

Abstract
Neoplastic cellularity contributes to the analytic sensitivity of most present technologies for mutation detection, such that they underperform when stroma and inflammatory cells dilute a cancer specimen’s variant fraction. Thus, tumor purity assessment by light microscopy is used to determine sample adequacy before sequencing and to interpret the significance of negative results and mutant allele fraction afterwards. However, pathologist estimates of tumor purity are imprecise and have limited reproducibility. With the advent of massively parallel sequencing, large amounts of molecular data can be analyzed by computational purity algorithms. We retrospectively compared tumor purity of 3 computational algorithms with neoplastic cellularity using hematoxylin and eosin light microscopy to determine which was best for clinical evaluation of molecular profiling. Data were analyzed from 881 cancer patients from a clinical trial cohort, LCCC1108 (UNCseq), whose tumors had targeted massively parallel sequencing. Concordance among algorithms was poor, and the specimens analyzed had high rates of algorithm failure partially due to variable tumor purity. Computational tumor purity estimates did not add value beyond the pathologist’s estimate of neoplastic cellularity microscopy. To improve present methods, we propose a semiquantitative, clinically applicable strategy based on mutant allele fraction and copy number changes present within a given specimen, which when combined with the morphologic tumor purity estimate, guide the interpretation of next-generation sequencing results in cancer patients.
Key Words: next-generation sequencing, light microscopy, computer tumor purity analysis
Traditional molecular testing methods have relatively high thresholds for limits of detection. Sanger sequencing can detect >20% variant allele fraction (VAF) or 40% neoplastic cellularity, while pyrosequencing detects >5% VAF or 10% neoplastic cellularity.1 It is a standard practice to estimate neoplastic cellularity using hematoxylin and eosin (H&E) light microscopy estimates before testing to ensure that the minimum limit of detection can be met for a certain test method and if necessary, to perform macrodissection to enrich the tumor content of the analyzed tissue. However, it has previously been shown that there is limited reproducibility of estimating tumor cell content by microscopy among pathologists.2–5
Targeted testing for single mutations with methods such as quantitative or allele-specific polymerase chain reaction can achieve analytic sensitivity for detecting mutations to <1%.6 With massively parallel sequencing (MPS), multiple targets may be interrogated to theoretical sensitivities of <5% or less, with a concurrent increase in detection of subclonal populations.7
The demand for clinical testing of small tissue samples and needle biopsies is increasing. Such samples may not be amenable to microdissection. If these samples are held to traditional levels of minimum neoplastic cellularity, they might be rejected even though mutations could be detected by newer molecular techniques. For example, samples taken from pancreatic adenocarcinomas usually have a low neoplastic cellularity.8 In another situation, samples from small volume biopsies may go directly for molecular assays without the possibility of light microscopy review. However, determination of whether a negative result is a true negative, rather than an artifact of low tumor input may be difficult to determine without a frame of reference. Therefore, it is imperative to be able to distinguish false-negative from true-negative results in patients. While the concept of informatically assessed “tumor purity” has attempted to address this issue, such algorithms have not been widely applied to clinical data sets. For example, most methods have been evaluated in the large data sets of The Cancer Genome Atlas that excluded >50% of all samples submitted based on tumor cellularity <60%.9 These algorithms10–12 aim to estimate the amount of neoplastic cells in a sample relative to nontumor cells such as stroma and inflammatory cells. However, unlike the traditional purity estimate of light microscopy, such algorithms utilize the genetic information contained within a tumor. We compared the application of computational tumor purity estimates to neoplastic cellularity estimates performed by light microscopy to determine their applicability in clinical analysis of tumor sequencing and to provide a framework for analysis of these complex data sets in determining whether a sample submitted for sequencing was appropriate for testing. As a complement, we also propose a semiquantitative, clinically applicable strategy based on mutant allele fraction (MAF) and copy number changes present within a given specimen, which when combined with the morphologic tumor purity estimate, help guide the interpretation of next-generation sequencing (NGS) results in cancer patients.
MATERIALS AND METHODS
Cohort and Trial (1108)
Subjects for the current analysis were consented between 18 November 2011 and 6 August 2014 on the clinical trial LCCC1108: Development of a Tumor Molecular Analyses Program and Its Use to Support Treatment Decisions (NCT01457196), which has been previously described.13 Briefly, inclusion criteria for the study were broad such that most patients treated for cancer at the University of North Carolina (UNC) were eligible for inclusion. Subjects were either referred by their treating physicians in settings where the physician felt the sequencing might be potentially beneficial or alternatively patients were consented as part of an ongoing cancer registry trial. Tumor tissue and matched normal controls were acquired for all study subjects. In the first phase of the study ∼400 patients whose tumors were available as fresh frozen material after banking on a research protocol were enrolled. As the sequencing technology allowed, the protocol transitioned nearly uniformly to rely on surplus paraffin-embedded tumor material. Normal control DNA was largely harvested in the form of lymphocytes from peripheral blood, although in isolated cases cells from a buccal swab were collected. Tumor and normal samples were sequenced using protocols that depended on Agilent Sure Select Custom Targeted panel of ∼250 genes and the Illumina HiSeq platform in either single end or paired end formats of 75 and 100 bases per end. Average depth of coverage for target sequences was between 500 and 1000 reads. Raw sequencing images were converted to FASTQ file format using Illumina CASAVA v1.8.2 software (http://support.illumina.com/sequencing/sequencing_software/casava.html). The sequence reads were aligned to the genome using the bwa mem algorithm (v0.7.4) with the default parameters. Realignment was performed simultaneously for tumor and normal pairs using ABRA (v0.46) with the default parameters. Variants were called using FreeBayes and somatic mutations using Strelka. Copy number assessments were performed using custom software. Quality statistics were generated with Picard and include measures of fragment length, sequence content, alignment, capture bias and efficiency, coverage, and variant call metrics. Final somatic calls were filtered to require a Strelka assigned quality of at least 30. All variants were annotated in their gene specific context using SnpEff (v3.1) and crossed with COSMIC and internal databases that capture the information from the UNC Clinical Committee for Genomic Research.13 The subset of patients with frozen material available were subjected to analysis using the Genome-Wide Human SNP Array 6.0 microarray (Affymetrix, Santa Clara, CA) for detecting copy number variation (CNV) using methods that have been previously described.14
Computational Tumor Purity Analysis
For the purposes of assessing tumor purity a variety of previously published tools were applied to the sequencing data and single nucleotide polymorphism (SNP) arrays. To identify representative and widely adopted approaches, the literature was reviewed in May 2014 by searching PubMed for algorithms associated with tumor purity, MPS, and SNP chip/copy number data. On the basis of the results of this search we identified multiple algorithms including ABSOLUTE,10 ASCAT,11 ExPANdS,15 PurityEst,16 PurBayes,17 and THetA2.12 In addition, we attempted to determine if robust comparisons of these algorithms suggested any which would lead us to select it as the standard method. Meta-analysis of these methods was lacking to justify selection of one as superior to the others. While objective measures are lacking to select the optimal algorithms, we determined that the following approaches were representative of the field. The ABSOLUTE algorithm was selected because it is applicable to both SNP chip data and NGS data and has been used in large cancer cohort studies such as The Cancer Genome Atlas.18 ABSOLUTE therefore allowed for analysis of the relative ability to assess tumor purity by different molecular assays (SNP chip and NGS) but using the same computational framework and same DNA isolation for a subset of samples. In order to assess the impact of a competing computational approach on SNP chip tumor purity estimates we applied the allele-specific copy number analysis of tumors (ASCAT) algorithm to the SNP chip data set.11 In order to assess the impact of a competing computational approach on NGS tumor purity estimates we applied the THetA2 algorithm to the NGS data set.12 In order to ensure that each algorithm was applied in the manner most consistent with optimal performance, we obtained the original data and software from the manuscripts reporting ABSOLUTE, ASCAT, and THetA2 and repeated the original reports as published by the authors (results not shown). We then applied the software to our novel data set. All the SNP and sequencing data were preprocessed to be suitable to run each computational algorithm. For SNP chips, we used Affymetrix Power Tools (v1.15.1) and HAPSEG (v1.1.1) to run ABSOLUTE (v1.0.6), used PennCNV to run ASCAT (v2.3). For sequencing data, we used an internal UNC pipeline to run ABSOLUTE and used ExomeCNV to run THeTA2 (v0.62). Subsequently, all algorithms were run in a default setting and parameters. After application, ABSOLUTE and ASCAT are intended to return a “failed” result in a fraction of samples they run as described in the publications for samples that fail to conform to the model parameters. The THetA2 algorithm does not issue a “fail” call but does report a warning flag which for the purposes of this study were considered the same as a “fail.”
Light Microscopy
A paraffin section immediately adjacent to that from the block providing the tumor sample was obtained for each patient. In cases where the tumor was obtained from frozen material, the tumor was bisected before freezing and divided such that a paraffin block immediately adjacent to the tumor could be generated. This block was sectioned and served as the source of morphologic review, including percent tumor nuclei. In cases where paraffin material was used as the source of DNA, DNA was obtained from sections immediately adjacent from one which provided the source of morphologic review of the tumor. For all subjects in the study, a 5-µm section was obtained and prepared in the usual manner to generate an H&E stain for review by light microscopy. Each slide was given a tumor purity by a single pathologist reported as the percentage of viable malignant nuclei in respect to total nuclei. Factors such as necrosis and immune infiltration were not graded independently. For the purposes of assessing interobserver variability for light microscopy estimates of tumor purity a random subset of 100 cases was selected for review by a second pathologist by the same criteria.
Statistical Analysis
All statistical analysis was performed using R 3.0.1 software (http://cran.r-project.org) unless otherwise stated. Pearson correlation coefficient (R) was used to assess linear relationship on tumor purity estimate among data type, different algorithms, and light microscopy. Root mean square deviation (RMSD) was used to measure the difference between the estimates mentioned above.
RESULTS
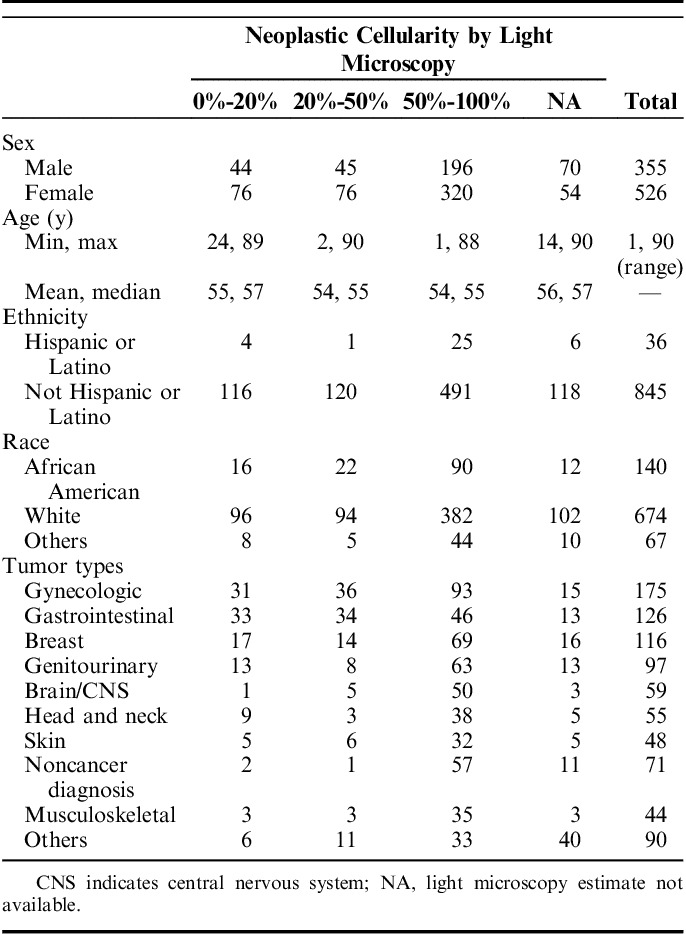
Patients included in the cohort represented a broad cross-section of cancer histologies and fractional tumor nuclei (Table 1). The mean tumor purity across all tumors was 57% (SD±27). Fourteen percent of patients fell into the <20% tumor category that would generally have been excluded from molecular characterization by standard methods. An additional 14% contained <50% tumor and would generally also be excluded from large cohort studies of molecular characterization. These data suggest that in clinical practice a significant proportion of patients (28% in our series) have tumor cellularity below the level by which computational algorithms would have been investigated in prior studies, a level generally set at 50% to 70%. The remaining 72% of samples would have been more consistent with tumors characterized in most reported research data sets such as The Cancer Genome Atlas.9,18 In addition, we note that our series included tumors of many different organs with gynecologic tumors being the most common (20%), followed by gastrointestinal (14%), and breast cancers (13%). As expected, percent tumor varied across the tumor types. The tumors represented in our cohort were a function of patients referred to the study, which we noted had a lower fraction of lung cancers and malignant hematology specimens. We suspect this was due to the fact that these patients had molecular profiling done as standard of care and were therefore less likely to be referred to the trial.
TABLE 1.
Patient and Tumor Characteristics Subdivided by Light Microscopy Neoplastic Cellularity Estimates
We analyzed the concordance of different estimates of tumor purity based on 3 data sets (pathologist review of light microscopy, SNP chip copy number data, and NGS sequencing data) and 3 computational methods [ABSOLUTE (SNP and NGS), THetA2 (NGS only), and ASCAT (SNP only)]. This produced 8 estimates of concordance using the RMSD methodology, such as the concordance between SNP and NGS using ABSOLUTE, SNP Absolute and SNP ASCAT, and SNP LM (Table 2). As SNP chips and secondary pathology reviewer were only obtained for a subset of cases, the last column of Table 2 provides the absolute number of samples available for the specified comparison.
TABLE 2.
Concordance of Purity Estimates by Computational Methods and Pathologist Interpretations of Hematoxylin and Eosin Light Microscopy

The first striking observation was the high failure rate of the computational methods, especially at the lower range of tumor cellularity. The failure rates for each algorithm can be seen in comparison to light microscopy (data rows 4 to 7 in Table 2) as all failures in those comparisons are attributable to the computational methodology. Overall, of the 3 algorithms, ABSOLUTE (SNP/SEQ) and ASCAT had similar failure rates (18%/25% and 21%, respectively). The overall THETA failure rate was 34% for samples. On the lower end of tumor purity, failure rates reach up to 65% using ASCAT. Even in tumors of high purity, the failure rate never fell below 15%. It is clearly an advantage of light microscopy that pathologists were able to assign sample purity to every case.
We then assessed concordance for each of the 8 possible comparisons using RMSD both overall and as a function of tumor cellularity. RMSD was most favorable in the low purity samples assessed by repeat light microscopy by independent pathologists at 0.12 (highest agreement). The lowest agreement was in low purity samples comparing ASCAT to light microscopy, 0.73. As expected, RMSD increased with decreasing purity samples in each individual computational algorithm as compared with light microscopy. Interobserver light microscopy was the most concordant methodology in every comparison. When comparing RMSD of SNP methods (SNP ABS vs. SNP ASCAT) across the cellularity range, no pattern was clearly seen. Interestingly, when comparing RMSD of sequencing methods (SEQ ABS vs. SEQ THetA2) across the cellularity range, the RMSD did not vary. In the comparison of SNP versus sequencing using ABSOLUTE, there appeared to be an improvement in RMSD with higher purity samples.
We conclude that existing computational methods have failure rates that render them difficult to rely upon in a clinical setting even at high purity samples. Likewise, when compared with light microscopy or to other computational methods, the overall high RSMD for all molecular estimates did not support these as robust estimates of tumor purity in clinical practice and failed to suggest parameters for the settings in which they would be most useful. RSMD was high in both low and high cellularity cases and failures were common. We therefore considered alternative approaches, including semiquantitative and integrated approaches based on our experience in reviewing the first 880 cases (Table 3).
TABLE 3.
Integrated Interpretation of Neoplastic Cellularity and Sequencing Results (IINCaSe) Guidelines

The Integrated Interpretation of Neoplastic Cellularity and Sequencing Results (IINCaSe) Approach
We propose an empiric and semiquantitative framework—IINCaSe (Integrated Interpretation of Neoplastic Cellularity and Sequencing Results) for tumor purity that incorporates expected relationships that may be seen during review of copy number and minor allele frequencies in individual samples, and incorporates the expectation of variants commonly associated with the tumor under review. These patterns are classifiable based upon assumptions about the expected patterns of mutation in a gene based upon its mechanism of action in tumor biology. For example, in oncogenes such as KRAS, a known activating mutation on a single allele (heterozygous mutation) is enough to support tumor growth, whereas tumor suppressor genes such as APC may attain either a heterozygous or homozygous loss of function state, either through mechanisms of loss of heterozygosity or compound heterozygous mutations or epigenetic silencing.
When analyzing an individual sample, VAF provide support for characterizing the relationships of mutations in a sample. The majority of samples demonstrated the presence of groupings when VAF was plotted against mutation density (Figs. 1A, B). The VAF distributions usually fell into a primary group characterized by mutations in oncogenes such as IDH1 and MET, which are expected to occur as heterozygous mutation. When a canonical mutation in an oncogene (like IDH1 R132H or MET M1268T) occurs, it is assumed to be heterozygous and a likely driver mutation, present in all neoplastic cells. Heterozygous mutation VAF will therefore equal approximately half of the tumor purity (tumor purity=heterozygous mutation allele frequency×2, Fig. 2A).
FIGURE 1.
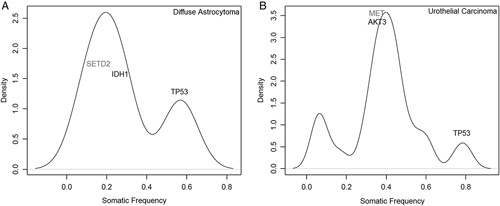
Sample variant allele frequency graphs demonstrating density of mutations occurring at specified variant allele frequency, with genes of therapeutic or biological significance noted in 2 individuals tumors (A, B). Mutation densities were calculated using a kernel density estimate from R package with default parameters. Genes listed in black have protein altering mutations and genes listed in gray have nonprotein altering mutations, such as intronic, 3′UTR, and synonymous changes.
FIGURE 2.
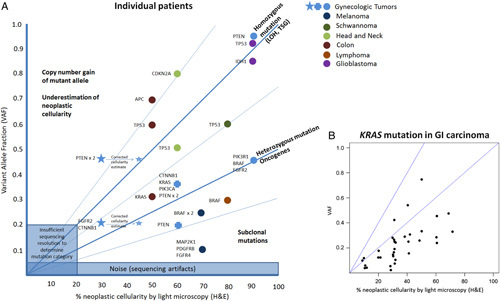
Variant allele fraction versus neoplastic cellularity. A, Individual samples plotted by percent neoplastic cellularity and variant allele frequency of mutations in specified genes. Reported mutant allele fraction from sequencing data may be used to adjust the percent neoplastic cellularity, as shown by the gynecologic tumor represented by a blue star. B, Grouped sample analysis for all KRAS mutations in gastrointestinal carcinomas. GI indicates gastrointestinal; H&E, hematoxylin and eosin; LOH, loss of heterozygosity; TSG, tumor suppressor gene.
Two common additional groupings of mutations may also be identifiable. The first occurs at a VAF approximately twice that of the heterozygous group, and is characterized by mutation in tumor suppressor genes such as TP53 (Figs. 1A, B). These types of mutations are expected to occur as part of a “double-hit” resulting in complete inactivation of protein product in tumor cells by mutations in both alleles or more commonly by deletion of one allele and mutation of the other. Of interest when analyzing these 2 tumor suppressor genes is the observation that PTEN mutations more commonly occurred in a heterozygous state, while TP53 mutations occurred at both heterozygous and homozygous frequencies in tumors. This difference in mutation patterns is supported by the known presence of gain-of-function and inactivating mutations in TP53.19
The second grouping of mutations is most often seen if a sample has a high tumor burden, where mutations may be seen at VAF less than that of the primary oncogene peak (Fig. 1B). These mutations may represent subclonal populations of neoplastic cells or reductions in allele frequency due to complex genomes. The interpretation of subclonal mutations may be more complex for several reasons.
First we consider the case in which a tumor is homogenous in its clonality, but complex in its genome such as hyperdiploid (>2 copies of each chromosome) tumors and aneuploidy tumors. In such tumors, mutations may occur after genome doubling such that heterozygous mutations occur in <50% of the alleles. For example, it is not uncommon to see colorectal tumors with 3 APC mutations with one at 50% VAF, and the second and third mutations at 25% each. In this case, the driver APC mutation occurred before genome doubling when the tumor was 2n. After genome doubling the original APC mutation is found on 2 copies of chromosome and 2 independent and additional APC mutations occur on the third and fourth copies of chromosome 5 in the tumor cell containing 46 chromosomes. A hyperdiploid genome can also explain situations such as a PTEN mutation is present at 100% VAF and KRAS mutation is present at 25%. In this case, there may have been an initial mutation of PTEN followed by chromosome 10 loss, then genome doubling and then mutation of 1 of 4 copies of chromosome 12p at the KRAS locus. Subclonal mutations may also occur in regions of more complex copy number alteration such as is often seen for EGFR. Mutant EGFR is often present at many copies per genome above the baseline ploidy of the sample, such that the MAF may approach 100% of all alleles in the sample, deviating from the expectation that oncogenes usually occur in the heterozygous state. Alternatively, the oncogene may occur in a region of copy number gain, but not be present in all of the mutated alleles. Our experience suggests that the gene PIK3CA, which is often mutated in squamous tumors in a region of amplification on chromosome 3q often demonstrates this phenomenon. In such a case, the mutational allele frequency in this oncogene will occur at <50% of the estimate of tumor purity.
The interpretation of subclonal mutations is more complex in the setting of tumor cellular heterogeneity. In samples in which the population of malignant cells is heterogenous with different clones under the influence of different driver mutations, it is more challenging to propose the rules by which we might estimate the tumor purity. Such mutations would not be expected to follow the periodicity described in the sections above. There is little data to inform the estimation of tumor heterogeneity although it might be reasonable to expect that in most cases tumor samples would be comprised of one dominant clone with subclones present at low fractions. In this case, mutation detection in subclones becomes a more daunting task with many heterozygous mutations expected in the low single digits for their fractional component. Unless special molecular techniques are employed (single molecule bar-coding) mutations at allele fractions <<5% are difficult to interpret even if the quality and coverage of the variant allows the automated algorithm to call the variant as somatically mutated. The challenges and tools to overcome them in mutation calling at low allele frequency are beyond the scope of this report. Ultimately clinical judgment must be used in such situations such that some variants with high pretest probability (KRAS codon 12/13 mutations in lung cancer) are likely to be true whereas variants of unknown significance (especially missense) or that are otherwise unexpected should be viewed more skeptically.
Our experience suggests that tumor heterogeneity does not present a common dilemma in clinical interpretation for the most common purpose of driver gene characterization in cancer. Our conclusion is supported by most reports of tumor heterogeneity in which the vast majority of subclonal mutations are “passenger” noncoding or low impact mutations. By contrast, in heterogenous tumors most driver mutations will be present in all clones. Again, expert knowledge should guide interpretation of exceptions in cases such as patients pretreated with therapies known to induce selective pressure. Specific clinical situations are known to result in clinically relevant clonal evolution such as in endocrine therapy for breast and prostate cancer and targeted therapies in lung cancer, gastrointestinal stromal tumors, and chronic myelogenous leukemia. Likewise, the clinician should remain alert of unusual situations such as the melanoma case seen in our trial population, where a patient whose tumor was highly pure in tumor content had a BRAF V600E mutation detected at a low MAF by standard of care targeted sequencing. He progressed upon treatment with a BRAF inhibitor and was later found to have an NF1 mutation at a MAF concordant with his reported tumor purity on our UNCseq assay.
We consider one final category of common single nucleotide variant that the clinician will encounter commonly in this setting. These are variants that appear either at ∼50% or ∼100%. While allele frequencies around 50% or 100% occur commonly as somatic variants, unappreciated germline variants may also appear commonly in this distribution. This is particularly a challenge in cases where tumor is sequenced without matched germline DNA, although for technical reasons matched germline DNA does not completely eliminate the concern. Unappreciated germline variants are often identified by referring to public databases of germline polymorphisms, although there remain some limitations of this approach. The 2 primary concerns are that some pathogenic variants are present in the germline databases at a frequency that approaches common normal variants. Until a catalogue of such pathologic variants is completed, this creates a challenge for automatic exclusion of variants present in germline databases from mutation reports. The second challenge is that at the present time it is estimated that 10% of all single nucleotide variants in the germline of any given patient are not yet annotated in public databases.
As a final note to the complexity of sequence variant interpretation, the rules leveraging VAF to estimate tumor purity do not apply to the variant class called “insertion/deletions” or “indels.” Indel mutations detected in sequencing data are more difficult to align in the computational pipeline that ultimately determines the VAF. As such, the sequencing reads containing indels are more likely to be excluded as “questionable sequencing” compared with reads from the alleles without these mutations. This creates the situation in which the indel read is undercounted relative to its presence in the data, and a falsely low VAF for this class of mutations.
By following the rules described above, with an emphasis on the canonical variants, we are able to demonstrate that the VAF across tumor types tracks very closely with the pathologists’ estimates of percent tumor nuclei. This is true across tumor types and variants (Fig. 2A) and within a single tumor type such as colorectal cancer (Fig. 2B). The individual assumptions about gene VAF biology are supported in grouped sample analysis. For example, when all mutations in KRAS occurring in gastrointestinal carcinomas were plotted against estimated VAF by light microscopy, the majority of mutations trended along a line most consistent with heterozygous presence of KRAS mutation within tumor cells (Fig. 2B). This may be compared with PTEN in gynecologic cancers, where mutations occur in the compound heterozygous and homozygous states, as well as in subclonal populations, as endometrial tumors are known to be heterogenous. Individual case examples are plotted against the expected category of mutation in Figure 2A.
In some cases, however, canonical mutations are absent or confusing in their interpretation. In such cases, we have relied on copy number changes (Fig. 3) to characterize the tumor. When expected mutations are not detected, for example KRAS or APC in a colorectal cancer, the presence of expected copy number changes such as amplification of chromosome 13 offer some confidence that sufficient tumor is present to offer confidence in the negative result. By contrast, in the same situation with no APC mutation and none of the expected copy number changes generates concern that a false-negative is more likely. Certain tumors, such as sarcomas, are characterized by cytogenetic changes with less frequent single nucleotide mutations such that the presence of segmental copy number change by massive parallel sequencing may be sufficient to prove that adequate tumor (usually >30% tumor burden) was sequenced. Again, this allows greater confidence in sequencing results, including reporting that important genes are truly wild type. While segmental (whole chromosome arm) single copy gains and losses are difficult to detect as percent tumor falls much below 30%, focal gene-level amplifications may be detectable even at low percentage of tumor nuclei (>10% tumor burden). When present, genes such as ERRB2 and MYC may appear abnormal in the context of high levels of amplification, with affected cells possessing >20 copies of the gene in question.20 In addition, comparison of expected copy number with VAF can provide information to help resolve unexpected patterns of mutations.
FIGURE 3.

Copy number variation (CNV) changes providing evidence for tumor presence. A, CNV plot demonstrating genomically stable tumor with focal loss of CDKN2a on chromosome 9. B, CNV plot demonstrating genomically unstable tumor with segmental copy number changes and amplification of EGFR on chromosome 7.
DISCUSSION
No computational method for estimating tumor purity emerged as a “gold standard,” especially as algorithms did not produce a discrete estimation of “tumor purity” a significant proportion of the time. While useful for curated research cohorts, we feel this excludes currently available computational purity algorithms from use in decision trees for clinical settings.
The most concordant analyses were those made between the 2 pathologists by light microscopy. This is likely due to the fact that while tumors may appear morphologically homogenous, there is often significant genetic heterogeneity within a single lesion.7 While imperfect, this allows for a level of reproducibility that supports its continued use.
In clinical settings, microscopic estimation of tumor purity is unique in that it is the only method available before sequencing, and may therefore be used to quickly disqualify inappropriate samples for analysis. In addition, clinical samples are formalin-fixed and paraffin-embedded, and must have H&E slides made for clinical purposes. This enables earlier identification of optimal samples for analysis and faster return of patient results to physicians. However, disqualification of samples at pathologist review should be done judiciously as MPS with increased sensitivity now enables mutation detection at allele frequencies approaching 1%. With the high yield of information and relatively low cost of sequencing,20 as well as lowering limits of detection, usable data can be obtained from nearly all samples with tumor present, including those which were previously considered unusable samples for molecular testing (neoplastic cellularity <20%).
When applied in the research setting, the ability to determine whether a sample had sufficient tumor present for sequencing based on sequencing data alone may facilitate faster turnaround of specimens and ultimate cost savings as the need for pathologist review and macrodissection may be reduced. In addition, there may be savings in terms of specimen quantity/purity, as non–paraffin-embedded specimens do not have to be further processed to create slides.
By appropriate combination of the estimates of tumor purity by light microscopy with the VAF, knowledge of the specific gene involved (tumor suppressor vs. oncogenic), the class of mutation [missense variant of unknown significance, missense canonical mutation, silencing mutation (nonsense, most splice site mutations, most indels)], and other expert knowledge, pertinent clinical value can be added to the sequencing report. The utilization of these tools are important in oncology with the added complexity of additional genomic data supplied by NGS. For example, while the biological importance of TP53 has long been known, its clinical relevance in patient care has been largely absent. Recent months, however, have seen an explosion of reports as to the emerging clinical use of TP53 mutation in myelodysplastic syndrome, germ cell tumors, and acute leukemia.21–23 To characterize this tumor suppressor gene we will be increasingly challenged to confidently call both mutations and wild type across a range of tumor purities with a confidence that has been absent before the advent of NGS.
In summary, the neoplastic cellularity estimate by a pathologist may give rough guidance for interpreting clonality as evidenced by VAF within the sample (Table 3). If no mutations are found, interpretation of this result should be done in light of data on tumor purity. In cases having an acceptable but marginally low percent cellularity (<20%), the report should indicate that a negative result does not fully imply a lack of mutation, and state that the specimen may not have contained enough malignant cells (false-negative result). For specimens with higher amount of neoplastic cellularity (>50%), the pathologist can state with a high level of confidence that no mutation was detected in the gene regions encompassed by the assay (true negative result). Correlation of VAFs within the sample with estimated tumor purity light microscopy can be incorporated into interpretations addressing the likelihood that specific mutations are primary or secondary (subclonal) events. This is especially important given documented reports of intratumor heterogeneity for known driver mutations with implications for targeted therapy, such as BRAF V600E in melanoma.24
Footnotes
N.M.P. and H.J. contributed equally.
N.M.P., H.J., D.A.E., J.E.G.-O., M.K.S., and D.N.H.: participated in data analysis, writing, and review of the paper.
The authors declare no conflict of interest.
REFERENCES
- 1.Davidson JD, Todd J, Hunter J, et al. Improving the limit of detection for Sanger sequencing: a comparison of methodologies for KRAS variant detection. Biotechniques. 2012;53:182–188. [DOI] [PubMed] [Google Scholar]
- 2.Viray H, Li K, Long TA, et al. A prospective, multi-institutional diagnostic trial to determine pathologist accuracy in estimation of percentage of malignant cells. Arch Pathol Lab Med. 2013;137:1545–1549. [DOI] [PubMed] [Google Scholar]
- 3.Dudley J, Tseng L-H, Rooper L, et al. Challenges posed to pathologists in the detection of KRAS mutations in colorectal cancers. Arch Pathol Lab Med. 2015;139:211–218. [DOI] [PubMed] [Google Scholar]
- 4.Smits AJJ, Kummer JA, de Bruin PC, et al. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod Pathol. 2014;27:168–174. [DOI] [PubMed] [Google Scholar]
- 5.Grilley-Olson JE, Hayes DN, Moore DT, et al. Validation of interobserver agreement in lung cancer assessment: hematoxylin-eosin diagnostic reproducibility for non–small cell lung cancer: the 2004 World Health Organization Classification and Therapeutically Relevant Subsets. Arch Pathol Lab Med. 2012;137:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorello P, Cazzaniga G, Alberti F, et al. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia. 2006;20:1103–1108. [DOI] [PubMed] [Google Scholar]
- 7.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Cancer Genome Atlas Research Network. TCGA Tissue Sample Requirements: High Quality Requirements Yield High Quality Data. Cancer Genome Atlas—Natl. Cancer Inst. Available at: https://cancergenome.nih.gov/cancersselected/biospeccriteria. Accessed February 10, 2017.
- 10.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Loo P, Nordgard SH, Lingjærde OC, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci. 2010;107:16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oesper L, Satas G, Raphael BJ. Quantifying tumor heterogeneity in whole-genome and whole-exome sequencing data. Bioinformatics. 2014;30:3532–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X, Wang A, Walter V, et al. Combined targeted DNA sequencing in non-small cell lung cancer (NSCLC) using UNCseq and NGScopy, and RNA sequencing using UNCqeR for the detection of genetic aberrations in NSCLC. PLoS ONE. 2015;10:e0129280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkerson MD, Yin X, Walter V, et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE. 2012;7:e36530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andor N, Harness JV, Muller S, et al. EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics. 2014;30:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su X, Zhang L, Zhang J, et al. PurityEst: estimating purity of human tumor samples using next-generation sequencing data. Bioinforma. 2012;28:2265–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larson NB, Fridley BL. PurBayes: estimating tumor cellularity and subclonality in next-generation sequencing data. Bioinforma. 2013;29:1888–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Cancer Genome Atlas Research Network. Comprehensive characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller PAJ, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. [DOI] [PubMed] [Google Scholar]
- 20.Wetterstrand K. DNA sequencing costs: data from the NHGRI Genome sequencing program (GSP). 2013. Available at: www.GenomeGovsequencingcosts. Accessed January 1, 2017.
- 21.Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376:536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor-Weiner A, Zack T, O’Donnell E, et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature. 2016;540:114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yancovitz M, Litterman A, Yoon J, et al. Intra- and inter-tumor heterogeneity of BRAFV600E mutations in primary and metastatic melanoma. PLoS ONE. 2012;7:e29336. [DOI] [PMC free article] [PubMed] [Google Scholar]