

Abstract
The TAIRE family of kinases are an understudied branch of the CDK kinase family, that have been implicated in a number of cancers. This manuscript describes the design, synthesis and SAR of covalent CDK14 inhibitors, culminating in identification of FMF-04-159-2, a potent, covalent CDK14 inhibitor with a TAIRE kinase biased selectivity profile.
Keywords: Covalent kinase inhibitor, CDK14, TAIRE kinase, CDK15, CDK16, CDK17, CDK18, CDK inhibitor, mitosis, cell cycle
CDK14 is a member of the understudied TAIRE subfamily of cyclin-dependent kinases, named after the “TAIRE” sequence motif in their cyclin binding site, and comprising of CDKs 14-18.(1,)(2,)(3) CDK14 overexpression has been reported in numerous cancers including colorectal cancer(4), ovarian cancer(5) and gastric cancer(6) However, selective tool compounds to interrogate the pharmacological consequences of CDK14 inhibition were, until recently, unavailable.
We recently reported the identification and characterization of FMF-04-159-2 (100), the first covalent CDK14 inhibitor with TAIRE-kinase bias(7) Off-target CDK2 activity was identified, and experimental conditions to minimize CDK2 engagement and maximize CDK14 engagement were reported.(7) Here we describe the structure activity relationships of a series of 4-amino-1H-pyrazole analogs for CDK14 biochemical and cellular potency, and measure their effects on HCT116 proliferation. We show cellular engagement of CDK14 by lead molecules, and demonstrate their covalent nature by MS/MS studies. This data is of interest, as 4-amino-1H-pyrazole is a widely used kinase inhibitor scaffold.(8) This SAR analysis aids development of further, improved CDK14 inhibitors, and in addition, provides insight into how CDK14 activity can be removed from 4-amino-1H-pyrazole analogs targeted towards other kinases.
In search of chemical leads for CDK14, we performed a CDK14 cellular engagement screen of a library of reported CDK inhibitors using a biotin JNK-IN-7 pulldown assay.(9) This identified the multi-targeted CMCG kinase inhibitor AT7519 as an efficient CDK14 inhibitor at 1 μM.(10) This result was confirmed using a LanthaScreen CDK14 binding assay (Table 1).
Table 1.
Single ring R1 analogs of AT7519. A) CDK14 IC50s were measured using a LanthaScreen binding assay. IC50s were calculated as the average of three replicates, and are reported ± the standard errorB) Antiproliferative activity against the HCT116 cell line was measured using a CellTiter-Glo assay. IC50s were calculated as the average of three replicates, and are reported ± the standard error.
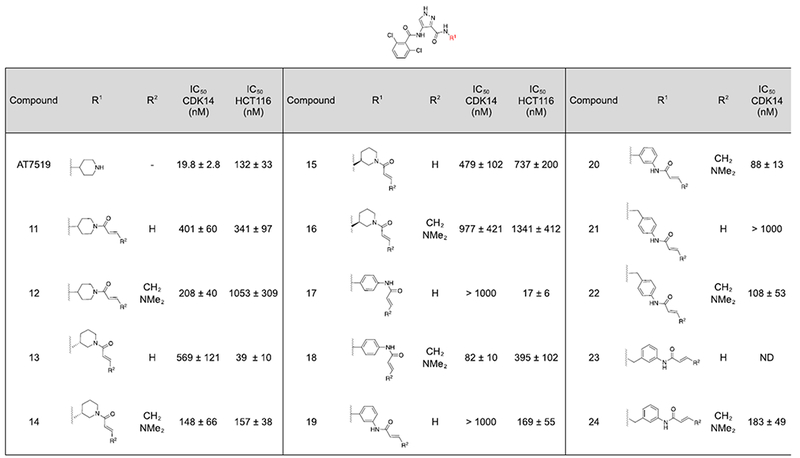 |
SiRNA mediated CDK14 knockdown does not cause significant proliferation defects in the HCT116 cell line, unlike knockdown of other CDK kinases.(11) A selective CDK14 inhibitor is consequently also expected to have mild to insignificant effects on HCT116 cell growth. Therefore we used potency in a CellTiter-Glo (Promega) proliferation assay as a first pass approximation of compound selectivity for CDK14 when prioritizing molecules for progress through further rounds of characterization (Supporting Figure 1). As previously reported, AT7519 is a potent inhibitor of HCT116 cell proliferation, with an IC50 of 132 nM.(7)
CDK14 contains a uniquely placed cysteine, C218, located at the beginning of the αD helix, proximal to the ATP pocket(12) In order to improve the potency and selectivity of AT7519 towards CDK14, we sought to design a covalent inhibitor.
Examination of the co-crystal structure of CDK2 in complex with AT7519 (PDB: 2VU3) revealed that the 4-aminopiperidine is oriented towards the αD helix in CDK2(10) Assuming a conserved binding mode, this substituent (R1) should provide a suitable vector for targeting CDK14 C218.
Therefore analogs containing varied R1 substituents incorporating acrylamide and (E)-4-(dimethylamino)but-2-enamide warheads (R2) were synthesized according to Scheme 1.
Scheme 1.
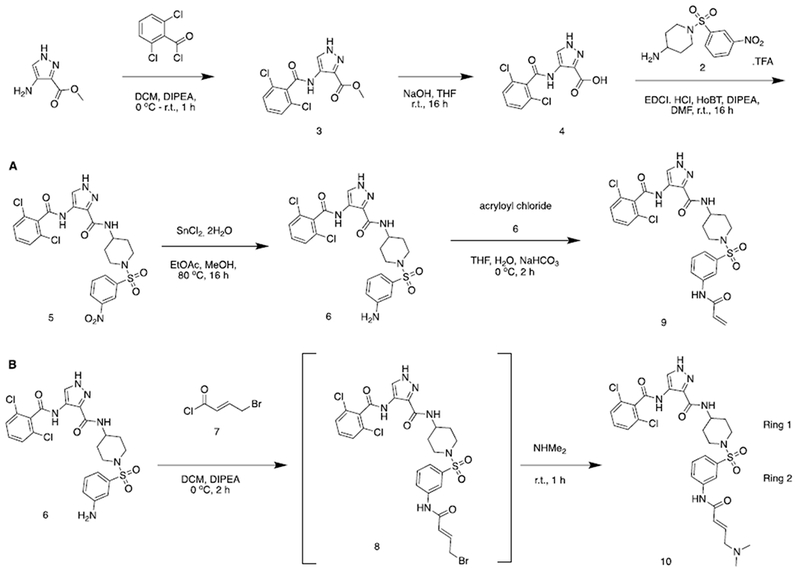
Representative synthesis of 4-amino-1H-pyrazole analogs. See also supporting information.
Initially, molecules containing a single saturated or unsaturated ring were synthesized (Table 1, compounds 11-24).
These analogs lost significant potency relative to AT7519, potentially due to the loss of a hydrogen bonding interaction with the piperidine NH seen in CDK2 (PDB:2VU3), which was not compensated for by covalent inhibition.
JNK3 contains a cysteine in an equivalent region of the kinase to CDK14. Examination of the structure of JNK3 in complex with JNK-IN-7 (PDB: 3V6S) indicated that a longer distance between the hinge binding motif and the acrylamide is required in order to successfully form a covalent bond.(9) Therefore analogs were prepared containing two linked cyclic aliphatic or aromatic rings decorated with acrylamide or (E)-4-(dimethylamino)but-2-enamide warheads (Table 2, 25-70). Docking studies into a CDK14 homology model built from the X-ray structure of CDK12 (PDB: 4NST) predicted that a range of linked two ring systems, with 1,4-regiochemistry in ring 1 and a 1,3-regiochemistry in ring 2 would best allow for C218 covalent engagement (Figure 1).(7) (13)
Table 2:
Extended R1 analogs of AT7519. A) CDK14 IC50s were measured using a LanthaScreen binding assay. IC50s were calculated as the average of three replicates, and are reported ± the standard errorB) Antiproliferative activity against the HCT116 cell line was measured using a CellTiter-Glo assay. IC50s were calculated as the average of three replicates, and are reported ± the standard error. –R is used to denote reversible control compounds (without an alkene group).
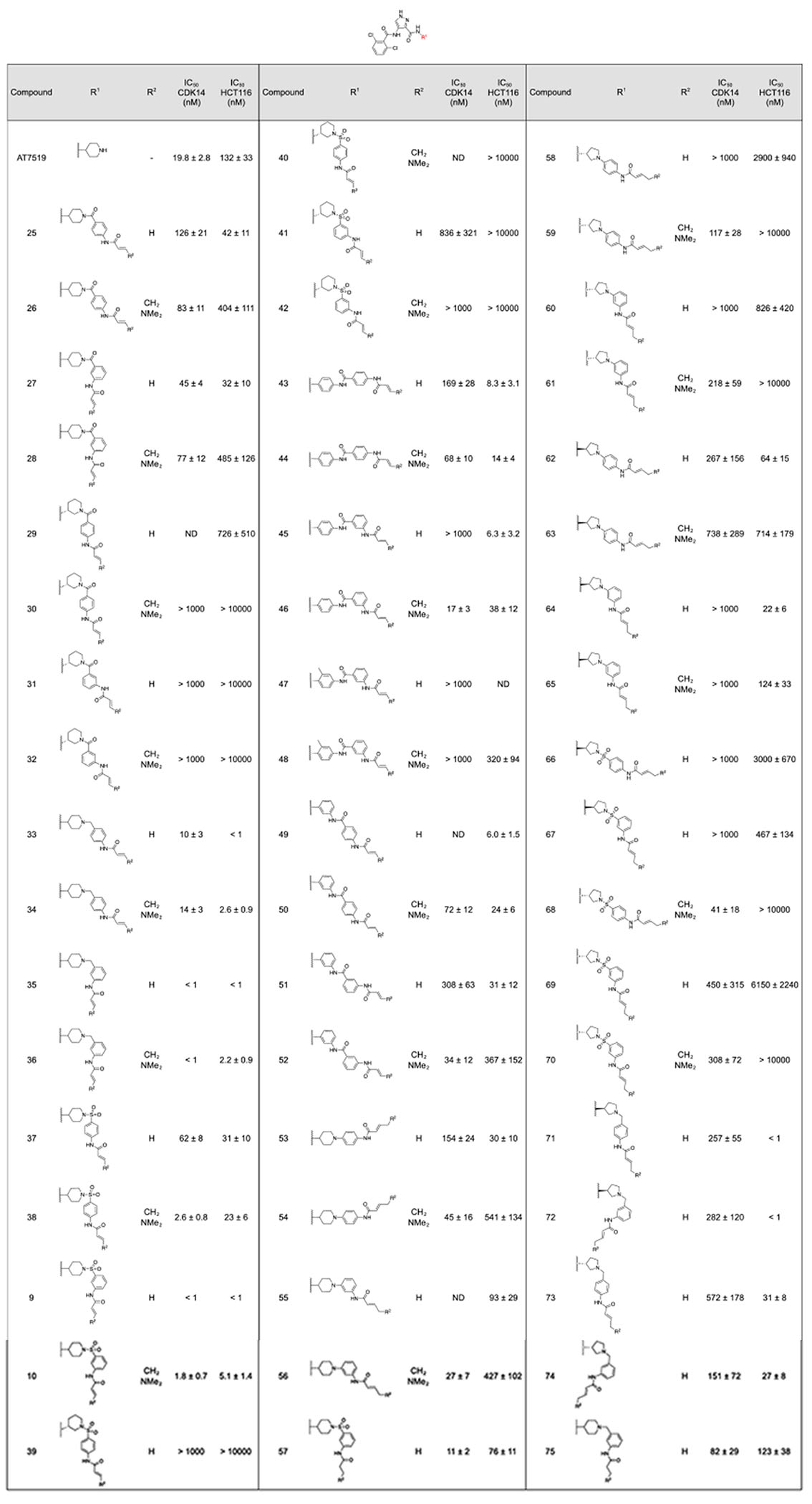 |
Figure 1:
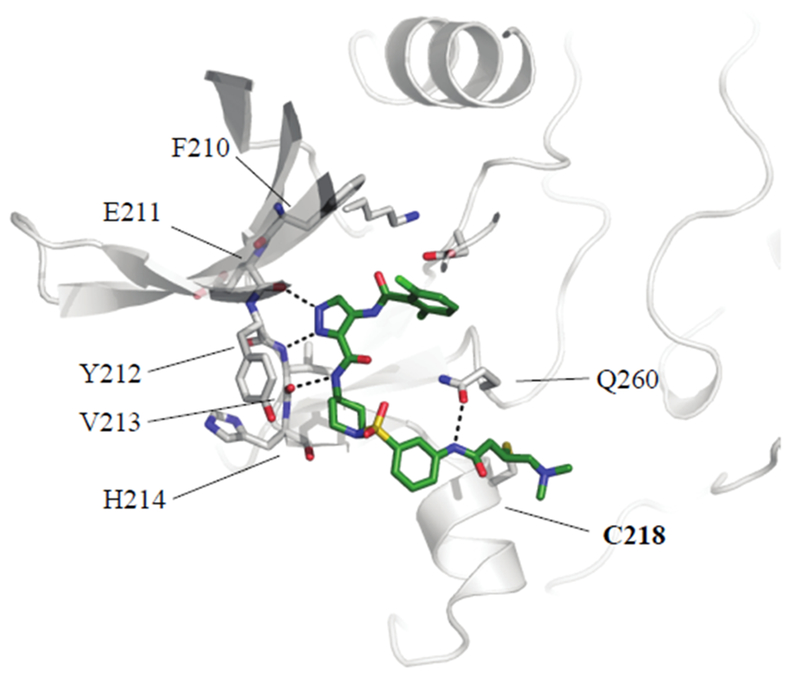
Docking of compound 10 into a homology model of CDK14. View from above.
Molecules containing a piperidine linked to an aminobenzamide at R1 displayed reduced potency for CDK14 relative to AT7519 (Table 2, 25-32). 1,4 aminopiperidine regiochemistry at R1 is strongly preferred, with 1,3 aminopiperidine regiochemistry not well tolerated at this position (e.g. 27 vs 31). Preference for a 1,3 aminobenzamide regiochemistry of the second ring was observed with the most potent analogs, 27 and 28, combining these two regiochemistries, in line with computational docking results.
Replacing the aminobenzamide (28) with an aminobenzylamine (36) dramatically increased potency against CDK14. Again, a strong preference for a 1,4 aminopiperidine regiochemistry and a 1,3 aminobenzylamine regiochemistry was observed (Table 2, 36 vs 34). The most potent analogs, 35 and 36, inhibited CDK14 with IC50s below 1 nM in the LanthaScreen biochemical assay. 36 was chosen for follow-up studies due to its lower toxicity in the cell titer glo assay, relative to 35. Both compounds show increased toxicity relative to AT7519, indicating off-target activity. Compounds containing an aminobenzylsulfonamide followed similar trends to the aminobenzamide series. Compound 36 is highly potent against CDK14, and also displays increased toxicity against HCT116 cells.
Inspired by the acrylamide bearing substituent in JNK-IN-7, amide linked biphenyls were introduced to R1 (Table 1 compounds 43–52).(9) Although these compounds displayed reduced affinity, they also exhibited the same regiochemical preferences. Interestingly in this series, the E)-4-(dimethylamino)but-2-enamide warhead was preferred to the acrylamide warhead. 46 was the most potent of this subseries against CDK14, but also had increased toxicity relative to AT7519.
In an attempt to improve selectivity in a manner analogous to JNK-IN-8, compounds were synthesized with ortho-methylation of the 1,4-diaminoaniline in ring 1 (47, 48). Unfortunately, this methylation was not tolerated by CDK14.
Substitution of the ring 1 piperidine group for a pyrrolidine resulted in compounds with a less favorable CDK14 : HCT116 potency profile (e.g. 72 vs 35) with the exception of 68, which was taken forward for validation.
Removal of the linking atoms between rings 1 and 2 afforded 3(piperidine-1-yl)anilines 53-56. This series maintained acceptable CDK14 potency and the dimethylamino-substituted analogs 54 and 56 had low HCT116 toxicity. 56 was chosen for follow up studies.
Finally, the propyl amide analogs of the most potent molecules were synthesized to examine the effects of removal of the covalent warhead. 57 was ~ 10-fold less potent against CDK14 compared to 9 and 10, and displayed similar toxicity against HCT116 cells to 10. Compound 75 was significantly less potent against both CDK14 and HCT116 cells than 35 and 36. This indicated that the covalent binding component improved binding towards CDK14, but also towards off-targets that alter cell proliferation.
To verify that the 6 lead compounds and two reversible control compounds identified in Table 2 were able to engage CDK14 in cells we performed a pull-down experiment. Cells were treated for 4 h with compounds at various doses, and then lysed and treated with biotinylated JNK-IN-7(9), followed by streptavidin coated beads. CDK14 capture was assayed by western blot. (Table 3, SI Figure 1). Of these compounds, 10 was able to potently block CDK14 pulldown at 50 nM, and 36, 46 and 27 also showed effects at 500 – 1000 nM concentrations (Supporting Figure 2). Compounds 56 and 68 were not active, or weakly active in the cellular assay (Supporting Figure 2). The reversible control molecules also showed no activity in this assay, indicating a dependency on covalent bond formation for activity in cells. Covalent bond formation by 10 and 36 was verified by incubating compounds with purified, recombinant CDK14 followed by MS/MS analysis. Both compounds achieved complete labeling of CDK14 when incubated for 3 h at r.t., at a 10:1 molar ratio of compound to CDK14/Cyclin Y protein. Digest experiments followed by MS analysis showed that C218 was exclusively labeled.
Table 3:
Cellular target engagement of lead compounds in biotin pull-down assay, estimated based on testing of three compound concentrations.
| Compound | IC50 CDK14 (nM) | Compound | IC50 CDK14 (nM) |
|---|---|---|---|
| 9 | 1000 | 46 | 1000 |
| 36 | 500 | 10 | 50 |
| 75 | > 1000 | 57 | > 1000 |
| 56 | 1000 | 68 | > 1000 |
To evaluate the cellular targets of 10 and 36 more broadly, we performed KiNativ profiling at 1 μM compound concentration (Figure 1)(14) (7) This revealed that whilst both compounds were potent against CDK14, they also inhibited a large number of other kinases.
As hinge binding is a common feature of Type I kinase inhibitors, analogs of FMF-03-198-2 with altered hinge binding motifs predicted to reduce hydrogen bonding interactions were synthesized, in an attempt to increase the selectivity for CDK14 by reducing the reversible binding affinity, but maintaining the ability to bind covalently (Table 4, 76-87).
Table 4:
FMF-03-198 analogs with a modified heterocyclic core. A) CDK14 IC50s were measured using a LanthaScreen binding assay. IC50s were calculated as the average of three replicates, and are reported ± the standard error. B) Antiproliferative activity against the HCT116 cell line was measured using a CellTiter-Glo assay. IC50s were calculated as the average of three replicates, and are reported ± the standard error.
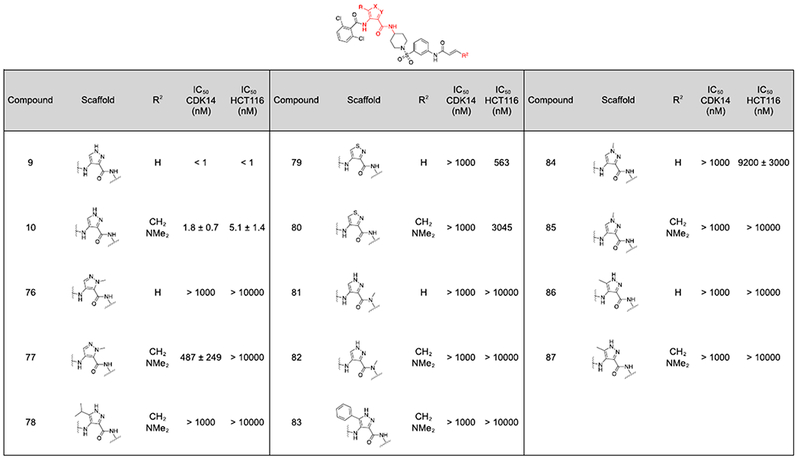 |
Unfortunately, none of these analogs demonstrated potent inhibition in the CDK14 binding assay (Table 4). Therefore, the 2,6-dichlorobenzamide substituent (R3) was varied in an effort to improve selectivity (Table 5). In the co-crystal structure of AT7519 in complex with CDK2, the 2,6-benzamide group fills a small hydrophobic pocket, and prior studies have demonstrated that CDK2 binding activity can be tuned by altering it’s substituents, by introducing steric clashes with the back pocket.(10) Substitution of the 2,6-dichloro phenyl ring of 9 and 10 for a more polar 2,6-difluoro phenyl ring (88, 89) yielded a modest reduction in toxicity and maintained potent CDK14 inhibition.
Table 5:
FMF-03-198 analogs with varied R3 substituents. A) CDK14 IC50s were measured using a LanthaScreen binding assay. IC50s were calculated as the average of three replicates, and are reported ± the standard error. B) Antiproliferative activity against the HCT116 cell line was measured using a CellTiter-Glo assay. IC50s were calculated as the average of three replicates, and are reported ± the standard error. –R is used to denote reversible control compounds (without an alkene group).
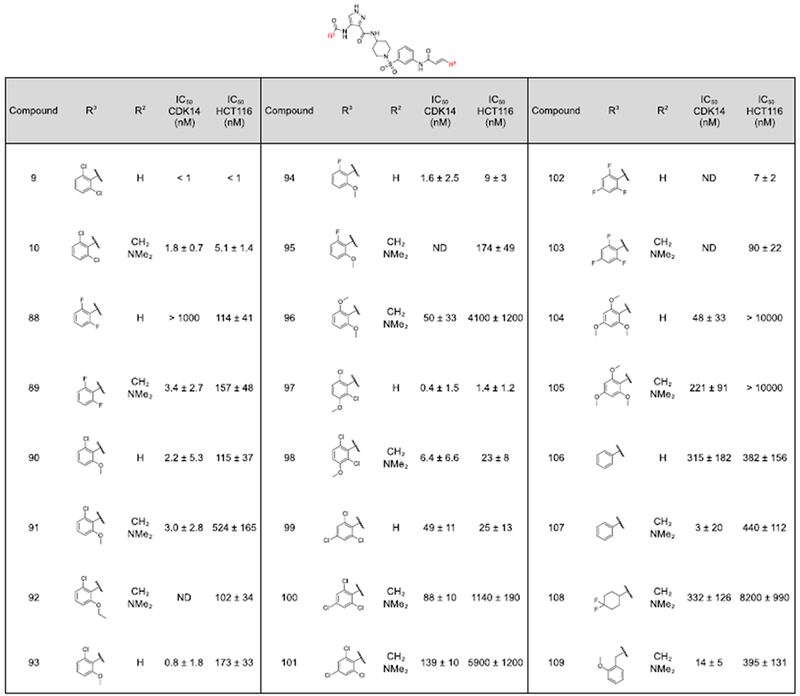 |
The bulkier 2-chloro, 6-methoxy substitution yielded a further reduction in toxicity, whilst maintaining potency for CDK14 (90, 91). The reversible propyl amide 93 maintained comparable potency towards CDK14 and comparable potency against HCT116 cells as 90, indicating this effect was primarily driven by changes in the reversible binding profile of the compounds. However, further increase in the bulk of the 6-substituent to an ethoxy group afforded a more toxic compound (92), indicative of a narrow SAR window at the 6-position. 2,6-dimethoxy substitution (96), 2,4,6-methoxy substitution (104) and 2,4,6-chloro substitution (100) patterns resulted in reduced CDK14 potency, but also dramatically reduced HCT116 toxicity.
Addition of a methoxy group to the 3-position of 10 to afford 97 didn’t significantly affect CDK14 binding or toxicity. Finally, the un-substituted compound 107 exhibited potent CDK14 inhibition and reduced HCT116 toxicity. The active compounds from this series were taken forward for cellular target engagement studies (Table 6, Supporting Figure 2). Of these, only 91 and 100 were able to completely inhibit CDK14 pull-down at concentrations below 1 μM.
Table 6.
Cellular target engagement of additional lead compounds in biotin pull-down assay, estimated based on testing of three compound concentrations.
| Compound | IC50 CDK14 (nM) | Compound | IC50 CDK14 (nM) |
|---|---|---|---|
| 91 | 50 | 96 | > 1000 |
| 100 | 500 | 104 | > 1000 |
| 107 | 1000 | 101 | > 1000 |
Compound 91 and 100 were both evaluated using the KiNativ platform for cellular selectivity at concentrations of 1 μM and compared to the profiles of 10 and 36. Compound 91 demonstrated a comparable number of targets compared to 10 and 36, despite its reduced cytotoxicity. However, compound 100 showed a favorable profile, with a dramatically reduced number of targets.
Of the 6 targets inhibited at > 75%, 4 were TAIRE kinases (CDK14, CDK16, CDK17, CDK18). These kinases have been reported to display functional redundancy, and thus the pan-TAIRE activity of 100 may aid interrogation of the biology of these understudied kinases.(15) The other targets of 100 were CDK2 and CDK10. Despite binding to these off-targets, 100 was demonstrated to engage CDK14 in cellular context much more potently than CDK2, with an IC50 of 39.6 nM for CDK14 compared to 256 nM for CDK2, as measured by the NanoBRET assay.(7) This improved cellular potency for CDK14 over CDK2, and other off-targets of compound 100 offered an improvement in the relative cellular selectivity for CDK14 compared to AT7519. Future work is required to remove activity against these targets from CDK14 / TAIRE kinase probes, therefore use of washout conditions is recommended when using 100 to probe the pharmacological consequences of CDK14 inhibition.(7) Compound 100 represents a significant advance towards developing chemical probes for the TAIRE kinase family, and is the first targeted covalent CDK14 inhibitor.
Supplementary Material
Figure 2:
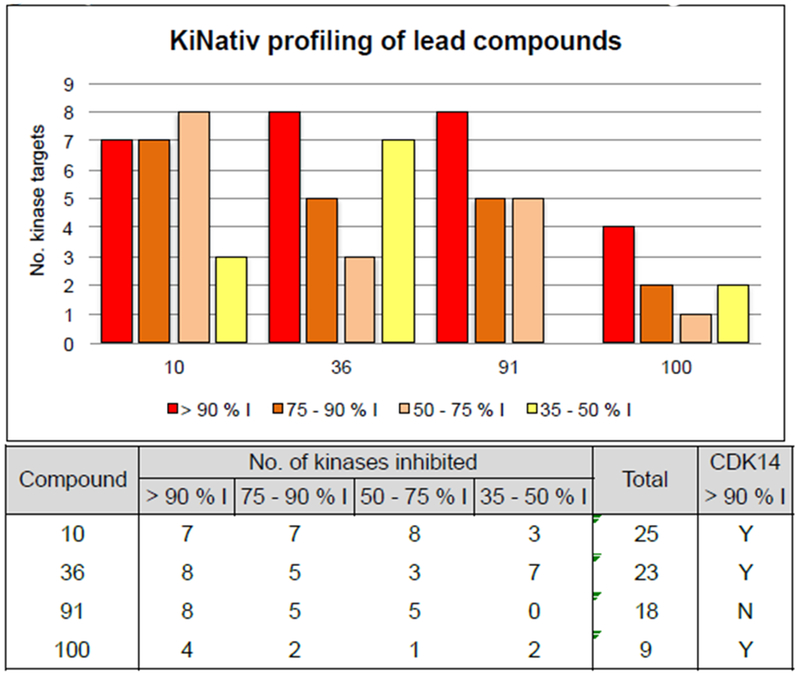
KiNativ profiling results of lead compounds at 1 μM compound concentration. % I, percent inhibition relative to DMSO only control.
Acknowledgements
This work was funded by the following grants; R21CA188881, R01CA219850, DFCI Strategic Research Initiative
Disclosures
J.A.M. is a member of the SAB of Devices. N.S.G. is a founder, SAB member, and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Her2llc, Deerfield, and Sanofi. N.S.G., F.M.F., and Z.M.D. are inventors on a patent application covering chemical matter in this publication owned by Dana-Farber Cancer Institute. S.B.F, N.D.K. and T.S. have no conflicts to declare
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fedorov O, Muller S, and Knapp S (2010) The (un)targeted cancer kinome, Nature chemical biology 6, 166–169. [DOI] [PubMed] [Google Scholar]
- 2.Fleuren ED, Zhang L, Wu J, and Daly RJ (2016) The kinome ‘at large’ in cancer, Nature reviews. Cancer 16,83–98. [DOI] [PubMed] [Google Scholar]
- 3.Malumbres M (2014) Cyclin-dependent kinases, Genome biology 15,122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu J, Liu C, Liu F, Wang Y, and Zhu M (2016) Knockdown of PFTAIRE Protein Kinase 1 (PFTK1) Inhibits Proliferation, Invasion, and EMT in Colon Cancer Cells, Oncology research 24,137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W, Liu R, Tang C, Xi Q, Lu S, Chen W, Zhu L, Cheng J, Chen Y, Wang W, Zhong J, and Deng Y (2016) PFTK1 regulates cell proliferation, migration and invasion in epithelial ovarian cancer, International journal of biological macromolecules 85, 405–416. [DOI] [PubMed] [Google Scholar]
- 6.Yang L, Zhu J, Huang H, Yang Q, Cai J, Wang Q, Zhu J, Shao M, Xiao J, Cao J, Gu X, Zhang S, and Wang Y (2015) PFTK1 Promotes Gastric Cancer Progression by Regulating Proliferation, Migration and Invasion, PloS one 10, e0140451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson FM, Doctor ZM, Ficarro SB, Browne CM, Marto JA, Johnson JL, Yaron TM, Cantley LC, Kim ND, Sim T, Berberich MJ, Kalocsay M, Sorger PK, and Gray NS (2019) Discovery of Covalent CDK14 Inhibitors with Pan-TAIRE Family Specificity, Cell chemical biology 26, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimova D, and Bajorath J (2017) Assessing Scaffold Diversity of Kinase Inhibitors Using Alternative Scaffold Concepts and Estimating the Scaffold Hopping Potential for Different Kinases, Molecules (Basel, Switzerland) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang T, Inesta-Vaquera F, Niepel M , Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, Laughlin JD, Park H, LoGrasso PV, Patricelli M, Nomanbhoy TK, Sorger PK, Alessi DR, and Gray NS (2012) Discovery of potent and selective covalent inhibitors of JNK, Chemistry & biology 19, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wyatt PG, Woodhead AJ, Berdini V, Boulstridge JA, Carr MG, Cross DM, Davis DJ, Devine LA, Early TR, Feltell RE, Lewis EJ, McMenamin RL, Navarro EF, O’Brien MA, O’Reilly M, Reule M, Saxty G, Seavers LC, Smith DM, Squires MS, Trewartha G, Walker MT, and Woolford AJ (2008) Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design, Journal of medicinal chemistry 51, 4986–4999. [DOI] [PubMed] [Google Scholar]
- 11.McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, Golji J, Kao I, Li L, Megel P, Perkins TA, Ramadan N, Ruddy DA, Silver SJ, Sovath S, Stump M, Weber O, Widmer R, Yu J, Yu K, Yue Y, Abramowski D, Ackley D, Barrett R, Berger J, Bernard JL, Billig R, Brachmann SM, Buxton F, Caothien R, Caushi JX, Chung FS, Cortes-Cros M, deBeaumont RS, Delaunay C, Desplat A, Duong W, Dwoske DA, Eldridge RS, Farsidjani A, Feng F, Feng J, Flemming D, Forrester W, Galli GG, Gao Z, Gauter F, Gibaja V, Haas K, Hattenberger M, Hood T, Hurov KE, Jagani Z, J enal M, Johnson JA, Jones MD, Kapoor A, Korn J, Liu J, Liu Q, Liu S, Liu Y, Loo AT, Macchi KJ, Martin T, McAllister G, Meyer A, Molle S, Pagliarini RA, Phadke T, Repko B, Schouwey T, Shanahan F, Shen Q, Stamm C, Stephan C, Stucke VM, Tiedt R, Varadarajan M, Venkatesan K, Vitari AC, Wallroth M, Weiler J, Zhang J, Mickanin C, Myer VE, Porter JA, Lai A, Bitter H, Lees E, Keen N, Kauffmann A, Stegmeier F, Hofmann F, Schmelzle T, and Sellers WR (2017) Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening, Cell 170, 577–592 e510. [DOI] [PubMed] [Google Scholar]
- 12.Chaikuad A, Koch P, Laufer SA, and Knapp S (2018) The Cysteinome of Protein Kinases as a Target in Drug Development, Angewandte Chemie (International ed. in English) 57, 4372–4385. [DOI] [PubMed] [Google Scholar]
- 13.Bosken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, and Geyer M (2014) The structure and substrate specificity of human Cdk12/Cyclin K, Nature communications 5, 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, and Kozarich JW (2011) In situ kinase profiling reveals functionally relevant properties of native kinases, Chemistry & biology 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, and Niehrs C (2009) Cell cycle control of wnt receptor activation, Developmental cell 17, 788–799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.