learning phenotypes using a cross-modal detection task. Further, utilizing optogenetic approaches to selectively excite corticothalamic or thalamocortical projections, the team identified that loss of ErbB4 in TRN neurons causes selective strengthening of cortical excitatory synaptic input onto the TRN and consequently an increased IPSC-EPSC ratio in the thalamus. While the biological mechanisms for deficits in reversal learning can be difficult to determine, perturbed PFC function (and by extension, frontally-connected thalamic circuits) can result in difficulty with rule reversal. As such, reversal learning phenotypes observed in the TRN-specific ErbB4 deletion mice may be attributed to deficits associated with frontally-projecting thalamic circuits output in an appropriate manner.
These studies demonstrate how functional circuit manipulations in animal models can help tie human genetics to circuits and behavior and hopefully back to a complement of human behaviors, to begin to build a picture of a TRN circuit endophenotype. Current human genetic studies are only able to clearly support the involvement of relatively few genes in the TRN at this time. However additional work is likely to support many more candidates in the near future. The studies discussed above offer a roadmap for how models based on genetic lesions can clearly tie together circuits and behaviors.
The quest for thalamic biomarker in disease
Because of the lack of non-invasive measures of neuronal activity at the cellular level, malfunction at the circuit level is in general difficult to identify in humans. Larger scale electrophysiological activity measures, such as electroencephalographic (EEG) recordings, or imaging techniques such as magnetoencephalography (MEG) or magnet resonance imaging (MRI) usually cannot provide a sensitive measure for neuronal activity restricted to a specific circuit. The case of the TRN might present an exception. A large number of studies suggest a causal relationship between TRN activity and cortical spindles (Barthó et al., 2014; Halassa et al., 2011; Huguenard and McCormick, 2007; Steriade et al., 1986; Wimmer et al., 2012), a highly characteristic and conserved EEG signal consisting of a short (2–12 s) event characterized by a 9–15 Hz waxing and waning oscillation that occurs specifically during light non-rapid-eye-movement (NREM) sleep. In anesthetized cats, a preferred preparation for classical visual neuroscience, intracellular recordings of TRN neurons showed that they exhibit oscillatory activity that correlates with cortical ‘spindles’ (Steriade et al., 1985, 1987). In mice, brief optogenetic activation of TRN is sufficient to trigger cortical spindles (Barthó et al., 2014; Halassa et al., 2011). However, further experiments including for example closed-loop optogenetic TRN inactivation are needed to fully establish the causal contribution of TRN to spindle generation.
Extensive characterization of spindle activity has revealed multiple features that if disrupted could eventually be informative of underlying circuit contributions. For instance, different frequencies of spindle oscillation are observed at different brain regions (Andrillon et al., 2011) as well as both locally restricted and globally propagating spindle activity (Nir et al., 2011). Spindles oscillations can also be coupled with other characteristic oscillations, such as hippocampal ripples (Novitskaya et al., 2016). Additionally, the variability in spindle engagement across TRN subnetworks (Halassa et al., 2014) could indicate a potential for establishing a correspondence between surface recorded spindle variables (e.g, amplitude, power, coherence) and TRN subnetwork parameters (individual subsets or functional interactions between them).
Thus spindle dynamics are a promising readout for TRN activity. Furthermore there is evidence for changes in spindles across disorders. Global suppression of spindle activity is observed in SCZ and is predictive of psychotic symptoms. Using high density EEG to monitor sleep, medicated patients suffering from SCZ were found to have a reduction in sigma power between 13.75 and 15 Hz as well as a decrease in sleep spindle number, amplitude and duration during the first sleep episode (Ferrarelli et al., 2007, 2010). Importantly reduced spindle density and amplitude, and correspondingly, sigma power was also seen in early-course unmedicated schizophrenics and their first degree relatives compared to controls (Manoach et al., 2014). A decrease in sleep spindle number has also been observed in children (Tessier et al., 2015) and adults with ASD (Limoges et al., 2005).
Disease-linked genetic evidence may provide one of the clearest arguments for a major involvement of the thalamus and particularly TRN in SCZ. The intrinsic rebound bursting properties of TRN neurons have been shown to be a determining factor for the generation of sleep spindles (Steriade et al., 1985, 1987) and requires the interplay of a characteristic low-voltage activated calcium current (T-current), which has different properties in the TRN compared to the rest of the thalamus (Huguenard and Prince, 1992) and a small-conductance calcium-activated potassium channels (SK-channels) (Astori et al., 2011; Cueni et al., 2008; Wimmer et al., 2012). The enriched expression of the voltage-activated calcium channel Cacna1i in the TRN long implicated these channels in the generation of characteristic TRN bursting (Liu et al., 2011; Talley et al., 1999). This was confirmed in Cacna1i mouse knockouts that demonstrate a lack of repetitive TRN bursting and a global deficit in EEG spindle activity (Astori et al., 2011; Lee et al., 2014). These findings help demonstrate that Cacna1i function is important for TRN neuronal properties. However, as is the case for most GWAS hits, the polymorphism found in the Cacnai locus is not exonic and is thus difficult to link to a mechanistic change at the moment. As mentioned above, a more direct link between disease and TRN function comes from a de novo amino acid change in CACNA1i identified in a SCZ proband (Gulsuner et al., 2013) which leads to disruption of TRN bursting dynamics in a mouse model (Ghoshal et al., SfN poster 173.15). Ongoing efforts will reveal whether a change in spindle oscillations can be detected in this model. Overall, such a mechanistic, circuit-specific interrogation of a gene genetically implicated in SCZ supports the role of a change in TRN neuronal kinetics and spindle oscillation in these disorders.
More evidence for the possibility of TRN dysfunction resulting in measureable spindle deficits in the case of disorders comes from the Ptchd1 mouse model (Wells et al., 2016). Patients with PTCHD1 mutations were also found to have disruptions in sleep as well as deficits in attention and hyperactivity (Chaudhry et al., 2015). In line with the human findings, PtchD1 knockout mice showed disrupted, fragmented sleep and reduced spindle number (Wells et al., 2016). Furthermore, these deficits mapped onto TRN-specific expression of PtchD1.
Do such findings from mouse models indicate that spindle activity will be a biomarker for TRN dysfunction? This is likely to be an oversimplification. As mentioned, the TRN is heterogeneous, composed of functional sub-networks with differential engagement in relation to spindle activity. Therefore, deficits in particular TRN sub-networks may also be differentially (or not at all) reflected in altered cortical spindle activity. However, spindle deficits in combination with other metrics including carefully assessed attentional and sensory processing readouts could represent a reliable way to diagnose a TRN dysfunction.
Perspective
Neurodevelopmental disorders present multiple diagnostic challenges, not least of which is their heterogeneity and wide range of presentation, which has led to a relatively arbitrary drawing of classic disorder boundaries. In this review, we step away from these classifications and instead consider what a diagnostic framework that is more mechanism focused would look like. As we discuss explicitly with the example of the TRN, individual circuits engage in multiple computations and correspondingly contribute to multiple behaviors. Therefore, impacting a particular circuit is unlikely to give rise to a single symptom but instead a group of readouts that derive from the physiological function of that circuit. We refer to such a group of biologically related readouts as a circuit endophenotype and suggest it as a relevant mechanistic basis for integration into future diagnosis. Of note, even a single genetic mutation can affect multiple circuits and generally many genes are involved in a disorder (Figure 1B). Therefore, it is likely that multiple circuits will be affected in each disorder, manifesting in multiple circuit endophenotypes.
Relatively detailed knowledge about the function of a particular neural circuit can lead to improved design of behavioral testing. Considering better-defined individual readouts within the context of a circuit endophenotype can help narrow down individual symptoms’ source and identity as well as helping narrow in on a specific diagnosis. The addition of a directly measureable correlate of a particular circuit activity could further serve as a biomarker to imply a disruption of this circuit in disease. In the case of the TRN, non-invasive measures of surface recorded sleep spindles might represent such a biomarker. However, more experimental evidence is needed to address to what extent spindle activity maps onto TRN dysfunction or whether it may rather be a more general marker for thalamocortical network dysfunction. Given the recent finding that MEG may be a more sensitive method to detect spindle activity (Dehghani et al., 2011) potentially allowing for a more isolated spindle profile, combining this technique with a more refined analysis of spindle activity, including their spatio-temporal dynamics and relationship with other ongoing oscillatory brain activity, could narrow down the extent of TRN or even TRN sub-network involvement. Beyond spindle activity, recent advances in combining imaging techniques with transcranial magnetic stimulation (TMS) might be helpful in the search for such biomarkers. TMS in combination with EEG or fMRI is increasingly used to stimulate restricted brain areas while simultaneously studying the evoked responses in functionally connected brain regions (Ruff et al., 2009). This approach can be used to probe for specific connectivity deficits in the context of disease. For example, using TMS to stimulate frontal cortex resulted in reduced thalamic activation in patients with SCZ as measured using fMRI (Guller et al., 2012), providing direct evidence for reduced functional cortico-thalamic connectivity in SCZ.
As more and more genetic features of these disorders are revealed, genetics will play an increasing role in understanding these disorders. Eventually, genetics may also be an important diagnostic component. However, the genetics underlying these disorders presents its own challenges. In very few cases is the gene to phenotype relationship as simple as the examples presented here that have led to animal models. On the one hand the genetic contribution and heritability of these disorders is firmly established both from twin and family studies as well as from recent large-scale genomic efforts, which have identified specific genetic associations. On the other hand, these studies have revealed the polygenic and heterogeneous nature of these disorders. In most cases, multiple genes of small effect size contribute to disorder in an individual. Additionally, any two individuals will likely have largely different underlying genetics. Both to identify which genes of small effect combine to lead to specific behavioral changes as well as identifying which individuals have genuinely similar disorders, it will be critical to achieve the ‘best’ grouping possible among genetic contributions. Part of this grouping will likely be cell biological in nature, i.e. what cellular process is affected such as DNA regulation or synapse structure. However, as we have discussed for understanding the broader function of the brain, identifying the affected circuit-level biological substrates of computations is likely to be of particular importance. Circuit analyses based on spatio-temporal convergence of risk gene expression and functional circuit readouts may lead to prediction of circuit endophenotypes, which can then be tested in animal models and human patients. If there is enough convergence on a circuit from the small effect changes (Figure 1D), then analysis of the same circuit endophenotype in animal models of the larger effect mutations would be informative and could help to design translational biomarkers for analysis in human patients.
At the simplest level, where and when genes act within and between individuals is a starting point for identifying affected circuits (State and Šestan, 2012). Thus the spatio-temporal convergence of multiple risk gene expression in a specific circuit would implicate the circuit’s involvement in the disorder. Indeed, several studies have taken such an approach to begin to identify which regions and cell types in humans are enriched in expression of risk alleles, using publicly available gene expression information from BrainSpan: Atlas of the Developing Human Brain. Using several different strategies to identify risk genes and pathways implicated in ASD, ASD risk genes were found to be enriched specifically in layer 5 and layer 6 cortical projection neurons during mid-fetal development (Parikshak et al., 2013; Willsey et al., 2013; Xu et al., 2014), as well as layer 2/3 neurons and more weakly 5/6 neurons in adults (Parikshak et al., 2013) and striatal neurons (Xu et al., 2014). A similar approach taken in a SCZ study identified fetal prefrontal cortex as the area where co-expression of de novo mutations found in patients was enriched compared to controls (Gulsuner et al., 2013). As better human gene expression data becomes available, such as from single cell RNA sequencing, and more risk genes are identified, our ability to identify relevant cell types and circuits should increase.
The extent of a particular circuit endophenotype will require updating as additional information becomes available about circuit function both through creating animal models of disease-relevant genetic lesions as well as basic systems neurobiology. A better understanding of circuit function can then be translated into better diagnostic tools in patients. Additionally, such improved diagnostics could lead to stronger genetic associations. However, testing of any new hypotheses as best as possible in humans will be critical as the limit of animal models for understanding neural dysfunction has been repeatedly demonstrated in the failure of countless clinical trials. An iterative process between basic science and the clinic will be necessary to hopefully give rise to a better understanding of biological processes underlying these disorders.
Box 1.
Endophenotype
A heritable symptom, a component of the overall disorder, ideally more quantifiable than traditional diagnoses in psychiatric disorders. Different manifestations of the same disorder can consist of different collections of endophenotypes.
Circuit endophenotype
A group of correlated network-level readouts associated with the dysfunction of a particular neural circuit.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler LE, Waldo MC, Freedman R. Neurophysiologic studies of sensory gating in schizophrenia: comparison of auditory and visual responses. Biol Psychiatry. 1985;20:1284–1296. doi: 10.1016/0006-3223(85)90113-1. [DOI] [PubMed] [Google Scholar]
- Ahrens S, Jaramillo S, Yu K, Ghosh S, Hwang GR, Paik R, Lai C, He M, Huang ZJ, Li B. ErbB4 regulation of a thalamic reticular nucleus circuit for sensory selection. Nat Neurosci. 2015;18:104–111. doi: 10.1038/nn.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airan RD, Meltzer LA, Roy M, Gong Y, Chen H, Deisseroth K. High-speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science. 2007;317:819–823. doi: 10.1126/science.1144400. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Andrade A, Hope J, Allen A, Yorgan V, Lipscombe D, Pan JQ. A rare schizophrenia risk variant of CACNA1I disrupts CaV3.3 channel activity. Sci Rep. 2016;6:34233. doi: 10.1038/srep34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrillon T, Nir Y, Staba RJ, Ferrarelli F, Cirelli C, Tononi G, Fried I. Sleep Spindles in Humans: Insights from Intracranial EEG and Unit Recordings. J Neurosci. 2011;31:17821–17834. doi: 10.1523/JNEUROSCI.2604-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. 2012;21:4781–4792. doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anney RJL, Ripke S, Anttila V, Grove J, Holmans P, Huang H, Klei L, Lee PH, Medland SE, Neale B, Robinson E, Weiss LA, Zwaigenbaum L, Yu TW, Wittemeyer K, Willsey AJ, Wijsman EM, Werge T, Wassink TH, Waltes R, Walsh CA, Wallace S, Vorstman JAS, Vieland VJDM. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 2017;8:21. doi: 10.1186/s13229-017-0137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astori S, Wimmer RD, Prosser HM, Corti C, Corsi M, Liaudet N, Volterra A, Franken P, Adelman JP, Luthi A. The Ca(V)3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci U S A. 2011;108:13823–13828. doi: 10.1073/pnas.1105115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthó P, Slézia A, Mátyás F, Faradzs-Zade L, Ulbert I, Harris KD, Acsády L. Ongoing network state controls the length of sleep spindles via inhibitory activity. Neuron. 2014;82:1367–1379. doi: 10.1016/j.neuron.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore SJ, Tavassoli T, Calò S, Thomas RM, Catmur C, Frith U, Haggard P. Tactile sensitivity in Asperger syndrome. Brain Cogn. 2006;61:5–13. doi: 10.1016/j.bandc.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Bolkan SS, Stujenske JM, Parnaudeau S, Spellman T, Rauffenbart C, Abbas AI, Harris AZ, Gordon JA, Kellendonk C. Thalamic projections sustain prefrontal activity during working memory maintenance. Nat Neurosci. 2017;20:987–996. doi: 10.1038/nn.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buia CI, Tiesinga PH. Role of Interneuron Diversity in the Cortical Microcircuit for Attention. J Neurophysiol. 2008;99:2158–2182. doi: 10.1152/jn.01004.2007. [DOI] [PubMed] [Google Scholar]
- Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, Duncan L, Perry JRB, Patterson N, Robinson EB, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush G. Cingulate, Frontal, and Parietal Cortical Dysfunction in Attention-Deficit/Hyperactivity Disorder. Biol Psychiatry. 2011;69:1160–1167. doi: 10.1016/j.biopsych.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenhead KS, Light GA, Geyer MA, Braff DL. Sensory Gating Deficits Assessed by the P50 Event-Related Potential in Subjects With Schizotypal Personality Disorder. Am J Psychiatry. 2000;157:55–59. doi: 10.1176/ajp.157.1.55. [DOI] [PubMed] [Google Scholar]
- Carandini M, Heeger DJ. Normalization as a canonical neural computation. Nat Rev Neurosci. 2011;13:51–62. doi: 10.1038/nrn3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio C, McGlone F, Folger S, Tannan V, Baranek G, Pelphrey KA, Essick G. Tactile Perception in Adults with Autism: a Multidimensional Psychophysical Study. J Autism Dev Disord. 2008;38:127–137. doi: 10.1007/s10803-007-0370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Noor A, Degagne B, Baker K, Bok LA, Brady AF, Chitayat D, Chung BH, Cytrynbaum C, Dyment D, et al. Phenotypic spectrum associated with PTCHD1 deletions and truncating mutations includes intellectual disability and autism spectrum disorder. Clin Genet. 2015;88:224–233. doi: 10.1111/cge.12482. [DOI] [PubMed] [Google Scholar]
- Chen Z, Wimmer RD, Wilson MA, Halassa MM. Thalamic Circuit Mechanisms Link Sensory Processing in Sleep and Attention. Front Neural Circuits. 2016;9:83. doi: 10.3389/fncir.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasca F, Rubio-Garrido P, Jabaudon D. Unveiling the diversity of thalamocortical neuron subtypes. Eur J Neurosci. 2012;35:1524–1532. doi: 10.1111/j.1460-9568.2012.08033.x. [DOI] [PubMed] [Google Scholar]
- Cohen JD, Servan-Schreiber D. Context, cortex, and dopamine: A connectionist approach to behavior and biology in schizophrenia. Psychol Rev. 1992;99:45–77. doi: 10.1037/0033-295x.99.1.45. [DOI] [PubMed] [Google Scholar]
- Corbett BA, Constantine LJ. Autism and attention deficit hyperactivity disorder: assessing attention and response control with the integrated visual and auditory continuous performance test. Child Neuropsychol. 2006;12:335–348. doi: 10.1080/09297040500350938. [DOI] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Lüthi A. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci. 2008;11:683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Dehaene S, Changeux JP. A hierarchical neuronal network for planning behavior. Proc Natl Acad Sci. 1997;94:13293–13298. doi: 10.1073/pnas.94.24.13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehaene S, Charles L, King JR, Marti S. Toward a computational theory of conscious processing. Curr Opin Neurobiol. 2014;25:76–84. doi: 10.1016/j.conb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghani N, Cash SS, Halgren E. Emergence of synchronous EEG spindles from asynchronous MEG spindles. Hum Brain Mapp. 2011;32:2217–2227. doi: 10.1002/hbm.21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin MJ, Weissman MM, Xu D, Bansal R, Hao X, Liu J, Warner V, Peterson BS. Identification of a circuit-based endophenotype for familial depression. Psychiatry Res. 2012;201:175–181. doi: 10.1016/j.pscychresns.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erp TGM, Hibar DP, Rasmussen JM, Glahn DC, Pearlson GD, Andreassen OA, Agartz I, Westlye LT, Haukvik UK, Dale AM, et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry. 2016;21:547–553. doi: 10.1038/mp.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarelli F, Huber R, Peterson MJ, Massimini M, Murphy M, Riedner BA, Watson A, Bria P, Tononi G. Reduced sleep spindle activity in schizophrenia patients. Am J Psychiatry. 2007;164:483–492. doi: 10.1176/ajp.2007.164.3.483. [DOI] [PubMed] [Google Scholar]
- Ferrarelli F, Peterson MJ, Sarasso S, Riedner BA, Murphy MJ, Benca RM, Bria P, Kalin NH, Tononi G. Thalamic dysfunction in schizophrenia suggested by whole-night deficits in slow and fast spindles. Am J Psychiatry. 2010;167:1339–1348. doi: 10.1176/appi.ajp.2010.09121731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher TG, Alitto HJ, Usrey WM. Retinal and Nonretinal Contributions to Extraclassical Surround Suppression in the Lateral Geniculate Nucleus. J Neurosci. 2017;37:226–235. doi: 10.1523/JNEUROSCI.1577-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Timpson N, Munafò M. Assessing the utility of intermediate phenotypes for genetic mapping of psychiatric disease. Trends Neurosci. 2014;37:733–741. doi: 10.1016/j.tins.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston KJ, Frith CD. Schizophrenia: a disconnection syndrome? Clin Neurosci. 1995;3:89–97. [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M, Moran JL, Purcell SM, Sklar P, Sullivan PF, et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci. 2016;19:1433–1441. doi: 10.1038/nn.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Flint J. Genetics and genomics of psychiatric disease. Science (80-) 2015;349:1489–1494. doi: 10.1126/science.aaa8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirardi L, Brikell I, Kuja-Halkola R, Freitag CM, Franke B, Asherson P, Lichtenstein P, Larsson H. The familial co-aggregation of ASD and ADHD: a register-based cohort study. Mol Psychiatry. 2017 doi: 10.1038/mp.2017.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold JM, Fuller RL, Robinson BM, Braun EL, Luck SJ. Impaired top-down control of visual search in schizophrenia. Schizophr Res. 2007;94:148–155. doi: 10.1016/j.schres.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden EC, Graff-Radford J, Jones DT, Benarroch EE. Mediodorsal nucleus and its multiple cognitive functions. Neurology. 2016;87:2161–2168. doi: 10.1212/WNL.0000000000003344. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The Endophenotype Concept in Psychiatry: Etymology and Strategic Intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Grafton ST. The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol. 2015;7:a021691. doi: 10.1101/cshperspect.a021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano MSA, Aflalo TN. Mapping behavioral repertoire onto the cortex. Neuron. 2007;56:239–251. doi: 10.1016/j.neuron.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Greenwood TA, Swerdlow NR, Gur RE, Cadenhead KS, Calkins ME, Dobie DJ, Freedman R, Green MF, Gur RC, Lazzeroni LC, et al. Genome-wide linkage analyses of 12 endophenotypes for schizophrenia from the consortium on the genetics of schizophrenia. Am J Psychiatry. 2013;170:521–532. doi: 10.1176/appi.ajp.2012.12020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzadzinski R, Dick C, Lord C, Bishop S. Parent-reported and clinician-observed autism spectrum disorder (ASD) symptoms in children with attention deficit/hyperactivity disorder (ADHD): implications for practice under DSM-5. Mol Autism. 2016;7:7. doi: 10.1186/s13229-016-0072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW, Sherman SM. Thalamic relay functions and their role in corticocortical communication: Generalizations from the visual system. Neuron. 2002;33:163–175. doi: 10.1016/s0896-6273(01)00582-7. [DOI] [PubMed] [Google Scholar]
- Guller Y, Tononi G, Postle BR. Conserved Functional Connectivity but Impaired Effective Connectivity of Thalamocortical Circuitry in Schizophrenia. Brain Connect. 2012;2:311–319. doi: 10.1089/brain.2012.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Nimgaonkar VL, Go RCP, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Acsady L. Thalamic Inhibition: Diverse Sources, Diverse Scales. Trends Neurosci. 2016;39:680–693. doi: 10.1016/j.tins.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Kastner S. Thalamic functions in distributed cognitive control. Nat Neurosci. 2017;20:1669–1679. doi: 10.1038/s41593-017-0020-1. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Siegle JH, Ritt JT, Ting JT, Feng G, Moore CI. Selective optical drive of thalamic reticular nucleus generates thalamic bursts and cortical spindles. Nat Neurosci. 2011;14:1118–1120. doi: 10.1038/nn.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Chen Z, Wimmer RD, Brunetti PM, Zhao S, Zikopoulos B, Wang F, Brown EN, Wilson MA. State-Dependent Architecture of Thalamic Reticular Subnetworks. Cell. 2014;158:808–821. doi: 10.1016/j.cell.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MH, Taylor G, Sham P, Schulze K, Rijsdijk F, Picchioni M, Toulopoulou T, Ettinger U, Bramon E, Murray RM, et al. The early auditory gamma-band response is heritable and a putative endophenotype of schizophrenia. Schizophr Bull. 2011;37:778–787. doi: 10.1093/schbul/sbp134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrisberger F, Smieskova R, Vogler C, Egli T, Schmidt A, Lenz C, Simon AE, Riecher-Rossler A, Papassotiropoulos A, Borgwardt S. Impact of polygenic schizophrenia-related risk and hippocampal volumes on the onset of psychosis. Transl Psychiatry. 2016;6:e868. doi: 10.1038/tp.2016.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WD, Davies G, Liewald DC, McIntosh AM, Deary IJ. Age-Dependent Pleiotropy Between General Cognitive Function and Major Psychiatric Disorders. Biol Psychiatry. 2016;80:266–273. doi: 10.1016/j.biopsych.2015.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffaker SJ, Chen J, Nicodemus KK, Sambataro F, Yang F, Mattay V, Lipska BK, Hyde TM, Song J, Rujescu D, et al. A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat Med. 2009;15:509–518. doi: 10.1038/nm.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–3817. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y, Narzisi G, Leotta A, et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa EB, Wang X. Altered Neural Responses to Sounds in Primate Primary Auditory Cortex during Slow-Wave Sleep. J Neurosci. 2011;31:2965–2973. doi: 10.1523/JNEUROSCI.4920-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito HT, Zhang SJ, Witter MP, Moser EI, Moser MB. A prefrontal–thalamo–hippocampal circuit for goal-directed spatial navigation. Nature. 2015;522:50–55. doi: 10.1038/nature14396. [DOI] [PubMed] [Google Scholar]
- Jadi MP, Sejnowski TJ. Cortical oscillations arise from contextual interactions that regulate sparse coding. Proc Natl Acad Sci U S A. 2014;111:6780–6785. doi: 10.1073/pnas.1405300111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Doneshka P, Zylberman I, Ritter W, Vaughan HGJ. Impairment of early cortical processing in schizophrenia: an event-related potential confirmation study. Biol Psychiatry. 1993;33:513–519. doi: 10.1016/0006-3223(93)90005-x. [DOI] [PubMed] [Google Scholar]
- Jazayeri M, Afraz A. Navigating the Neural Space in Search of the Neural Code. Neuron. 2017;93:1003–1014. doi: 10.1016/j.neuron.2017.02.019. [DOI] [PubMed] [Google Scholar]
- John B, Lewis KR. Chromosome variability and geographic distribution in insects. Science. 1966;152:711–721. doi: 10.1126/science.152.3723.711. [DOI] [PubMed] [Google Scholar]
- Kanai R, Komura Y, Shipp S, Friston K. Cerebral hierarchies: predictive processing, precision and the pulvinar. Philos Trans R Soc Lond B Biol Sci. 2015;370 doi: 10.1098/rstb.2014.0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keehn B, Müller RA, Townsend J. Atypical attentional networks and the emergence of autism. Neurosci Biobehav Rev. 2013;37:164–183. doi: 10.1016/j.neubiorev.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime Prevalence and Age-of-Onset Distributions of Distributions of DSM-IV Disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Ketz NA, Jensen O, O’Reilly RC. Thalamic pathways underlying prefrontal cortex-medial temporal lobe oscillatory interactions. Trends Neurosci. 2015;38:3–12. doi: 10.1016/j.tins.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Kim H, Ährlund-Richter S, Wang X, Deisseroth K, Carlén M, Jenvay S, Miyamichi K, Luo L, Dan Y, Boyden ES, et al. Prefrontal Parvalbumin Neurons in Control of Attention. Cell. 2016;164:208–218. doi: 10.1016/j.cell.2015.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komura Y, Nikkuni A, Hirashima N, Uetake T, Miyamoto A. Responses of pulvinar neurons reflect a subject’s confidence in visual categorization. Nat Neurosci. 2013;16:749–755. doi: 10.1038/nn.3393. [DOI] [PubMed] [Google Scholar]
- Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, Karczewski KJ, Cutler DJ, Devlin B, Roeder K, et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet. 2017;49:504–510. doi: 10.1038/ng.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47:582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperberg GR, Broome MR, McGuire PK, David AS, Eddy M, Ozawa F, Goff D, West WC, Williams SCR, van der Kouwe AJW, et al. Regionally localized thinning of the cerebral cortex in schizophrenia. Arch Gen Psychiatry. 2003;60:878–888. doi: 10.1001/archpsyc.60.9.878. [DOI] [PubMed] [Google Scholar]
- Larsson HJ, Eaton WW, Madsen KM, Vestergaard M, Olesen AV, Agerbo E, Schendel D, Thorsen P, Mortensen PB. Risk Factors for Autism: Perinatal Factors, Parental Psychiatric History, and Socioeconomic Status. Am J Epidemiol. 2005;161:916–925. doi: 10.1093/aje/kwi123. [DOI] [PubMed] [Google Scholar]
- Lee SE, Lee J, Latchoumane C, Lee B, Oh SJ, Saud ZA, Park C, Sun N, Cheong E, Chen CC, et al. Rebound burst firing in the reticular thalamus is not essential for pharmacological absence seizures in mice. Proc Natl Acad Sci U S A. 2014;111:11828–11833. doi: 10.1073/pnas.1408609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, Witte JS, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lener MS, Wong E, Tang CY, Byne W, Goldstein KE, Blair NJ, Haznedar MM, New AS, Chemerinski E, Chu KW, et al. White matter abnormalities in schizophrenia and schizotypal personality disorder. Schizophr Bull. 2015;41:300–310. doi: 10.1093/schbul/sbu093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppa VM, Kravitz SN, Martin CL, Andrieux J, Le Caignec C, Martin-Coignard D, DyBuncio C, Sanders SJ, Lowe JK, Cantor RM, et al. Rare Inherited and De Novo CNVs Reveal Complex Contributions to ASD Risk in Multiplex Families. Am J Hum Genet. 2016;99:540–554. doi: 10.1016/j.ajhg.2016.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF, Hultman CM. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoges E, Mottron L, Bolduc C, Berthiaume C, Godbout R. Atypical sleep architecture and the autism phenotype. Brain. 2005;128:1049–1061. doi: 10.1093/brain/awh425. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG. Low-threshold calcium channel subunit Ca(v) 3.3 is specifically localized in GABAergic neurons of rodent thalamus and cerebral cortex. J Comp Neurol. 2011;519:1181–1195. doi: 10.1002/cne.22567. [DOI] [PubMed] [Google Scholar]
- Mangeot SD, Miller LJ, McIntosh DN, McGrath-Clarke J, Simon J, Hagerman RJ, Goldson E. Sensory modulation dysfunction in children with attention-deficit-hyperactivity disorder. Dev Med Child Neurol. 2001;43:399–406. doi: 10.1017/s0012162201000743. [DOI] [PubMed] [Google Scholar]
- Manoach DS, Demanuele C, Wamsley EJ, Vangel M, Montrose DM, Miewald J, Kupfer D, Buysse D, Stickgold R, Keshavan MS. Sleep spindle deficits in antipsychotic-naïve early course schizophrenia and in non-psychotic first-degree relatives. Front Hum Neurosci. 2014;8:762. doi: 10.3389/fnhum.2014.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco EJ, Hinkley LBN, Hill SS, Nagarajan S. Sensory processing in autism: A review of neuropsychologic findings. Pediatr Res. 2011;69:48–54. doi: 10.1203/PDR.0b013e3182130c54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35. doi: 10.1038/ng.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlonan K, Cavanaugh J, Wurtz RH. Attentional modulation of thalamic reticular neurons. J Neurosci. 2006;26:4444–4450. doi: 10.1523/JNEUROSCI.5602-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlonan K, Cavanaugh J, Wurtz RH. Guarding the gateway to cortex with attention in visual thalamus. Nature. 2008;456:391–394. doi: 10.1038/nature07382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Hyman SE. Progress in the genetics of polygenic brain disorders: Significant new challenges for neurobiology. Neuron. 2013;80:578–587. doi: 10.1016/j.neuron.2013.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol. 1961;34:103–116. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- Mei L, Nave KA. Neuregulin-ERBB Signaling in the Nervous System and Neuropsychiatric Diseases. Neuron. 2014;83:27–49. doi: 10.1016/j.neuron.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micoulaud-Franchi JA, Lopez R, Vaillant F, Richieri R, El-Kaim A, Bioulac S, Philip P, Boyer L, Lancon C. Perceptual abnormalities related to sensory gating deficit are core symptoms in adults with ADHD. Psychiatry Res. 2015a;230:357–363. doi: 10.1016/j.psychres.2015.09.016. [DOI] [PubMed] [Google Scholar]
- Micoulaud-Franchi JA, Vaillant F, Lopez R, Peri P, Baillif A, Brandejsky L, Steffen ML, Boyer L, Richieri R, Cermolacce M, et al. Sensory gating in adult with attention-deficit/hyperactivity disorder: Event-evoked potential and perceptual experience reports comparisons with schizophrenia. Biol Psychol. 2015b;107:16–23. doi: 10.1016/j.biopsycho.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Minshew NJ, Keller TA. The nature of brain dysfunction in autism: functional brain imaging studies. Curr Opin Neurol. 2010;23:124–130. doi: 10.1097/WCO.0b013e32833782d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan A, Anney RJL, O’Regan M, Chen W, Butler L, Fitzgerald M, Buitelaar J, Steinhausen HC, Rothenberger A, Minderaa R, et al. Autism symptoms in Attention-Deficit/Hyperactivity Disorder: a familial trait which correlates with conduct, oppositional defiant, language and motor disorders. J Autism Dev Disord. 2009;39:197–209. doi: 10.1007/s10803-008-0621-3. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Halassa MM. Thalamic control of functional cortical connectivity. Curr Opin Neurobiol. 2017;44:127–131. doi: 10.1016/j.conb.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir Y, Staba RJ, Andrillon T, Vyazovskiy VV, Cirelli C, Fried I, Tononi G. Regional Slow Waves and Spindles in Human Sleep. Neuron. 2011;70:153–169. doi: 10.1016/j.neuron.2011.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir Y, Vyazovskiy VV, Cirelli C, Banks MI, Tononi G. Auditory responses and stimulus-specific adaptation in rat auditory cortex are preserved across NREM and REM sleep. Cereb Cortex. 2015;25:1362–1378. doi: 10.1093/cercor/bht328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitskaya Y, Sara SJ, Logothetis NK, Eschenko O. Ripple-triggered stimulation of the locus coeruleus during post-learning sleep disrupts ripple/spindle coupling and impairs memory consolidation. Learn Mem. 2016;23:238–248. doi: 10.1101/lm.040923.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dushlaine C, Rossin L, Lee PH, Duncan L, Parikshak NN, Newhouse S, Ripke S, Neale BM, Purcell SM, Posthuma D, et al. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18:199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jönsson B. The economic cost of brain disorders in Europe. Eur J Neurol. 2012;19:155–162. doi: 10.1111/j.1468-1331.2011.03590.x. [DOI] [PubMed] [Google Scholar]
- Pappa I, St Pourcain B, Benke K, Cavadino A, Hakulinen C, Nivard MG, Nolte IM, Tiesler CMT, Bakermans-Kranenburg MJ, Davies GE, et al. A genome-wide approach to children’s aggressive behavior: The EAGLE consortium. Am J Med Genet B Neuropsychiatr Genet. 2016;171:562–572. doi: 10.1002/ajmg.b.32333. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet. 2015;16:441–458. doi: 10.1038/nrg3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Pinault D, Deschênes M. Projection and innervation patterns of individual thalamic reticular axons in the thalamus of the adult rat: A three-dimensional, graphic, and morphometric analysis. J Comp Neurol. 1998;391:180–203. doi: 10.1002/(sici)1096-9861(19980209)391:2<180::aid-cne3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portas CM, Krakow K, Allen P, Josephs O, Armony JL, Frith CD. Auditory processing across the sleep-wake cycle: simultaneous EEG and fMRI monitoring in humans. Neuron. 2000;28:991–999. doi: 10.1016/s0896-6273(00)00169-0. [DOI] [PubMed] [Google Scholar]
- Posthuma D, Polderman TJC. What have we learned from recent twin studies about the etiology of neurodevelopmental disorders? Curr Opin Neurol. 2013;26:111–121. doi: 10.1097/WCO.0b013e32835f19c3. [DOI] [PubMed] [Google Scholar]
- Prasad KM, Keshavan MS. Structural Cerebral Variations as Useful Endophenotypes in Schizophrenia: Do They Help Construct “Extended Endophenotypes”? Schizophr Bull. 2008;34:774–790. doi: 10.1093/schbul/sbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, Sato D, Rickaby J, Lu C, Szatmari P, Roberts W, et al. A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder. G3 Genes, Genomes, Genet. 2012;2:1665–1685. doi: 10.1534/g3.112.004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kähler A, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N. Autism spectrum disorders and childhood-onset schizophrenia: clinical and biological contributions to a relation revisited. J Am Acad Child Adolesc Psychiatry. 2009;48:10–18. doi: 10.1097/CHI.0b013e31818b1c63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reus LM, Shen X, Gibson J, Wigmore E, Ligthart L, Adams MJ, Davies G, Cox SR, Hagenaars SP, Bastin ME, et al. Association of polygenic risk for major psychiatric illness with subcortical volumes and white matter integrity in UK Biobank. Sci Rep. 2017;7:42140. doi: 10.1038/srep42140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, Lee P, Bulik-Sullivan B, Collier DA, Huang H, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovo Z, Ulbert I, Acsady L. Drivers of the primate thalamus. J Neurosci. 2012;32:17894–17908. doi: 10.1523/JNEUROSCI.2815-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff CC, Blankenburg F, Bjoertomt O, Bestmann S, Weiskopf N, Driver J. Hemispheric differences in frontal and parietal influences on human occipital cortex: direct confirmation with concurrent TMS-fMRI. J Cogn Neurosci. 2009;21:1146–1161. doi: 10.1162/jocn.2009.21097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saalmann YB, Kastner S. Cognitive and Perceptual Functions of the Visual Thalamus. Neuron. 2011;71:209–223. doi: 10.1016/j.neuron.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sable JJ, Kyle MR, Knopf KL, Schully LT, Brooks MM, Parry KH, Diamond RE, Flink LA, Stowe R, Suna E, et al. The Sensory Gating Inventory as a potential diagnostic tool for attention-deficit hyperactivity disorder. Atten Defic Hyperact Disord. 2012;4:141–144. doi: 10.1007/s12402-012-0079-1. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, Uruno K, Kumada S, Nishiyama K, Nishimura A, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- Saxena S, Funk M, Chisholm D. World health assembly adopts comprehensive mental health action plan 2013–2020. Lancet. 2013;381:1970–1971. doi: 10.1016/S0140-6736(13)61139-3. [DOI] [PubMed] [Google Scholar]
- Schmitt LI, Halassa MM. Interrogating the mouse thalamus to correct human neurodevelopmental disorders. Mol Psychiatry. 2017;22:183–191. doi: 10.1038/mp.2016.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt LI, Wimmer RD, Nakajima M, Happ M, Mofakham S, Halassa MM. Thalamic amplification of cortical connectivity sustains attentional control. Nature. 2017;545:219–223. doi: 10.1038/nature22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelley AM, Ward PB, Catts SV, Michie PT, Andrews S, McConaghy N. Mismatch negativity: an index of a preattentive processing deficit in schizophrenia. Biol Psychiatry. 1991;30:1059–1062. doi: 10.1016/0006-3223(91)90126-7. [DOI] [PubMed] [Google Scholar]
- Sherman SM. Thalamic relays and cortical functioning. Prog Brain Res. 2005;149:107–126. doi: 10.1016/S0079-6123(05)49009-3. [DOI] [PubMed] [Google Scholar]
- Sherman SM. Thalamus plays a central role in ongoing cortical functioning. Nat Neurosci. 2016;16:533–541. doi: 10.1038/nn.4269. [DOI] [PubMed] [Google Scholar]
- Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, Suvisaari J, Chheda H, Blackwood D, Breen G, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–577. doi: 10.1038/nn.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoller JW, Kendler K, Craddock N, Writing JW, Smoller N, Craddock K, Kendler PH, Lee BM, Neale JI, Nurnberger S, et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smucny J, Rojas DC, Eichman LC, Tregellas JR. Neural Effects of Auditory Distraction on Visual Attention in Schizophrenia. PLoS One. 2013;8:e60606. doi: 10.1371/journal.pone.0060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, Šestan N. The Emerging Biology of Autism Spectrum Disorders. Science (80-) 2012;337:1301 LP–1303. doi: 10.1126/science.1224989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Deschenes M, Domich L, Mulle C. Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol. 1985;54:1473–1497. doi: 10.1152/jn.1985.54.6.1473. [DOI] [PubMed] [Google Scholar]
- Steriade M, Domich L, Oakson G. Reticularis thalami neurons revisited: activity changes during shifts in states of vigilance. J Neurosci. 1986;6:68–81. doi: 10.1523/JNEUROSCI.06-01-00068.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Domich L, Oakson G, Deschenes M. The deafferented reticular thalamic nucleus generates spindle rhythmicity. J Neurophysiol. 1987;57:260–273. doi: 10.1152/jn.1987.57.1.260. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Kendler KS, Neale MC, S M, T P, J L, L P, JA W, SW L, PC S, et al. Schizophrenia as a Complex Trait. Arch Gen Psychiatry. 2003;60:1187. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Nakahachi T, Komatsu S, Ogino K, Iida Y, Kamio Y. Hyperreactivity to weak acoustic stimuli and prolonged acoustic startle latency in children with autism spectrum disorders. Mol Autism. 2014;5:23. doi: 10.1186/2040-2392-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier S, Lambert A, Chicoine M, Scherzer P, Soulières I, Godbout R. Intelligence measures and stage 2 sleep in typically-developing and autistic children. Int J Psychophysiol. 2015;97:58–65. doi: 10.1016/j.ijpsycho.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Tomchek SD, Dunn W. Sensory processing in children with and without autism: a comparative study using the short sensory profile. Am J Occup Ther Off Publ Am Occup Ther Assoc. 2007;61:190–200. doi: 10.5014/ajot.61.2.190. [DOI] [PubMed] [Google Scholar]
- Trampush JW, Yang MLZ, Yu J, Knowles E, Davies G, Liewald DC, Starr JM, Djurovic S, Melle I, Sundet K, et al. GWAS meta-analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol Psychiatry. 2017;22:336–345. doi: 10.1038/mp.2016.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005;76:1–23. doi: 10.1016/j.schres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Ung DC, Iacono G, Méziane H, Blanchard E, Papon MA, Selten M, van Rhijn JR, Montjean R, Rucci J, Martin S, et al. Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Mol Psychiatry. 2017:1–12. doi: 10.1038/mp.2017.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usrey WM, Alitto HJ. Visual Functions of the Thalamus. Annu Rev Vis Sci. 2015;1:351–371. doi: 10.1146/annurev-vision-082114-035920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaghi MM, Hampshire A, Fineberg NA, Kaser M, Bruhl AB, Sahakian BJ, Chamberlain SR, Robbins TW. Hypoactivation and Dysconnectivity of a Frontostriatal Circuit During Goal-Directed Planning as an Endophenotype for Obsessive-Compulsive Disorder. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2:655–663. doi: 10.1016/j.bpsc.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit J, Hakim R, Jadi MP, Sejnowski TJ, Adesnik H. Cortical gamma band synchronization through somatostatin interneurons. Nat Neurosci. 2017 doi: 10.1038/nn.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinck M, Womelsdorf T, Buffalo EA, Desimone R, Fries P. Attentional modulation of cell-class-specific gamma-band synchronization in awake monkey area v4. Neuron. 2013;80:1077–1089. doi: 10.1016/j.neuron.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineskos AN, Felsky D, Wheeler AL, Rotenberg DJ, Levesque M, Patel S, Szeszko PR, Kennedy JL, Lencz T, Malhotra AK. Limited Evidence for Association of Genome-Wide Schizophrenia Risk Variants on Cortical Neuroimaging Phenotypes. Schizophr Bull. 2016;42:1027–1036. doi: 10.1093/schbul/sbv180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukadinovic Z. NMDA receptor hypofunction and the thalamus in schizophrenia. Physiol Behav. 2014;131:156–159. doi: 10.1016/j.physbeh.2014.04.038. [DOI] [PubMed] [Google Scholar]
- Weidmann S. Effect of current flow on the membrane potential of cardiac muscle. J Physiol. 1951;115:227–236. doi: 10.1113/jphysiol.1951.sp004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, Samocha KE, Goldstein JI, Okbay A, Bybjerg-Grauholm J, et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet. 2017;49:978–985. doi: 10.1038/ng.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells MF, Wimmer RD, Schmitt LI, Feng G, Halassa MM. Thalamic reticular impairment underlies attention deficit in Ptchd1Y/− mice. Nature. 2016;532:58–63. doi: 10.1038/nature17427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, Charlson FJ, Norman RE, Flaxman AD, Johns N, et al. Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet. 2013;382:1575–1586. doi: 10.1016/S0140-6736(13)61611-6. [DOI] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Coexpression Networks Implicate Human Midfetal Deep Cortical Projection Neurons in the Pathogenesis of Autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer RD, Astori S, Bond CT, Rovó Z, Chatton JY, Adelman JP, Franken P, Lüthi A. Sustaining sleep spindles through enhanced SK2-channel activity consolidates sleep and elevates arousal threshold. J Neurosci. 2012;32:13917–13928. doi: 10.1523/JNEUROSCI.2313-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer RD, Schmitt LI, Davidson TJ, Nakajima M, Deisseroth K, Halassa MM. Thalamic control of sensory selection in divided attention. Nature. 2015;526:705–709. doi: 10.1038/nature15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womelsdorf T, Fries P, Mitra PP, Desimone R. Gamma-band synchronization in visual cortex predicts speed of change detection. Nature. 2006;439:733–736. doi: 10.1038/nature04258. [DOI] [PubMed] [Google Scholar]
- Xu W, Sudhof TC. A Neural Circuit for Memory Specificity and Generalization. Science (80-) 2013;339:1290–1295. doi: 10.1126/science.1229534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wells AB, O’Brien DR, Nehorai A, Dougherty JD. Cell Type-Specific Expression Analysis to Identify Putative Cellular Mechanisms for Neurogenetic Disorders. J Neurosci. 2014;34:1420–1431. doi: 10.1523/JNEUROSCI.4488-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh LF, Watanabe M, Sulkes-Cuevas J, Johansen JP. Dysregulation of aversive signaling pathways: a novel circuit endophenotype for pain and anxiety disorders. Curr Opin Neurobiol. 2017;48:37–44. doi: 10.1016/j.conb.2017.09.006. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XJ, Xu XX, He S, He J. Change detection by thalamic reticular neurons. Nat Neurosci. 2009;12:1165–1170. doi: 10.1038/nn.2373. [DOI] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Pathways for emotions and attention converge on the thalamic reticular nucleus in primates. J Neurosci. 2012;32:5338–5350. doi: 10.1523/JNEUROSCI.4793-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaigenbaum L, Bryson S, Rogers T, Roberts W, Brian J, Szatmari P. Behavioral manifestations of autism in the first year of life. Int J Dev Neurosci. 2005;23:143–152. doi: 10.1016/j.ijdevneu.2004.05.001. [DOI] [PubMed] [Google Scholar]