

Abstract
The linker of nucleoskeleton and cytoskeleton (LINC) complex mediates intracellular cross talk between the nucleus and the cytoplasm. In striated muscle, the LINC complex provides structural support to the myocyte nucleus and plays an essential role in regulating gene expression and mechanotransduction. A wide range of cardiac and skeletal myopathies have been linked to mutations in LINC complex proteins. Studies utilizing tissue-specific knockout and mutant mouse models have revealed important insights into the roles of the LINC complex in striated muscle. In this chapter, we describe several feasible approaches for generating striated muscle-specific gene knockout and mutant mouse models to study LINC complex protein function in cardiac and skeletal muscle. The experimental procedures used for phenotyping and analysis of LINC complex knockout mice are also described.
Keywords: LINC complex, Nuclear envelope, Knockout mouse, Knock-in mouse, Cre/loxP, CRISPR/Cas9, Striated muscle, Cardiac muscle, Skeletal muscle
1. Introduction
The application of gene targeting technology to create modifications in a tissue-specific manner and at a precise stage in development is a powerful tool to elucidate the functions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins in vivo. Targeting specific genes for modification by homologous recombination in embryonic stem (ES) cells has become a routine procedure [1, 2]. Over the past decade, we have routinely reported the use ES cell gene targeting technology to generate null or point mutation alleles in the study of LINC complex protein function [3–6]. More recently, a growing body of studies has utilized clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology to generate genetically modified mice with extraordinary simplicity and speed [7–11]. Here, we briefly describe the methodology of traditional gene targeting (Fig. 1) and report in detail a cloning-free method to successfully generate floxed as well as single-amino-acid-substituted mice that can be used for striated muscle-specific gene targeting of LINC complex proteins. The general approaches and techniques described here can also be applied to establish other gene knockout and mutant mouse models.
Fig. 1.
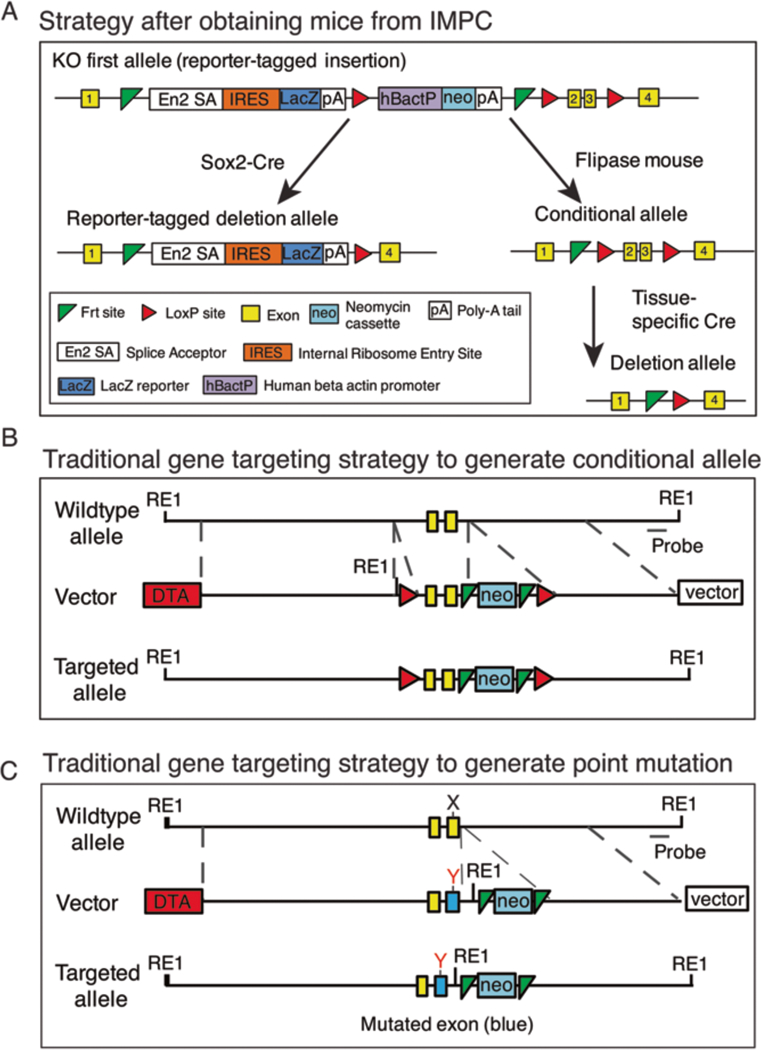
Mouse generation strategies. (A) Mice obtained from the International Mouse Phenotype Consortium (IMPC) can be crossed with Sox2-Cre mice to generate global knockout mice. Alternatively, they can be crossed with flippase deleter mice to generate the conditional allele followed by a tissue-specific Cre deletion in the relevant tissue or cell type. (B) The traditional gene targeting approach inserts two 34 base pair sequences referred to as loxP sites (red triangles) that flank either side of a critical exon in a gene of interest and a neomycin cassette for embryonic stem (ES) cell selection. Diptheria toxin A gene is incorporated outside of the homology arms to select against ES colonies that have not taken up the insert by homologous recombination. A new restriction enzyme site is introduced in the mutant construct to aid screening with Southern blotting. (C) Traditional gene targeting approach to generate the substitution mutation from nucleotide X to Y in an exon (blue)
1.1. Generation of Floxed Mice
Conditional knockout of a target gene in mice is based on generating a floxed allele by inserting two 34 base pair (bp) sequences of DNA referred to as loxP sites that flank either side of a critical exon in a gene of interest. The loxP sites are recognized by a Cre recombinase, which mediates recombination to excise the floxed exon and achieve gene inactivation [3, 12–14]. Many of the genes encoding the LINC complex are available as floxed alleles on the International Mouse Phenotype Consortium (IMPC) website (https://www.mousephenotype.org/data/search), which greatly expedites the process of generating conditional null alleles. Generally, the IMPC mouse lines contain the desired floxed allele with LacZ and neomycin cassettes still present (Fig. 1A) (see Note 1), which are removed by crossing the mice with a flippase (FLP) deleter mouse [15]. Alternatively, global or conditional tissue-specific knockout mice can be generated by crossing the IMPC mice with a desired Cre deleter mouse (Fig. 1A). For mouse lines unavailable through the IMPC website, two approaches can be used to generate floxed alleles as outlined below.
In traditional gene targeting, a conditional construct is first generated (Fig. 1B) (see Notes 2 and 3), linearized with a restriction enzyme, and electroporated into ES cells. Targeted ES cells are identified by Southern blot analysis. ES cells from a homologous recombinant clone are then microinjected into mouse blastocysts. Male chimeras are bred with female breeder mice to generate germline-transmitted heterozygous mice with a neomycin cassette, which are confirmed by PCR or Southern blot analysis of mouse tail DNA [3–6, 16].
In this chapter, we focus on the clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system as a powerful tool to generate striated muscle-specific gene knockout and mutant mouse models to study LINC complex protein function in cardiac and skeletal muscle. The CRISPR/Cas9 system consists of a Cas9 nuclease and two small RNAs: CRISPR RNA (crRNA), which acts as a guide for gene targeting, and trans-activating crRNA (tracrRNA), which binds to crRNA and forms a ribonucleoprotein complex with Cas9 to direct sequence-specific double-stranded breaks (DSBs) [17–19]. Recently, we developed a cloning-free method to generate floxed alleles by pronuclear injection of a commercial Cas9 protein:crRNA:tracrRNA:single-strand oligodeoxynucleotide (ssODN) complex into mouse zygotes [9] (Fig. 2A). In this method, two crRNAs are designed to direct the Cas9 to target the upstream and downstream introns of the target exon, along with corresponding loxP site oligos with 60 bp homology sequences on either side surrounding each Cas9-mediated DSB (Fig. 2B).
Fig. 2.
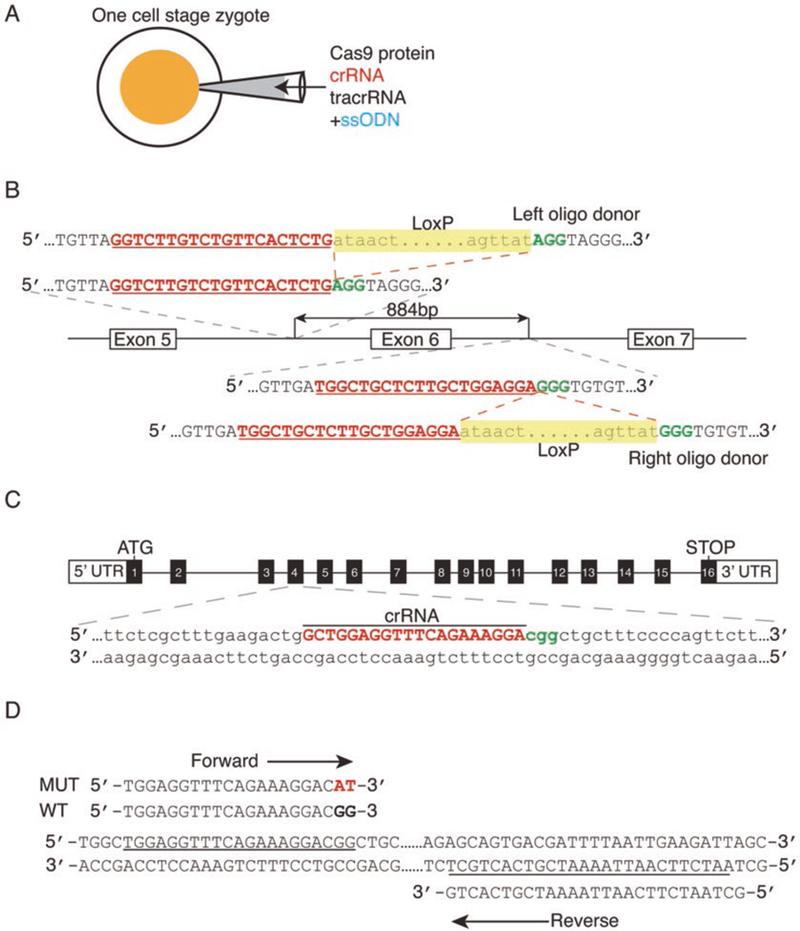
CRISPR/Cas9-mediated gene manipulation to generate floxed and point mutation alleles. (A) A cloning-free CRISPR/Cas9 system by pronuclear injection into a one-cell stage zygote of commercial Cas9 protein combined with chemically synthesized CRISPR RNA (crRNA), trans-activating crRNA (tracrRNA), and single-strand oligodeoxynucleotides (ssODNs). (B) Schematic illustration to generate a conditional allele by insertion of two loxP sites. In this method, two crRNAs are designed to direct the Cas9 to target the upstream and downstream introns of the target exon, along with corresponding loxP site oligos with 60 bp homology sequences on either side surrounding each Cas9-mediated double-strand break. The protospacer adjacent motif (PAM) is shown in green. In the oligo donor sequence, the loxP site is highlighted in yellow. (C) To generate point mutation mice, the sequence of the crRNA (red) targets the Cas9 nuclease to the desired mutation-specific region (in this example, CGG to CAT) of the target gene. The PAM is shown in green. (D) Design of the mutation-specific primers with a site-specific variant sequence at the 3’ terminus of the forward primer to screen for correctly targeted mice. Figure modified from [9]
1.2. Generation of Single-Amino-Acid-Substituted Mutant Mice
Whereas conditional knockout strategies are ideal for understanding protein function in a tissue-specific manner, the generation of mouse models that recapitulate human disease-causing single amino acid substitutions using knock-in strategies is critical in the study of disease pathophysiology. Indeed, missense mutations in LINC complex proteins are a major cause of striated muscle disorders in humans [20–29]. As for the conditional knockout strategies, two main strategies exist to generate single-amino-acid-substituted mutant mice (Fig. 1C).
Similar to the gene targeting approach to generate knockouts described above, a targeting construct can be used, except that the vector does not contain loxP sites, but instead contains the mutated codon(s) [30–32]. Alternatively, similar to the generation of floxed alleles described above, a mixture of Cas9 protein, crRNA, tracrRNA, and ssODN is injected into the pronuclei of zygotes (Fig. 2A), except that the sequence of the crRNA targets the Cas9 nuclease to the desired mutation-specific region of the target gene (Fig. 2C) [9]. Mutation-specific primers with a site-specific variant sequence at the 3’ terminus of the forward primer are used to screen for correctly targeted mice (Fig. 2D).
1.3. Generation of Striated Muscle-Specific Knockout or Mutant Mice by Cre Recombination
Tissue-specific knockout is achieved by crossing floxed mice with Cre mouse lines that express Cre recombinase under the control of a tissue-specific promoter or enhancer [3, 12–14]. The selection of an appropriate Cre-expressing mouse line is a crucial step in the process of generating a striated muscle selective LINC complex protein knockout or mutant mice. Three criteria need to be considered: (1) tissue and/or cell selectivity of Cre expression, (2) timing and duration of Cre expression, and (3) efficiency of the Cre recombinase. Here, we discuss a number of Cre mouse lines used for the generation of striated muscle-specific knockout or mutant mice by Cre recombination.
There are many cardiac-specific Cre lines available for ablating gene expression in cardiomyocytes (see Table 1 and references therein) [33]. Here we describe those we have used in our studies of cardiac function. The most widely used Cre lines for inducing gene ablation in cardiomyocytes are those utilizing promoter regions from the alpha (α)-myosin heavy chain (MHC) gene [34–36]. However, a potential caveat of using certain αMHC lines is that those with high levels of myocardial expression of Cre recombinase lead to dilated cardiomyopathy [36]. In contrast and in our hands, the αMHC-Cre line developed by Dale Abel’s laboratory has shown no evidence of Cre toxicity [32].
Table 1.
Cardiac and skeletal muscle-specific Cre lines
| Tissue specificity |
Cre line | Expression Temporal |
Expression Spatial |
Efficiency | Laboratory source |
Original reference |
Potential caveats |
|---|---|---|---|---|---|---|---|
| Cardiac constitutive |
Alpha MHC | E10.5 | CMs | 70% excision in adult CMs |
Abel | [34] | - |
| Alpha MHC | Low levels E8.0–E10.0 |
CMs | 70–80% excision by 3 weeks |
Schneider | [35] | High expression level leads to DCM |
|
| Beta MHC | E12.5 | CMs | 70% excision by E17.5 |
Yutzey and Molkentin |
[66] | Expressed in somites and soleus muscle |
|
| Nkx2.5 | E8.0-E8.5 | CMs | 88% excision in adult CMs |
Olson | [37] | - | |
| Nkx2.5 | E7.75–E8.0 | CMs, pharyngeal endoderm |
High | Harvey | [38] | - | |
| Nkx2.5 | E7.5 | CMs | 100% excision by PI |
Schwarz | [39] | Development of heart abnormalities |
|
| cTnT | E7.5–E10.5 | CMs | High | Hogun | [42] | Breeding problems in older mice |
|
| XMLC2 | E7.5– adulthood |
CMs | High | Mohun | [44] | - | |
| MLC2v | E8.5 | Ventricular CMs | Low | Chen | [45] | - | |
| Inducible cardiac |
MerCreMer-Alpha MHC |
N/A | CMs | >80% excision after 5 days |
Molkentin | [48] | Cre toxicity observed at high levels |
| MerCreMer-cTnT | N/A | CMs | High | Cai | [55] | - | |
| Tet-TnT | N/A | CMs | Not reported | Zhou | [56] | - | |
| Constitutive skeletal |
HSA | E9.5 | Skeletal muscle, some evidence in heart |
High | Melki | [57] | Low level expression in the heart |
| Myo | E8.5 | Skeletal muscle lineage | >90% excision | Olson | [60] | - | |
| Inducible skeletal |
HSA-MerCreMer | - | Skeletal muscle, some evidence in heart |
High | Esser | [58] | Low level expression in the heart |
| HSA-Tet | - | Skeletal muscle | Not reported | Monks | [59] | - | |
| CreRT2 lines | - | Various | Lepper | [61] | - |
CMs: cardiomyocytes, E: embryonic day, MHC: myosin heavy chain, Nkx2.5: Nk2 homeobox 5, cTnT: cardiac troponin T, XMLC2: Xenopus myosin light chain 2, MLC2V: myosin light chain 2 ventricular, MerCreMer: mutated estrogen receptor, Dox: doxycycline, HSA: human alpha skeletal actin, DCM: dilated cardiomyopathy
As one of the earliest markers of heart progenitor cells, the regulatory regions of Nkx2.5 have been used to generate several Cre lines for inducing gene ablation during early cardiogenesis [37–39]. In our experience, the Nkx2.5-Cre transgenic line developed by the Olson laboratory reduced the RNA level of nesprin 1 by 88% in adult cardiomyocytes [3, 37]. The Nkx2.5-Cre developed by the Harvey laboratory is also highly efficient; however, it is expressed in derivatives of the pharyngeal endoderm, which include the stomach, spleen, pancreas, and liver [38]. The Schwarz laboratory developed an Nkx2.5 knock-in Cre line, which is heterozygous null for Nkx2.5 [39]. However, owing to the importance of the Nkx2.5 gene, these mice develop cardiac abnormalities [40, 41].
The cardiac troponin T (cTnT)-Cre line generated by Brigid Hogan’s laboratory, in which Cre is driven by the rat cTnT promoter, induces early recombination at E7.5 with high efficiency in cardiomyocytes [42]. In our experience of using this line, we achieved ~80% reduction at the protein level of the two floxed alleles, Numb and NumbL, from whole hearts at E10.5 [43]. For this line, it is important to maintain male breeders of a young age (<6 months of age) as older breeders develop a shorter stature and don’t reproduce as well (unpublished observations).
The Mohun laboratory has developed a Xenopus myosin light chain 2 promoter-driven Cre recombinase (XMLC2-Cre) that is functional in all myocardial cells throughout embryonic development and adulthood [44]. Importantly, the onset of recombinase activity occurs very early in cardiogenesis at the cardiac crescent stage. Given that the XMLC2-Cre mouse line has high efficiency, precise tissue specificity, and is early onset, it is a powerful tool to achieve gene ablation in a myocardial-specific manner in both embryos and adults.
Our laboratory generated a ventricle-restricted Cre line by knocking-in Cre into the myosin light chain 2v (MLC2v) locus [45]. This MLC2v-Cre mouse is heterozygous for MLC2v, but the mice are normal, display no morphogenic defects, and express normal levels of MLC2v protein [46]. The Cre recombinase expresses at the earliest stages of ventricular chamber specification; however, the efficiency of the recombinase is relatively low in embryonic ventricular cardiomyocytes [45, 47].
Temporal regulation of gene excision is highly desirable for studying gene function in the adult heart, especially if ablation during development results in embryonic lethality. This can be achieved through using inducible tamoxifen- or tetracycline-driven Cre lines (Table 1). The αMHC-MerCreMer transgenic line, in which the cardiac-specific αMHC promoter directs expression of tamoxifen-inducible Cre recombinase (MerCreMer) [48], has been used extensively. We and others have shown that Cre activity is tightly regulated and that Cre-mediated recombination occurs only in response to the injection of tamoxifen [49, 50]. There have been reports of tamoxifen treatment induced phenotypes using this line; therefore, we recommend using αMHC-MerCreMer mice alone and/or αMHC-MerCreMer mice with the heterozygous floxed allele as controls [51–54].
Recently, a tamoxifen-inducible cTnT-Cre was developed by introducing the MerCreMer cassette upstream of the first exon of the cTnT gene [55]. Minimal Cre “leakiness” prior to tamoxifen injection and robust recombination in cardiomyocytes was reported 24 h after administration. Tamoxifen administration alone may cause behavioral alterations; therefore, dosage optimization is pertinent when using tamoxifen-inducible mouse lines [54]. As an alternative to tamoxifen, for cardiac-specific ablation we have used the tetracycline-inducible system developed by the Zhou laboratory. This approach uses the rat troponin T promoter to express the reverse tetracycline-controlled transactivator, which in turn drives Cre expression via a tetracycline-responsive promoter [43, 56].
A number of constitutive and inducible skeletal muscle-specific Cre mice have also been developed (Table 1). The human α-skeletal actin (HSA) promoter has been used to drive constitutive [57], tamoxifen-inducible [58], and tetracycline-inducible [59] Cre expression. HSA-Cre expression is largely restricted to skeletal muscle; however, there is evidence of mosaicism between different skeletal muscle types and evidence of low-level expression in the heart. Myogenin-Cre transgenic mice were developed by the Olson laboratory [60] in which Cre expression is under the control of the mouse myogenin promoter and mouse myocyte enhancer factor 2C (MEF2C) enhancer region. There are an extensive number of tamoxifen-inducible skeletal muscle knock-in Cre lines developed by the Lepper laboratory, which include Pax3, MyoD, Myog, Myf6, and Myl1, and have been well characterized [61].
If global knock-in homozygous mice are not viable or unable to reproduce, tissue-specific mutant mice strategies can be employed to circumvent this. The Kontaridis laboratory generated Cre-dependent conditional knock-in mice harboring a Y279C mutant of the Ptpnll gene [62]. In this case, the Y279C mutant is only expressed after Cre-mediated excision. Therefore, a tissue-specific Cre could be used to drive expression only in the tissue of interest. As a simpler alternative, we crossed conditional knockout mice (f/f; Cre+) with heterozygous mutants (m/+) to generate conditional mutant mice (f/m; Cre+) mice [32]. Conditional mutant mice were subsequently crossed with floxed (f/f) mice to generate relevant control littermates (Fig. 3) (see Note 4).
Fig. 3.
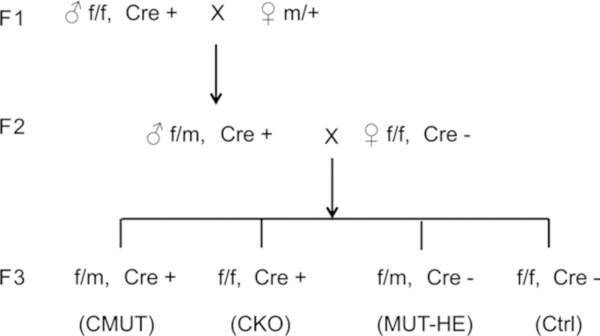
Breeding strategy of conditional knock-in mice. See text for description
2. Materials
2.1. CRISPR/Cas9-Mediated Gene Manipulation to Create Floxed and Point Mutation Mice with a Cloning-Free Method
2.1.1. CRISPR/Cas9 Injection
Mice. C57BL/6 J female mice at age 3–4 weeks and male mice at 9–24 weeks for zygote collection. ICR (CD-1) mice for pseudopregnant mother and vasectomized males.
Recombinant Cas9 proteins.
CrRNA and tracrRNA: chemically synthesized and RNase-free HPLC purified.
Single-strand ODN: chemically synthesized and standard desalted.
1× Tris-EDTA (TE) solution.
Pregnant mare serum gonadotropin (PMSG).
Human chorionic gonadotropin (hCG).
KSOM medium.
Reagents and equipment used for routine pronuclear microinjection are provided in the protocol on the University of California, San Diego Transgenic Mouse Core website (https://healthsciences.ucsd.edu/research/moores/shared-resources/transgenic-core/services/Pages/pronuclear-injection.aspx).
2.1.2. Genotyping and Screening of Targeted Alleles
Primers.
50 mM NaOH.
1 M Tris–HCl (pH 8.0).
Taq PCR Kit.
Agarose.
Tris-acetate-EDTA (TAE) buffer.
Zero Blunt TOPO PCR cloning kit.
LB plates containing 50 μg/mL kanamycin.
2.2. Maintenance of Mouse Stains and Generation of Conditional Knockout and Mutant Mice
2.3. Induction of Cre Expression
2.3.1. Tamoxifen
Chow containing 0.4–1 g/kg tamoxifen.
Water containing 0.5–1 mg/mL tamoxifen.
Sesame oil containing 10 mg/mL tamoxifen.
2.3.2. Doxycycline
Water containing at 1–2 mg/mL doxycycline.
2.4. Analysis of Hearts
2.4.1. Echocardiographic Analysis of Cardiac Function
Electric razor.
Hair removal cream (Nair).
Echocardiography machine (FujiFilm Visualsonics 2.0) with heating pad and electrodes.
Echo software.
Echo gel.
Isofluorane.
2.4.2. Sample Preparation
Heparin (1000 U/mL).
Ketamine (100 mg/kg).
Xylazine (10 mg/kg).
Insulin needles.
Fine scissors.
Extra fine Graefe forceps.
Ice cold phosphate-buffered saline (PBS).
2.4.3. Measurement of Heart Weight/Tibia Length and Heart Weight/Body Weight Ratios
70% ethanol.
Medium-size forceps.
Vernier calipers (Kingsmart 6 in. LCD digital/micrometer gage 150 mm).
2.4.4. Immunofluorescence Anaysis
Cryostat.
Cryostat blades.
Wax pen.
Superfrost Plus slides.
Humidified slide-incubation chamber.
Thickness 1.5 coverslips.
Inverted confocal microscope.
PBS.
Acetone.
4% paraformaldehyde (PFA) in PBS (wt/vol).
Optimal cutting temperature (OCT) compound.
Sucrose.
Dry ice.
3% bovine serum albumin (99.5% pure) in PBS.
Normal donkey serum.
DAKO mounting medium.
Clear nail polish.
Vectashield mounting medium.
0.2% Triton-X100 in PBS (PBS/T).
Relevant antibodies (see Table 2).
Table 2.
Recommended antibodies for detecting LINC complex proteins and sarcomeres
| Protein | Company/source | Cat. No./clone | Application | Epitope | Host species and nature |
Validated with knockout tissue? | Reference |
|---|---|---|---|---|---|---|---|
| Nesprin 1 | Glenn E Morris | 7A12 | WB and IF | Human Nesprin lα2 |
Rabbit monoclonal | Yes | [67] |
| SUN1 | Millipore | ABT285 | WB | Mouse C-terminus |
Rabbit polyclonal | - | [3] |
| SUN1 | Abeam | abl03021 | IF | Mouse AAs200–300 |
Rabbit polyclonal | - | [5] |
| SUN2 | Abeam | EPR6557 | WB and IF | Human C-terminus |
Rabbit monoclonal | - | [5] |
| LUMA | Abeam | EPR15378 | WB and IF | Human AAs 100–200 |
Rabbit monoclonal | Yes | [69] |
| Emeriti | Novocastra | NCL-EMERIN (4G5) |
IF | Human N-terminus |
Mouse monoclonal | - | [68] |
| Emeriti | Santa Cruz | sc-15378 (FL-254) | WB | Human AAs 3–254 |
Rabbit polyclonal | - | [5] |
| LEM2 | Sigma | HPAO17340 | WB and IF | Human AAs 243–361 |
Rabbit polyclonal | Yes | [69] |
| Sarcomeric α-actinin |
Sigma | A7811 | WB and IF | Rabbit skeletal α-actinin |
Rabbit polyclonal | - | [30] |
WB: Western blot, IF: immunofluorescence
2.4.5. Histological Preparation for Chemical Staining
Tissue cassettes.
Wide-necked Erlenmeyer flask.
Glass histology staining chambers.
Microtome.
Water bath.
Oven.
Ethanol.
Xylene.
10% neutral buffered formalin.
4% PFA in PBS.
Paraffin.
Hematoxylin and eosin stain.
Masson’s trichrome stain.
Brightfield microscope.
2.4.6. RNA Extraction
RNA extraction kit.
cDNA synthesis kit.
Quantitative real-time PCR machine.
Primer pairs (see Table 3).
Table 3.
Sequences of qRT-PCR primers used to detect fetal, pro-fibrotic, early activator, and reference genes
| Forward | Reverse | |
|---|---|---|
| Fetal genes | ||
| ANP | GATAGATGAAGGCAGGAAGCCGC | AGGATTGGAGCCCAGAGTGGACTAGG |
| BNP | TGTTTCTGCTTTTCCTTTATCTGTC | CTCCGACTTTTCTCTTATCAGCTC |
| AMHC | CTGCTGGAGAGGTTATTCCTCG | GGAAGAGTGAGCGGCGCATCAAGG |
| BMHC | TGCAAAGGCTCCAGGTCTGAGGGC | GCCAACACCAACCTGTCCAAGTTC |
| Pro-fibrotic genes | ||
| COL1A1 | TCACCAAACTCAGAAGATGTAGGA | GACCAGGAGGACCAGGAAG |
| COL3A1 | ACAGCAGTCCAACGTAGATGAAT | TCACAGATTATGTCATCGCAAAG |
| Early activator genes | ||
| Egr-1 | CCTATGAGCACCTGACCACA | TCGTTTGGCTGGGATAACTC |
| lex-1 | TTATAGGGTCGGTAAGACAGAGTTG | GACGGAGTGTTACCCCTAATCTTAT |
| c-fos | AGCCCCTGTGTACTCCCGTG | GCCTTGCCTTCTCTGACTGC |
| c-jun | TTCCTCCAGTCCGAGAGCG | TGAGAAGGTCCGAGTTCTTGG |
| c-myc | ATGCCCCTCAACGTGAACTTC | GTCGCAGATGAAATAGGGCTG |
| Reference genes | ||
| 18S | GGAAGGGCACCACCAGGAGT | TGCAGCCCCGGACATCTAAG |
| GAPDH | CTCAAGATTGTCAGCAATGCATCC | CCAGTGGATGCAGGGATGATGTTC |
Primers are in 5’ to 3’ orientation
2.4.7. Protein Extract Preparation
Pestle and mortar.
Sonicator.
Spatula.
Western blotting apparatus.
Liquid nitrogen.
Lysis buffer: 8 M urea, 2 M thiourea, 3% SDS (wt/vol), 75 mM DTT, 0.03% bromophenol blue (wt/vol), 50 mM Tris–HCl pH 6.8.
2.5. Analysis of Skeletal Muscle
2.5.1. Preparation of Tools for TA Muscte Fiber Isolation
P200 tips.
Bunsen burner.
Razor blade.
0.2% Triton-X100 in PBS (PBS/T).
Superfrost Plus slides.
Wax pen.
2.5.2. Extraction of TA Muscle from Leg
Dissection microscope.
Lightsource.
Dumont #5 fine forceps.
Spring scissors.
Dumont AA polished forceps.
4% PFA in PBS.
2.5.3. Analysis of Myonuclear Positioning and Immunofluorescence Staining
Thermomixer (Eppendorf Thermomixer F).
Superfrost Plus slides. n
Thickness 1.5 coverslips.
DAKO mounting medium.
Wax pen.
10 M NaOH.
PBS.
4’,6-diamidine-2’ -phenylindole dihydrochloride (DAPI).
3% bovine serum albumin (99.5% pure) in PBS.
Normal donkey serum.
0.2% Triton-X100 in PBS (PBS/T).
Relevant antibodies (see Table 2).
3. Methods
3.1. CRISPR/Cas9-Mediated Gene Manipulation to Create Floxed and Point Mutation Mice with a Cloning-Free Method
3.1.1. CRISPR/Cas9 Targeting Design
For design of the guide CrRNA, follow the protocol described by the Zhang laboratory [63]. Briefly, input the target genomic DNA sequence into the online CRISPR design tool: http://crispr.mit.edu/. The CRISPR design tool takes an input sequence (e.g., a 1 kb genomic fragment from the region of interest), identifies and ranks suitable target sites, and computationally predicts off-target sites for each intended target. Alternatively, one can manually select guide sequences by identifying the 20 bp sequence directly upstream of a proto-spacer adjacent motif (PAM) sequence (5’-NGG) [9] (Fig. 2B).
For conditional allele generation, two sgRNAs are designed to elicit DSBs that flank the sequence to be deleted. In addition, an ssDNA oligo is designed that contains the corresponding loxP site flanked by two 40- to 60-base homology arms that correspond to the sequence surrounding each sgRNA-mediated DSB (Fig. 2B).
To introduce a point mutation, in addition to the sgRNA targeting the site of interest, an ssDNA oligo is designed that contains the desired alteration flanked on each side by 40–60 bases that are homologous to the sequences directly upstream and downstream of the DSB.
3.1.2. Zygote Preparation and Microinjection
Inject 12–15 female C57BL/6 J (3–4 weeks old) mice with PMSG (5 IU) at 1:00–2:00 p.m. on day 1.
After 48 h, inject female mice with hCG (5 IU). After hCG injection, house female mice with C57BL/6 J male mice overnight.
Prepare the medium for embryo culture. Place several drops (30–50 μL for each drop) of KSOM medium on a 6-cm dish and cover with mineral oil; place the dish into a 37 °C incubator for at least 30 min before use.
At 20–21 h after hCG injection, euthanize the mice and collect zygote-cumulus complexes from the oviduct where it is most swollen.
Move the zygote-cumulus complexes into M2 + Hy medium; gently triturate three times with a P200 tip.
Gently aspirate complexes with a transfer pipette, wash three times in M2 medium, and place the embryos into KSOM medium at 37 °C in a 5% CO2 incubator.
Prepare the injection mix (Cas9 protein, crRNA, and tracrRNA, with or without ssODN (experiment dependent)) and mix in TE buffer to a working concentration of 30 ng/μL, 0.6 pmol/μL, 0.6 pmol/μL, and 20 ng/μL, respectively. Incubate the mixture at 37 °C for 5 min (Fig. 2A).
The standard protocol describing pronuclear microinjection is provided on the University of California, San Diego Transgenic Mouse Core website (https://healthsciences.ucsd.edu/research/moores/shared-resources/transgenic-core/services/Pages/pronuclear-injection.aspx).
3.1.3. Primer Design, Genotyping, and Screening of Targeted Alleles
Primer design: To screen for correctly targeted point mutation mice, a mutation-specific forward primer is designed with the site-specific variant sequence at the 3’ end, and a generic wild-type reverse primer is used [9] (Fig. 2D). To detect floxed alleles, a loxP-specific and wild-type primer pair are designed.
- Genomic DNA preparation:
-
(a)Collect tail biopsies from 3-week-old mice and add 300 μL of 50 mM NaOH.
-
(b)Incubate at 98 °C for 30 min; then add 50 μL of 1 M Tris–HCl (pH 8.0). 2 μL of genomic DNA sample is used for genotyping PCR.
-
(a)
- Genotyping PCR of F0 founders:
-
(a)Amplify the extracted DNA using gene-specific primers under the following conditions: 30 cycles at 94 °C for 20 s, 60 °C for 20 s, and 72 °C for 30 s.
-
(b)Run the PCR product on a 2% (wt/vol) agarose gel in TAE buffer to verify that the product is unique and of the expected size.
-
(a)
- PCR for sequencing validation of mutated alleles of F0 founders:
-
(a)PCR amplify the extracted genomic DNA using primers flanking the targeted region.
-
(b)Clone the PCR product using a Zero Blunt TOPO PCR cloning kit according to manufacturer’s instructions.
-
(c)To validate mutant alleles, extract plasmid DNA from six individual colonies and sequence (see Note 5).
-
(a)
3.2. Maintenance of Mouse Stains and Generation of Conditional Knockout and Mutant Mice
Breed F0 mutant mice with wild-type C57BL/6 J mice.
The first-generation offspring (F1) from each founder are subjected to genotyping using PCR and are sequenced to confirm germline transmission (as described in Subheading 3.1.3) (see Note 6).
Cross F1 mutant heterozygous mice with wild-type C57/B6J mice to maintain the mutant line.
Floxed (f/f) females are mated with Cre-positive (+/+; Cre+) males to generate f/+; Cre+ mice. Floxed females are then mated with f/+; Cre+ males to generate tissue-specific knockout mice (f/f; Cre+) (CKO) and their control littermates (f/f) and (f/+; Cre+) (see Notes 7 and 8).
Heterozygous mutant (m/+) females are mated with CKO (f/f; Cre+) males to generate cell-selective specific mutant mice (f/m; Cre+) (CMUT). CMUT males are bred with homozygous floxed (f/f) females to generate CMUT (f/m, Cre+) and littermate controls (Fig. 3) [32].
3.3. Induction of Cre Expression
Tamoxifen can be administered via food (custom-made chow containing 0.4–1 g/kg tamoxifen, Harlan), water intake (0.5–1 mg/mL), or 1–5 consecutive days of intraperitoneal injection (0.03–09 mg/g body weight) [48, 53]. Doxycycline is administered in drinking water at 1–2 mg/mL [56, 59].
3.4. Analysis of Hearts
3 4 1. Echocardiographic Analysis of Cardiac Function
Weigh mouse.
Shave the middle and the left-hand side of the chest on the underside of the mouse with a small electric razor (see Fig. 4A).
Using a Q-tip, place a small amount of hair removal cream on the chest around the center and to the left of the rib cage on the underside of the mouse (see Note 9). Leave for 10–20 s, and then using a surgical swab, wipe off the hair removal cream and hair. Wipe the chest with a wet surgical swab to remove the excess hair removal cream and hair.
Place mouse in supine position on pre-warmed echo pad at 39 °C.
Place one electrode on the right leg and another on the right arm to measure heart rate.
Tape the limbs to the pad.
Anaesthetize the mouse using a quick burst of 5% isofluorane in oxygen for a few seconds.
Once anesthetized, reduce to 0.5% isofluorane to ensure the heart rate is maintained above 500 beats per minute (see Note 10).
Squeeze a 4 mm cylinder of conductive echo gel on the probe to be used for echocardiography (use a 45 MHz probe).
Place the probe on the chest and locate the heart (see Note 11).
For a four-chamber (or long axis) view using B-mode, place the probe perpendicular to the chest at approximately a 45° angle to make an imaginary line between the right arm and middle of the left side of the abdomen (see Fig. 4A, left).
For a two-chamber (or short axis) view or short axis using M-mode, rotate the probe by 90° to make an imaginary line between the left arm and middle of the right side of the abdomen (see Fig. 4A, right).
Images are taken at the level of the papillary muscles, which are very prominent, echogenic regions (appear white) in mouse hearts.
Record three consecutive cycles of diastole (during heart relaxation) and systole (heart contraction).
Remove the excess echo gel and place the mouse back in cage.
Download the echo images and perform analysis on a computer with Vevo 2100 software installed.
Measure the left ventricle chamber sizes, called the left ventricle internal dimension (LVID), and interventricular septum (IVS) wall thickness and left ventricle posterior wall (LVPW) thickness in diastole and systole, and then calculate the mean values between the three beats (see Fig. 4B) (see Note 12).
Left ventricle systolic function is measured using the parameter called fractional shortening, which is measured as a percentage of the amount the chamber sizes change between systole and diastole divided by the chamber size in diastole (FS = [LVIDd-LVIDs]/LVIDd).
Fig. 4.
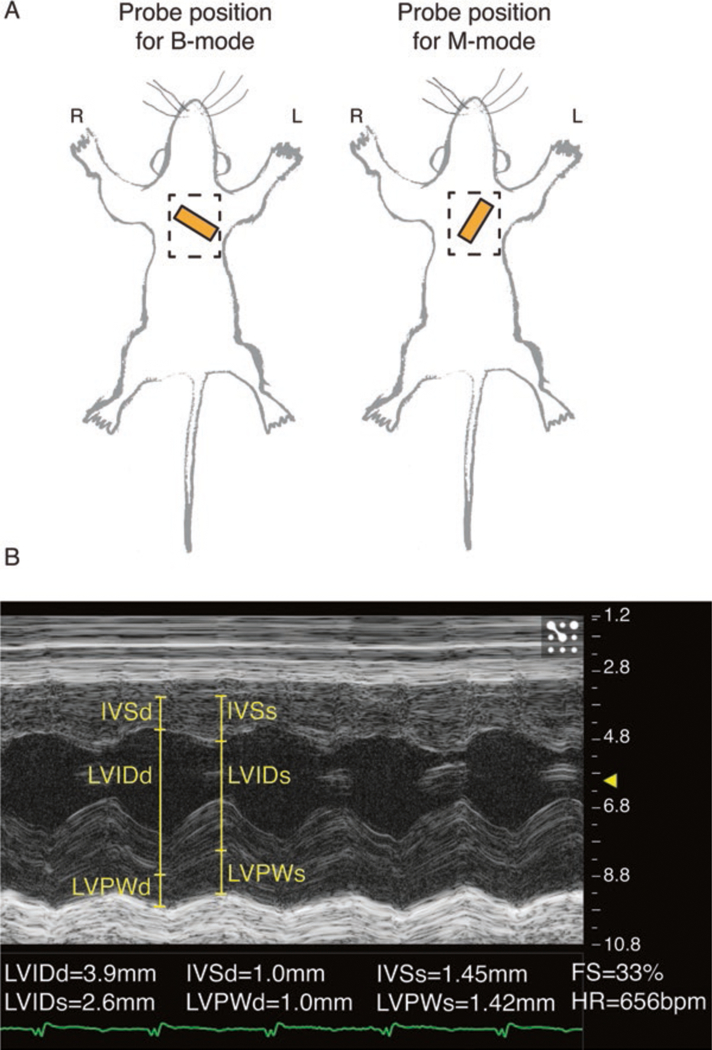
Echocardiography recording and analysis. (A) Approximate positioning of echocardiography probe (orange rectangle) for a 2D long-axis B-mode view (left) and short-axis M-mode view (right). Dashed rectangle indicates the recommended area from which to remove hair. (B) Parasternal short-axis M-mode with left ventricular functional measurements. Post capture, left ventricle internal dimension in diastole/systole (LVIDd/s); left ventricle posterior wall thickness in diastole/systole (LVPWd/s); interventricular septum thickness in diatole/systole (IVSd/s); fractional shortening (FS); and heart rate (HR) are measured
3.4.2. Sample Preparation
Weigh the animal.
Intraperitoneal inject 0.2 mL heparin (1000 U/mL) (see Note 13).
Open up the chest by cutting around the edge of the rib cage to avoid cutting the heart.
Using forceps grip the base of the heart and with sharp scissors excise the heart above the aorta to avoid cutting the atria.
Rinse heart in ice cold PBS to remove excess blood.
Remove the aorta and other non-cardiac tissue.
Briefly dry the heart using a surgical swab.
Weigh the heart on a fine balance.
Using a scalpel blade, dissect the heart into four pieces (see Fig. 5A, B) (see Notes 14 and 15).
Place the middle section in a glass vial containing 5 mL 10% neutral buffered formalin and fix at 4 °C overnight for histology (see Subheading 3.4.5).
Snap freeze the two apical halves in liquid nitrogen (for RNA and protein analysis, see Subheadings 3.4.6 and 3.4.7).
Take tail biopsy for genotype confirmation and snap freeze.
Fig. 5.
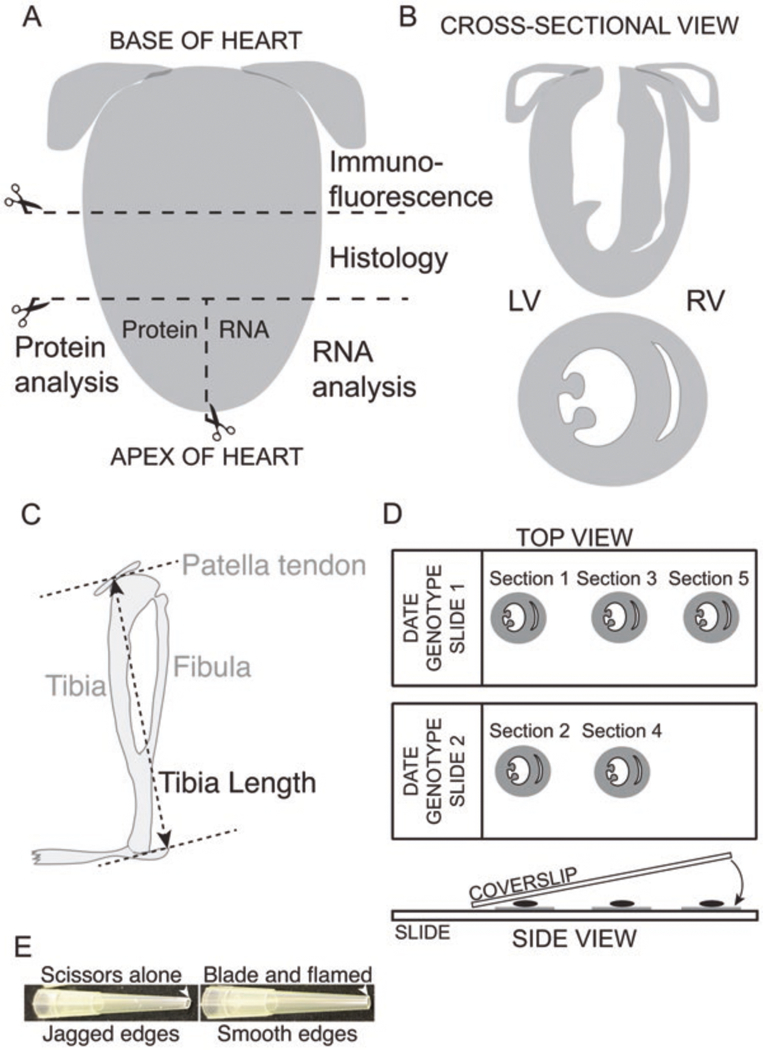
Processing of heart for analysis. (A) Post-extraction, the heart is cut into three pieces: The base is used for immunofluorescence and the midsection for histology; the apex is divided into two parts, for RNA and protein extraction. (B) Cross-sectional view of the heart showing all four chambers (top) and two chambers (bottom). (C) Schematic of a mouse tibia with measurement points indicated between the bottom of the tibia and the patella tendon. (D) Top view: After sectioning the heart for immunofluorescence analysis, sequential sections are placed on consecutive slides to allow direct comparison of different antibodies at similar spatial locations. Side view: Post-antibody staining and wash steps, a small amount of mounting medium is placed directly on the section, and the coverslip is slowly lowered using forceps to avoid generating air bubbles. Depending on the mounting medium, coverslips are either left to dry overnight or sealed with clear nail polish. (E) Preparation of P200 tips to pipette isolated myofibers. Note the importance of using a razor blade and gentle Bunsen burner flame to generate smooth edges on the pipette tip to minimize sample loss
3.4.3. Measurement of Heart Weight/Tibia Length and Heart Weight/Body Weight Ratios
Spray the lower legs with 70% ethanol to dampen the hair.
Using medium-size forceps or between fingers, grab one of the feet
With a pair of medium-size sharp-ended forceps, pierce and grab the skin above the foot and pull skin over the front of the knee joint to expose the patella tendon (shiny, almost metallic-looking strip that is approx. 1 mm wide by 5 mm long) that runs over the front of the knee (see Note 16).
Using Vernier calipers, measure the distance between the middle of the exposed patella tendon and the bottom of the ankle (see Fig. 5B).
Divide heart weight by tibia length or body weight for global indices of cardiac hypertrophy (see Note 17).
3.4.4. Immunofluorescence Analysis
Place the base of the heart in isopentane to dehydrate (see Note 18).
Remove after 30–60 s and place in a small pool of OCT compound on a sectioning mold.
Place the mold on a block of dry ice, and let the tissue/OCT slowly freeze into place.
Slowly fill the mold with OCT, making sure the orientation of the heart does not change until the mold is filled with OCT.
Use immediately or store at −80 °C until ready for sectioning.
Before sectioning, place the mold in the cryostat at −20 °C to warm up the section to −20 °C (see Note 19).
Label slides with date, genotype, slide number.
Using a new blade, cut 10 μm sections sequentially using a separate slide for each section cut (see Fig. 5D) (see Notes 20 and 21).
Allow sections to dry.
Place in −80 °C freezer.
Remove sections and immediately place in −20 °C acetone at −20 °C to fix sample for 5 min (see Note 22).
Remove and wash section with PBS.
Permeabilize sections with PBS containing 0.2% Triton-X100 at room temperature for 15 min.
Use wax pen to draw around individual sections to allow for use of multiple antibodies/slide.
Incubate antibodies overnight in PBS containing 3% BSA and 2% normal donkey serum at 4 °C in humidified slide-incubation chamber (see Note 23).
Wash 3× for 10 min with PBS.
Incubate with relevant secondary antibodies for 1 h at room temperature in a humidified slide-incubation chamber.
Wash 3× for 10 min with PBS.
Stain with DAPI for 5 min at room temperature.
Wash 3× for 10 min with PBS.
Add Vectashield dropwise to sections (see Note 24).
Carefully place coverslip (type 1.5 high tolerance) down at one end of the slide holding the other end up with forceps; then slowly lower the forceps (see Fig. 5D) (to avoid generating air bubbles).
Let solidify overnight at 4 °C.
Image on confocal microscope/deconvolution microscope.
3.4.5. Histological Preparation for Chemical Staining
After fixing the heart overnight at 4 °C wash 3× in PBS.
Label the tissue cassette using a pencil (see Note 25).
Prepare molten paraffin wax by placing paraffin in a bottle in an incubator set to 55–65 °C.
Remove tissue from glass vial and transfer to tissue cassette.
Place cassettes in an Erlenmeyer flask (with a neck wide enough to slot the cassettes in) or beaker containing 70% ethanol (filled as necessary to completely cover the cassettes) and wait 20 min.
Replace 70% ethanol with 95% ethanol and wait 20 min.
Remove 95% ethanol and replace with fresh 95% ethanol and wait 20 min.
Remove 95% ethanol and replace with 100% ethanol and wait 20 min.
Remove 100% ethanol and replace with fresh 100% ethanol and wait 20 min.
Remove 100% ethanol and replace with xylene and wait 20 min.
Remove xylene and replace with fresh xylene.
Open cassette and pour molten paraffin wax into the cassette.
Carefully adjust the orientation of the heart to ensure a two-chamber view.
Allow paraffin to set around tissue.
Turn on water bath at 35–40 °C.
Mount hardened section on microtome with new blade attached.
Cut 5 μm sections and place sections on the surface of the water in the bath.
Immerse the slide below the floating section and scoop up the section onto the slide.
Place slide in an oven at 65 °C for 10–15 min to melt the paraffin and mount the section to the slide.
Remove slide.
Stain slides with commercially available staining kits for hematoxylin and eosin (nuclei, dark blue/violet; tissue, red/pink) or Masson’s trichrome (nuclei, dark red/purple; tissue, red/pink; collagen, blue).
3.4.6. RNA Extraction
3.4.7. Protein Extract Preparation
Pre-chill pestle and mortar with liquid nitrogen.
Take half of the apical section of the heart (Fig. 5A) out of liquid nitrogen.
Weigh tissue.
Calculate volume of lysis buffer needed to achieve 50 μL/mg tissue (see Note 26).
For tissue weighing >6 mg, place frozen tissue in mortar and add 300 μL lysis buffer (the lysis buffer will freeze, you want to keep everything frozen during this process, and more liquid nitrogen can be added gently if necessary).
For tissue weighing <6 mg, add the calculated volume of lysis buffer and continue as below.
Homogenize tissue and lysis buffer with pestle and mortar into a fine powder.
Use a spatula to scrape the powdered lysate into a 2.0 mL tube.
Put sample on ice.
Sonicate sample to shear DNA, until the lysate is easy to triturate.
Add remaining lysis buffer to top up to the calculated volume necessary for 50 μL/mg.
Triturate 3× to mix lysis buffer.
Spin at 17,000 G for 15 min.
Aspirate supernatant.
Aliquot into working aliquots of 300 μL and snap freeze in liquid nitrogen, snap freeze the remaining lysate.
Use for Western blot or other downstream analysis.
3.5. Analysis of Skeletal Muscle
3.5.1. Preparation of Tools for TA Muscle Fiber Isolation
Before starting, prepare some pipette tips to handle the fibers and a “dissection slide” for the dissection of individual fibers. These steps are important to avoid losing a lot of fibers in the pipette tip and also to avoid contaminating your samples with plastic contaminants.
Cut the end from a P200 tip using a razor blade (not scissors), and then under a gentle Bunsen burner flame, flame the edges of the cut tip (see Fig. 5E) (see Note 27). Rinse the P200 tip with PBS/Triton mix to prevent fibers sticking to the inside of the tip.
Prepare a “dissection slide” by drawing a square in the middle of a glass slide with a wax pen and leave to dry. This slide will be used to tease apart the individual fibers in a pool of PBS/T.
3.5.2. Extraction of TA Muscle from Leg
Cut the whole legs from an E18.5 or neonatal pup and place each in 1 mL of 4% PFA.
Fix the samples overnight at 4 °C.
Wash legs with PBS 3× to remove the PFA.
Remove the skin from the leg using fine forceps.
Using a dissection microscope, remove the fascia (thin membrane) surrounding the TA muscle by piercing it and dragging it away from the muscle with sharp, fine forceps.
Slide the fine forceps in between the tibia bone and the TA muscle at the front of the shin.
Cut/remove the tendons (shiny structures connecting the end of the muscle to bone) with fine scissors (see Note 28).
3.5.3. Analysis of Myonuclear Positioning
Place a 0.5 mm × 2 mm strip of the TA muscle in 200 μL of 10 M NaOH in a 1.5 mL microcentrifuge tube.
Place in a thermomixer set to 1000 rpm at 20 °C for 20–30 min depending on the size of the TA fragment. Alternatively, a regular vortex can be used, and pulsed until the fibers start to separate.
Inspect the fibers every 10 min to look at the progress of fiber separation.
Allow fibers to settle by gravity.
Wash 5× with 1 mL PBS to remove the NaOH.
Resuspend in 200 μL of PBS and add DAPI for 5 min.
Wash 3× for 5 min with PBS.
Using a wax pen, draw around the edges of the slide and leave to dry (so that you won’t lose any fibers when you squash the coverslip down on the fibers).
Remove as much PBS as possible leaving ~20 μL.
Resuspend fibers in an equal volume of DAKO mounting medium.
Pipette the fibers directly onto the slide, spreading them along the length of the slide.
Lower the coverslip and seal with nail polish.
Image on a confocal or wide-field deconvolution microscope.
Nuclear lengths and internuclear distances were measured using ImageJ software. In brief, for internuclear distances, a line was drawn between the nuclear centroid to centroid; for nuclear lengths, a line was drawn along the axis of the myofiber between the shortest widths of the nuclei [5].
3.5.4. Immunofluorescence Staining
Pipette a pool of PBS/T into the square on the “dissection slide.” Place the fiber in the center of the square containing PBS/T.
Using two very small/fine forceps, tease apart the fibers into the smallest bundles as best you can. It will take about 1 h per TA muscle, which will yield plenty of fibers for staining; therefore, only about half of the TA muscle will be required and three to four per genotype.
It is important to keep the fibers wet during this process, as they will dry up if left.
Once you have dissected a good number of fibers, using the pipette tips prepared earlier, transfer the fibers into a 1.5 mL microcentrifuge tube.
The amount of fibers should come to about the 200 μL marker line on the side of the tube. This will provide enough fibers for up to six antibody stainings.
Resuspend the fibers in an equal volume of PBS/T (~400 μL total including fibers).
Aliquot the fibers according to the number of antibody stainings required. You may want to store a number of dissected fibers in the fridge in case problems are encountered or it requires optimization.
Dilute the antibodies in 4% BSA/PBS/T at double the concentration to the recommended dilution.
Add the antibody mix to an equal volume of the fibers (this will lead to the recommended dilution).
Put on rotator overnight at 4 °C.
In the morning, wash with PBS/T 3× for 5 min. For the washes, do not spin the fibers in a centrifuge, let them settle by gravity.
Add secondary antibodies for 1 h and DAPI using the same procedure as primary antibodies.
Wash 3× for 5 min with PBS.
Using a wax pen, draw around the edges of the slide and leave to dry (so that you won’t lose any fibers when you squash the coverslip down on the fibers).
Resuspend the fibers in an equal volume of DAKO mounting medium (about 10–15 μL/slide).
Pipette the stained fibers on to the center of the slide (about 10–15 μL/slide).
Separate the fibers around the slide (you want to avoid clustering in the center of the slide).
Lower a large, rectangular coverslip onto the fibers avoiding bubbles.
Seal the coverslip with nail varnish.
Image using a confocal microscope or wide-field deconvolution microscope (see Note 29).
4. Notes
It is important to check for LacZ expression on a mouse line-by-line basis, as some of the mutant alleles don’t express LacZ.
For homologous recombination to occur, a minimum of 2 kb of sequence homology is required [64]. Five to fourteen kilo base of sequence homology is typical for targeting constructs. The availability of suitable restriction enzymes within the locus of interest and the sizes of inserted DNA fragments are common limitations of conventional cloning strategies. The following restriction enzymes are suitable for digesting genomic DNA of ES cells for Southern blotting: BamHI, HindIII, Acc65I/KpnI, EcoRV, SpeI, StuI, BglI.
Upon deciding which exon to flox, ideally, the loxP site should be more than 250 bp away from splice donor and acceptor sites. We also try and avoid floxing exon 1 and exons that are divisible by 3 (so to avoid potential frameshift mutations).
Note that the conditional mutant mice have the genotype “−/m” in the tissue that the Cre is specifically expressed in, and “f/m” genotype in other tissues.
CRISPR/Cas9 injection in mouse zygotes may introduce mosaicism as the Cas9 enzyme may be active after the single-cell stage [65]. However, in our cloning-free method, there were no cases of mosaicism observed [9].
It is important to be aware that the CRISPR/Cas9 approach may introduce off-target mutations. Therefore, in addition to sequencing the predicted off-target loci, mutant mice should be backcrossed with wild-type mice to dilute potential off-target effects, and at least two independently generated lines should be used per genotype.
Depending on the mouse background, litter sizes can vary. Therefore, the number of animals set up for breeding should be adjusted according to preliminary breeding data.
To avoid the unexpected (non-specific or broader expression) pattern of Cre, we utilize male Cre carriers crossed with floxed females.
It is important to remove as much hair as possible as hair can interfere with the echo signal.
The cardiac function measurement will be dramatically reduced due to the artifact of a low heart rate.
Because results can vary between operators, we recommend using a trained sonographer who is blinded to the genotypes and the mice be randomized prior to performing the echo. This will ensure better reproducibility, which is essential for robust examination of cardiac function in a serial study.
For echocardiography physiological measurements, we usually observe standard deviation of 10% between mice of the same genotype. With this assumption, 6–12 mice per group will be needed to measure a 14–22% change in heart function, to ensure robust analysis of statistical significance (P < 0.05 using two-tailed t-test, with a power of a90%). A useful resource for calculating numbers required for physiological analyses: http://www.3rs-reduction.co.uk/html/6_power_and_sample_size.html.
Heparin is used to prevent blood clotting in the heart.
To make full use of the animals in line with the 3Rs policy, once the defined end point for cardiac function has been performed, hearts can be processed for Western blotting, histology, immunofluorescence, and RNA analyses.
It is important to always check where your molecule of interest is expressed in the heart by performing immunofluorescence analysis on a wild-type four-chamber view heart section.
To measure tibia lengths, it is also possible to dissolve the whole leg at room temperature over several days in 10 M NaOH. However, this involves handling caustic NaOH and produces an unpleasant odor and therefore should be performed in a fume cupboard.
Measuring tibia length is preferred to body weight for measuring cardiac hypertrophy owing to greater fluctuations in body weight over time.
It is also possible to perform sucrose gradients to dehydrate the heart. Start with 10% sucrose (wt/vol) followed by 12%, 16%, and 20% sucrose. For each step, incubate in sucrose for a sufficient amount of time until the heart sinks to the bottom of the tube. For the final step, incubate briefly with a 1:1 ratio of 20% sucrose/OCT before embedding on dry ice.
Place OCT/heart block in the cryostat set to −20 °C prior to sectioning to soften the section; otherwise, sections can be brittle if sectioned immediately after removal from a −80 °C freezer.
For sectioning, an anti-roll plate or fine paint brush can be used to prevent the section from folding back upon itself.
By putting sequential sections on sequential slides, it allows comparison of multiple antibodies at approximately the same region of heart (within 10 μm of each other).
It is also possible to fix with 4% PFA, but this is antibody dependent.
Incubation times can be varied as required, but we found the antibodies listed in Table 2 work best with overnight incubation at 4 °C.
Other mounting media can be used at this step. The benefit of using Vectashield is that it has a well-defined refractive index of 1.45, which is important to know for super-resolution imaging.
Use pencil to mark slides as markings from most pens will be erased by further processing steps.
Heart tissue is very proteinaceous and therefore requires well-diluted samples to run on SDS-PAGE gels.
It is important to prepare P200 tips in this manner as it will minimize the myofibers from becoming stuck inside the tip and therefore loss of precious material.
Removing the contaminating tendon will make teasing the muscle fibers apart much easier.
If no sarcomeric marker is available (such as sarcomeric alpha actinin), be sure to take a differential interference contrast or phase/contrast image.
References
- 1.Muller U (1999) Ten years of gene targeting: targeted mouse mutants, from vector design to phenotype analysis. Mech Dev 82(1–2):3–21 [DOI] [PubMed] [Google Scholar]
- 2.van der Weyden L, Adams DJ, Bradley A (2002) Tools for targeted manipulation of the mouse genome. Physiol Genomics 11:133–164 [DOI] [PubMed] [Google Scholar]
- 3.Banerjee I, Zhang J, Moore-Morris T et al. (2014) Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet 10(2):e1004114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman MA, Zhang J, Banerjee I et al. (2014) Disruption of both nesprin 1 and desmin results in nuclear anchorage defects and fibrosis in skeletal muscle. Hum Mol Genet 23(22):5879–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stroud MJ, Feng W, Zhang J et al. (2017) Nesprin 1alpha2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. J Cell Biol 216(7):1915–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Felder A, Liu Y et al. (2010) Nesprin 1 is critical for nuclear positioning and anchorage. Hum Mol Genet 19(2):329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aida T, Chiyo K, Usami T et al. (2015) Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol 16:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto M, Yamashita Y, Takemoto T (2016) Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Dev Biol 418(1):1–9 [DOI] [PubMed] [Google Scholar]
- 9.Ma X, Chen C, Veevers J et al. (2017) CRISPR/Cas9-mediated gene manipulation to create single-amino-acid-substituted and floxed mice with a cloning-free method. Sci Rep 7:42244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa Y, Sakuma T, Nishimichi N et al. (2016) Ultra-superovulation for the CRISPR-Cas9-mediated production of gene-knockout, single-amino-acid-substituted, and floxed mice. Biol Open 5(8):1142–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Yang H, Shivalila CS et al. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4):910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajewsky K, Gu H, Kuhn R et al. (1996) Conditional gene targeting. J Clin Invest 98(3):600–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhn R, Schwenk F, Aguet M et al. (1995) Inducible gene targeting in mice. Science 269(5229):1427–1429 [DOI] [PubMed] [Google Scholar]
- 14.Kilby NJ, Snaith MR, Murray JA (1993) Site-specific recombinases: tools for genome engineering. Trends Genet 9(12):413–421 [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez CI, Buchholz F, Galloway J et al. (2000) High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 25(2):139–140 [DOI] [PubMed] [Google Scholar]
- 16.Liang X, Zhou Q, Li X et al. (2005) PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes. Mol Cell Biol 25(8):3056–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinek M, Chylinski K, Fonfara I et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deltcheva E, Chylinski K, Sharma CM et al. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340):602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chylinski K, Le Rhun A, Charpentier E (2013) The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol 10(5):726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bione S, Maestrini E, Rivella S et al. (1994) Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet 8(4):323–327 [DOI] [PubMed] [Google Scholar]
- 21.Bione S, Small K, Aksmanovic VM et al. (1995) Identification of new mutations in the Emery-Dreifuss muscular dystrophy gene and evidence for genetic heterogeneity of the disease. Hum Mol Genet 4(10):1859–1863 [DOI] [PubMed] [Google Scholar]
- 22.Christensen AH, Andersen CB, Tybjaerg-Hansen A et al. (2011) Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin Genet 80(3):256–264 [DOI] [PubMed] [Google Scholar]
- 23.Haque F, Mazzeo D, Patel JT et al. (2010) Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem 285(5):3487–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodgkinson KA, Connors SP, Merner N et al. (2013) The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin Genet 83(4):321–331 [DOI] [PubMed] [Google Scholar]
- 25.Malhotra R, Mason PK (2009) Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Curr Opin Cardiol 24(3):203–208 [DOI] [PubMed] [Google Scholar]
- 26.Merner ND, Hodgkinson KA, Haywood AF et al. (2008) Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 82(4):809–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puckelwartz MJ, Kessler EJ, Kim G et al. (2010) Nesprin-1 mutations in human and murine cardiomyopathy. J Mol Cell Cardiol 48(4):600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamada T, Kobayashi T (1996) A novel emerin mutation in a Japanese patient with Emery-Dreifuss muscular dystrophy. Hum Genet 97(5):693–694 [DOI] [PubMed] [Google Scholar]
- 29.Zhang Q, Bethmann C, Worth NF et al. (2007) Nesprin-1 and −2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet 16(23):2816–2833 [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Stroud MJ, Zhang J et al. (2015) Normalization of Naxos plakoglobin levels restores cardiac function in mice. J Clin Invest 125(4):1708–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheikh F, Ouyang K, Campbell SG et al. (2012) Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest 122(4):1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang X, Bogomolovas J, Wu T et al. (2017) Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest 127(8):3189–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis J, Maillet M, Miano JM et al. (2012) Lost in transgenesis: a user’s guide for genetically manipulating the mouse in cardiac research. Circ Res 111(6):761–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abel ED, Kaulbach HC, Tian R et al. (1999) Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 104(12):1703–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agah R, Frenkel PA, French BA et al. (1997) Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest 100(1):169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buerger A, Rozhitskaya O, Sherwood MC et al. (2006) Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J Card Fail 12(5):392–398 [DOI] [PubMed] [Google Scholar]
- 37.McFadden DG, Barbosa AC, Richardson JA et al. (2005) The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 132(1):189–201 [DOI] [PubMed] [Google Scholar]
- 38.Stanley EG, Biben C, Elefanty A et al. (2002) Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3’UTR-ires-Cre allele of the homeobox gene Nkx2–5. Int J Dev Biol 46(4):431–439 [PubMed] [Google Scholar]
- 39.Moses KA, DeMayo F, Braun RM et al. (2001) Embryonic expression of an Nkx2–5/Cre gene using ROSA26 reporter mice. Genesis 31(4):176–180 [DOI] [PubMed] [Google Scholar]
- 40.Jay PY, Harris BS, Maguire CT et al. (2004) Nkx2–5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest 113(8):1130–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biben C, Weber R, Kesteven S et al. (2000) Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2–5. Circ Res 87(10):888–895 [DOI] [PubMed] [Google Scholar]
- 42.Jiao K, Kulessa H, Tompkins K et al. (2003) An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev 17(19):2362–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirai M, Arita Y, McGlade CJ et al. (2017) Adaptor proteins NUMB and NUMBL promote cell cycle withdrawal by targeting ERBB2 for degradation. J Clin Invest 127(2):569–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breckenridge R, Kotecha S, Towers N et al. (2007) Pan-myocardial expression of Cre recombinase throughout mouse development. Genesis 45(3):135–144 [DOI] [PubMed] [Google Scholar]
- 45.Chen J, Kubalak SW, Chien KR (1998) Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 125(10):1943–1949 [DOI] [PubMed] [Google Scholar]
- 46.Minamisawa S, Gu Y, Ross J Jr et al. (1999) A post-transcriptional compensatory pathway in heterozygous ventricular myosin light chain 2-deficient mice results in lack of gene dosage effect during normal cardiac growth or hypertrophy. J Biol Chem 274(15): 10066–10070 [DOI] [PubMed] [Google Scholar]
- 47.Shai SY, Harpf AE, Babbitt CJ et al. (2002) Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res 90(4):458–464 [DOI] [PubMed] [Google Scholar]
- 48.Sohal DS, Nghiem M, Crackower MA et al. (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89(1):20–25 [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Mu Y, Veevers J et al. (2016) Postnatal loss of Kindlin-2 leads to progressive heart failure. Circ Heart Fail 9(8):e003129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakai A, Yamaguchi O, Takeda T et al. (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13(5):619–624 [DOI] [PubMed] [Google Scholar]
- 51.Lexow J, Poggioli T, Sarathchandra P et al. (2013) Cardiac fibrosis in mice expressing an inducible myocardial-specific Cre driver. Dis Model Mech 6(6):1470–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bersell K, Choudhury S, Mollova M et al. (2013) Moderate and high amounts of tamoxifen in alphaMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis Model Mech 6(6):1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koitabashi N, Bedja D, Zaiman AL et al. (2009) Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res 105(1):12–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vogt MA, Chourbaji S, Brandwein C et al. (2008) Suitability of tamoxifen-induced mutagenesis for behavioral phenotyping. Exp Neurol 211(1):25–33 [DOI] [PubMed] [Google Scholar]
- 55.Yan J, Sultana N, al ZL (2015) Generation of a tamoxifen inducible Tnnt2MerCreMer knock-in mouse model for cardiac studies. Genesis 53(6):377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu B, Zhou B, Wang Y et al. (2010) Inducible cardiomyocyte-specific gene disruption directed by the rat Tnnt2 promoter in the mouse. Genesis 48(1):63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miniou P, Tiziano D, Frugier T et al. (1999) Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res 27(19):e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCarthy JJ, Srikuea R, Kirby TJ et al. (2012) Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet Muscle 2(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rao P, Monks DA (2009) A tetracycline-inducible and skeletal muscle-specific Cre recombinase transgenic mouse. Dev Neurobiol 69(6):401–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li S, Czubryt MP, McAnally J et al. (2005) Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci U S A 102(4):1082–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Southard S, Low S, Li L et al. (2014) A series of Cre-ER(T2) drivers for manipulation of the skeletal muscle lineage. Genesis 52(8):759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marin TM, Keith K, Davies B et al. (2011) Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest 121(3):1026–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ran FA, Hsu PD, Wright J et al. (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11):2281–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melton DW (2002) Gene-targeting strategies. Methods Mol Biol 180:151–173 [DOI] [PubMed] [Google Scholar]
- 65.Yen ST, Zhang M, Deng JM et al. (2014) Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev Biol 393(1):3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parsons SA, Millay DP, Wilkins BJ et al. (2004) Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J Biol Chem 279(25):26192–26200 [DOI] [PubMed] [Google Scholar]
- 67.Randles KN, Lam le T, Sewry CA et al. (2010) Nesprins, but not sun proteins, switch isoforms at the nuclear envelope during muscle development. Dev Dyn 239(3):998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huber MD, Guan T, Gerace L (2009) Overlapping functions of nuclear envelope proteins NET25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol Cell Biol 29(21):5718–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stroud Matthew J, Xi Fang, Zhang Jianlin et al. (2018) Luma is not essential for murine cardiac development and function, Cardiovascular Research 114(3):378–388. 10.1093/cvr/cvx205PMID:29040414 [DOI] [PMC free article] [PubMed] [Google Scholar]