

Abstract
Colorectal cancer (CRC) is the third most prevalent cancer worldwide causing an estimated 700 000 deaths annually. Different types of treatment are available for patients with advanced metastatic colorectal cancer, including targeted biological agents, such as cetuximab, a monoclonal antibody that targets EGFR. We have previously reported a study indicating multiple levels of interaction between metallopeptidase inhibitor 1 (TIMP‐1) and the epidermal growth factor (EGF) signaling axis, which could explain how TIMP‐1 levels can affect the antitumor effects of EGFR inhibitors. We also reported an association between TIMP‐1‐mediated cell invasive behavior and KRAS status. To gain insight into the molecular mechanisms underlying the effects of TIMP‐1 in CRC, we examined by transcriptomics, proteomics, and kinase activity profiling a matched pair of isogenic human CRC isogenic DLD‐1 CRC cell clones, bearing either an hemizygous KRAS wild‐type allele or KRAS G13D mutant allele, exposed, or not, to TIMP‐1. Omics analysis of the two cell lines identified the receptor tyrosine kinase c‐Kit, a proto‐oncogene that can modulate cell proliferation and invasion in CRC, as a target for TIMP‐1. We found that exposure of DLD‐1 CRC cells to exogenously added TIMP‐1 promoted phosphorylation of c‐Kit, indicative of a stimulatory effect of TIMP‐1 on the c‐Kit signaling axis. In addition, TIMP‐1 inhibited c‐Kit shedding in CRC cells grown in the presence of exogenous TIMP‐1. Given the regulatory roles that c‐Kit plays in cell proliferation and migration, and the realization that c‐Kit is an important oncogene in CRC, it is likely that some of the biological effects of TIMP‐1 overexpression in CRC may be exerted through its effect on c‐Kit signaling.
Keywords: colorectal cancer, c‐Kit, cetuximab, predictive biomarkers, omics, invasion
We identified a proto‐oncogene, c‐Kit, as a potential mediator of metallopeptidase inhibitor 1 (TIMP‐1) function in colorectal cancer (CRC) cells. Given the role that c‐Kit plays in regulation of cell proliferation and migration, it is likely that some of the effects of TIMP‐1 overexpression in CRC may be exerted through its effect on c‐Kit signaling.
Abbreviations
- CE
cellular extracts
- CM
conditioned media
- CRC
colorectal cancer
- DAPI
4′,6‐diamidino‐2‐phenylindole
- ECM
extracellular matrix
- EGF
epidermal growth factor
- MAPK
mitogen‐activated protein kinase
- MMP
matrix metalloproteinase
- MS
mass spectrometry
- PLA
proximity ligation assay
- RTK
receptor tyrosine kinase
- STK
serine/threonine kinase
- TIMP‐1
metallopeptidase inhibitor 1
- TK
tyrosine kinase
- TRIM59
tripartite motif‐containing 59
- Y2H
yeast two‐hybrid
1. Introduction
Metallopeptidase Inhibitor 1 (TIMP‐1) was originally discovered as a serum protein responsible for collagenase inhibition and growth of erythroid progenitor cells and is one of the four known endogenous human metallopeptidase inhibitors (TIMP‐1 through TIMP‐4). TIMP‐1 is expressed in various tissues, including colon, and as the name suggests, it inhibits matrix metalloproteinases (MMPs), a family of peptidases regulating extracellular matrix (ECM) turnover. TIMPs and MMPs regulate tumor invasion, migration, and progression through tissue remodeling. Accordingly, one would expect TIMP‐1 to exert mainly a beneficial effect on cancer outcomes, by inhibiting remodeling of the ECM and therefore blocking invasion and metastasis. However, regulatory effects on cell proliferation and survival have also been reported for TIMP‐1, with both positive and negative effects on tumor growth and progression reported (Hornebeck et al., 2005). Available evidence suggests TIMP‐1 to work in a dual way, regulating cellular functions by both MMP‐dependent and MMP‐independent mechanisms. Structurally, TIMP‐1 consists of two distinct and independent structural domains: an N‐terminal domain, spanning the MMP‐inhibitory activity, and a C‐terminal domain thought to hold some of the MMP‐independent cytokine‐like activities of TIMP‐1 (Chesler et al., 1995; Gasson et al., 1985; Herszenyi et al., 2012; Stetler‐Stevenson, 2008). The localization of TIMP‐1 may be crucial to explain its dual effects: Extracellular TIMP‐1 confined to the ECM prevents metastasis by inhibiting nearby MMPs, whereas intracellular TIMP‐1 promotes cancer cell proliferation and survival by effecting different signaling pathways. These different effects may also be modulated by different interaction partners. TIMP‐1 interacts with a complex of the tetraspanin cluster of differentiation 63 (CD63) and integrin β1 at the cell membrane (Jung et al., 2006), and CD63 down‐regulation decreases TIMP‐1 presence at the cell surface and reduces cell survival in breast cancer cells. The TIMP‐1–CD63–integrin β1 complex can also stimulate Src phosphorylation, followed by activation of yes‐associated protein and transcriptional co‐activator with PDZ binding domain, which in turn promote cancer cell proliferation (Ando et al., 2018). TIMP‐1 also interacts with a complex of the cell surface glycoprotein cluster of differentiation 44 and pro‐MMP‐9, and these mediate TIMP‐1′s antiapoptotic effects in erythroid cells (Lambert et al., 2009). In addition, TIMP‐1 can also up‐regulate RAS and MAPK activity, possibly through activation of receptor tyrosine kinases (RTKs) (Akahane et al., 2004; Wang et al., 2002; Yamashita et al., 1996).
Recently, we found that TIMP‐1 plasma levels predicted benefit from EGFR‐inhibition therapy in KRAS‐mutated metastatic colorectal cancer patients. These results were supported by data from cellular models showing that exposure of colorectal cancer (CRC) cells to exogenously added recombinant TIMP‐1 promoted a more aggressive behavior in these cells, specifically in KRAS‐mutated cells (Tarpgaard et al., 2016). We added recombinant TIMP‐1 to a matched set of isogenic DLD‐1 CRC cell clones, in which either the endogenous KRAS wt allele (from here on referred to as DLD‐1 G13D cell line) or the KRAS(G13D) mutant allele (from here on referred to as DLD‐1 wt cell line) has been inactivated by targeted homologous recombination (Yun et al., 2009), respectively. Whereas the DLD‐1 wt cells showed no significant difference in invasive potential, DLD‐1 G13D cells responded to the presence of TIMP‐1 in the growth medium by becoming significantly more invasive (Tarpgaard et al., 2016).
To address the effect of TIMP‐1 on the invasive potential of CRC cells, we took a three‐pronged approach: We performed a comparative proteomic analysis of the DLD‐1 G13D and DLD‐1 wt CRC cells to identify differentially expressed proteins. We also analyzed the transcriptome of DLD‐1 G13D and DLD‐1 wt cells at baseline and following exposure to TIMP‐1 to identify changes caused by exposure to TIMP‐1. Finally, we performed a broad kinase activity assay study of the DLD‐1 G13D and DLD‐1 wt cells exposed to TIMP‐1, to identify differential effects on RTKs in these cells. As a result, we identified the RTK c‐Kit as a target of TIMP‐1 in these cells. KIT encodes the human homolog of the proto‐oncogene c‐Kit, the cellular homolog of the transforming oncogene of the Hardy–Zuckerman 4 feline sarcoma virus (Yarden et al., 1987). c‐Kit is a RTK, and aberrant expression or mutational activation of c‐Kit is associated with a variety of diseases in humans, including cancer (Heinrich et al., 2002; Lennartsson et al., 2005). We found that TIMP‐1 increased phosphorylation of c‐Kit, as well as corresponding targets, and inhibited c‐Kit shedding. In summary, our data provide some further mechanistic insight into the effects of TIMP‐1 on CRC cells.
2. Materials and Methods
2.1. Cell culture
A set of isogenic colorectal adenocarcinoma cell lines, derived from the DLD‐1 CRC line by targeted homologous recombination of the KRAS locus, were used in this study (Yun et al., 2009): The KRAS‐mutated (KRASG13D/−) DLD‐1 cell line (referred to as DLD‐1 G13D) was generated by knockout of the wild‐type KRAS allele in heterozygous DLD‐1 parental cells (KRASG13D/+), whereas the KRAS wild‐type (KRAS+/−) DLD‐1 cell line (referred to as DLD‐1 wt) was generated by knockout of the mutant KRAS allele, respectively. The cell lines were kindly provided by Bert Vogelstein (Johns Hopkins University, USA). All cell lines were grown under sterile conditions at 37 °C and 5% CO2 in McCoy’s 5A medium (Invitrogen, Carlsbad, CA, USA). Growth media were supplemented with 10% FBS (Invitrogen), unless stated otherwise. To investigate the role of TIMP‐1, cells were stimulated with 1 or 5 µg·mL−1 N‐glycosylated recombinant His6‐tagged human TIMP‐1, as described (Vinther et al., 2014). As control, cells were stimulated with vehicle (PBS) or with an equivalent amount of BSA, as appropriate (Roche Diagnostics, Basel, Switzerland). The murine pro‐B cell line Ba/F3 was maintained in RPMI 1640 medium supplemented with 10% heat‐inactivated FBS and 10 ng·mL−1 recombinant murine IL‐3 (Prospec Tany, Rehovot, Israel) (Pedersen et al., 2009). The identity of all cell lines used was verified by short tandem repeat loci profiling (IdentiCell, Aarhus, Denmark).
2.2. Western blot analysis
For protein extractions, we used the Mammalian Protein Extraction Reagent (M‐PER; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with Pierce Protease and Phosphatase Inhibitor Mini Tablets (Thermo Fisher Scientific). The lysates were centrifuged at 14 000 g for 10 min to remove cell debris, and protein concentrations were determined using Thermo Scientific Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). The samples were diluted with Laemmli sample buffer (Sigma‐Aldrich, St. Louis, MI, USA) to contain ~ 20 µg protein in 25 µL total volume or 15 µg protein in 15 µL total volume, depending on the use of either 10‐ or 15‐well gels (4–15% Mini‐PROTEAN® TGC™ gel; Bio‐Rad, Hercules, CA, USA). Samples were incubated at 70 °C for 10 min prior to loading. The protein samples were resolved on a gel using Bio‐Rad SDS System (Bio‐Rad) and blotted onto a 0.2‐µm nitrocellulose membrane (Trans‐Blot® Turbo™ Midi Nitrocellulose Transfer Pack; Bio‐Rad). Membranes were blocked with 5% skim milk powder (Merck Life Sciences, Darmstadt, Germany) or 5% BSA fraction V (Roche Diagnostics) in TBS‐T, before being incubated with primary antibodies followed by a horseradish peroxidase ‐conjugated secondary antibody in blocking buffer. Protein bands were developed using either Clarity Western ECL Substrate (Bio‐Rad) or Amersham ECL Select Western Blotting Detection Reagent (GE Healthcare Life Sciences, Piscataway, NJ, USA), as appropriate. Images were acquired with UVP BioSpectrum Imaging System (UVP, Upland, CA, USA), and bands were quantified using the software (imagej version 1.49, U. S. National Institutes of Health, Bethesda, MD, USA).
2.3. Immunostaining
Cells were washed, trypsinized, and then fixed with 4% buffered formalin (Sigma‐Aldrich). Following extensive washings with PBS, cells were centrifuged to form a pellet, which was kept in 70% EtOH (Sigma‐Aldrich) until embedded in paraffin. Before use, embedded cells were freshly cut into 3‐µm sections, deparaffinized in xylene, and rehydrated through an alcohol gradient. Sections were exposed to heat‐induced antigen retrieval and placed in EnVision™ FLEX Target Retrieval Solution, low pH (Dako, Glostrup, Denmark) diluted 1 : 50 in MilliQ H2O and boiled. For some of the experiments, cells were grown on coverslips or in Permanox chamber slides (Lab‐Tek Chamber Slide System; Thermo Fisher Scientific) in order to avoid trypsin treatment or cell scraping. The surfaces were coated with poly‐l‐lysine (Sigma‐Aldrich) prior to seeding. Cells were washed twice with ice‐cold PBS prior to 20 min of fixation with 4% buffered formalin. Cells were permeabilized with 0.05% Triton X‐100 in Tris‐buffered saline (TBS; 20 mm Tris pH 7.6, 150 mm NaCl) for 5 min at RT and washed with 1× TBS. Coverslips and chamber slides were kept in humidity chambers during all incubations. Prior to incubation of primary antibody, sections were blocked with 25% BSA in TBS before incubation with primary antibodies. Sections were then incubated with secondary Alexa Fluor antibodies (Invitrogen) diluted in 25% BSA in TBS. Slides were mounted with ProLong Gold Antifade Reagent with 4',6‐diamidino‐2‐phenylindole (DAPI; Invitrogen) and examined by confocal microscopy. All antibodies used in this study, and the dilutions used, are listed in Table S1.
2.4. Proximity ligation assay
Proximity ligation assay (PLA) (Duolink; Olink Bioscience, Uppsala, Sweden) was performed according to the manufacturer’s instructions. Briefly, cell pellets mounted on glass slides were blocked with Duolink blocking buffer for 30 min at 37 °C in humidity chamber. Blocking buffer was tapped off, and primary antibodies were diluted in Duolink Antibody Diluent (TIMP‐1: 1 : 4000 and c‐Kit: 1 : 250). Slides were mounted with cover plates, placed in cassettes or in humidity chamber (for embedded cells vs chamber slides), then incubated with either one of the antibodies as control or both, and left to incubate at 4 °C overnight. The following day slides were washed with wash buffer A (Duolink) 2 × 5 min before released from cassettes and incubated for 1 h at 37 °C with both PLA probes diluted 1 : 5 in Antibody Diluent. PLA probes were tapped off, and slides were washed twice for 5 min with wash buffer A. Ligation stock was diluted 1 : 5 in MilliQ water, and ligase was diluted in the mixture 1 : 40. Ligation–ligase was added to slides, which were then incubated 30 min at 37 °C and then tapped off. Sections were washed 2 × 2 min with wash buffer A. Amplification stock was diluted 1 : 5, and polymerase was diluted 1 : 80 in this mixture. Amplification–polymerase mixture was added to the slides followed by incubation for 100 min at 37 °C. Amplification‐polymerase was tapped off, and slides were washed once either for 10 min in wash buffer B or for 5 min in wash buffer B, followed by 15‐min incubation with Atto488 Phalloidin (Sigma‐Aldrich) diluted 1 : 100 in wash buffer B, and subsequently washed with wash buffer B for 10 min. Slides were washed 1 min in 0.01× wash buffer B and dried in the dark prior to mounting with Duolink In Situ Mounting Medium with DAPI. Edges were sealed with nail polish, and slides were imaged with confocal microscopy.
2.5. Kinase activity assay
DLD‐1 wt and DLD‐1 G13D cells were serum‐starved for 4 h prior to 15‐, 30‐, and 120‐min stimulations with 5 µg·mL−1 TIMP‐1 or vehicle. Cells were washed with 3× cold PBS and lysed with M‐PER lysis buffer. Protein concentrations were measured, and samples were diluted to equal concentration. Kinase activity assay was performed using PamChip tyrosine kinase (TK) and serine/threonine kinase (STK) arrays (PamGene, Hertogenbosch, the Netherlands). STK arrays consist of 140 ser/thr‐containing peptides. TK arrays consist of 144 peptides with known phosphorylation sites, representing 100 different proteins. Each peptide represents a 15‐amino‐acid sequence of which 13 residues are derived from a putative phosphorylation site present in human proteins. Peptide sequences are derived from literature and are a cognate target of one or multiple kinases. Activation status of represented kinases is derived from the phosphorylation levels of its combined cognate targets. Incubation, washing, dispensing of reagents, and imaging of the PamChip were performed automatically using the PamStation 12. PamGene’s Bionavigator software was used for image quantification, quality control, statistical analysis, visualization, and interpretation. Downstream data analysis was done using MetaCore pathway analysis software (Thomson Reuters, Toronto, Canada).
2.6. c‐Kit shedding ELISA kit
To measure TIMP‐1’s effect on shedding of c‐Kit to the media, c‐Kit (Soluble) (Human) ELISA Kit (Nordic Biosite, Copenhagen, Denmark) was employed. This kit measures the levels of soluble c‐Kit as readout for shedding of c‐Kit. The experiments were performed in accordance with the manufacturer’s instructions.
2.7. Gene expression assays
DLD‐1 wt and DLD‐1 G13D cells were cultured in the presence, or absence, of TIMP‐1 (5 µg·mL−1) for 4 h, washed thrice, and collected by trypsinization in three independent, consecutive (passages 28, 29, and 30) experiments. The RNeasy Mini Kit (Qiagen, Venlo, The Netherlands) was used to isolate RNA from cells as described in the manufacturer’s protocol. In order to optimize samples for gene expression assay, samples were treated with RQ1 RNase‐free DNase (Promega, Madison, Wisconsin, WI, USA); 1 µL DNase was used per µg RNA, samples were mixed with 1 µL reaction buffer, and RNase‐free water was added to a volume of 10 µL. Samples were incubated at 37 °C for 30 min before 1 µL of DNase stop solution was added followed by 10 min of incubation at 65 °C to terminate the reaction. Samples were stored at −80 °C before processing.
Samples were analyzed at the Multi‐Assay Core Facility at the Danish Technical University (Center for Biological Sequence Analysis, DTU) using Agilent arrays. Briefly, samples were labeled using Low Input Quick Amp Labeling Kits (One‐Color; Agilent Technologies, Santa Clara, CA, USA) in accordance with the manufacturer’s instructions. Arrays were processed and data were collected using Agilent Feature Extraction Software (Agilent Technologies). Data analysis was done using the partek genomics suite v7.0 software package (Partek, St. Louis, MO, USA). Results are presented as Log2FoldChange of DLD‐1 wt vs DLD‐1 G13D, and TIMP‐1‐treated vs TIMP‐1‐untreated cells, genes were considered significantly different expressed if P‐value < 0.0075 and Log2FoldChange>+/‐1. Multiple testing correction of P‐values was performed using the Benjamini–Hochberg method (Benjamini and Hochberg, 1995).
2.8. In vitro wound healing assay
DLD‐1 wt cells were plated at a density of 5 × 104 cells/well in 2‐well silicone inserts (ibidi GmbH, Gräfelfing, Germany) and incubated at 37 °C for 24 h. Then, inserts were removed leaving a 500‐µm gap without any cells. Dishes were cultured for 24 h in McCoy’s 5A medium (Invitrogen), supplemented with 10% FBS (Invitrogen), and in the presence, or absence, of TIMP‐1 (5 µg·mL−1), imatinib mesylate (Sigma‐Aldrich), or both, and imaged for gap size measurement. Three nonoverlapping representative images from each of the gapped areas were acquired to estimate the relative migration of cells. Each condition was examined in triplicate experiments.
2.9. Mass spectrometry (MS)‐based proteomic analysis
Cells were processed according to the in stage tip sample preparation method (Kulak et al., 2014). Briefly, 250 µL of the reducing and alkylating sodium deoxycholate buffer (PreOmics) was added to the samples before protein denaturation at 99 °C and 1500 g for 10 min. Samples were further solubilized using the bioruptor for 10 min and digested by trypsin and Lys‐C (1 : 100) overnight at 37 °C and up to 2000 g. Following peptide desalting using SDB‐RPS, samples were measured on an quadrupole–Orbitrap mass spectrometer (Scheltema et al., 2014; Kelstrup et al., 2014) (Q Exactive HF; Thermo Fisher Scientific, Rockford, IL, USA) coupled to an EASYnLC 1200 ultra‐high‐pressure system (Thermo Fisher Scientific). About 0.5 µg of peptides were loaded on a 45‐cm HPLC column (75 μm inner diameter; in‐house‐packed using ReproSil‐Pur C18‐AQ 1.9 µm silica beads; Dr. Maisch GmbH, Ammerbuch, Germany). Peptides were separated using a linear 100 min gradient from 3% to 23% B in 82 min and stepped up to 40% in 8 min at 350 nL per min where solvent A was 0.1% formic acid in water and solvent B was 80% acetonitrile and 0.1% formic acid in water. Column temperature was kept at 60 °C. The mass spectrometer was operated in ‘top‐15’ data‐dependent mode, collecting MS spectra in the Orbitrap mass analyzer (60 000 resolution, 300–1650 m/z range) with a maximum ion injection time of 25 ms and an automatic gain control (AGC) target of 3E6. The most intense ions from the full scan were isolated with a width of 1.4 m/z. Following higher‐energy collisional dissociation with a normalized collision energy of 27%, MS/MS spectra were collected in the Orbitrap (15 000 resolution) with a maximum ion injection time of 28 ms and an AGC target of 1E5. Precursor dynamic exclusion was enabled with a duration of 30 s. Spectra were searched against the 2015 UniProt human databases (UP000005640_9606 and UP000005640_9606_additional) using MaxQuant version 1.5.3.34 with a 1% FDR at the peptide and protein level and default settings. The ‘match between runs’ feature was on with a matching time window of 0.7 min and an alignment time window of 20 min. Label‐free quantification was performed using the MaxLFQ algorithm with a minimum ratio count of 1. Following bioinformatic analyses were performed using the perseus software (Tyanova et al., 2016) (version 1.5.5.0). Proteins that were identified in the decoy reverse database or only by site modification were removed for data analysis. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2016) partner repository and are available upon request.
3. Results
3.1. Transcriptome profiling of DLD‐1 wt and DLD‐1 G13D cells
To address the molecular mechanisms underlying the differential invasive phenotype we had observed in KRAS‐mutated cells exposed to TIMP‐1 (Tarpgaard et al., 2016), we performed genome‐wide mRNA expression profiling of the DLD‐1 wt and DLD‐1 G13D cell lines (dataset provided as Data S1). Given that the two cell lines are isogenic, only differing in their KRAS mutational status, we first determined whether we could find any biologically relevant linear dimensions in the expression dataset that would differentiate between the two cell lines. Principal component analysis (PCA) of our dataset showed that the two cell lines could be separated on the basis of their three main principal components (PC‐#1, PC‐#2, and PC‐#3, respectively; Fig. 1A). We then compared the two cell lines’ mRNA profiles and found only rather moderate differences in mRNA expression between the two lines. The 25 most highly up‐ or down‐regulated transcripts are presented in Table 1. Two differentially expressed genes (Table 1) called our attention: the KIT proto‐oncogene RTK (c‐Kit), a key regulator of cell survival, proliferation, and migration (Lennartsson and Rönnstrand, 2012), and tripartite motif‐containing 59 (TRIM59) protein, which is known to affect CRC proliferation and metastasis (Sun et al., 2017). These two genes stood out, not only because they are known to affect CRC cell invasion, but also, importantly, because they were present in a set of 58 hits we discovered in a yeast two‐hybrid (Y2H) screening for TIMP‐1 potential interaction partners (M. Hoeberg, personal communication).
Figure 1.
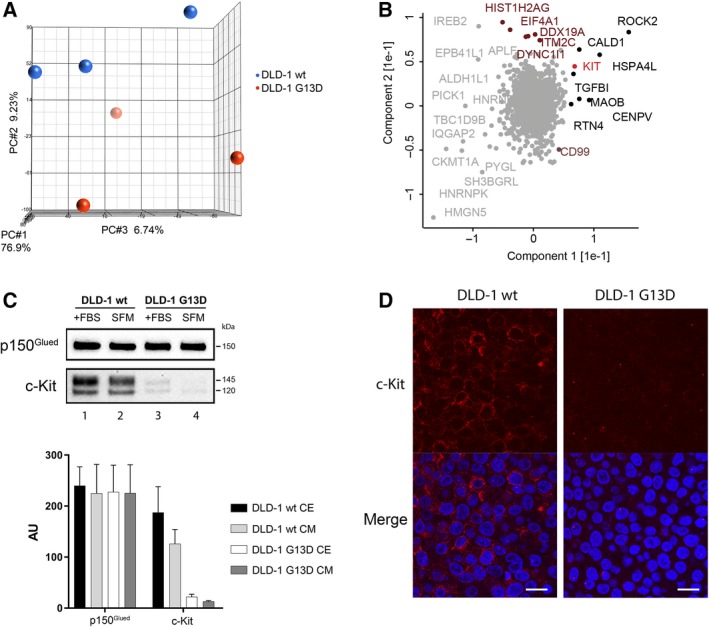
(A) PCA plot of expression microarray data of DLD‐1 wt (red circles) and DLD‐1 G13D (blue circles) cell lines. The percentages of the total variation that are accounted for by the 1st, 2nd, and 3rd principal components are shown on the x‐, y‐, and z‐axes labels, respectively. (B) PCA plot of comparative analysis between DLD‐1 wt and DLD‐1 G13 protein expression data, respectively. Proteins within the GO class ‘mitotic cell cycle’ are highlighted (magenta). c‐Kit is also pinpointed (red). (C) Upper panel: immunoblot analysis of c‐Kit and p150Glued (normalizing control) protein expression in DLD‐1 wt and DLD‐1 G13D cells grown in media with serum (+FBS) and sereum free media (SFM), respectively, lower panel: graph depicting pooled densitometry measurements of c‐Kit levels relative to those of p150Gluedin DLD‐1 wt and DLD‐1 G13D CE and CM, respectively. Data points are presented as mean ± SEM of triplicate experiments. (D) c‐Kit expression was examined in both DLD‐1 wt and DLD‐1 G13D cells by immunocytochemistry. Sections were immunofluorescently stained for c‐Kit (red) and the cell nucleus (DAPI, blue), respectively. Scale bar = 20 µm.
Table 1.
List of top 25 genes, which showed differential expression between DLD‐1 wt and DLD‐1 G13D, ranked by P‐value, with cut off Log2 FoldChange>+/‐1.
| Gene | Log2FoldChange | P‐value | ||||||
|---|---|---|---|---|---|---|---|---|
| Symbol | Name | wt vs G13D | wt vs wt + TIMP‐1 | G13D vs G13D + TIMP‐1 | wt vs G13D | wt vs wt + TIMP‐1 | G13D vs G13D + TIMP‐1 | |
| 1 | S100P | S100 calcium binding protein P | 3.30 | 0.10 | −0.40 | 0.00000 | 0.691 | 0.138 |
| 2 | PDZK1IP1 | PDZK1 interacting protein 1 | −1.72 | 0.10 | 0.30 | 0.00001 | 0.657 | 0.209 |
| 3 | SH3BGRL | SH3 domain binding glutamate‐rich protein‐like | 1.50 | −0.33 | 0.09 | 0.00003 | 0.168 | 0.699 |
| 4 | KBTBD11 | Kelch repeat and BTB (POZ) domain‐containing 11 | 1.68 | 0.09 | −0.39 | 0.00004 | 0.742 | 0.172 |
| 5 | HAGH | Hydroxyacylglutathione hydrolase | −1.74 | −1.56 | 0.39 | 0.00006 | 0.000 | 0.198 |
| 6 | KLK7 | Kallikrein‐related peptidase 7 | 1.74 | −0.17 | −0.50 | 0.00006 | 0.558 | 0.104 |
| 7 | NPPB | Natriuretic peptide B | 1.90 | 0.12 | 0.49 | 0.00006 | 0.715 | 0.138 |
| 8 | CYB5A | Cytochrome b5 type A (microsomal) | 1.55 | 0.20 | 0.51 | 0.00008 | 0.465 | 0.075 |
| 9 | TRIM59 | Tripartite motif‐containing 59 | −1.46 | 0.26 | −0.43 | 0.00009 | 0.326 | 0.114 |
| 10 | PPT2 | palmitoyl‐protein thioesterase 2 | 1.82 | 0.47 | −0.13 | 0.00018 | 0.188 | 0.714 |
| 11 | KIT | v‐Kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog | −1.33 | 0.33 | −0.08 | 0.00025 | 0.221 | 0.771 |
| 12 | KRT20 | keratin 20, type I | 1.99 | 0.35 | −1.07 | 0.00026 | 0.376 | 0.017 |
| 13 | GFRA4 | GDNF family receptor alpha 4 | −1.96 | −1.68 | 0.20 | 0.00028 | 0.001 | 0.605 |
| 14 | SEZ6L2 | Seizure‐related 6 homolog (mouse)‐like 2 | 1.25 | 0.26 | 0.11 | 0.00029 | 0.320 | 0.651 |
| 15 | ICAM2 | intercellular adhesion molecule 2 | −1.76 | −0.07 | 0.73 | 0.00035 | 0.852 | 0.063 |
| 16 | LKAAEAR1 | LKAAEAR motif‐containing 1 | −1.89 | −1.50 | 0.38 | 0.00042 | 0.002 | 0.350 |
| 17 | KIAA1211L | KIAA1211‐like | −1.28 | −1.02 | 0.17 | 0.00046 | 0.002 | 0.541 |
| 18 | B3GNT9 | UDP‐GlcNAc:betaGal beta‐1,3‐N‐acetylglucosaminyltransferase 9 | −1.23 | 0.11 | 0.39 | 0.00055 | 0.693 | 0.158 |
| 19 | DENND2D | DENN/MADD domain‐containing 2D | 1.21 | 0.01 | 0.07 | 0.00057 | 0.957 | 0.800 |
| 20 | PDE4D | Phosphodiesterase 4D, cAMP‐specific | 2.60 | 0.29 | 1.59 | 0.00057 | 0.615 | 0.015 |
| 21 | FOXB1 | Forkhead box B1 | −1.58 | −1.37 | 0.81 | 0.00059 | 0.002 | 0.035 |
| 22 | LOXL1 | Lysyl oxidase‐like 1 | 1.11 | 0.19 | 0.43 | 0.00066 | 0.447 | 0.100 |
| 23 | LAGE3 | L antigen family, member 3 | 1.75 | 0.49 | 1.06 | 0.00075 | 0.226 | 0.018 |
| 24 | NMB | Neuromedin B | 1.15 | −0.12 | −0.39 | 0.00086 | 0.654 | 0.159 |
| 25 | TNNT1 | Troponin T type 1 (skeletal, slow) | 1.24 | −0.14 | 0.58 | 0.00086 | 0.638 | 0.062 |
3.2. Proteome profiling of DLD‐1 wt and DLD‐1 G13D cells
To further address the molecular mechanisms underlying the TIMP‐1‐dependent differential invasive phenotype we observed in KRAS‐mutated cells, we performed MS‐based proteomic analysis of the DLD‐1 wt and DLD‐1 G13D cell lines. We quantified a total of 5861 proteins (Data S2). Again, PCA along the two main principal components segregated the two cell lines, with the main component driving the separation being proteins within the GO class ‘mitotic cell cycle’. This observation is consistent with the presence of the oncogenic KRAS G13D allele in DLD‐1 G13D cells, and a higher proliferative potential than DLD‐1 wt cells. Among the proteins identified as being differentially expressed between the two cell lines, we identified c‐Kit (Fig. 1B), which was concordant with our mRNA expression data (Table 1). TRIM59 was not present in our protein dataset, so we could not confirm its deregulation. We confirmed the decreased c‐Kit expression in DLD‐1 G13D cells compared to wt cells by western blot analysis of cellular extracts (CE; Fig. 1C) and immunofluorescence (Fig. 1D). As the extracellular domain of c‐Kit can be shed to the extracellular environment following proteolytic cleavage, it was conceivable that the difference in cellular expression we had observed was due to substantially increased protein shedding to the extracellular space, rather than true differential expression (Yee et al., 1994; Yee et al., 1993). Analysis of conditioned media (CM) showed this not to be the case (CM; Fig. 1C), as the amount of extracellular c‐Kit present in CM of DLD‐1 G13D cells was markedly lower than the levels observed in CM of DLD‐1 wt cells. In both cases, the extracellular expression of c‐Kit was proportional to the cellular levels of c‐Kit (Fig. 1C; compare c‐Kit in DLD‐1 wt CE to CM and DLD‐1 G13D CE to CM, respectively).
3.3. TIMP‐1 interacts with the cellular membrane in DLD‐1 wt and DLD‐1 G13D cells
Previous studies indicated that the cellular effects of TIMP‐1 may be mediated by direct binding to a cell surface receptor, an assumption emphasized by the identification of the tetraspanin CD63 as a TIMP‐1 cell surface interacting protein in a complex with β1 integrin (Jung et al., 2006; Toricelli et al., 2013; Wilk et al., 2013). Furthermore, uptake of TIMP‐1 in breast cancer cells has also been reported in co‐culture systems, suggesting that cancer cells can bind and internalize exogenous TIMP‐1 (Kuvaja et al., 2012; Ritter et al., 1999). We investigated whether DLD‐1 wt cells or DLD‐1 G13D cells can bind and internalize exogenously added human recombinant TIMP‐1 (Vinther et al., 2014), and whether the profile of cellular internalization of TIMP‐1 differed between the two cell lines. TIMP‐1 was added to the culture media of DLD‐1 wt and DLD‐1 G13D cells, and samples were collected at different time points and analyzed (Fig. 2). We found that both DLD‐1 wt cells and DLD‐1 G13D cells could bind TIMP‐1, although the interaction differed between the two cell lines. In both cases, TIMP‐1 binding showed a bimodal behavior (Fig. 2A,B): a fast (1 min) but transient interaction in a first step, followed by a slower second step where we observed continuously increasing interaction of TIMP‐1 until a stable state was reached sometime between 2 and 4 h, indicating the binding/internalization process had reached equilibrium (Fig. 2B). The only detectable difference between the two cell lines concerned the first interaction, where levels of bound TIMP‐1 protein were significantly higher in DLD‐1 G13D than in DLD‐1 wt (Fig. 2A).
Figure 2.
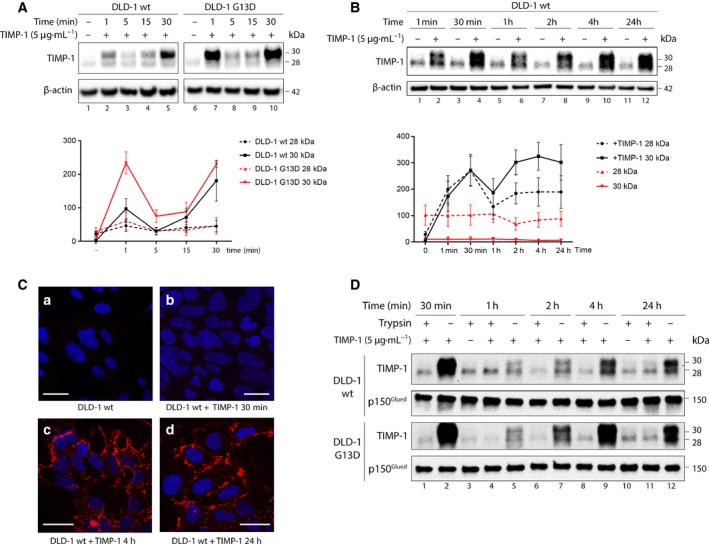
Presence and localization of exogenously added TIMP‐1 to DLD‐1 wt and G13D cells. (A, B) DLD‐1 wt and G13D cells were grown in the presence (or not) of TIMP‐1 (5 µg·mL−1) for the indicated amount of time. Cells were washed extensively, and whole‐cell extracts were examined by western blotting. Upper panels: immunoblots showing levels of TIMP‐1 protein present in whole‐cell extracts. β‐Actin was used as normalizing control. Lower panels: graphs depicting pooled densitometry measurements of TIMP‐1 levels relative to those of β‐actin. Data points are presented as mean ± SEM of triplicate experiments. (C) Immunostaining of DLD‐1 wt and G13D cells grown in the presence of TIMP‐1. Cells were grown in chamber slides and exposed to 5 µg·mL−1 TIMP‐1 for the indicated amount of time. Controls were grown in the absence of TIMP‐1. Cells were fixed and immunofluorescently stained for TIMP‐1 (red) and the cell nucleus (DAPI, blue), respectively. Scale bars, 20 µm. (D) DLD‐1 wt and G13D cells were grown in the presence of TIMP‐1 (5 µg·mL−1) for the indicated amount of time. Cells were washed extensively and trypsinized prior lysis and analysis of TIMP‐1 presence in whole‐cell extracts by western blotting. P150Glued was used as normalizing control.
Given that this assay does not discriminate between TIMP‐1 bound to a receptor on the cell surface and protein that has been internalized, we performed immunolocalization of TIMP‐1. We found that exogenously added TIMP‐1 localized mainly to the membrane (Fig. 2C). Trypsinization of cells prior to analysis of interacting TIMP‐1 revealed that most of the exogenous TIMP‐1 protein that interacted with DLD‐1 G13D or DLD‐1 wt cells, respectively, could be removed by trypsinization of cells prior to analysis. These observations suggest that in the case of DLD‐1 G13D and DLD‐1 wt cells, exogenous TIMP‐1 could bind to the cell surface but was not internalized, at least at any detectable rate (Fig. 2D).
3.4. TIMP‐1 has different effects on DLD‐1 wt and DLD‐1 G13D cells
Having established a gene expression baseline for DLD‐1 wt and DLD‐1 G13D cells, we examined what changes took place upon cell exposure to exogenous TIMP‐1. DLD‐1 wt and DLD‐1 G13D cells were cultured in the presence, or absence, of TIMP‐1 (5 µg·mL−1) for 4 h, and their transcriptomes were profiled as before. For the analysis of the gene expression dataset, Log2FoldChange and statistical significance were calculated (P < 0.05, t‐test with correction for multiple hypothesis testing) (Benjamini and Hochberg, 1995). However, this analysis gave no significant hits, and we repeated the analysis with a lower stringency (P < 0.0075, t‐test with no multiple hypothesis correction). We found that only seven genes were significantly changed by TIMP‐1 exposure in DLD‐1 wt cells (Table 2), whereas 1183 genes were affected in DLD‐1 G13D cells (Table 3). Clearly, the extent of the overall effect of TIMP‐1 was much greater in the KRAS‐mutated cells than in the KRAS wt cells, and it was conceivable that the cumulative effect of the changes brought about by exposure to TIMP‐1 could account for the differential effect of TIMP‐1 on the KRAS‐mutated cells. Noteworthy, neither c‐Kit nor TRIM59 expression were affected by TIMP‐1 (Table 1). Interestingly, we found that five genes, which were significantly up‐regulated upon TIMP‐1 stimulation in DLD‐1 G13D cells but showed no change in DLD‐1 wt cells, mapped to the Ras‐Raf‐MEK‐Erk/JNK signaling axis (Table 4), indicating that TIMP‐1 may sustain increased activity in the Ras signaling axis in cells expressing oncogenic Ras, which may result in reinforced signaling through this pathway.
Table 2.
Significantly deregulated genes in DLD‐1 wt cells exposed to TIMP‐1.
| Symbol | Name | Log2FoldChange | P‐value | |
|---|---|---|---|---|
| 1 | HAGH | hydroxyacylglutathione hydrolase | −1.56 | 0.00016 |
| 2 | GFRA4 | GDNF family receptor alpha 4 | −1.68 | 0.00091 |
| 3 | FOXB1 | forkhead box B1 | −1.37 | 0.00176 |
| 4 | LKAAEAR1 | LKAAEAR motif‐containing 1 | −1.50 | 0.00235 |
| 5 | KIAA1211L | KIAA1211‐like | −1.02 | 0.00246 |
| 6 | ARTN | Artemin | −2.21 | 0.00599 |
| 7 | PTH2 | parathyroid hormone 2 | −1.03 | 0.00735 |
Table 3.
List of top 25 genes, ranked by P‐value, affected by TIMP‐1 in DLD‐1 G13D cells.
| Symbol | Description | Log2FoldChange | P‐value | |
|---|---|---|---|---|
| 1 | EVX1 | Even‐skipped homeobox 1 | −1.32 | 0.0018 |
| 2 | STX10 | Syntaxin 10 | 1.81 | 0.0020 |
| 3 | ZNF692 | Zinc finger protein 692 | 2.17 | 0.0020 |
| 4 | MC1R | Melanocortin 1 receptor (alpha melanocyte‐stimulating hormone receptor) | 1.91 | 0.0021 |
| 5 | RASL10A | RAS‐like, family 10, member A | 1.72 | 0.0021 |
| 6 | LZTS2 | Leucine zipper, putative tumor suppressor 2 | 1.60 | 0.0023 |
| 7 | EREG | Epiregulin | −1.84 | 0.0024 |
| 8 | FAM173A | Family with sequence similarity 173, member A | 1.85 | 0.0025 |
| 9 | PPIE | Peptidylprolyl isomerase E (cyclophilin E) | 1.55 | 0.0026 |
| 10 | CRYBA2 | Crystallin, beta A2 | 2.11 | 0.0026 |
| 11 | TCIRG1 | T‐cell, immune regulator 1, ATPase, H + transporting, lysosomal V0 subunit A3 | 1.76 | 0.0028 |
| 12 | GGA1 | Golgi‐associated, gamma adaptin ear‐containing, ARF binding protein 1 | 2.07 | 0.0028 |
| 13 | EDF1 | Endothelial differentiation‐related factor 1 | 1.61 | 0.0028 |
| 14 | GYLTL1B | Glycosyltransferase‐like 1B | 1.87 | 0.0028 |
| 15 | SQLE | Squalene epoxidase | −3.22 | 0.0028 |
| 16 | TELO2 | Telomere maintenance 2 | 2.09 | 0.0028 |
| 17 | C14orf80 | Chromosome 14 open reading frame 80 | 2.25 | 0.0029 |
| 18 | WDR6 | WD repeat domain 6 | 1.76 | 0.0029 |
| 19 | CD151 | CD151 molecule (Raph blood group) | 2.05 | 0.0029 |
| 20 | ASIC1 | acid sensing (proton gated) ion channel 1 | 1.37 | 0.0029 |
| 21 | DBET | D4Z4 binding element transcript (nonprotein coding) | 1.93 | 0.0029 |
| 22 | USP20 | Ubiquitin specific peptidase 20 | 1.44 | 0.0029 |
| 23 | RECQL4 | RecQ protein‐like 4 | 1.63 | 0.0029 |
| 24 | HMG20B | High mobility group 20B | 1.84 | 0.0030 |
| 25 | CKS2 | CDC28 protein kinase regulatory subunit 2 | ‐2.61 | 0.0030 |
Table 4.
Selected hits from the Ras‐Raf‐MEK‐Erk/JNK signaling axis significantly up‐regulated upon TIMP‐1 stimulation in DLD‐1 G13D cells.
| Log2FoldChange | P‐value | |||||
|---|---|---|---|---|---|---|
| Gene | wt vs G13D | wt vs wt + TIMP‐1 | G13D vs G13D + TIMP‐1 | wt vs G13D | wt vs wt + TIMP‐1 | G13D vs G13D + TIMP‐1 |
| RAF1 | −0.05 | 0.01 | 1.93 | 0.9291 | 0.9834 | 0.0068 |
| MAP3K1 | −0.58 | 0.05 | 2.20 | 0.3576 | 0.9340 | 0.0036 |
| MAP3K3 | −0.21 | −0.09 | 1.64 | 0.6709 | 0.8587 | 0.0056 |
| MAP3K6 | −0.40 | 0.03 | 2.10 | 0.5012 | 0.9612 | 0.0035 |
| MAP4K2 | 0.18 | 0.22 | 1.63 | 0.7054 | 0.6521 | 0.0047 |
3.5. Kinome profiling of DLD‐1 wt and DLD‐1 G13D cells exposed to TIMP‐1
TIMP‐1 has been shown to effect several distinct signaling pathways, including the mitogen‐activated protein kinase (MAPK) pathway and cAMP–protein kinase A pathway, and to be associated with activation of RAS and ERK, as well as PI3‐K induced Cyclin D expression. TIMP‐1 has also been reported to increase AKT activity (Akahane et al., 2004; Lu et al., 2011; Stetler‐Stevenson, 2008). Given that multiple associations with signaling pathways have been reported for TIMP‐1 and that these appear to be contextual, we performed a screening for kinases that may specifically be involved in TIMP‐1‐mediated CRC cellular signaling (Chirco et al., 2006; Stetler‐Stevenson, 2008). Taking the patterns of association of exogenous TIMP‐1 with DLD‐1 G13D and wt cells in consideration (Fig. 2), we analyzed the kinome of DLD‐1 wt and DLD‐1 G13D cells, respectively, immediately before addition of exogenous TIMP‐1 to the growth medium, and after 15, 30, and 120 min of adding TIMP‐1 (Fig. 3D). Significant hits are reported in Fig. 3. TIMP‐1 stimulation had quite distinct effects on the two different cell lines (Fig. 3). For both TKs (Fig. 3A) and STKs (Fig. 3B), a general increase in kinase activity appeared in DLD‐1 wt cells exposed to exogenous TIMP‐1 for 30 min. For TKs only, a subsequent decrease was detected after 120 min compared to control cells (Fig. 3A). This pattern was different for DLD‐1 G13D, where significant inhibition of TK and STK activity, rather than activation, was observed upon 15 min of TIMP‐1 exposure followed by minor, nonsignificant, increases in TK activity after 30 and 120 min. This suggests KRAS‐dependent differences in TIMP‐1 signaling dynamics. Table 5 shows the top 10 hits for the ranking of active kinases derived from the STK and TK array analysis for A) protein TKs and B) STKs. Ranking is based on the sum of sensitivity (significance of the difference between samples) and specificity (specificity of the mapping to specific upstream kinases). One hit was particularly suggestive: c‐Kit, which we had found in our transcriptome and proteome profiling of DLD‐1 wt and DLD‐1 G13D cells to be up‐regulated in DLD‐1 wt cells compared to DLD‐1 G13D cells, both at the mRNA (Table 1) and at the protein levels (Fig. 1B), was on the list of kinases (Table 3). MetaCore pathway mapping of kinase hits, based on the enrichment distribution of the kinase dataset, identified the c‐Kit canonical signaling pathway as the highest significant [−log(P‐value) = 11] (Fig. 3C).
Figure 3.
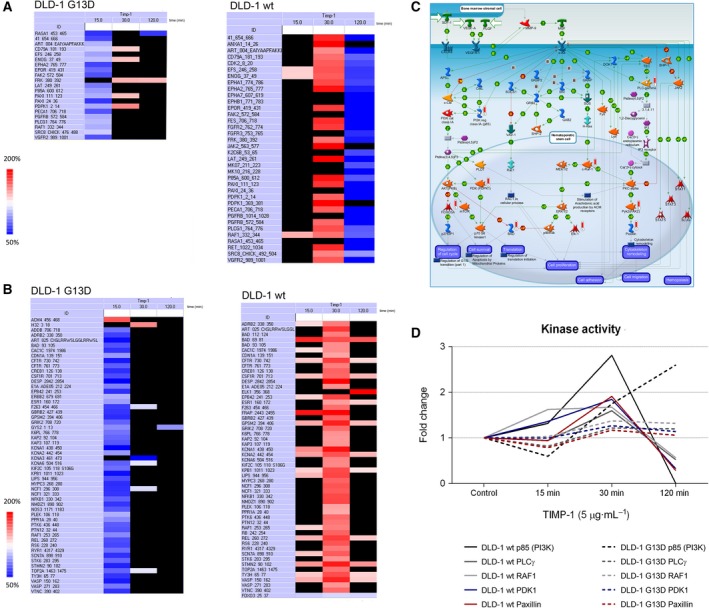
Kinase activity regulation results from PamGene analysis. DLD‐1 wt and DLD‐1 G13D cells were exposed (or not) with 5 μg·mL−1 TIMP‐1 for 15, 30, and 120 min and analyzed with PamGene kinase activity assay. The tables show significantly (P < 0.05) TIMP‐1‐regulated kinase activities of (A) protein TKs and (B) STKs (Red and blue are significant, P < 0.05; red denotes up‐regulation, and blue down‐regulation, respectively). Statistical tests were performed with PamGene’s Bionavigator software. (C) MetaCore pathway map visualization of PamGene kinase array data for the c‐Kit pathway. Kinases identified in our analysis are indicated by a histogram next to a network object with activity denoted by bar height (D) Analysis of kinase activity of kinases linked to the c‐Kit signaling axis upon exposure to TIMP‐1. Kinase activity raw data were extracted for c‐Kit‐related kinases (PI3K, PLCγ, RAF1, PDK1, and paxilin). The figure shows the mean fold change between the results of the control and TIMP‐1‐stimulated samples. Kinase activity was significantly down‐regulated upon 120 min in DLD‐1 wt, except for RAF1, which was borderline significant (P < 0.10).
Table 5.
Kinase analysis of TIMP‐1‐mediated effects.
| Tyrosine kinases | Serine/threonine kinases |
|---|---|
| 1 Insulin receptor (INSR) | 1 Spleen tyrosine kinase (SYK) |
| 2 Insulin‐like growth factor 1 receptor (IGF1R) | 2 B lymphocyte kinase (BLK) |
| 3 Insulin receptor‐related protein (INSRR) | 3 Hepatocyte growth factor receptor (MET) |
| 4 NT‐3 growth factor receptor (NTRK2) | 4 Fibroblast growth factor receptor 3 (FGFR3) |
| 5 High‐affinity nerve growth factor receptor (NTRK1) | 5 Abelson murine leukemia viral oncogene homolog 1 (ABL) |
| 6 v‐Kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog (c‐Kit) | 6 Receptor tyrosine‐protein kinase erbB‐3 (ERBB3) |
| 7 Fibroblast growth factor receptor 4 (FGFR4) | 7 Serine/arginine repetitive matrix protein 1 (SRMS) |
| 8 Proto‐oncogene tyrosine‐protein kinase Src (SRC) | 8 Gardner–Rasheed feline sarcoma viral (v‐fgr) oncogene homolog (FGR) |
| 9 Tyrosine‐protein kinase Lyn (LYN) | 9 Macrophage‐stimulating protein receptor (MST1R/RON) |
| 10 Lymphocyte‐specific protein tyrosine kinase (LCK) | 10 Related to receptor tyrosine kinase (RYK) |
3.6. TIMP‐1 increases c‐Kit activity
Given that the Ras/MAPK and PI3K/Akt signaling modules are effectors of the c‐Kit signaling axis, and immunoprecipitation studies have shown that c‐Kit is associated with the tetraspanin CD63, a TIMP‐1 interactor, at the cellular membrane (Anzai et al., 2002; Termini and Gillette, 2017), we investigated further a possible connection between c‐Kit and TIMP‐1. We performed co‐immunoprecipitation studies with c‐Kit and TIMP‐1, but were unable to detect any interaction, neither with a TIMP‐1‐specific antibody nor with a tagged version of TIMP‐1 (Fig. S1A,B, respectively), under the conditions assayed. PLA, a method that allows one to visualize protein–protein interactions in situ, allowed us to confirm that c‐Kit was in physical proximity of TIMP‐1, as we detected a number of fluorescent signals in DLD‐1 wt cells (Fig. S1C, panel a, white arrows), suggestive of an interaction between c‐Kit and TIMP‐1, whereas very few signals were detected in DLD‐1 G13D cells, which express c‐Kit at much lower levels (Fig. S1C, panel b, white arrows, and Fig. 1B).
Given that c‐Kit was among the top hits in our kinase analysis, we decided to investigate c‐Kit activity upon TIMP‐1 stimulation. c‐Kit phosphorylation was used as a measure of c‐Kit activity. Phosphorylation of all tested c‐Kit phosphorylation sites increased significantly upon exposure of DLD‐1 wt or DLD‐1 G13D cells to TIMP‐1 stimulation (Fig. 4A–D, respectively). To investigate whether TIMP‐1‐increased phosphorylation of c‐Kit is followed by phosphorylation downstream targets of c‐Kit, p‐ERK levels were also examined. We found increased (twofold) p‐ERK levels upon 5 min of TIMP‐1 stimulation in DLD‐1 wt (Fig. 4B).
Figure 4.
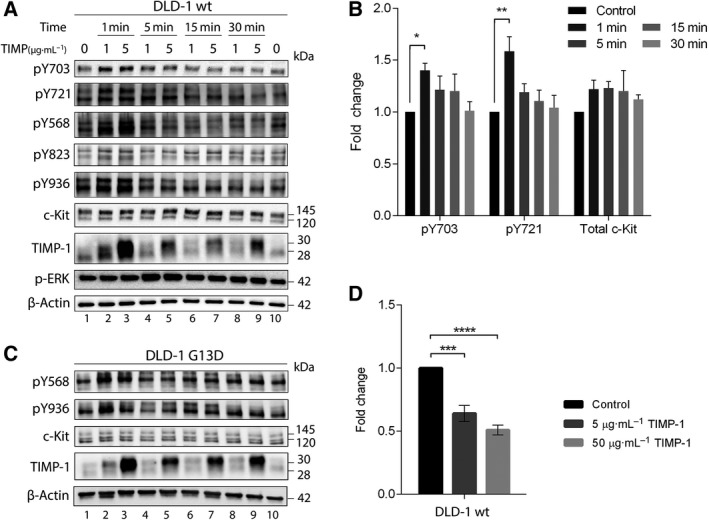
Phosphorylation of c‐Kit upon TIMP‐1 exposure. (A and C) DLD‐1 wt and G13D cells were stimulated with either 1 or 5 µg·mL−1 TIMP‐1 for the indicated time periods. Controls were treated with vehicle (PBS) for 30 min. Western blots were repeatedly stripped and probed for the different anti‐phospho‐c‐Kit antibodies recognizing various phosphorylated residues (Y703, Y721, Y568, Y823, and Y936 for DLD‐1 wt and Y568 and Y936 for DLD‐1 G13D, respectively) and finally for total c‐Kit. Blots were additionally blotted for pERK1/2 and TIMP‐1. β‐Actin was used as loading control. (B) Quantification of phosphorylated c‐Kit and total c‐Kit levels upon TIMP‐1 exposure in DLD‐1 wt cells. Quantification was performed with ImageJ. The mean fold change of c‐Kit pY703, c‐Kit pY721, and total c‐Kit levels between unstimulated and TIMP‐1‐treated samples is presented (±SEM). * indicates P < 0.05, and ** indicates P < 0.01, a two‐way ANOVA test with Dunnett’s correction for multiple comparison was used for significance. (D) Analysis of c‐Kit shedding upon exposure to TIMP‐1. Cells were stimulated for 24 h with 5 or 50 µg·mL−1 TIMP‐1 prior to collection of conditioned medium. Soluble c‐Kit levels in the medium were determined by a c‐Kit shedding‐specific ELISA kit. Bars represent the mean fold change of soluble c‐Kit levels between control and TIMP‐1‐stimulated samples (±SEM). No shed c‐Kit was detected for DLD‐1 G13D. *** and **** indicate P < 0.001 and P < 0.0001, respectively. Significance was determined by one‐way ANOVA test with Dunnett’s correction for multiple comparison.
Another level of regulation of c‐Kit function includes proteolytic extracellular cleavage allowing release of a surface protein ectodomain (‘shedding’) that produces a soluble form of c‐Kit. This reduces cell surface expression as well as produces a soluble c‐Kit polypeptide, which can competitively bind the c‐Kit ligand SCF. Stimulating DLD‐1 wt with either 5 or 50 μg·mL−1 TIMP‐1 significantly decreased soluble c‐Kit levels in the media down to ~ 60% and ~ 50%, respectively (Fig. 4D, black vs gray bars). These data are consistent with a previous observed decreased levels of soluble c‐Kit upon incubation of mast cells with TIMP‐1 and TIMP‐3 (Cruz et al., 2004). To determine whether the effect of TIMP‐1 on c‐Kit activation we observed was a DLD‐1 wt cell‐specific effect, we examined levels of pY703 c‐Kit in Ba/F3 cells upon exposure to TIMP‐1. Ba/F3 is a murine, interleukin (IL)‐3 dependent, hematopoietic cell line (Palacios and Steinmetz, 1985). We found that TIMP‐1 induced activation of c‐Kit, measured by increased phosphorylation of pY703 (Fig. 5A).
Figure 5.
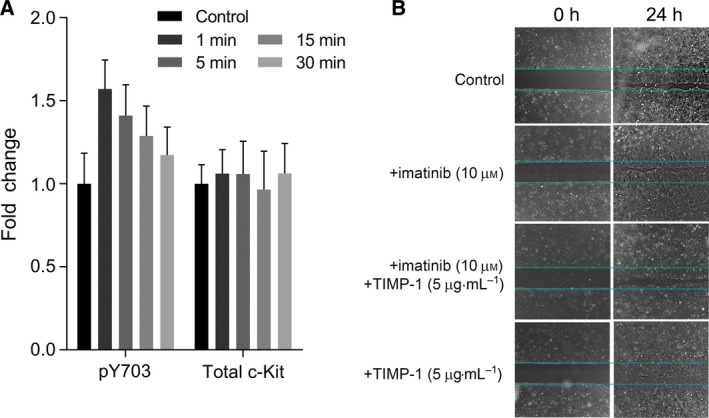
TIMP‐1 effects on (A) Ba/F3 pro‐B murine cells and (B) DLD‐1 wt cells. (A) Ba/F3 cells were stimulated with 5 µg·mL−1 TIMP‐1 for the indicated time periods. Samples were analyzed for phospho‐ (Y703) and total c‐Kit levels by western blot analysis. β‐Actin was used as loading control. Quantification was performed with imagej. The mean fold change of c‐Kit pY703, and total c‐Kit levels between unstimulated and TIMP‐1‐treated samples are presented (±SEM); a two‐way ANOVA test with Dunnett’s correction for multiple comparison was used for significance. (B) Light micrographs of wounded cell monolayers showing effect of TIMP‐1 on wound closure of DLD‐1 wt cells at 24 h. Cells were exposed to imatinib mesylate (10 µm), TIMP‐1 (5 µg·mL−1), or a combination of both. Representative images are shown.
4. Discussion
Several studies have reported that plasma levels of TIMP‐1 have prognostic value in CRC (Birgisson et al., 2010; Nielsen et al., 2010). Although TIMP‐1 expression is up‐regulated in colorectal tumor tissue (Lu et al., 1991), the molecular mechanisms underlying the prognostic effect of TIMP‐1 in CRC remain unclear. We previously reported a differential cellular effect of TIMP‐1 on CRC cell invasion, dependent on KRAS status (Tarpgaard et al., 2016). A similar observation has also been reported for pancreatic ductal adenocarcinoma, where exogenous addition of human recombinant TIMP‐1 significantly increased proliferation of pancreatic ductal cells, but only in KRAS‐transformed cells (Botta et al., 2013). To gain some insight into the molecular mechanisms underlying the KRAS‐dependent effects of TIMP‐1 in CRC, we examined a matched pair of isogenic human CRC DLD‐1 cell line clones (KRAS G13D and wild‐type) exposed, or not, to TIMP‐1. We performed genome‐wide mRNA expression profiling of the DLD‐1 wt and DLD‐1 G13D cell lines (Table 1 and Fig. 1) and, from the limited number of differentially expressed genes between the two cell lines, identified two, c‐Kit and TRIM59, that could conceivably play a role in the effects of TIMP‐1 on CRC cells. These two genes stood out, not only because they are known to affect CRC cell invasion, but also, importantly, because they were present in a set of 58 hits we discovered in a Y2H screening for TIMP‐1 potential interaction partners (M. Hoeberg, personal communication). We complemented the gene expression analysis of the DLD‐1 wt and DLD‐1 G13D cell lines with MS‐based proteome profiling and phosphopeptide array‐based kinome profiling. We found that all levels of our analyses linked c‐Kit, expression, and pathway activation, with exposure of cells to TIMP‐1. In fact, targeted analysis of c‐Kit activation showed that TIMP‐1 significantly increased the phosphorylation of c‐Kit docking sites for GRB2 and P85 (pY703 and pY719) in DLD‐1 wt. GRB2 has a major role in being the adapter protein linking the scaffolding protein GAB2 to c‐Kit. Activation of c‐Kit leads to phosphorylation of GAB2 and the recruitment of SHP2, which leads to RAS/ERK pathway activation, as well as p85, which in turn leads to PI3‐K pathway activation (Sun et al., 2008). In addition, the increased phosphorylation of c‐Kit observed in DLD‐1 G13D suggests that TIMP‐1 causes c‐Kit phosphorylation independently of KRAS mutational status, though the levels of phosphorylated c‐Kit still differed, presumably due to the difference in total c‐Kit levels. These results showed that TIMP‐1 increased phosphorylation, and therefore c‐Kit activity, as well as increased the activity of c‐Kit canonical targets (Fig. 4). Activation of c‐Kit by TIMP‐1 was not a cell‐line‐specific event, as we observed the same effect in Ba/F3 pro‐B murine cells (Fig. 5A). These observations raise two major questions, how does TIMP‐1 cause activation of c‐Kit and is there a biological significance for this activation?
Addressing the first question, protein interaction analysis, be it co‐immunoprecipitation or pull‐down assays, failed to show any physical interaction between TIMP‐1 and c‐Kit (Fig. S1A). We did detect a number of interactions using an in situ assay (Fig. S1B), but the number of interaction was very low compared to the presence of c‐Kit at the membrane (Fig. 1D) and the availability of TIMP‐1 (Fig. 2C). With the caveat of a very transient interaction between TIMP‐1 and c‐Kit, our data were suggestive of physical proximity rather than a direct interaction between these two proteins. TIMP‐1 interacts with a CD63/β1‐integrin receptor at the cell membrane (Jung et al., 2006); c‐Kit is also associated with the tetraspanin CD63 (Anzai et al., 2002). Tetraspanin‐enriched microdomains (TEM) are functional platforms, capable of regulating multiple cellular processes, including ectodomain ‘shedding’ and signaling (Termini and Gillette, 2017; Yanez‐Mo et al., 2011). We could, in fact, detect a significant effect of TIMP‐1 on shedding of c‐Kit (Fig. 4D), supporting the idea that proximity of TIMP‐1 and c‐Kit mediated by association within a TEM may be responsible for the activation of c‐Kit we observed.
Concerning the second question, on whether there is a biological significance for the TIMP‐1‐mediated activation of c‐Kit, we investigated the ability of TIMP‐1 to stimulate migration and proliferation of DLD‐1 wt cells, using an in vitro wound healing assay (Fig. 5B). We found that wound closure in DLD‐1 wt cells stimulated with TIMP‐1 was complete (100% confluency in the wound area) whereas in DLD‐1 wt cells, where no TIMP‐1 was added or where c‐Kit signaling was inhibited wound closure was delayed (~ 75% confluency in the wound area). These data suggest a TIMP‐1‐mediated effect on cell migration and proliferation in c‐Kit‐expressing CRC cells. These data are congruent with results from a recent study that showed that a subpopulation of endogenous cardiac stem cells, which express c‐Kit and CD63 (CD63+ve/c‐Kit+ve), was driven to proliferate, express cardiogenic factors, and differentiate into major cardiac cell types upon stimulation with TIMP‐1 (Abdelli and Singla, 2015).
5. Conclusions
In conclusion, we identified c‐Kit as a potential mediator of TIMP‐1 function in CRC cells and showed that TIMP‐1 can mediate activation of c‐Kit and prevent ectodomain shedding. Given the growing attention that c‐Kit has been gaining as an important oncogene in CRC (Attoub et al., 2002; Zhang et al., 2014) and the role it plays in regulation of cell proliferation and migration, it is likely that some of the biological effects of TIMP‐1 overexpression in CRC may be exerted through its effect on c‐Kit signaling. However, the KRAS‐associated effect of TIMP‐1 on CRC cell invasion cannot be explained by its potentiation of c‐Kit signaling alone, as this took place independently of RAS status. We did find KRAS status‐associated differences between the two cell lines both in terms of association with TIMP‐1 where levels of bound TIMP‐1 protein were significantly higher in DLD‐1 G13D than in DLD‐1 wt (Fig. 2A), and in terms of expression of genes affected by TIMP‐1 where we found that only seven genes were significantly changed by TIMP‐1 exposure in DLD‐1 wt cells (Table 2), whereas 1183 genes were affected in DLD‐1 G13D cells (Table 3). Five genes in particular, which were significantly up‐regulated upon TIMP‐1 stimulation in DLD‐1 G13D cells but showed no change in DLD‐1 wt cells, were interesting as they all mapped to the Ras‐Raf‐MEK‐Erk/JNK signaling axis (Table 4). It is conceivable that TIMP‐1 may cause increased activity in the Ras signaling axis in cells expressing oncogenic Ras, resulting in persistent enhanced signaling through this pathway, which would explain the worse outcome we observed in CRC patients in association with TIMP‐1 expression (Tarpgaard et al., 2016), but further studies will be needed to clarify these observations.
Conflict of interest
The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data as well as in writing of the manuscript. The authors declare no conflict of interest.
Author contributions
SF, JS, LR, MM, and JMAM: Study design and conceptualization; CN, SD, ALSAM, MH, and JUK: Methodology and data acquisition; CN, SD, ALSAM, MH, JUK, and SF: Data analysis and interpretation; CN, SD, JS, LR, MM, and JMAM: Writing and editing the manuscript.
Supporting information
Fig. S1. Interaction analysis of TIMP‐1 and c‐Kit. (A) DLD‐1 wt and DLD‐1 G13D cells were transfected with GFP or GFP‐TIMP‐1 24 h prior to lysis. Immunoprecipitation of lysates was performed with GFP‐TRAPs. TIMP‐1 and c‐Kit were examined in samples of untransfected cells (Ctrl), input (IN), unbound (UB), and immunoprecipitated fraction (IP) using western blot analysis. (B) DLD‐1 wt and DLD‐1 G13D cells were exposed to 5 µg·mL−1 TIMP‐1 or vehicle for 30 min prior to lysis. Co‐IP of lysates was performed with protein G sepharose and either mouse anti‐IgG control or VT4 (anti‐TIMP‐1 antibody). TIMP‐1 and c‐Kit were examined in input, unbound and immunoprecipitated (Bound) fraction using western blot analysis. P150Glued was examined to ensure true immunoprecipitation. The immunoprecipitation of TIMP‐1 was successful, however, c‐Kit was not detected in the immunoprecipitated fraction. (C) PLA of c‐Kit and TIMP‐1. DLD‐1 wt (a) and DLD‐1 G13D (b) cells were stimulated with 5 μg·mL−1 TIMP‐1 for 30 min prior to fixing and embedding. Embedded cell sections were examined for TIMP‐1 and c‐Kit proximity, detected as red dots by PLA assay, and cell nucleus were stained (DAPI, blue). (Representative pictures are shown, scale bars 20 μm).
{kind=link}
Table S1. Antibodies used in this study.
Data S1. Gene expression data.
Data S2. Mass spectrometry proteomics data.
Acknowledgements
The authors would like to thank Annette Bartels and Signe Lykke Nielsen for expert technical assistance. The authors would also like to thank PamGene, in particular Piet Boender, for excellent support and providing early access to their technological platform. The study was supported by grants from The Kathrine og Vigo Skovgaards Foundation, Agnes og Poul Friis Foundation, and Sawmill Owner Jeppe Juhl and Wife Ovita Juhl Memorial Foundation.
References
- Abdelli LS and Singla DK (2015) A CD63(+ve)/c‐kit(+ve) stem cell population isolated from the mouse heart. Mol Cell Biochem 406, 101–109. [DOI] [PubMed] [Google Scholar]
- Akahane T, Akahane M, Shah A and Thorgeirsson UP (2004) TIMP‐1 stimulates proliferation of human aortic smooth muscle cells and Ras effector pathways. Biochem Biophys Res Commun 324, 440–445. [DOI] [PubMed] [Google Scholar]
- Ando T, Charindra D, Shrestha M, Umehara H, Ogawa I, Miyauchi M and Takata T (2018) Tissue inhibitor of metalloproteinase‐1 promotes cell proliferation through YAP/TAZ activation in cancer. Oncogene 37, 263–270. [DOI] [PubMed] [Google Scholar]
- Anzai N, Lee Y, Youn BS, Fukuda S, Kim YJ, Mantel C, Akashi M and Broxmeyer HE (2002) C‐kit associated with the transmembrane 4 superfamily proteins constitutes a functionally distinct subunit in human hematopoietic progenitors. Blood 99, 4413–4421. [DOI] [PubMed] [Google Scholar]
- Attoub S, Rivat C, Rodrigues S, Van Bocxlaer S, Bedin M, Bruyneel E, Louvet C, Kornprobst M, Andre T, Mareel M et al (2002) The c‐kit tyrosine kinase inhibitor STI571 for colorectal cancer therapy. Cancer Res 62, 4879–4883. [PubMed] [Google Scholar]
- Benjamini Y and Hochberg J (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57, 289–300. [Google Scholar]
- Birgisson H, Nielsen HJ, Christensen IJ, Glimelius B and Brunner N (2010) Preoperative plasma TIMP‐1 is an independent prognostic indicator in patients with primary colorectal cancer: a prospective validation study. Eur J Cancer 46, 3323–3331. [DOI] [PubMed] [Google Scholar]
- Botta GP, Reichert M, Reginato MJ, Heeg S, Rustgi AK and Lelkes PI (2013) ERK2‐regulated TIMP1 induces hyperproliferation of K‐Ras(G12D)‐transformed pancreatic ductal cells. Neoplasia 15, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler L, Golde DW, Bersch N and Johnson MD (1995) Metalloproteinase inhibition and erythroid potentiation are independent activities of tissue inhibitor of metalloproteinases‐1. Blood 86, 4506–4515. [PubMed] [Google Scholar]
- Chirco R, Liu XW, Jung KK and Kim HR (2006) Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev 25, 99–113. [DOI] [PubMed] [Google Scholar]
- Cruz AC, Frank BT, Edwards ST, Dazin PF, Peschon JJ and Fang KC (2004) Tumor necrosis factor‐alpha‐converting enzyme controls surface expression of c‐Kit and survival of embryonic stem cell‐derived mast cells. J Biol Chem 279, 5612–5620. [DOI] [PubMed] [Google Scholar]
- Gasson JC, Golde DW, Kaufman SE, Westbrook CA, Hewick RM, Kaufman RJ, Wong GG, Temple PA, Leary AC, Brown EL et al (1985) Molecular characterization and expression of the gene encoding human erythroid‐potentiating activity. Nature 315, 768–771. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Blanke CD, Druker BJ and Corless CL (2002) Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT‐positive malignancies. J Clin Oncol 20, 1692–1703. [DOI] [PubMed] [Google Scholar]
- Herszenyi L, Hritz I, Lakatos G, Varga MZ and Tulassay Z (2012) The behavior of matrix metalloproteinases and their inhibitors in colorectal cancer. Int J Mol Sci 13, 13240–13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornebeck W, Lambert E, Petitfrere E and Bernard P (2005) Beneficial and detrimental influences of tissue inhibitor of metalloproteinase‐1 (TIMP‐1) in tumor progression. Biochimie 87, 377–383. [DOI] [PubMed] [Google Scholar]
- Jung KK, Liu XW, Chirco R, Fridman R and Kim HR (2006) Identification of CD63 as a tissue inhibitor of metalloproteinase‐1 interacting cell surface protein. EMBO J 25, 3934–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelstrup CD, Jersie-Christensen RR, Batth TS, Arrey TN, Kuehn A, Kellmann M and Olsen JV (2014) Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field orbitrap mass spectrometer. J Proteome Res 13, 6187–6195. 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N and Mann M (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods 11, 319–324. [DOI] [PubMed] [Google Scholar]
- Kuvaja P, Hulkkonen S, Pasanen I, Soini Y, Lehtonen S, Talvensaari‐Mattila A, Paakko P, Kaakinen M, Autio‐Harmainen H, Hurskainen T et al (2012) Tumor tissue inhibitor of metalloproteinases‐1 (TIMP‐1) in hormone‐independent breast cancer might originate in stromal cells, and improves stratification of prognosis together with nodal status. Exp Cell Res 318, 1094–1103. [DOI] [PubMed] [Google Scholar]
- Lambert E, Bridoux L, Devy J, Dasse E, Sowa ML, Duca L, Hornebeck W, Martiny L and Petitfrere‐Charpentier E (2009) TIMP‐1 binding to proMMP‐9/CD44 complex localized at the cell surface promotes erythroid cell survival. Int J Biochem Cell Biol 41, 1102–1115. [DOI] [PubMed] [Google Scholar]
- Lennartsson J, Jelacic T, Linnekin D and Shivakrupa R (2005) Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells 23, 16–43. [DOI] [PubMed] [Google Scholar]
- Lennartsson J and Rönnstrand L (2012) Stem cell factor receptor/c‐Kit: from basic science to clinical implications. Physiol Rev 92, 1619–1649. [DOI] [PubMed] [Google Scholar]
- Lu XQ, Levy M, Weinstein IB and Santella RM (1991) Immunological quantitation of levels of tissue inhibitor of metalloproteinase‐1 in human colon cancer. Cancer Res 51, 6231–6235. [PubMed] [Google Scholar]
- Lu Y, Liu S, Zhang S, Cai G, Jiang H, Su H, Li X, Hong Q, Zhang X and Chen X (2011) Tissue inhibitor of metalloproteinase‐1 promotes NIH3T3 fibroblast proliferation by activating p‐Akt and cell cycle progression. Mol Cells 31, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen HJ, Christensen IJ and Brunner N (2010) A novel prognostic index in colorectal cancer defined by serum carcinoembryonic antigen and plasma tissue inhibitor of metalloproteinases‐1. Scand J Gastroenterol 45, 200–207. [DOI] [PubMed] [Google Scholar]
- Palacios R and Steinmetz M (1985) Il‐3‐dependent mouse clones that express B‐220 surface antigen, contain Ig genes in germ‐line configuration, and generate B lymphocytes in vivo . Cell 41, 727–734. [DOI] [PubMed] [Google Scholar]
- Pedersen M, Ronnstrand L and Sun J (2009) The c‐Kit/D816V mutation eliminates the differences in signal transduction and biological responses between two isoforms of c‐Kit. Cell Signal 21, 413–418. [DOI] [PubMed] [Google Scholar]
- Ritter LM, Garfield SH and Thorgeirsson UP (1999) Tissue inhibitor of metalloproteinases‐1 (TIMP‐1) binds to the cell surface and translocates to the nucleus of human MCF‐7 breast carcinoma cells. Biochem Biophys Res Commun 257, 494–499. [DOI] [PubMed] [Google Scholar]
- Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A and Mann M (2014) The Q exactive HF, a benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field orbitrap analyzer. Mol Cell Proteomics 13, 3698–3708. 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler‐Stevenson WG (2008) Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase‐independent biological activities. Sci Signal 1, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pedersen M and Ronnstrand L (2008) Gab2 is involved in differential phosphoinositide 3‐kinase signaling by two splice forms of c‐Kit. J Biol Chem 283, 27444–27451. [DOI] [PubMed] [Google Scholar]
- Sun Y, Ji B, Feng Y, Zhang Y, Ji D, Zhu C, Wang S, Zhang C, Zhang D and Sun Y (2017) TRIM59 facilitates the proliferation of colorectal cancer and promotes metastasis via the PI3K/AKT pathway. Oncol Rep 38, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpgaard LS, Orum‐Madsen MS, Christensen IJ, Nordgaard C, Noer J, Guren TK, Glimelius B, Sorbye H, Ikdahl T, Kure EH et al (2016) TIMP‐1 is under regulation of the EGF signaling axis and promotes an aggressive phenotype in KRAS‐mutated colorectal cancer cells: a potential novel approach to the treatment of metastatic colorectal cancer. Oncotarget 7, 59441–59457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termini CM and Gillette JM (2017) Tetraspanins function as regulators of cellular signaling. Front Cell Dev Biol 5, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toricelli M, Melo FH, Peres GB, Silva DC and Jasiulionis MG (2013) Timp1 interacts with beta‐1 integrin and CD63 along melanoma genesis and confers anoikis resistance by activating PI3‐K signaling pathway independently of Akt phosphorylation. Mol Cancer 12, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M and Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Vinther L, Lademann U, Andersen EV, Hojrup P, Thaysen‐Andersen M, Krogh BO, Viuff B, Brunner N, Stenvang J and Moreira JM (2014) Purification and characterization of bioactive his6‐tagged recombinant human tissue inhibitor of metalloproteinases‐1 (TIMP‐1) protein expressed at high yields in mammalian cells. Protein Expr Purif 101, 157–164. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del‐Toro, N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yamashita K, Iwata K and Hayakawa T (2002) Both tissue inhibitors of metalloproteinases‐1 (TIMP‐1) and TIMP‐2 activate Ras but through different pathways. Biochem Biophys Res Commun 296, 201–205. [DOI] [PubMed] [Google Scholar]
- Wilk CM, Schildberg FA, Lauterbach MA, Cadeddu RP, Frobel J, Westphal V, Tolba RH, Hell SW, Czibere A, Bruns I et al (2013) The tissue inhibitor of metalloproteinases‐1 improves migration and adhesion of hematopoietic stem and progenitor cells. Exp Hematol 41, 823–831.e2. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Suzuki M, Iwata H, Koike T, Hamaguchi M, Shinagawa A, Noguchi T and Hayakawa T (1996) Tyrosine phosphorylation is crucial for growth signaling by tissue inhibitors of metalloproteinases (TIMP‐1 and TIMP‐2). FEBS Lett 396, 103–107. [DOI] [PubMed] [Google Scholar]
- Yanez‐Mo M, Gutierrez‐Lopez MD and Cabanas C (2011) Functional interplay between tetraspanins and proteases. Cell Mol Life Sci 68, 3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, Kuang WJ, Yang‐Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U and Ullrich A (1987) Human proto‐oncogene c‐kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 6, 3341–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee NS, Hsiau CW, Serve H, Vosseller K and Besmer P (1994) Mechanism of down‐regulation of c‐kit receptor. Roles of receptor tyrosine kinase, phosphatidylinositol 3'‐kinase, and protein kinase C. J Biol Chem 269, 31991–31998. [PubMed] [Google Scholar]
- Yee NS, Langen H and Besmer P (1993) Mechanism of kit ligand, phorbol ester, and calcium‐induced down‐regulation of c‐kit receptors in mast cells. J Biol Chem 268, 14189–14201. [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S et al (2009) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S et al. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Interaction analysis of TIMP‐1 and c‐Kit. (A) DLD‐1 wt and DLD‐1 G13D cells were transfected with GFP or GFP‐TIMP‐1 24 h prior to lysis. Immunoprecipitation of lysates was performed with GFP‐TRAPs. TIMP‐1 and c‐Kit were examined in samples of untransfected cells (Ctrl), input (IN), unbound (UB), and immunoprecipitated fraction (IP) using western blot analysis. (B) DLD‐1 wt and DLD‐1 G13D cells were exposed to 5 µg·mL−1 TIMP‐1 or vehicle for 30 min prior to lysis. Co‐IP of lysates was performed with protein G sepharose and either mouse anti‐IgG control or VT4 (anti‐TIMP‐1 antibody). TIMP‐1 and c‐Kit were examined in input, unbound and immunoprecipitated (Bound) fraction using western blot analysis. P150Glued was examined to ensure true immunoprecipitation. The immunoprecipitation of TIMP‐1 was successful, however, c‐Kit was not detected in the immunoprecipitated fraction. (C) PLA of c‐Kit and TIMP‐1. DLD‐1 wt (a) and DLD‐1 G13D (b) cells were stimulated with 5 μg·mL−1 TIMP‐1 for 30 min prior to fixing and embedding. Embedded cell sections were examined for TIMP‐1 and c‐Kit proximity, detected as red dots by PLA assay, and cell nucleus were stained (DAPI, blue). (Representative pictures are shown, scale bars 20 μm).
Table S1. Antibodies used in this study.
Data S1. Gene expression data.
Data S2. Mass spectrometry proteomics data.