

Abstract
Erdheim-Chester disease (ECD) is a rare histiocytosis that may affect the central nervous system (CNS). Infiltration by the disease occurs throughout the neuroaxis, usually involving the dentate nucleus and the pons, manifested as a pyramido-cerebellar syndrome. CNS involvement is an adverse prognostic factor which warrants prompt evaluation and treatment. BRAF mutation occurs in more than half of the cases and has become central in the therapeutic approach. There is rapidly growing evidence that BRAF inhibitors such as vemurafenib or dabrafenib are effective in treating CNS-spread disease. We present a patient with BRAF-V600E-mutant ECD with a classical pyramido-ataxic onset of disease who improved after prompt diagnosis with vemurafenib treatment as first-line therapy.
Keywords: brain stem / cerebellum, haematology (incl blood transfusion)
Background
Erdheim-Chester disease (ECD), a histiocytosis of the L group according to the latest classification,1 is a rare multisystemic disease that may involve the CNS,2 commonly manifesting as a pyramido-cerebellar syndrome. ECD has characteristic findings on brain MRI, bone X-ray and skin biopsy. Currently, interferon-α (IFN-α) is the first-line therapy in ECD.3 In patients with BRAF-mutated ECD, vemurafenib as second-line therapy has achieved a high grade of responses, although few cases have been treated with this drug as first-line therapy. Prompt diagnosis and treatment may halt disease progression before substantial disability occurs.
Case presentation
A 31-year-old woman with a 1-year history of unstable gait was admitted to our hospital. She had a history of hypercholesterolaemia and central diabetes insipidus and a skin biopsy that revealed ‘diffuse xanthomatosis’. On neurological examination she presented prominent gait ataxia with widespread hyper-reflexia, double vision caused by spontaneous nystagmus and mild dysarthria. Multiple skin xanthomas were observed.
Investigations
A brain and spinal cord MRI showed diffuse alteration of fluid-attenuated inversion recovery (FLAIR) and T2 signal affecting the deep white matter of both the cerebellar hemispheres and peduncles, the pons, and the lateral aspect of the spinal cord symmetrically, from C3 to D5. No diffusion restriction or gadolinium enhancement was observed (figure 1A,B). Whole-body positron emission tomography-CT showed cerebellar hypometabolism. Complete blood and cerebrospinal fluid analyses were unrevealing. Nerve conduction studies + electromyography and a cervico-thoraco-abdomino-pelvic CT scan were normal.
Figure 1.
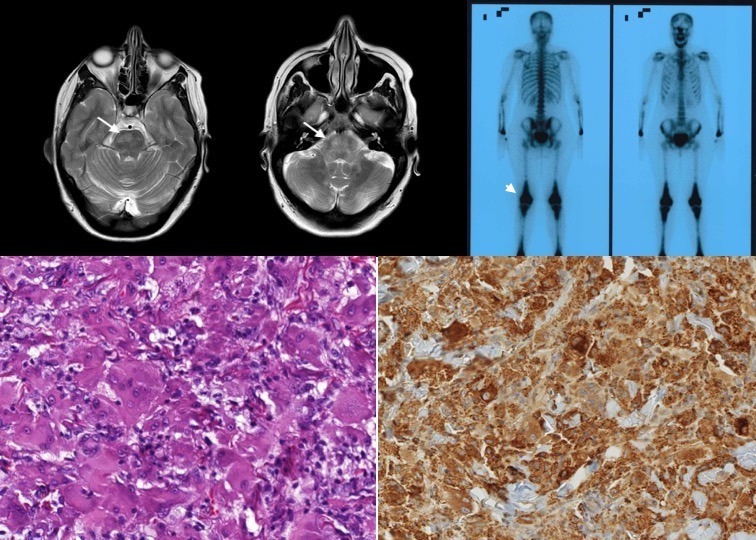
(A) Cerebral MRI showing diffuse alteration of T2 signal affecting the deep white matter of both the cerebellar hemispheres, extending along both the cerebellar peduncles and protuberance (marked by arrows). (B) No gadolinium enhancement on cerebral MRI. (C) Bone gammagraphy showing bilateral diafiso-metafisarial corticomedullar sclerosis (marked by arrow head). (D) H&E 20× showing multinucleated xanthomatous-like cellularity. (E) Immunohistochemistry showing diffuse CD68 staining.
Differential diagnosis
Normal cholestanol, sterol levels and long-chain fatty acids in serum ruled out cerebrotendinous xanthomatosis. CYP27A1 and spinocerebellar ataxia mutations were negative. The skin biopsy was recovered and reviewed, and showed a proliferation of xanthomatous-like cellularity, with a negative immunohistochemical profile for S100 and CD1a staining. Thus, the possibility of an ECD arose. Further staining for CD68 and factor XIII was positive, as well as BRAF-V600E mutation (figure 1D,E). X-ray of the long bone and bone gammagraphy supported the diagnosis of ECD (figure 1C).
Treatment
Treatment with vemurafenib 480 mg–0–480 mg was started. Photosensitivity has been a bothersome complication of the treatment; therefore, vemurafenib dose had to be lowered to 480 mg–0–240 mg.
Outcome and follow-up
During the first 10 months, there was partial clinical improvement after starting treatment with vemurafenib, and 3 years later disease progression has been halted (clinically and on MRI). The patient is able to walk short distances unassisted.
Discussion
ECD, a histiocytosis of the L group,1 is a rare disease that may frequently involve the CNS. The age of patients with ECD ranges from 16 to 80 years, with a mean age of 51 years at symptom onset,2 and shows a strong male predominance (72%).2
ECD is a multisystemic disease where skeletal involvement is almost always present (80%), with characteristic bilateral osteosclerotic lesions of the long bones of the lower extremities (figure 1). Of the cases, 37% show CNS involvement, being more frequent in BRAF-V600E mutated patients. CNS involvement is also an adverse prognostic factor,2 especially in the presence of paraneoplastic inflammatory lesions. Histiocytic infiltration or inflammatory lesions can occur throughout the neuroaxis,2 4 and while intra-axial lesions usually involve the dentate nucleus or the pons (10%–17%) (figure 1A,B), commonly manifested as a pyramido-cerebellar syndrome, extra-axial involvement such as meningeal mass lesions (23%) is also common.3 Spinal cord involvement is very rare.5 A quarter of patients may have preceding central diabetes insipidus, as in this case.2 3 Other neurological symptoms include headaches, seizures, cognitive impairment, cranial nerve palsies and neuropsychiatric manifestations. As previously mentioned, ECD is a multisystemic disease that can also cause cardiovascular (53%–64%), retroperitoneal (58%), lung (35%), skin (33%) and retro-orbital (22%) involvement in the largest series.2
The diagnosis of ECD is made in the appropriate clinical, radiological and histopathological context.3 The latter is characterised by foamy or lipid-laden histiocytes proliferating as polymorphic xanthomatous-like cellularity. Immunohistochemical staining is positive for CD68, CD163 and factor XIIIa (figure 1), whereas it is negative for CD1a and Langerin/CD207. S100 staining may sometimes be mildly positive.3 Furthermore, BRAF-V600E mutation should be studied, since as high as 66% of patients are BRAF mutation-positive according to the latest series using picodroplet digital PCR. This explains why patients with ECD improve with vemurafenib, a selective BRAF-V600E inhibitor, as recently shown.6 In wild-type BRAF, other mutations of genes implicated in the mitogen-activated protein kinases (MAPK) pathway have been found.7 8
Currently, IFN-α is the first-line therapy, although standard doses may be ineffective in severe cases (by means of CNS or cardiac involvement).3 Other treatment alternatives are anakinra, an interleukin-1 antagonist which at standard doses has not demonstrated to improve CNS symptoms, and vemurafenib, a BRAF inhibitor with increasing evidence of efficacy in treating CNS-spread disease. Common toxicities for this latter drug are arthralgias, photosensitivity and multiple skin complications.2 Future treatment options continue to emerge,9 and include other BRAF inhibitors such as dabrafenib in BRAF-mutated patients.10 Recently, a very high response (89%) with cobimetinib, a mitogen-activated protein kinase kinase (MEK) inhibitor, has been observed in wild-type BRAF patients.8 The median survival in ECD is 162 months, with a 5-year survival of 82.7%.2
In summary, ECD should be included in the differential diagnosis of a patient presenting with a pyramido-cerebellar syndrome and xanthomatosis. This case report should make neurologists aware of screening for BRAF-V600E mutation in patients with ECD and consider vemurafenib as a possible treatment, even as first-line therapy in selected cases.
Learning points.
Erdheim-Chester disease (ECD) is a rare histiocytosis commonly presenting as a pyramido-cerebellar syndrome with gait ataxia.
ECD has characteristic findings on brain MRI, bone X-ray, bone gammagraphy and skin biopsy.
CNS involvement must be taken into account when choosing the most appropriate therapy.
Early vemurafenib treatment in BRAF-V600E mutated patients may halt disease progression before substantial disability occurs.
Footnotes
Contributors: GF-E and MU contributed by filing and acquiring all the information and figures, as well as writing the manuscript. AM-L contributed by editing the information. IR contributed with anatomopathological figures and descriptions.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127:2672–81. 10.1182/blood-2016-01-690636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen-Aubart F, Emile J-F, Carrat F, et al. Phenotypes and survival in Erdheim-Chester disease: results from a 165-patient cohort. Am J Hematol 2018;93:E114–7. 10.1002/ajh.25055 [DOI] [PubMed] [Google Scholar]
- 3. Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood 2014;124:483–92. 10.1182/blood-2014-03-561381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sedrak P, Ketonen L, Hou P, et al. Erdheim-Chester disease of the central nervous system: new manifestations of a rare disease. AJNR Am J Neuroradiol 2011;32:2126–31. 10.3174/ajnr.A2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lachenal F, Cotton F, Desmurs-Clavel H, et al. Neurological manifestations and neuroradiological presentation of Erdheim-Chester disease: report of 6 cases and systematic review of the literature. J Neurol 2006;253:1267–77. 10.1007/s00415-006-0160-9 [DOI] [PubMed] [Google Scholar]
- 6. Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012;120:2700–3. 10.1182/blood-2012-05-430140 [DOI] [PubMed] [Google Scholar]
- 7. Emile J-F, Diamond EL, Hélias-Rodzewicz Z, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood 2014;124:3016–9. 10.1182/blood-2014-04-570937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019;567:521–4. 10.1038/s41586-019-1012-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haroche J, Cohen-Aubart F, Rollins BJ, et al. Histiocytoses: emerging neoplasia behind inflammation. Lancet Oncol 2017;18:e113–25. 10.1016/S1470-2045(17)30031-1 [DOI] [PubMed] [Google Scholar]
- 10. Cohen Aubart F, Emile J-F, Carrat F, et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood 2017;130:1377–80. 10.1182/blood-2017-03-771873 [DOI] [PubMed] [Google Scholar]