

Abstract
Severe alcoholic hepatitis (SAH) has high mortality. Dysregulated lipid transport and metabolism in liver/macrophages contributes to disease pathophysiology. Paraoxonase/arylesterase 1 (PON1), a liver‐specific enzyme, inhibits oxidation of phospholipids and prevents lipid‐mediated oxidative damage. However, its functional contribution in macrophage‐mediated hepatic injury warrants elucidation. Plasma proteome of patients with SAH (n = 20), alcoholic cirrhosis (n = 20), and healthy controls was analyzed. Dysregulated pathways were identified, validated, and correlated with severity and outcomes in 200 patients with SAH. Tohoku‐Hospital‐Pediatrics‐1 (THP1)‐derived macrophages were treated with plasma from study groups in the presence/absence of recombinant PON1 and the phenotype; intracellular lipid bodies and linked functions were evaluated. In patients with SAH, 208 proteins were >1.5 fold differentially regulated (32 up‐regulated and 176 down‐regulated; P < 0.01).Validation studies confirmed lower levels of lipid transporter proteins (Pon1, apolipoprotein [Apo]B, ApoA1, ApoA2, and ApoC3; P < 0.01). Low PON1 levels inversely correlated with severity and mortality (r2 > 0.3; hazard ratio, 0.91; P < 0.01) and predicted nonsurvivors (area under the receiver operating characteristic curve, 0.86; cut‐off, <18 μg/mL; log rank, <0.01). Low PON1 levels corroborated with increased oxidized low‐density lipoprotein levels, intracellular lipid bodies, lipid uptake, lipid metabolism, biosynthesis, and alternative macrophage activation genes in nonsurvivors (P < 0.01). Importantly, in vitro recombinant PON1 treatment on THP1 macrophages reversed these changes (P < 0.01), specifically by alteration in expression of clusters of differentiation 36 (CD36) and adenosine triphosphate‐binding cassette subfamily A1 (ABCA1) receptor on macrophages. Conclusion: Lipid transport proteins contribute to the pathogenesis of SAH, and low PON1 levels inversely correlate with the severity of alcoholic hepatitis and 28‐day mortality. Restitution of circulating PON1 may be beneficial and needs therapeutic evaluation in patients with SAH.
Lipid transport proteins contribute to the pathogenesis of SAH and low Paraoxonase 1 levels inversely correlate with the severity of alcoholic hepatitis and 28 day mortality. Restitution of circulating Paraoxonase‐1 may be beneficial and needs therapeutic evaluation in SAH patients.
Abbreviations
- ABCA1
adenosine triphosphate‐binding cassette subfamily A1
- AC
alcoholic cirrhosis
- Apo
apolipoprotein
- AUROC
area under the receiver operating characteristic curve
- CD
clusters of differentiation
- CTP
Child‐Turcotte‐Pugh
- DF
discriminant function
- FABP1
fatty acid‐binding protein
- FAS
Fas cell surface death receptor
- FC
fold change
- GO
Gene Ontology
- HC
healthy control
- HDL
high‐density lipoprotein
- HLA‐DR
human leukocyte antigen DR isotype
- HR
hazard ratio
- IFN
interferon
- IHC
immunohistochemistry
- IL‐6
interleukin‐6
- INR
international normalized ratio
- ITGAM
integrin subunit alpha M
- LDL
low‐density lipoprotein
- LDLR
low‐density lipoprotein receptor
- LPL
lipoprotein lipase
- LXR
liver X receptor
- MDM
monocyte‐derived macrophage
- MELD
Model for End‐Stage Liver Disease
- MMP
matrix metalloproteinase
- NOD1
nucleotide‐binding oligomerization domain‐containing protein 1
- NR1H3
nuclear receptor subfamily 1 group H member 3
- PBMC
peripheral blood mononuclear cell
- PON1
paraoxonase/arylesterase 1
- PPARD
peroxisome proliferator activated receptor delta
- PPARG
peroxisome proliferator activated receptor gamma
- rePON1
recombinant paraoxonase
- SAH
severe alcoholic hepatitis
- SOFA
sequential organ failure assessment
- SRA/B1
scavenger receptor class A/B type 1
- SREBF1
sterol regulatory element binding transcription factor 1
- TG
triglyceride
- TGM2
transglutaminase 2
- THP1
Tohoku‐Hospital‐Pediatrics‐1
- TG
triglyceride
- TLC
total leukocyte count
- TNF
tumor necrosis factor
- TRAIL
tumor necrosis factor‐related apoptosis‐inducing ligand
- VLDL
very low‐density lipoprotein
Severe alcoholic hepatitis (SAH) is a progressive ailment with high mortality and limited treatment options.1 Systemic inflammatory responses, necrosis, fatty degeneration, and oxidative stress contribute to the progression of SAH.2, 3
Fatty degeneration is characterized by accumulation of lipid bodies in macrophages,2 which shift lipid homeostasis to lipid peroxidation, resulting in increased production of reactive oxygen species (ROS) and inflammation in SAH.4, 5 An increase in systemic inflammation induces the differentiation of circulating monocytes into M1 (inflammatory macrophages) or M2 (clusters of differentiation [CD]163+ alternatively activated) macrophages.6 Recently, we demonstrated a higher percentage of a circulating M2 phenotype in SAH.7, 8 Involvement of M2 phenotypes is known in lipid accumulation and differentiation.9
Lipid accumulation in macrophages is either through constitutive uptake by the surface receptors or by degradation of native/modified lipoproteins.10, 11 A distinguishable change in the circulating lipid content and/or alternate activation of the macrophages mediates lipid accumulation.12 In macrophages, lipid accumulates as cytoplasmic lipid droplets (esterified) to prevent cytotoxic effects or is exported through cholesterol efflux pathways.13, 14 In patients suffering from chronic alcoholic liver diseases, the former mechanism often precedes as a result of a continuous drop in high‐density lipoprotein (HDL) concentration (acceptor for cholesterol).15 Levels of circulating paraoxonase/arylesterase 1 (PON1) and HDL correlate to inflammation.7, 8 Moreover, oxidation of low‐density lipoprotein (LDL), cholesterol biosynthesis, and inflammatory response are interlinked biological processes, the deregulation of which plays a vital role in the progression of alcoholic liver diseases.12, 15
Liver plays a key role in cholesterol metabolism and transport,16 and alcohol consumption alters it. In patients with alcoholic cirrhosis (AC), LDL cholesterol, HDL cholesterol, and plasma triglycerides (TGs) are increased17, 18 Increased LDL cholesterol acts as a precursor molecule for the generation of immunogenic oxidized LDL,19 which promotes alternate activation of macrophages through activation of CD36.20
PON1 is a 384‐amino acid enzyme secreted by the liver.21, 22 Hepatic PON1 is similar to serum PON1 and has been corroborated with hepatic functions.23 Low PON1 levels and activity are associated with predisposition to hepatic damage.24, 25 PON1 inhibits the end product of lipid peroxidation (4‐hydroxy‐2‐noneal) and oxidation of phospholipids, thereby reducing production of monocyte chemoattractant protein 1.26, 27, 28 Animal studies have also shown a high degree of macrophage oxidative stress in PON1‐knockout mice.29 Thus, even in the absence of a hyperlipidemic state, PON1 deficiency promotes lipid accumulation and inflammatory and oxidative changes in monocytes/macrophages.30, 31 However, the correlation of circulating PON1 levels with the severity of liver diseases and short‐term mortality in SAH is not yet documented, and further data regarding the role of PON1 in the management of lipid‐laden macrophages and its functionality have not been investigated in SAH. We undertook the present study to identify markers of short‐term mortality in SAH and to characterize the phenotype of circulating monocytes/macrophages in SAH and in the presence or absence of PON1, which was correlated with the pathogenesis of patients with severe alcoholic hepatitis. This study put forward a compendium of proteomic alteration that is specific for patients with severe alcoholic hepatitis and can serve as a clinical resource. In addition, results of the study potentiate the need for therapeutic evaluation for recombinant PON1 in severe alcoholic hepatitis.
Patients and Methods
In this prospective study, 220 liver biopsy‐proven patients with SAH were enrolled between January 2014 and January 2016. Patients with hepatocellular carcinoma (n = 15) and associated with portal vein thrombosis (n = 5) were excluded. The diagnosis of SAH was based on histologic evidence and Maddery’s discriminant function (DF) of >32.32 The diagnosis of AC was based on a history of chronic heavy alcohol intake (with >1 month alcohol abstinence) and a combination of clinical, biochemical, endoscopic, and radiological criteria confirming presence of cirrhosis.33 Subjects enrolled as healthy controls (HCs) had no evidence of present/past liver disease. All the patient groups were managed according to the standard of care, which included intensive care monitoring, high‐caloric diet (35‐40 cal/kg/day), broad‐spectrum antibiotics, and intravenous albumin. None of the patients received corticosteroids before the samples were drawn for analysis. Laboratory staff were unaware of the clinical details of the study groups at the time of the experiments. Child‐Turcotte‐Pugh (CTP), Model for End‐Stage Liver Disease (MELD), sequential organ failure assessment (SOFA), and Maddrey’s DF were calculated to determine the severity of liver disease at initial presentation. All patients with SAH were followed up for a period of at least 1 month or until death. All blood samples were collected at fasting condition, and only baseline samples were analyzed and correlated with outcomes. The study was approved by the institutional ethical committee of the Institute of Liver and Biliary Science, New Delhi, India, and written informed consent was obtained in all cases.
Quantitative Proteomics
Discovery Phase
Plasma samples from age‐ and sex‐matched patients with SAH (n = 20), AC (n = 20), and HCs (n = 20) were depleted of the most abundant proteins and were trypsin digested overnight at room temperature. Peptides from each group were labelled using isobaric tags for relative and absolute quantitation, fractionated using strong cation exchange chromatography, and followed by mass spectrometry analysis (see details in Supporting Methods S1).
Validation Phase
The validation cohort consisted of 200 patients with SAH. Proteins differentially regulated and associated to lipid metabolism/transport were validated in this cohort using standard enzyme‐linked immunosorbent assay kits. The laboratory research scientists were blinded to patient identity and the results of the investigations.
Serum/Plasma Measurements
Plasma PON1 levels were measured in the study groups using a standard kit (cat. no. E90243hu; sensitivity, <1.39 ng/mL). Levels of other apolipoproteins (ApoA1 [EA5201‐1; sensitivity, <1.1 µg/mL), ApoA2 [EA5222‐1; sensitivity, <2.5 ng/mL], ApoC1 [EA8011‐1; sensitivity, <45 ng/mL], ApoC3 [EA8133‐1; sensitivity, <1.3 ng/mL], ApoB [EA7001‐1; sensitivity, <1.2 ng/mL], and ApoE [EA8003‐1; sensitivity, <5 ng/mL]) and oxidized LDL (STA‐388; sensitivity, ~150 ng/mL) were measured as per the manufacturer’s protocol and correlated to the biochemical profile of the patients.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on formalin‐fixed paraffin‐embedded liver tissues in 5 patients each with SAH and AC. Expression of PON1 (cat. no. PA5‐28997), LDL receptor (LDLR; cat. no. PA5‐22976), CD36 (cat. no. PA1‐16813), scavenger receptor class B type 1 (SRB1; cat. no. PA1‐16788), adenosine triphosphate‐binding cassette subfamily A1 (ABCA1; cat. no. PA5‐22906), ABCG1 (cat. no. PA5‐56757), CD68 (cat. no. MA513324), tumor necrosis factor alpha (TNF‐α; cat. no. PAA133Hu01), and nucleotide‐binding oligomerization domain‐containing protein 1 (NOD1; cat. no. PAK296Hu‐01) was estimated in the membrane and cytoplasmic space of the positive stained cells and counted in 10 consecutive high‐power fields (40×); relative quantitation as mean number of cells/10 high‐power field (40×) was recorded.
Flow Cytometry Analysis
Peripheral blood mononuclear cells (PBMCs) were isolated and incubated with Fc blocker (CD16/CD32; BD Biosciences, San‐Jose, CA) for 15 minutes to avoid nonspecific binding and were stained with a combination of Nile red fluorescein isothiocyanate, phycoerythrin (PE)‐human leukocyte antigen DR isotype (HLA‐DR), PE/cyanine 7/anti‐CD11b, and allophycocanin anti‐CD163 antibodies for 30 minutes at 4°C, washed with phosphate‐buffered saline, and fixed in 0.5% paraformaldehyde. The cells were analyzed on BD FACS VERSE (BD Biosciences).
Gene Expression Analysis
Total RNA from PBMCs of patients with SAH (survivors [n = 10] and nonsurvivors [n = 10]) was subjected to complementary DNA preparation and followed by reverse‐transcription polymerase chain reaction (RT‐PCR) analysis for genes linked to lipid uptake (CD36, SRA1, LDLR, and integrin subunit alpha M [ITGAM]), lipid metabolism/biosynthesis (Fas cell surface death receptor [FAS], sterol regulatory element binding transcription factor 1 [SREBF1], peroxisome proliferator activated receptor gamma, [PPARG], peroxisome proliferator activated receptor delta [PPARD], nuclear receptor subfamily 1 group H member 3 [NR1H3], NR1H2, and fatty acid‐binding protein [FABP1]), macrophage alternate activation (CD163, CD68, matrix metalloproteinase [MMP27], and transglutaminase 2 [TGM2]), and inflammation (TNF, interleukin‐6 [IL‐6]).
Immunofluorescence Analysis
Human monocyte‐derived macrophages (MDMs) were treated with plasma of patients with SAH, whereas Tohoku‐Hospital‐Pediatrics‐1(THP1)‐derived macrophages were treated with plasma of different study groups in the presence or absence of PON1. The density and number of lipid bodies stored in the macrophages was analyzed by Nile red staining.34
THP1 Stimulation Assay: Plasma Stimulation Assay
THP1 monocytes were differentiated into macrophages by 24‐hour incubation with 150 nM phorbol 12‐myristate 13‐acetate (Sigma), followed by 24‐hour incubation in Roswell Park Memorial Institute 1640 medium. A total of 106 THP1 cells in triplicate were treated with study plasma samples at a 10% concentration in the presence or absence of PON1 (cat. no. ENZ‐299; Prospec, Israel) at 2.5 units/mL (50 ng/mL) for the next 24 hours.35 One set of THP1‐derived macrophages was subjected to estimation of Nile red staining, HLA‐DR, CD11b, and CD163, as detailed above. Total RNA and proteins were isolated from the other set, and RT‐PCR analysis was performed for a panel of genes linked to lipid uptake, metabolism, biosynthesis, and inflammation. Protein samples were used for validation of expression of CD36 (cat. no. PA1‐16813), SRB1 (cat. no. PA1‐16788), ABCA1 (cat. no. PA5‐22906), ABCG1 (cat. no. PA5‐56757), and beta‐actin (cat. no. MA5‐15739). In a separate experiment, we used APOA1 (cat. no. CYT‐037; 100 ng/mL) and HDL (cat. no. MBS173147; 100 ng/mL) in combination with PON1, and expressions of genes linked to lipid uptake, metabolism, biosynthesis, and inflammation were evaluated.
Measurement of Oxidative Stress in THP1‐Derived Macrophages
Production of intracellular ROS in THP1‐derived macrophages treated with the plasma of study groups in the presence or absence of 2.5 units/mL PON1 was assessed using dihydrorhodamine‐123 (Sigma; Chemical Abstracts Service no. 109244‐58‐8), as described.36
THP1‐Derived Macrophage Proteomics
THP1‐derived macrophages were treated with study plasma samples in the presence or absence of PON1 (2.5 units/mL) for 24 hours and were subjected to eight‐plex proteomic analysis. The proteomic expression of the study groups was first normalized by the expression of untreated THP1 macrophages. Effect of plasma exposure on THP1 macrophages in each of the three groups was compared with or without addition of recombinant PON1. Differentially expressed proteins were subjected to Gene Ontology (GO) enrichment analysis using GOlorize‐Cytoscape37 and pathway analysis using FunRich38 and Enrichr39 (Supporting Methods S2).
Statistical Analyses
Statistical analyses were performed using SPSS, version 20. Analysis of variance was performed, and Bonferroni correction was used as post‐hoc analysis for normally distributed continuous variables; Mann‐Whitney U test and Kruskal‐Wallis tests were used for non‐normally distributed continuous variables. Spearman’s correlation was drawn comparing PON1 and lipid transporter proteins against severity assessment scores. Receiver operating characteristic curves were drawn for SAH to identify cut‐off values of PON 1 to predict mortality. Cox regression and Kaplan‐Meier survival analyses were performed to determine the significance of PON1 in outcome prediction.
Results
The clinical and demographic profile of the study groups (SAH, AC, and HC) is shown in Table 1. Patients with severe alcoholic hepatitis in the discovery and validation cohort had similar baseline characteristics (Table 1). Patients with SAH had more severe liver disease (higher MELD, CTP, DF, and SOFA scores) and increased levels of leucocyte counts, serum bilirubin, prothrombin time (PT)‐international normalized ratio (INR), serum cholesterol, HDL cholesterol, LDL cholesterol, very low‐density lipoprotein (VLDL) cholesterol, and TGs compared to AC (Table 1). In the validation cohort (n = 200), the nonsurvivors (63%) were clinically more severe and had higher CTP, MELD, DF, and SOFA scores compared to survivors (37%; Table 1).
Table 1.
Demographic Profile of the Study Cohort (Discovery and Validation Cohort)
| Parameters | SAH | AC | HC | P Value | P Value | P Value | — | — | — |
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | A vs. B | A vs. C | B vs. C | — | — | — | |
| Patient numbers | 20 | 20 | 20 | — | — | — | |||
| Median (min‐max) | Median (min‐max) | Median (min‐max) | — | — | — | ||||
| Discovery Set (Proteomics) | |||||||||
| Male‐female | 18‐02 | 18‐02 | 17−3 | 0.693 | 0.229 | 0.229 | — | — | — |
| Age (years) | 39 (24‐68) | 33 (17‐59) | 34 (31‐49) | 0.128 | 0.201 | 0.312 | — | — | — |
| TLC (×103/mm3) | 12.85 (3.5‐39) | 8.1 (3.3‐19) | 9.3 (4.2‐12) | 0.039 | 0.013 | 0.042 | — | — | — |
| Platelet (×103/mm3) | 134.8 (49.0‐269.0) | 165.0 (65.0‐309.0) | 159.0 (145.0‐400.0) | 0.014 | 0.015 | 0.038 | — | — | — |
| Bilirubin (mg/dL) | 20.2 (3.8‐30.9) | 1.5 (1.9‐2.9) | 0.9 (0.5‐1.5) | 0.000 | 0.000 | 0.047 | — | — | — |
| INR | 2.1 (1.6‐4.3) | 1.3 (0.9‐1.5) | 0.7 (0.5‐1.2) | 0.002 | 0.004 | 0.421 | — | — | — |
| ALT (IU/L) | 76.5 (10.0‐222.0) | 50.0 (33.0‐83.0) | 25.0 (17.0‐40.0) | 0.048 | 0.005 | 0.040 | — | — | — |
| Albumin (gm/dL) | 2.6 (1.5‐3.5) | 3.8 (2.9‐4.2) | 4.1 (3.4‐5.5) | 0.003 | 0.004 | 0.018 | — | — | — |
| CTP score | 11.0 (8.0‐12.0) | 6.0 (5.0‐8.0) | — | 0.004 | — | — | — | — | — |
| MELD score | 26.9 (18.0‐34.0) | 12.0 (8.0‐17.0) | — | 0.001 | — | — | — | — | — |
| DF score | 69.3 (34.0‐153.0) | 25.0 (21.0‐29.0) | — | 0.001 | — | — | — | — | — |
| SOFA score | 8.0 (5.0‐10.0) | 5.0 (4.0‐6.0) | — | 0.005 | — | — | — | — | — |
| Cholesterol (mg/dL) | 100.1 (75.0‐225.0) | 50.1 (45.0‐56.0) | 162.9 (155.1‐170.9) | 0.005 | 0.006 | 0.029 | — | — | — |
| TG (mg/dL) | 83.9 (29.5‐190.8) | 40.0 (15.0‐65.3) | 63.6 (52.1‐90.5) | 0.001 | 0.018 | 0.039 | — | — | — |
| HDL cholesterol (mg/dL) | 17.5 (3.3‐73.5) | 5.7 (3.1‐15.3) | 53.2 (45.7‐123.5) | 0.004 | 0.020 | 0.006 | — | — | — |
| LDL cholesterol (mg/dL) | 49.6 (20.9‐125.3) | 16.0 (14.9‐25.3) | 72.0 (65.0‐133.3) | 0.017 | 0.026 | 0.003 | — | — | — |
| VLDL cholesterol (mg/dL) | 52.8 (16.2‐77.0) | 18.3 (14.3‐30.9) | 20.5 (11.5‐33.9) | 0.024 | 0.024 | 0.479 | — | — | — |
| Parameters | SAH | AC | HC | P Value | P Value | P Value | Nonsurvivors | Survivors | P Value |
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | A vs. B | A vs. C | B vs. C | “D” | “E” | D vs. E | |
| Patient numbers | 200 | 20 | 20 | ||||||
| Median (min‐max) | Median (min‐max) | Median (min‐max) | 125 | 75 | |||||
| Median (min‐max) | Median (min‐max) | ||||||||
| Validation Set | |||||||||
| Male‐female | 191‐9 | 18‐02 | 16‐04 | 0.281 | 0.021 | 0.371 | 119‐6 | 72‐3 | 0.52 |
| Age (years) | 40 (25‐65) | 31 (18‐57) | 33 (30‐40) | 0.801 | 0.620 | 0.112 | 40 (25‐61) | 39 (30‐65) | 0.12 |
| TLC (×103/mm3) | 12.7 (3.8‐34.0) | 8.3 (3.1‐20.0) | 9.6 (4.0‐11.0) | 0.042 | 0.001 | 0.047 | 13.0 (3.8‐ 23.0) | 12.3 (5.0‐34.0) | 0.08 |
| Platelet (×103/mm3) | 135.5 (50‐265) | 171 (60‐300) | 155 (135‐400) | 0.014 | 0.002 | 0.049 | 143.0 (50.0‐186.0) | 128.0 (75.0‐265.0) | 0.08 |
| Bilirubin (mg/dL) | 19.9 (3.8‐32.0) | 2.1 (1.7‐3.1) | 0.7 (0.4‐1.5) | 0.000 | 0.000 | 0.037 | 20.0 (9.0‐32.0) | 16.0 (3.8‐21.0) | 0.00 |
| INR | 2.1 (1.8‐4.0) | 1.4 (1.0‐1.5) | 0.7 (0.6‐1.1) | 0.001 | 0.000 | 0.521 | 2.5 (2.1‐4.0) | 1.9 (1.8‐3.3) | 0.05 |
| ALT (IU/L) | 74.5 (15.0‐220.0) | 55.0 (33.0‐80.0) | 27.0 (17.5‐41.3) | 0.046 | 0.002 | 0.033 | 96.0 (21.0‐220.0) | 35 (15.0‐188.0) | 0.01 |
| Albumin (gm/dL) | 2.5 (1.7‐3.3) | 3.6 (3.2‐4.6) | 4.0 (3.7‐5.4) | 0.032 | 0.032 | 0.674 | 2.5 (1.7‐2.6) | 2.9 (2.1‐3.3) | 0.57 |
| CTP score | 11.0 (9.0‐12.0) | 7.0 (5.0‐9.0) | — | 0.002 | — | — | 11.0 (10.0‐12.0) | 9.0 (9.0‐11.0) | 0.00 |
| MELD score | 25.5 (19.0‐34.0) | 13.0 (9.0‐17.0) | — | 0.019 | — | — | 24.5 (23.0‐34.0) | 21 (19.0‐27.0) | 0.00 |
| DF score | 71.1 (34.0‐157.0) | 25.0 (20.0‐28.0) | — | 0.000 | — | — | 70.5 (56‐157) | 65.5 (34‐132) | 0.00 |
| SOFA score | 8.0 (5.0‐12.0) | 5.0 (4.0‐7.0) | — | 0.001 | — | — | 10 (8‐12) | 8 (5‐9) | 0.00 |
| PON1 (μg/mL) | 23.2 (2.3‐139.4) | 44.7 (25.8‐131) | 139.5 (46.6‐196.0) | 0.000 | <0.0001 | <0.0001 | 13.1 (2.3‐97) | 33.4 (5.7‐139.4) | <0.0001 |
| APOB (μg/mL) | 83.7 (3.7‐247.7) | 131.3 (35.4‐218.3) | 167.8 (36.4‐264.4) | 0.118 | 0.001 | 0.064 | 68.0 (4.5‐233.6) | 145.7 (3.7‐247.7) | 0.00 |
| APOE (μg/mL) | 245.2 (6.1‐449) | 126.4 (9.1‐277.5) | 323.4 (106‐540.3) | 0.000 | 0.000 | <0.0001 | 222.2 (7.0‐449.0) | 277.5 (6.1‐445.7) | 0.03 |
| APOA1 (μg/mL) | 55.7 (9.5‐134.8) | 117.7 (99‐124.2) | 119.5 (100.2‐130) | <0.0001 | <0.0001 | 0.404 | 35.6 (6.4‐132.5) | 64.2 (9.5‐134.8) | 0.52 |
| APOA2 (μg/mL) | 132.8 (0.7‐197.8) | 248.3 (173.4‐300) | 247.6 (214‐278.5) | <0.0001 | <0.0001 | 0.856 | 119.6 (0.75‐187.2) | 139.3 (7.9‐197.8) | 0.00 |
| APOC1 (μg/mL) | 43.5 (0.7‐54.5) | 28.76 (18.4‐42.4) | 34.2 (26.3‐44.1) | 0.046 | 0.671 | 0.015 | 40.9 (10.7‐54.5) | 43.9 (18.9‐54.5) | 0.04 |
| APOC3 (μg/mL) | 110.0 (10.6‐360.0) | 620.0 (120.0‐1680.0) | 720.0 (200.0‐2520.0) | <0.0001 | <0.0001 | 0.183 | 106.8 (12.0‐200.0) | 112.5 (40.0‐240.0) | 0.01 |
| PON1/APOB | 0.3 (0.01‐3.3) | 0.3 (0.1‐1.3) | 0.8 (0.2‐1.9) | 0.430 | 0.011 | 0.001 | 0.2 (0.01‐2.2) | 0.3 (0.02‐3.3) | 0.04 |
| PON1/APOE | 0.1 (0.01‐2.1) | 0.4 (0.14‐3.05) | 0.4 (0.14‐1.76) | <0.0001 | <0.0001 | 0.347 | 0.08 (0.01‐1.51) | 0.1 (0.03‐1.8) | 0.03 |
| PON1/APOA1 | 0.5 (0.1‐7.3) | 0.4 (0.2‐1.1) | 1.15 (0.4‐1.6) | 0.067 | 0.526 | <0.0001 | 0.4 (0.1‐5.2) | 0.6 (0.1‐4.6) | 0.03 |
| PON1/APOA2 | 0.2 (0.02‐4.5) | 0.2 (0.1‐0.7) | 0.8 (0.2‐1.1) | 0.326 | 0.061 | <0.0001 | 0.1 (0.02‐4.5) | 0.2 (0.03‐2.7) | 0.40 |
| PON1/APOC1 | 0.6 (0.04‐4.6) | 1.5 (0.6‐4.3) | 3.8 (1.5‐5.6) | 0.000 | <0.0001 | <0.0001 | 0.4 (0.04‐3.8) | 0.8 (0.2‐4.6) | 0.01 |
| PON1/APOC3 | 0.2 (0.01‐1.6) | 0.07 (0.02‐0.5) | 0.14 (0.05‐0.7) | 0.003 | 0.285 | 0.019 | 0.14 (0.01‐0.9) | 0.29 (0.0285‐1.686) | 0.00 |
| Cholesterol (mg/dL) | 98.0 (70.0‐220.0) | 49.5 (42.0‐56.0) | 160.9 (153.1‐174.7) | 0.000 | 0.000 | 0.031 | 108.0 (89.1‐220.0) | 90.0 (70.0‐189.3) | 0.04 |
| TG (mg/dL) | 83.5 (28.0‐193.0) | 44.0 (14.0‐62.0) | 60.7 (50.6‐90.9) | 0.005 | 0.000 | 0.022 | 88.3 (38.0‐193.0) | 81.1 (28.0‐153.6) | 0.04 |
| HDL cholesterol (mg/dL) | 17.3 (3.0‐74.5) | 5.8 (3.0‐14.8) | 55.5 (45.0‐125.3) | 0.004 | 0.004 | 0.033 | 16.8 (3.0‐54.5) | 23.3 (13.0‐74.5) | 0.03 |
| LDL cholesterol (mg/dL) | 48.4 (20.5‐123.3) | 18.0 (14.9‐23.2) | 70.0 (60.0‐135.2) | 0.002 | 0.020 | 0.004 | 58.2 (40.5‐123.3) | 40.4 (20.5‐103.3) | 0.04 |
| VLDL cholesterol (mg/dL) | 52.0 (13.2‐72.0) | 19.2 (14.0‐31.8) | 20.2 (10.9‐35.0) | 0.025 | 0.036 | 0.372 | 63.9 (22.5‐72.0) | 52.3 (13.2‐51.3) | 0.02 |
Values are expressed in medians and range unless stated otherwise; P < 0.05 is significant.
Abbreviation: ALT, alanine aminotransferase.
Discovery Phase
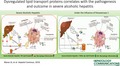
Quantitative proteomics identified 305 proteins (Supporting Table S1), of which 176 were down‐regulated and 32 were up‐regulated in patients with SAH compared to other groups (fold change [FC] >1.5; P < 0.05). Most significantly down‐regulated proteins showed lipid transporter activity (Fig. 1A) and were linked to lipoprotein metabolism, lipid transport, platelet activation, and homeostasis (P < 0.01; Fig. 1B; Supporting Tables S2 and S3). Of the up‐regulated proteins, only hemoglobin subunit gamma 1 (HBG1) was linked to lipid transport activity (Fig. 1A). Up‐regulated proteins were enriched for lipopolysaccharide (LPS) and toll‐like receptor (TLR) based on immune activation, complement activation, and IL‐6 signaling (Fig. 1B; Supporting Tables S2 and S3).
Figure 1.
Quantitative proteomic analysis identifies significantly down‐regulated lipid transporter proteins in SAH. (A) Partial list of proteins differentially regulated (>1.5‐fold; P < 0.05) between SAH, AC, HC, and its GO analysis, which documents enrichment of lipid transporters in SAH. (B) KEGG enrichment analysis of the up‐regulated (n = 32) and down‐regulated (n = 176) proteins in patients with SAH represents significant (P < 0.05) enrichment. (C) Plasma level of PON1 was down‐regulated in patients with SAH (median, 23.2; range, 2.3‐139.4) compared to those with AC (median, 44.7; range, 25.8‐131) and HC (median, 139.5; range, 46.6‐196.0) (P < 0.05). (D) Plasma level of apolipoproteins (APOB, APOE, APOA1, APOA2, APOC1, APOC3) in SAH compared to AC and HC (P < 0.05 is significant). (E) IHC showing expression of PON1, LDL, CD36, SRA1, ABCA1, and ABCG1 in SAH (n = 5) and AC (n = 5). For all IHC analyses, relative quantization of positively stained cells are expressed as mean number of positive cells/10 high‐power field (40×) and *P < 0.05. Abbreviations: COA, coenzyme A; HBG1, hemoglobin subunit gamma 1; KEGG, Kyoto Encyclopedia of Genes and Genomes; LBP, lipopolysaccharide binding protein; LPS, lipopolysaccharide; TLR4, toll‐like receptor 4;
Validation Phase
This cohort included 200 patients with SAH, with a dominance of male patients (95%). In this cohort, plasma levels of PON1, APOB, APOA1, APOA2, and APOC3 were significantly lower, whereas APOC1 and APOE levels were significantly higher compared to AC (P < 0.05; Fig. 1C,D; Table 1).Total cholesterol, TGs, HDL cholesterol, LDL cholesterol, and VLDL cholesterol were also significantly higher in patients with SAH (P < 0.05; Table 1). IHC results were concordant with plasma results and showed significantly lower levels of PON1, LDLR, and ABCA1, whereas the levels of CD36, SRB1, and ABCG1 were increased in the liver of patients with SAH (P < 0.05; Fig. 1E). Together, these results suggest that low circulating levels of lipid transporter proteins are associated with severity of liver diseases. Further, low tissue expression of PON1, LDLR, and ABCA1 and a higher expression of the lipid‐uptake receptor (CD36, SRB1) could relate to the observed steatosis in alcoholics.
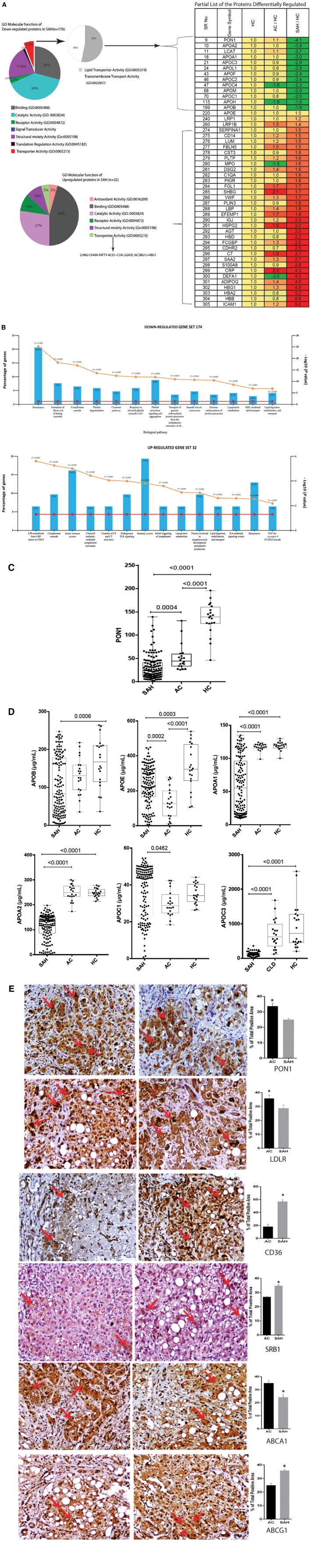
Dysregulated Lipid Transporter Proteins are Associated With Early Mortality in SAH
Circulating PON1 and other lipid transporters, such as APOB, APOE, APOA1, APOA2, APOC1, and APOC3 levels, were significantly low in nonsurvivors compared to survivors (P < 0.05; Fig. 2A,B). As PON1 directly interacts with lipid transporter proteins, the ratios of PON1:APOB, PON1:APOE, PON1:APOA1, PON1:APOC1, and PON1:APOC3 were studied and were found to be significantly lower in nonsurvivors (P < 0.05; Fig. 2C). Levels of circulating PON1, lipid transporter proteins (APOB, APOE, APOC1, APOC2, APOA2), and ratio of PON1:lipid transporter proteins inversely (r 2 > −0.3) correlated with disease severity and directly correlated with survival in patients with SAH (r 2 > 0.3; P < 0.05; Fig. 2D). Univariate Cox regression analysis identified significant parameters that correlated with short‐term mortality (28 days) in SAH (Table 2). Multivariate Cox regression analysis was performed for parameters with the original value (Model 1) and parameters with the derived value (Model 2). Model 1 highlighted significant associations of baseline PON1 levels (hazard ratio [HR], 0.934; range, 0.917‐0.952), APOB1, APOC3, platelet, total leukocyte count (TLC), and INR. Model 2 showed PON1/APOC1 (HR, 0.852), MELD score (HR, 1.072), CTP (HR, 1.133), and SOFA score (HR, 1.95) with short‐term mortality (28 days) in patients with SAH (Table 2). In addition, circulating PON1 levels showed the highest area under the receiver operating characteristic curve (AUROC) of 0.86 (P < 0.001), which was the greatest predictor of survival in patients with SAH (Fig. 2E). Based on the AUROC of PON1, a cut‐off value of 18 μg/mL or below showed 80% sensitivity and 85% specificity for mortality prediction in patients with SAH. The Kaplan‐Meier curve analysis elucidated short‐term (28‐day) mortality in patients with SAH with baseline PON1 <18 μg/mL (log rank, <0.01; Fig. 2F).
Figure 2.
Plasma levels of lipid transporter proteins correlate to severity and outcome in SAH. (A) Plasma level of PON1 was down‐regulated in nonsurvivors of SAH (median, 13.1; range, 2.3‐97) versus survivors (median, 33.4; range, 5.7‐139.4; P < 0.01). (B) Plasma levels of apolipoproteins (APOB, APOE, APOA1, APOA2, APOC1, and APOC3) in nonsurvivors compared to survivors. (C) PON1 interaction network and the levels of the ratios of PON1:lipid transporter proteins (PON1:APOB, PON1:APOE, PON1:APOA1, PON1:APOC1, PON1:APOC3) in nonsurvivors and survivors. (D) Correlation of PON1 and lipid transporter proteins with severity and outcome in SAH; pink represents P < 0.05. (E) Diagnostic efficacy of PON1 compared to other lipid transporter proteins in predicting severity and mortality in patients with SAH; P < 0.05. (F) Kaplan‐Meier survival analysis of patients with SAH based on PON1 levels of <18 μg/mL versus >18 μg/mL (log rank <0.01). Abbreviations: ALB, albumin; ALT, alanine aminotransferase; CC, correlation coefficient; CI, confidence interval; LB, lower bound; MTTP, microsomal triglyceride transfer protein; NS, not significant; Plat_B, platelet_baseline; Sig., significance; UB, upper bound.
Table 2.
Univariate and Multivariate Cox Regression Analysis of the Most Significant Parameters for the Determination of Mortality in Severe Alcoholic Hepatitis
| Variable | Significance* | HR | 95.0% CI for HR | |
|---|---|---|---|---|
| Lower | Upper | |||
| Univariate Analysis | ||||
| PON1 | 0.000 | 0.967 | 0.952 | 0.982 |
| APOB | 0.015 | 0.996 | 0.993 | 0.999 |
| APOE | 0.049 | 0.998 | 0.996 | 1.000 |
| APOA1 | 0.248 | 0.997 | 0.991 | 1.002 |
| APOA2 | 0.000 | 0.992 | 0.988 | 0.997 |
| APOC1 | 0.092 | 0.984 | 0.965 | 1.003 |
| APOC3 | 0.007 | 0.991 | 0.985 | 0.998 |
| PON1/APOB | 0.042 | 0.624 | 0.395 | 0.984 |
| PON1/APOE | 0.054 | 0.267 | 0.070 | 1.023 |
| PON1/APOA1 | 0.373 | 0.920 | 0.767 | 1.105 |
| PON1/APOA2 | 0.121 | 1.018 | 0.995 | 1.042 |
| PON1/APOC1 | 0.003 | 0.494 | 0.312 | 0.784 |
| PON1/APOC3 | 0.004 | 0.129 | 0.031 | 0.525 |
| Platelets | 0.001 | 0.481 | 0.309 | 0.748 |
| Sodium | 0.000 | 0.928 | 0.896 | 0.961 |
| TLC | 0.001 | 1.000 | 1.000 | 1.000 |
| Bilirubin | 0.102 | 1.021 | 0.996 | 1.046 |
| INR | 0.017 | 1.213 | 0.920 | 1.599 |
| CTP | 0.000 | 1.105 | 1.081 | 1.130 |
| MELD | 0.000 | 1.118 | 0.990 | 1.347 |
| SOFA | 0.000 | 1.469 | 1.347 | 1.603 |
| Variable | Significance | HR | 95.0% CI for HR | |
| Multivariate Analysis (parameters with original value) Model 1 | ||||
| PON1 | 0.000 | 0.934 | 0.917 | 0.952 |
| APOB | 0.001 | 0.995 | 0.992 | 0.998 |
| APOC3 | 0.007 | 0.995 | 0.991 | 0.999 |
| Platelet | 0.000 | 0.504 | 0.346 | 0.735 |
| TLC | 0.000 | 1.713 | 1.454 | 1.972 |
| INR | 0.024 | 1.282 | 1.033 | 1.591 |
| Variable | Significance | HR | 95.0% CI for HR | |
| Multivariate Analysis (parameters with derived values) Model 2 | ||||
| PON1/APOC1 | 0.047 | 0.852 | 0.796 | 0.895 |
| MELD | 0.000 | 1.072 | 1.033 | 1.112 |
| CTP | 0.000 | 1.133 | 1.098 | 1.169 |
| SOFA | 0.001 | 1.195 | 1.075 | 1.330 |
P < 0.05 is significant.
Abbreviation: CI, confidence interval.
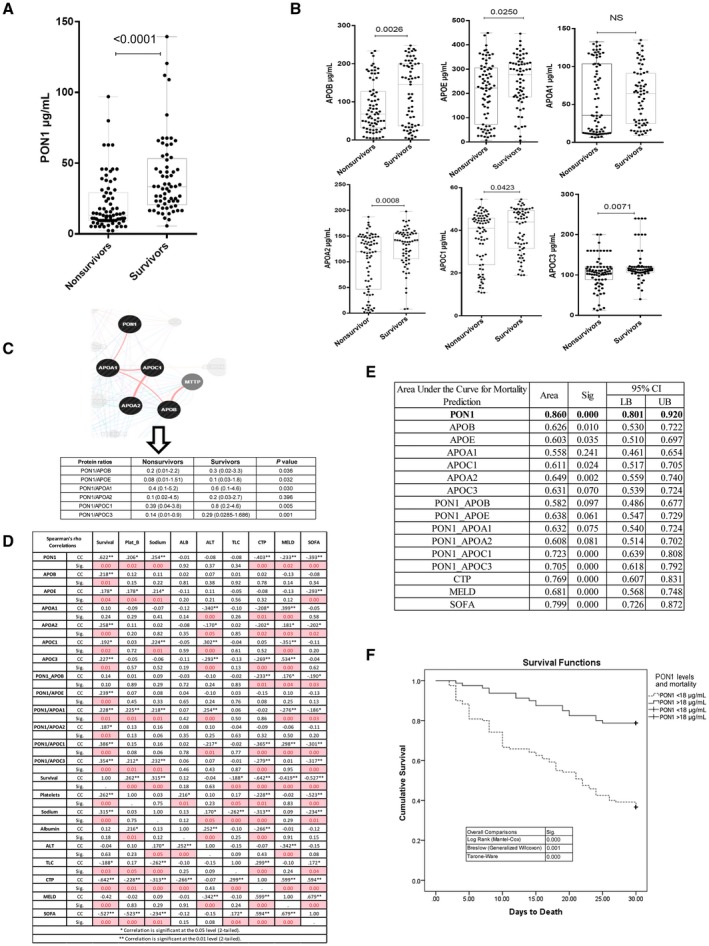
Low PON1 Level Promotes Oxidation of LDL and Macrophage Lipid Loading in Patients With SAH
Low PON1 level is associated with an increase in oxidation of LDL.20 The level of oxidized LDL was significantly higher in patients with SAH and more so in nonsurvivors (P < 0.05; Fig. 3A). To assess the association of the increase in oxidized LDL with the increase in the lipid body content of macrophages, we evaluated total lipid body content in circulating macrophages (CD11b+CD163+ and CD163+HLA‐DR+). Intracellular lipid body (Nile red staining) content was significantly higher in CD11b+CD163+ and CD163+HLA‐DR+ macrophages of nonsurvivors (P < 0.01; Fig. 3B,C). This increase in lipid bodies of macrophages was concordant with the expression of genes linked to lipid uptake (CD36, SRB1, LDLR, and ITGAM), lipid metabolism/biosynthesis (FAS, SREBF1, PPARG, PPARD, NR1H3, NR1H2, and FABP1), alternate activation (CD163, CD68, MMP27, and TGM2), and inflammation (TNF and IL‐6 with FC >2; P < 0.05; Fig. 3D), with no change in lipid‐export receptor (ABCA1, ABCG1) expression (Fig. 3D).
Figure 3.
Decreased PON1 levels associated with an increase in lipid oxidation and accumulation in macrophages in SAH. (A) Plasma oxidized LDL levels in SAH (median, 227; range, 150‐430) compared to AC (median, 175; range, 200‐100) and HC (median, 172; range, 185‐96), and nonsurvivors of SAH (median, 256; range, 183‐430) compared to survivors (median, 198; range, 155‐310); P < 0.05 is significant. (B) Accumulation of lipid bodies in CD11b+CD163+ macrophages in SAH (median, 4.6%; range, 1.01%‐22.7%) compared to AC (median, 3.07%, range, 1.02%‐4.51%) and HC (median, 1.85%; range, 1.01%‐2.22%), and accumulation of lipid bodies in nonsurvivors of SAH (median, 10.93%; range, 2.44%‐22.70%) compared to survivors (median, 2.24%; range, 0.85%‐7.62%); P < 0.05 is significant. (C) Accumulation of lipid bodies in CD11b+HLA‐DR+ macrophages in SAH (median, 24.55%; range, 4.65%‐68.60%) compared to AC (median, 6.38%; range, 3.79%‐8.41%) and HC (median, 0.87%; range, 0.10%‐2.22%), and accumulation of lipid bodies in nonsurvivors of SAH (median, 45.95%; range, 16.60%‐68.60%) compared to survivors (median, 18.70%; range, 4.65%‐39.00%); P < 0.05 is significant. (D) Relative expression of linked genes to lipid uptake, transport, metabolism, biosynthesis, macrophage activation, and inflammation in PBMCs of nonsurvivors versus survivors. (E) Immunofluorescent analysis of Nile red staining of healthy MDMs treated with plasma samples of nonsurvivors and survivors of SAH. Mean florescent intensity of Nile red staining represents the amount of lipid bodies stored. (F) IHC showing expression of CD68, TNF‐α, and NOD1 in SAH (n = 5) and AC (n = 5). For all IHC analyses, relative quantization of positively stained cells are expressed as mean number of positive cells/10 high‐power field (40×); *P < 0.05. Abbreviations: EPO, erythropoietin; FITC, fluorescein isothiocyanate; H&E, hematoxylin and eosin; LB, lipid body.
Healthy MDMs were treated with plasma samples of nonsurvivors and survivors of SAH, and the amount of lipid bodies stored in the macrophages was evaluated by Nile red staining. Mean florescent intensity of Nile red stain was significantly higher in macrophages treated with plasma of nonsurvivors (P < 0.001; Fig. 3E). Together, these results suggest that the increase in lipid bodies of the macrophages, more so in nonsurvivors, could be associated with an increase in oxidation of LDL and alternate activation of macrophages (increase in lipid‐uptake receptors CD36 and SRB1).
In order to document the association between lipid droplet accumulation and liver injury, liver biopsy specimens from 5 patients with SAH were compared to 5 patients with AC for expression of CD68 (macrophage marker),40 TNF‐α (marker for inflammation),41 and NOD1 (marker for metabolic inflammation).42 Results of our study showed a significant increase in inflammation around macrophages, as documented by hematoxylin and eosin staining (Fig. 3F). The size (vacuolated architecture, which is an indicator of lipid accumulation)43 and the number of CD68‐positive macrophages were significantly higher in the liver biopsy of patients with SAH compared to patients with AC (P < 0.05; Fig. 3F). Further, the level of TNF‐α and NOD1 was significantly higher in the liver biopsy of patients with SAH compared to patients with AC (P < 0.05; Fig. 3F). Together, these results suggest that lipid accumulation in SAH macrophages also contribute to metabolic inflammation and liver injury.
Resubstitution of PON1 Recalibrates Macrophage Lipid Homeostasis and Prevents Accumulation of Lipid Bodies in Patients With SAH
To further assess the role of PON1 in the recalibration of macrophage lipid homeostasis, THP1‐derived macrophages were treated with the study group plasma in the presence or absence of recombinant PON1 (rePON1) (Fig. 4A). SAH plasma treatment on THP1‐derived macrophages significantly increased the frequency of CD11b+CD163+ subtype and its lipid body content, which was significantly reduced with rePON1 treatment (P < 0.01; Fig. 4B). The frequency of the CD11b+HLA‐DR+ macrophage subset was unchanged, but the level of intracellular lipid bodies was significantly reduced with rePON1 treatment (P < 0.01; Fig. 4C). Treatment with rePON1 significantly reduced the intracellular lipid body, oxidative burst, and expression of genes linked to oxidative stress and inflammation (P < 0.01; Fig. 4D; Supporting Fig. S1). SAH plasma treatment on THP1 macrophages significantly induced the expression of CD36, SRB1 (lipid uptake), and FAS (lipid biosynthesis), whereas expression of ABCA1, ABCG1 (lipid export), and sterol regulatory element binding protein (SREBP1) were reduced (P < 0.01; Fig. 4E). Treatment with rePON1 solo or in combination with APOA1 or HDL (Supporting Fig. S2) significantly reduced the gene expression of lipid‐uptake receptors (CD36, SRB1), lipid biosynthesis (FAS, SREBP1, PPARG1, PPAR‐δ, liver X receptor [LXR]‐α, LXR‐β, and lipoprotein lipase [LPL]), and inflammation (IL‐6 and TNF‐α; P < 0.01) (Fig. 4E; Supporting Fig. S2). Protein levels of SRB1 and CD36 (lipid‐uptake receptor) were also reduced and ABCA1 was increased with rePON1 treatment (P < 0.05; Fig. 4F). Together, these results suggest that PON1 treatment alone or in combination with HDL and APOA1 reduces lipid body accumulation through inhibition of CD36 and SRB1 and induction of ABCA1 in macrophages.
Figure 4.
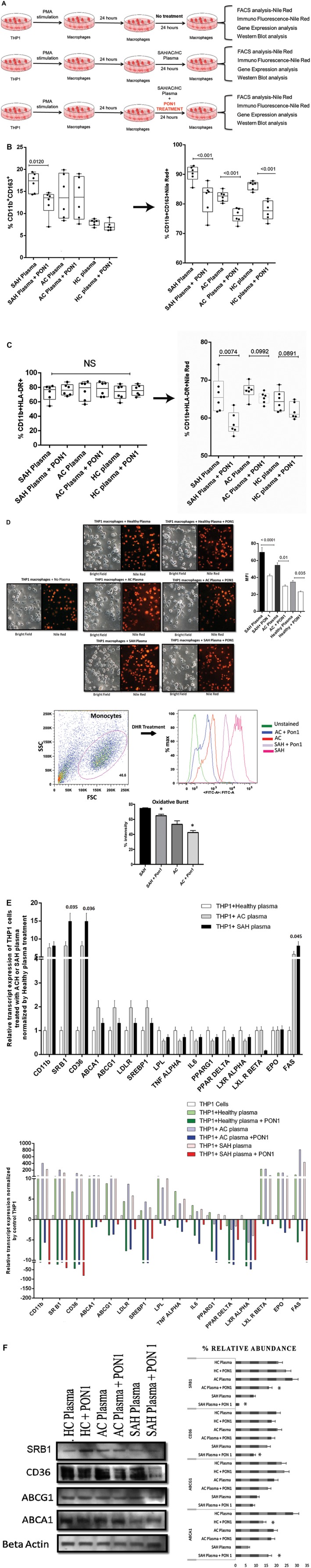
PON1 substitution modulates macrophage lipid homeostasis and prevents accumulation of lipid bodies in patients with SAH. (A) Workflow to estimate the effect of PON1 substitution in THP1 macrophages treated with plasma samples of SAH, AC, and HC. (B) Frequency of CD11b+CD163+ macrophages in SAH plasma (median, 17.41%; range, 14.30%‐19.40%) compared to SAH+PON1 (median, 13.55%; range, 6.98%‐14.70%), AC plasma (median, 13.55%; range, 8.39%‐19.90%) compared to AC+PON1 (median, 13.70%; range, 7.63%‐19.00%), and HC plasma (median, 8.20%; range, 6.79%‐8.82%) compared to HC+PON1 (median, 6.86%; range, 6.20%‐9.10%). Lipid body content:SAH plasma (median, 90%; range, 85.50%‐92.90%) compared to SAH+PON1 (median, 83.90%; range, 72.80%‐87.90%), AC plasma (median, 82.70%; range, 80.20%‐85.20%) compared to AC+PON1 (median, 76.05%; range, 72.40%‐78.60%), and HC plasma (median, 86.80%; range, 83.90%‐87.80%) compared to HC+PON1 (median, 77.65%; range, 73.30%‐81.80%); P < 0.05 is significant. (C) Frequency of CD11b+HLA‐DR+ macrophages was not significant in SAH plasma compared to SAH+PON1 or in AC plasma compared to AC+PON1 and HC plasma compared to HC+PON1. The lipid body content was SAH plasma (median, 67.35%; range, 63.6%‐70.3%) compared to SAH+PON1 (median, 57.7%; range, 55.2%‐63.4%), AC plasma (median, 65.55%; range, 63.1%‐67.3%) compared to AC+PON1 (median, 65.55%; range, 63.1%‐67.3%), and HC plasma (median, 64.35%; range, 61.6%‐68.8%) compared to HC+PON1 (median, 61%; range, 59.7%‐64.9%). (D) Validation of accumulation of lipid bodies by Nile red staining in macrophages treated with SAH, AC, or HC plasma in the presence and absence of PON1 (P < 0.05 is significant. Scale 20×.), and percentage intensity of oxidative burst calculated by DHR in THP1 macrophages treated with SAH and AC plasma in the presence and absence of PON1 (*P < 0.05). (E) Relative expression of linked genes to lipid uptake, transport, metabolism, biosynthesis, and inflammation in THP1 macrophages treated with SAH, AC, or HC plasma (upper panel). The effect of PON1 on the expression of these genes is shown in the lower panel (bright red, green, and blue are the expressions on PON1 treatment). (F) Relative protein expression of CD36, SRA1, ABCA1, and ABCG1 in THP1 macrophages treated with SAH, AC, or HC plasma in the presence or absence of PON1; *P < 0.05. Abbreviations: DHR, dihydrorhodamine; EPO, erythropoietin; FACS, fluorescence‐activated cell sorting; FITC, fluorescein isothiocyanate; FSC, forward scatter; LXL, luxate‐like; MFI, mean fluorescence intensity; NS, not significant; PMA, phorbol 12‐myristate 13‐acetate; SSC, side scatter.
Effect of PON1 Treatment on THP1‐Derived Macrophages Under SAH‐Like Condition: Macrophage Proteomic Analysis
To understand the effect of rePON1 treatment on THP1‐derived macrophages under an SAH‐like condition, an eight‐plex quantitative proteomic analysis was undertaken. A total of 1,817 proteins were identified, of which 406 were differentially regulated (FC >1.5; P < 0.01; Supporting Table S4) when THP1‐derived macrophages were exposed to study group plasma, with or without rePON1. Treatment with rePON1 significantly induced 64 proteins in HC plasma‐treated macrophages, 82 in AC plasma‐treated macrophages, and 29 in SAH plasma‐treated macrophages (Fig. 5A). Treatment with rePON1 significantly inhibited 126 proteins in HC plasma‐treated macrophages, 78 proteins in AC plasma‐treated macrophages, and 128 proteins in SAH plasma‐treated macrophages (Fig. 5A). Proteins induced by rePON1 treatment were linked to antigen processing, response to metal, transporter activity, and arginine/nitrogen metabolism (P < 0.01) (Fig. 5B; Supporting Table S5). Proteins suppressed by rePON1 treatment were linked to RAS signaling, insulin receptor signaling, and catalytic/hydrolase/acyl transferase activity (Fig. 5C; Supporting Table S5). Treatment of rePON1 significantly reduced the expression of proteins linked to lipid transport, lipoprotein metabolism, and inflammatory signaling pathways (IL‐1, interferon‐gamma [IFN‐γ], and TNF signaling) in SAH plasma‐treated macrophages (P < 0.01) (Fig. 5D; Supporting Table S6). rePON1 was also able to significantly reduce cell death by reducing FAS (CD95), activation of BAD, telomere extension, Rac family small guanosine triphosphase 1 (RAC1), TNF‐related apoptosis‐inducing ligand (TRAIL), Nef signaling, and alternate energy metabolism (glutamine degradation, glycerol‐3‐phosphate shuttle, glucokinase regulation) in SAH plasma‐treated macrophages (P < 0.01) (Fig. 5E; Supporting Table S6). These results were cross‐validated by quantifying the messenger RNA expression of the identified proteins, which showed similar expression trends (Supporting Fig. S1; Supporting Table S7). Taken together, these results validate the beneficial effects of rePON1 treatment on macrophages. Treatment of rePON1 reduces lipid storage and biosynthesis, reduces inflammation, reduces apoptosis, and alters energy metabolism in macrophages.
Figure 5.
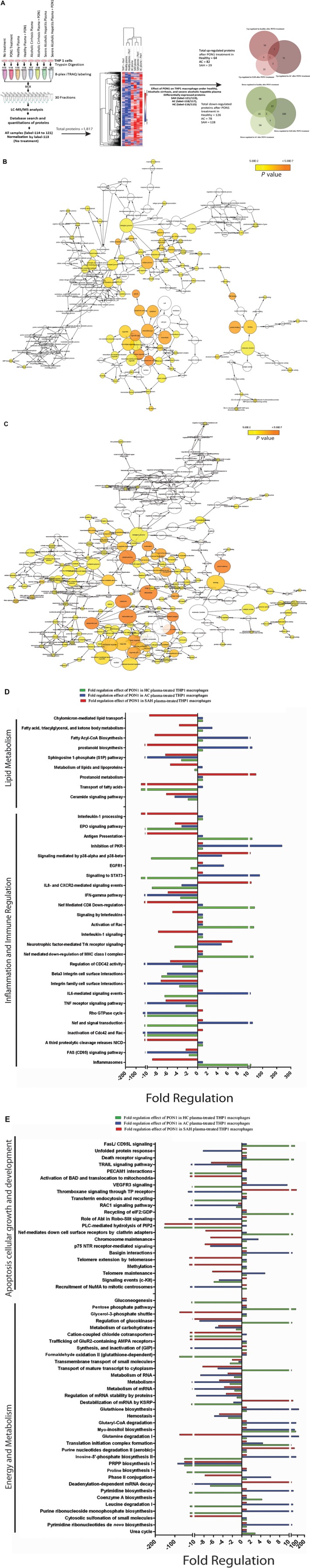
Macrophage proteomic analysis validates the decrease in lipid metabolism and biosynthesis after PON1 treatment in THP1‐derived macrophages. (A) Workflow documents eight‐plex proteomic analysis of THP1 macrophages treated with plasma samples of SAH, AC, and HC in the presence or absence of PON1. This was followed by clustering analysis of 1,817 proteins. To estimate the effect of PON1 expression of THP1 macrophages treated with SAH plasma+ PON1/SAH plasma treatment, THP1 macrophages treated with AC plasma+ PON1/AC plasma treatment, THP1 macrophages treated with HC plasma+ PON1/HC plasma treatment, PON1‐induced and PON1‐suppressed proteins were identified. Red venny diagram documents the integration of PON1‐induced genes in SAH, AC, and HC plasma, and green venny documents the integration of PON1‐suppressed genes. (B) Network of GO (biological, molecular, and cellular function) functional enrichment of the PON1‐induced genes in SAH, AC, and HC; significant enrichment is highlighted with shades of yellow with a darker shade having more significance (P < 0.05). (C) Network of GO (biological, molecular, and cellular function) functional enrichment of the PON1‐suppressed genes in SAH, AC, and HC; significant enrichment is highlighted with shades of yellow with a darker shade showing greater significance (P < 0.05). (D) Functional analysis based on the proteins induced or suppressed by PON1 treatment. Red bar documents fold regulation, the effect of PON1 in SAH plasma‐treated THP1 macrophages. Blue bar documents fold regulation, the effect of PON1 in AC plasma‐treated THP1 macrophages. Green bar documents fold regulation, the effect of PON1 in AC plasma‐treated THP1 macrophages for lipid metabolism and inflammation and immune regulation linked pathways. Fold regulation is calculated based on the percentage enrichment and mean expression of proteins in the pathways. (E) Functional analysis based on proteins induced or suppressed by PON1 treatment. Red bar documents fold regulation, the effect of PON1 in SAH plasma‐treated THP1 macrophages. Blue bar documents fold regulation, the effect of PON1 in AC plasma‐treated THP1 macrophages. Green bar documents fold regulation, the effect of PON1 in AC plasma‐treated THP1 macrophages for apoptosis, cellular growth and development, and energy metabolism linked pathways. Fold regulation is calculated based on the percentage enrichment and mean expression of proteins in the pathways. Abbreviations: Abl, albumin; AMPA, α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor; CoA, coenzyme A; CXCR2, chemokine (C‐X‐C motif) receptor 2; EGFR1, epidermal growth factor receptor1; GDP, Glyceraldehyde‐3‐phosphate dehydrogenase; GIP, Gastric inhibitory polypeptide; GluR2, Glutamate receptor 2; GTPase, guanosine triphosphatase; iTRAQ, isobaric tag for relative and absolute quantitation; KSRP, Far upstream element‐binding protein 2; LC‐MS/MS, liquid chromatography–tandem mass spectrometry; MHC, major histocompatibility complex; mRNA, messenger RNA; NICD, Neurogenic locus notch homolog protein 1; NuMA, Nuclear mitotic apparatus protein 1; PECAM1, Platelet endothelial cell adhesion molecule; PIP2, Phosphatidylinositol (4,5)‐bisphosphate; PKR, protein kinase R; PLC, Phospholipase C; PRPP, Phosphoribosyl pyrophosphate; STAT3, signal transducer and activator of transcription 3; TP, thromboxane receptor; VEGFR3, Vascular endothelial growth factor receptor 3.
Discussion
In the present study, we observed a significant decrease in the circulating levels of lipid transporter proteins in patients with severe alcoholic hepatitis that correlated with disease severity and outcome. Low plasma levels of PON1 promote oxidation of LDL and accumulation of lipids in the macrophages through induction of lipid‐uptake receptor (CD36, SRA1). This results in an increase in oxidative stress and inflammation that correlates with the pathophysiology of SAH. PON1 resubstitution was able to reverse these phenomena and was found to be beneficial for the macrophages in an in vitro setting. The plasma proteome of patients with SAH was distinct from that of AC and healthy controls, allowing us to identify that lipid transporters are decreased and lipid transport/metabolism is deranged in patients with SAH.
Our results show that of the 305 differentially expressed proteins in patients with SAH, 32 up‐regulated proteins are linked to nuclear factor (NF)‐κB signaling, toll‐like receptor signaling pathway, toll/IL‐1 receptor domain‐containing adaptor‐inducing IFN‐ (TRIF)‐mediated cell death, glycosaminoglycan metabolism, and caspase activation. The 176 down‐regulated proteins are linked to lipoprotein metabolism and HDL‐mediated lipid transport, biosynthesis of amino acids, glycolysis/gluconeogenesis, cell cycle, and intrinsic pathway for apoptosis. Many previous studies support our proteomic findings with respect to a significant increase in NF‐κB signaling, toll‐like receptor signaling pathway, and inflammation.3, 5, 7, 8 A seminal feature of our study is reduction in levels of plasma PON1 and other APO lipoproteins. This correlated with a reduction in tissue levels of PON1, LDL, and ABCA1 and an increase in tissue levels of CD36, SRA1, and ABCG1 in patients with SAH. These results potentiate that lipid metabolism and transport are decreased in patients with SAH. Further increases in the hepatocyte/macrophage expression of lipid‐uptake receptors (CD36, SRA1) and decreases in the lipid‐export receptor (ABCA1) indicate accumulation of lipid bodies in macrophages, as seen in severe alcoholic hepatitis.
To document an association of lipid transporter proteins with severity and outcome in patients with SAH, circulating levels of lipid transporter proteins were compared between nonsurvivors and survivors. Our data demonstrate that the level of PON1 and lipid transporter proteins (APOB, APOE, APOA1, APOA2, APOC1, and APOC3) and the ratio of PON1 to lipid transporter proteins were significantly low and inversely correlated to severity indices (MELD, SOFA, and CTP) and directly correlated to survival in patients with SAH. In addition, we also show that plasma PON1 levels <18 μg/mL at baseline correlate with 28‐day mortality in patients with SAH. This observation clearly suggests that PON1 levels could be used in addition to the MELD score for an accurate diagnosis of patients who have less likeness for survival in SAH.
PON1 is secreted from the liver23 and is known to work in association with HDL.35 As an enzyme, PON1 plays dual roles, one to protect the hepatocytes/macrophages from oxidative stress (antioxidant) and the other to reduce the plasma lipids (LDL) and channel their transportation for cholesterol metabolism.44 In our study, low PON1 levels in nonsurvivors was directly associated with the increase in plasma level of oxidized LDL. The decrease in circulating PON1 levels could be due to severe hepatic injury, altered HDL synthesis, or low lecithin to cholesterol acyl transferase (LCAT) levels.45, 46 A significant decrease in the plasma levels of LCAT was shown in our study (Fig. 1A), which could be one of the reasons for low PON1 levels observed in patients with SAH.47 An increase in the oxidation of circulating lipids (LDL) induces its uptake and processing by macrophages through micropinocytosis, phagocytosis, and scavenger receptor‐mediated pathways.47 Further, the ingested lipids are converted into cholesterol fatty acid esters and are stored in the cytosol as lipid droplets.48 In our study, LDL oxidation and lipid body accumulation was higher in CD11b+CD163+ and CD11b+HLADR+ macrophages and was significantly more so in nonsurvivors. This increase in lipid body content of macrophages was validated by Nile red staining49 and was in line with the increase in expression of lipid‐uptake receptors, lipid metabolism/biosynthesis, macrophage alternate activation, and inflammation genes.
We investigated the influence of PON1 resubstitution in the modulation of lipid homeostasis and lipid body accumulation. We subjected THP1‐derived macrophages to plasma taken from patients and HCs, in the presence or absence of rePON1. PON1 replenishment significantly reduced CD11b+CD163+ cell number as well as the amount of lipid bodies stored in them. We did not find any change in the number of CD11b+HLA‐DR+ macrophages, but the lipid bodies stored in them were significantly reduced. This was validated by Nile red staining. The addition of PON1 also reduced the level of oxidative burst and genes linked to oxidative stress in SAH plasma‐treated macrophages compared to AC‐treated macrophages.50
PON1 is known to inhibit monocyte to macrophage differentiation and CD36 expression in macrophages.51 In our study, THP1 macrophages treated with SAH plasma showed a significant induction of genes linked to lipid‐uptake receptors (CD36, SRB1) and lipid biosynthesis (FAS), which were reduced with PON1 treatment alone or in combination with APOA1 and HDL, respectively. PON1 treatment also reduced protein levels of lipid‐uptake receptors (SRB1, CD36) and increased expression of lipid exporter (ABCA1) in SAH plasma‐treated macrophages. This suggests that PON1 with or without HDL and APOA1 modulates macrophage oxidative stress and inflammation inherent in patients with SAH.
To investigate the beneficial effects of PON1, global proteomic analysis was performed; this showed that PON1 resubstitution in SAH or AC plasma‐treated macrophages significantly induced antigen processing, posttranslational regulation, response to metal, protein transporter activity, and arginine/nitrogen compound metabolism. Further, PON1 treatment significantly decreased lipid metabolism (chylomicron‐mediated lipid transport, fatty acid, triacylglycerol metabolism, free fatty acid biosynthesis, sphingosine 1‐phosphate, and ceramide signaling pathway), inflammation and immune regulation (IL‐1 signaling, IFN‐γ pathway, TNF receptor signaling, inflammasome), apoptosis and cell death (RAC1, TRAIL, and Nef‐signaling pathway; activation of BAD, translocation to mitochondria, telomere extension), and pathways linked to alternate energy metabolism (glutamine degradation, glycerol‐3‐phosphate shuttle, regulation of glucokinase) (Fig. 6).
Figure 6.
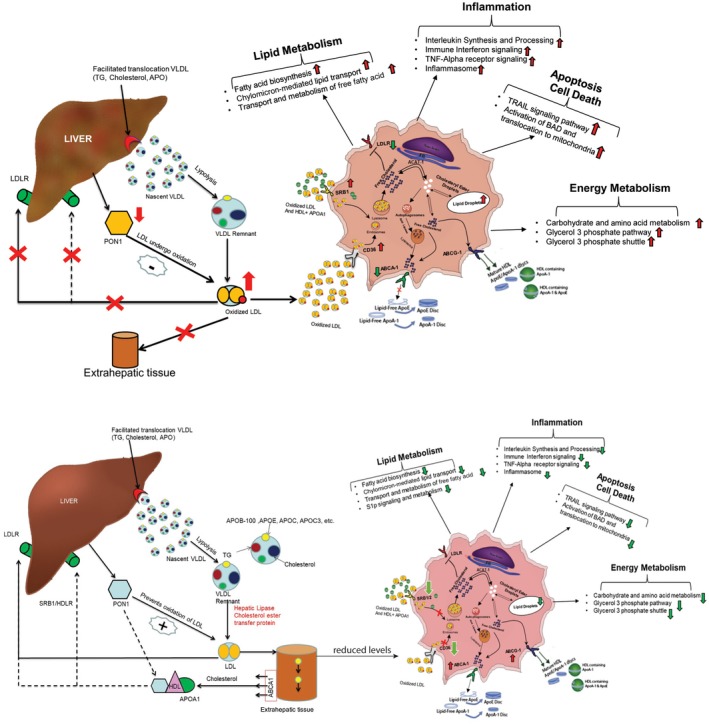
Causality for PON1‐mediated decrease in accumulation of lipid bodies, oxidative stress, and inflammation. Paradigm for the role of PON1 treatment in SAH plasma‐treated macrophages. In patients with SAH under low PON1, lipid transport and metabolism is perturbed. This is followed by an increase in plasma LDL oxidation, which significantly induces circulating macrophages for lipid uptake by increasing expression of CD36 and SRB1. Lipid bodies accumulate in the macrophages due to a decrease in lipid metabolism and lipid export receptor ABCA1. Increase in the content of lipid bodies increases inflammation and oxidative stress and cellular apoptosis (TRAIL, BAD‐mediated apoptosis) and shifts macrophages to alternate energy metabolism. Resubstitution of PON1 recalibrates the macrophage–lipid homeostasis by mediating a decrease in lipid uptake receptor and an increase in lipid export receptor, thereby neutralizing macrophage inflammation and oxidative stress, apoptosis, and a shift in energy metabolism. Abbreviation: ACAT, acyl‐coenzyme A:cholesterol acyltransferase.
The added value of this study is that by using advance mass spectrometry we were able to identify and validate circulating lipid transporter levels as a surrogate for prediction of short‐term mortality in severe alcoholic hepatitis. We also showed that a subtle change in the circulating levels of lipid transporters correlates to disease severity indicators, such as MELD, and also results in recalibration of macrophage lipid homeostasis, thereby decreasing its functionality. Thus, routine assessment of PON1 in clinics could aid traditionally used severity scores for the prediction of likelihood of mortality, and replenishing key lipid transporters (PON1) could also improve macrophage functionality, severity, and outcome in severe alcoholic hepatitis.
In summary, PON1 reduces lipid storage (through inhibiting CD36, SRB1) and biosynthesis, reduces hepatic inflammation and cellular apoptosis, and alters energy metabolism in SAH macrophages. These novel observations underline the potential therapeutic role of PON1 in the treatment of severe alcoholic hepatitis.
Supporting information
Supported by the Department of Science and Technology, Science and Engineering Research Board (EMR/2016/004829 to J.S.M.) and research funds (to S.K.S.).
Potential conflict of interest: Nothing to report.
Contributor Information
Jaswinder Singh Maras, Email: jassi2param@gmail.com, Email: shivsarin@gmail.com.
Shiv Kumar Sarin, Email: shivsarin@gmail.com.
REFERENCES
- 1. Forrest E, Mellor J, Stanton L, Bowers M, Ryder P, Austin A, et al. Steroids or pentoxifylline for alcoholic hepatitis (STOPAH): study protocol for a randomised controlled trial. Trials 2013;14:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uchida T, Kao H, Quispe‐Sjogren M, Peters RL. Alcoholic foamy degeneration–a pattern of acute alcoholic injury of the liver. Gastroenterology 1983;84:683‐692. [PubMed] [Google Scholar]
- 3. Celli R, Zhang X. Pathology of Alcoholic Liver Disease. J Clin Transl Hepatol 2014;2:103‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lundqvist A, Magnusson LU, Ullstrom C, Krasilnikova J, Telysheva G, Dizhbite T, et al. Oregonin reduces lipid accumulation and proinflammatory responses in primary human macrophages. Biochem Biophys Res Commun 2015;458:693‐699. [DOI] [PubMed] [Google Scholar]
- 5. Casanova J, Bataller R. Alcoholic hepatitis: prognosis and treatment. Gastroenterol Hepatol 2014;37:262‐268. [DOI] [PubMed] [Google Scholar]
- 6. Saha B, Bruneau JC, Kodys K, Szabo G. Alcohol‐induced miR‐27a regulates differentiation and M2 macrophage polarization of normal human monocytes. J Immunol 2015;194:3079‐3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maras JS, Das S, Sharma S, Sukriti S, Kumar J, Vyas AK, et al. Iron‐overload triggers ADAM‐17 mediated inflammation in severe alcoholic hepatitis. Sci Rep 2018;8:10264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maras JS, Maiwall R, Harsha HC, Das S, Hussain MS, Kumar C, et al. Dysregulated iron homeostasis is strongly associated with multiorgan failure and early mortality in acute‐on‐chronic liver failure. Hepatology 2015;61:1306‐1320. [DOI] [PubMed] [Google Scholar]
- 9. Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem 2012;287:11629‐11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell 2001;104:503‐516. [DOI] [PubMed] [Google Scholar]
- 11. Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol 2009;6:399‐409. [DOI] [PubMed] [Google Scholar]
- 12. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010;32:593‐604. [DOI] [PubMed] [Google Scholar]
- 13. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 2010;10:36‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol 2018;330:27‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trieb M, Horvath A, Birner‐Gruenberger R, Spindelboeck W, Stadlbauer V, Taschler U, et al. Liver disease alters high‐density lipoprotein composition, metabolism and function. Biochim Biophys Acta 2016;1861:630‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mardones P, Quinones V, Amigo L, Moreno M, Miquel JF, Schwarz M, et al. Hepatic cholesterol and bile acid metabolism and intestinal cholesterol absorption in scavenger receptor class B type I‐deficient mice. J Lipid Res 2001;42:170‐180. [PubMed] [Google Scholar]
- 17. Janicko M, Veseliny E, Lesko D, Jarcuska P. Serum cholesterol is a significant and independent mortality predictor in liver cirrhosis patients. Ann Hepatol 2013;12:581‐587. [PubMed] [Google Scholar]
- 18. Phukan JP, Sinha A, Deka JP. Serum lipid profile in alcoholic cirrhosis: a study in a teaching hospital of north‐eastern India. Niger Med J 2013;54:5‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alho H, Sillanaukee P, Kalela A, Jaakkola O, Laine S, Nikkari ST. Alcohol misuse increases serum antibodies to oxidized LDL and C‐reactive protein. Alcohol Alcohol 2004;39:312‐315. [DOI] [PubMed] [Google Scholar]
- 20. Rios FJ, Koga MM, Pecenin M, Ferracini M, Gidlund M, Jancar S. Oxidized LDL induces alternative macrophage phenotype through activation of CD36 and PAFR. Mediators Inflamm 2013;2013:198193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelso GJ, Stuart WD, Richter RJ, Furlong CE, Jordan‐Starck TC, Harmony JA. Apolipoprotein J is associated with paraoxonase in human plasma. Biochemistry 1994;33:832‐839. [DOI] [PubMed] [Google Scholar]
- 22. Leviev I, Negro F, James RW. Two alleles of the human paraoxonase gene produce different amounts of mRNA. An explanation for differences in serum concentrations of paraoxonase associated with the (Leu‐Met54) polymorphism. Arterioscler Thromb Vasc Biol 1997;17:2935‐2939. [DOI] [PubMed] [Google Scholar]
- 23. Gonzalvo MC, Gil F, Hernandez AF, Rodrigo L, Villanueva E, Pla A. Human liver paraoxonase (PON1): subcellular distribution and characterization. J Biochem Mol Toxicol 1998;12:61‐69. [DOI] [PubMed] [Google Scholar]
- 24. Zhang C, Peng W, Jiang X, Chen B, Zhu J, Zang Y, et al. Transgene expression of human PON1 Q in mice protected the liver against CCl4‐induced injury. J Gene Med 2008;10:94‐100. [DOI] [PubMed] [Google Scholar]
- 25. Ferre N, Camps J, Prats E, Vilella E, Paul A, Figuera L, et al. Serum paraoxonase activity: a new additional test for the improved evaluation of chronic liver damage. Clin Chem 2002;48:261‐268. [PubMed] [Google Scholar]
- 26. Parola M, Bellomo G, Robino G, Barrera G, Dianzani MU. 4‐Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal 1999;1:255‐284. [DOI] [PubMed] [Google Scholar]
- 27. Zamara E, Novo E, Marra F, Gentilini A, Romanelli RG, Caligiuri A, et al. 4‐Hydroxynonenal as a selective pro‐fibrogenic stimulus for activated human hepatic stellate cells. J Hepatol 2004;40:60‐68. [DOI] [PubMed] [Google Scholar]
- 28. Mackness B, Hine D, Liu Y, Mastorikou M, Mackness M. Paraoxonase‐1 inhibits oxidised LDL‐induced MCP‐1 production by endothelial cells. Biochem Biophys Res Commun 2004;318:680‐683. [DOI] [PubMed] [Google Scholar]
- 29. Rozenberg O, Rosenblat M, Coleman R, Shih DM, Aviram M. Paraoxonase (PON1) deficiency is associated with increased macrophage oxidative stress: studies in PON1‐knockout mice. Free Radic Biol Med 2003;34:774‐784. [DOI] [PubMed] [Google Scholar]
- 30. Ng DS, Chu T, Esposito B, Hui P, Connelly PW, Gross PL. Paraoxonase‐1 deficiency in mice predisposes to vascular inflammation, oxidative stress, and thrombogenicity in the absence of hyperlipidemia. Cardiovasc Pathol 2008;17:226‐232. [DOI] [PubMed] [Google Scholar]
- 31. Ikhlef S, Berrougui H, Kamtchueng Simo O, Zerif E, Khalil A. Human paraoxonase 1 overexpression in mice stimulates HDL cholesterol efflux and reverse cholesterol transport. PLoS ONE 2017;12:e0173385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015;372:1619‐1628. [DOI] [PubMed] [Google Scholar]
- 33. Kumar Acharya S, Kumar Sharma P, Singh R, Kumar Mohanty S, Madan K, Kumar Jha J, et al. Hepatitis E virus (HEV) infection in patients with cirrhosis is associated with rapid decompensation and death. J Hepatol 2007;46:387‐394. [DOI] [PubMed] [Google Scholar]
- 34. Chan L, Hong J, Pan J, Li J, Wen Z, Shi H, et al. Role of Rab5 in the formation of macrophage‐derived foam cell. Lipids Health Dis 2017;16:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosenblat M, Elias A, Volkova N, Aviram M. Monocyte‐macrophage membrane possesses free radicals scavenging activity: stimulation by polyphenols or by paraoxonase 1 (PON1). Free Radic Res 2013;47:257‐267. [DOI] [PubMed] [Google Scholar]
- 36. Das S, Maras JS, Hussain MS, Sharma S, David P, Sukriti S, et al. Hyperoxidized albumin modulates neutrophils to induce oxidative stress and inflammation in severe alcoholic hepatitis. Hepatology 2017;65:631‐646. [DOI] [PubMed] [Google Scholar]
- 37. Garcia O, Saveanu C, Cline M, Fromont‐Racine M, Jacquier A, Schwikowski B, et al. GOlorize: a Cytoscape plug‐in for network visualization with gene ontology‐based layout and coloring. Bioinformatics 2007;23:394‐396. [DOI] [PubMed] [Google Scholar]
- 38. Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015;15:2597‐2601. [DOI] [PubMed] [Google Scholar]
- 39. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90‐W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Movita D, Kreefft K, Biesta P, van Oudenaren A, Leenen PJ, Janssen HL, et al. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J Leukoc Biol 2012;92:723‐733. [DOI] [PubMed] [Google Scholar]
- 41. Zelova H, Hosek J. TNF‐alpha signalling and inflammation: interactions between old acquaintances. Inflamm Res 2013;62:641‐651. [DOI] [PubMed] [Google Scholar]
- 42. Chan KL, Tam TH, Boroumand P, Prescott D, Costford SR, Escalante NK, et al. Circulating NOD1 activators and hematopoietic NOD1 contribute to metabolic inflammation and insulin resistance. Cell Rep 2017;18:2415‐2426. [DOI] [PubMed] [Google Scholar]
- 43. Miyata K, Takaya K. Vacuoles in macrophages and reticular cells of regional lymph nodes of the rat after injection of large doses of steroids. Cell Tissue Res 1983;230:57‐65. [DOI] [PubMed] [Google Scholar]
- 44. Guns PJ, Van Assche T, Verreth W, Fransen P, Mackness B, Mackness M, et al. Paraoxonase 1 gene transfer lowers vascular oxidative stress and improves vasomotor function in apolipoprotein E‐deficient mice with pre‐existing atherosclerosis. Br J Pharmacol 2008;153:508‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Navab M, Hama‐Levy S, Van Lenten BJ, Fonarow GC, Cardinez CJ, Castellani LW, et al. Mildly oxidized LDL induces an increased apolipoprotein J/paraoxonase ratio. J Clin Invest 1997;99:2005‐2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kappelle PJ, de Boer JF, Perton FG, Annema W, de Vries R, Dullaart RP, et al. Increased LCAT activity and hyperglycaemia decrease the antioxidative functionality of HDL. Eur J Clin Invest 2012;42:487‐495. [DOI] [PubMed] [Google Scholar]
- 47. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res 2016;118:653‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature 2005;438:612‐621. [DOI] [PubMed] [Google Scholar]
- 49. Greenspan P, Mayer EP, Fowler SD. Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 1985;100:965‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rozenberg O, Shih DM, Aviram M. Paraoxonase 1 (PON1) attenuates macrophage oxidative status: studies in PON1 transfected cells and in PON1 transgenic mice. Atherosclerosis 2005;181:9‐18. [DOI] [PubMed] [Google Scholar]
- 51. Rosenblat M, Volkova N, Ward J, Aviram M. Paraoxonase 1 (PON1) inhibits monocyte‐to‐macrophage differentiation. Atherosclerosis 2011;219:49‐56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials