

Abstract
We have recently shown that loss of β‐catenin prevents the development of cholestatic liver injury and fibrosis after bile duct ligation (BDL) due to loss of the inhibitory farnesoid X receptor (FXR)/β‐catenin complex, which results in decreased hepatic bile acids (BAs) through activation of FXR. To further understand the role of Wnt/β‐catenin signaling in regulating BA metabolism and cholestasis, we performed BDL on mice in which hepatocyte Wnt signaling is deficient but β‐catenin is intact (low‐density lipoprotein receptor‐related protein [LRP]5/6 knockout [DKO]) as well as mice that have enhanced hepatocyte β‐catenin expression (serine 45 mutated to aspartic acid [S45D] transgenic [TG] mice). Despite decreased biliary injury after BDL, hepatic injury, fibrosis, and inflammation were comparable in DKO and wild‐type (WT) mice. Notably, the FXR/β‐catenin complex was maintained in DKO livers after BDL, coincident with significantly elevated hepatic BA levels. Similarly, TG mice did not display accelerated injury or increased mortality despite overexpression of β‐catenin. There was no augmentation of FXR/β‐catenin association in TG livers; this resulted in equivalent hepatic BAs in WT and TG mice after BDL. Finally, we analyzed the effect of BDL on β‐catenin activity and identified an increase in periportal cytoplasmic stabilization and association with T‐cell factor 4 that correlated with increased expression of distinct downstream target genes. Conclusion: Localization of β‐catenin and expression of Wnt‐regulated genes were altered in liver after BDL; however, neither elimination of Wnt/β‐catenin signaling nor overexpression of β‐catenin in hepatocytes significantly impacted the phenotype or progression of BA‐driven cholestatic injury.
β‐catenin plays a role in regulating bile acid metabolism through an inhibitory association with farnesoid X receptor (FXR), and β‐catenin loss protects mice from bile duct ligation (BDL)‐induced liver injury. In this study, we show that FXR/β‐catenin association and progression of injury is unaffected by either loss of Wnt signaling or overexpression of β‐catenin. We also show that localization of β‐catenin was altered in liver after BDL, and may be activating target genes that are involved in hepatocyte reprogramming.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BA
bile acid
- BDL
bile duct ligation
- BSEP
bile salt export pump
- CD
clusters of differentiation
- Cyp
cytochrome P450
- DKO
low‐density lipoprotein receptor‐related protein knockout
- FXR
farnesoid X receptor
- GS
glutamine synthetase
- IHC
immunohistochemistry
- IP
immunoprecipitation
- KO
conditional knockout
- LRP
low‐density lipoprotein receptor‐related protein
- MRP
multidrug resistance‐associated protein
- Myc
myelocytomatosis oncogene
- NTCP
sodium‐taurocholate cotransporting polypeptide
- OATP4
organic anion transporting polypeptide 4
- Ost
organic solute transporter
- panCK
pan cytokeratin
- RXR
retinoic X receptor
- S45D
serine 45 mutated to aspartic acid
- SHP
small heterodimer partner
- Sox17
sex‐determining region Y‐box 17
- TCF
T‐cell factor
- TG
transgenic
- WB
western blot
- WT
wild type
Cholestasis due to either genetic or environmental factors leads to accumulation of bile acids (BAs) in the liver. Because most BAs are cytotoxic, they can cause injury and inflammation that over time can lead to fibrosis and liver failure.1, 2 Animal models of “toxic bile,” such as bile duct ligation (BDL), a surgical obstruction that results in cholestasis and intrahepatic accumulation of BAs, are widely used to study the progression of cholestatic liver injury and peribiliary fibrosis.3, 4, 5 In this model, toxic BAs have been shown to induce hepatocyte death either directly or through an inflammatory response triggered by cytokine production from injured hepatocytes or cholangiocytes.6, 7, 8 Increased hepatic BA levels also activate adaptive compensatory responses, including decreased BA uptake and synthesis, up‐regulation of apical and basolateral membrane export pumps, and increased BA detoxification.9 Many of these effects are mediated through activation of farnesoid X receptor (FXR), which regulates expression of efflux transporters and also induces expression of small heterodimer partner (SHP). SHP in turn represses the cytochrome P450 (Cyp) enzyme Cyp7a1, which is the first step and also a rate‐limiting step in BA synthesis.10 For this reason, FXR agonists, which decrease BA biosynthesis and increase transport, have been touted as potential treatments for cholestatic liver disease.11, 12, 13 Recently, knockdown or ablation of β‐catenin has also been shown to regulate BA metabolism during BDL through activation of FXR, providing an additional mechanism to modulate this process and enhance therapeutic intervention.
β‐catenin is the chief downstream effector of canonical Wnt signaling. Over the past 2 decades, many laboratories have demonstrated an essential role of β‐catenin in liver physiology and pathology; its normal activity is essential for hepatic development, postnatal hepatic growth, hepatocyte polarity and cell–cell junctions, and hepatocyte maturation (reviewed in Monga14 and Russell and Monga15). Additionally, β‐catenin signaling promotes the development and progression of several liver diseases, including diet‐induced steatohepatitis, fibrosis, and cancer (reviewed in Monga16 and Thompson and Monga17). Of note, several recent studies have highlighted the role of β‐catenin in regulating bile duct morphology and BA metabolism; this may have important implications for the pathogenesis and progression of cholestatic liver disease.
Previous studies have shown that conditional knockout (KO) mice lacking β‐catenin in liver exhibited modestly higher basal hepatic BA than wild‐type (WT) mice; this was likely due to dilated biliary canaliculi and loss of tight junction proteins.18, 19 Despite this, loss of β‐catenin results in maintenance of homeostatic BA levels after BDL in contrast to WT mice, which have significantly higher BA levels.20 Thus, β‐catenin KO mice are protected from BA‐induced cholestatic injury, resulting in fewer bile infarcts, less ductular response and fibrosis, and increased overall survival. The decreased accumulation of BAs occurs because of enhanced FXR activation, which is negatively regulated by β‐catenin through direct binding. Lack of β‐catenin eliminates sequestration and corepression of FXR, resulting in decreased BA synthesis and increased elimination.20 The pool of β‐catenin‐bound FXR is responsive to BA stimulation, which causes transient dissociation of the complex and nuclear localization of FXR. However, the role of Wnt/β‐catenin signaling in regulating this association is unknown.
In order to verify that the activation of FXR after BDL is modulated by the presence of β‐catenin, we used a model of disrupted Wnt signaling in which β‐catenin is still present in the cell as well as a model in which β‐catenin is overexpressed in hepatocytes. We also assessed the activation status of β‐catenin and its downstream target expression after BDL to further understand the role of this pathway in cholestatic liver disease.
Materials and Methods
Animals and Surgery
All mouse studies were performed in accordance with the guidelines of the Institutional Animal Use and Care Committee at the University of Pittsburgh School of Medicine and the National Institutes of Health (protocol number 17071066). Mice were maintained in a temperature‐controlled environment with a 12‐hour light–dark cycle and fed ad libitum standard chow with free access to drinking water.
Liver‐specific low‐density lipoprotein receptor‐related protein (LRP)5/6 KO (DKO) mice under the albumin‐cre promoter were generated as described and maintained in a mixed background.21, 22 Mice overexpressing a stable form of β‐catenin, where serine 45 is mutated to aspartic acid (S45D), have been characterized and were bred for >8 generations into a C57BL6 background.23 Male C57BL6 mice were also purchased from Jackson Laboratories.
At >14 weeks of age, male C57BL6 mice, male and female DKO mice and their WT littermates, or male and female mice transgenic (TG) for S45D‐mutated β‐catenin in the liver and age‐matched WT controls were subjected to BDL by dissecting the common bile duct above the pancreas and cutting it between two 5‐0 silk ligatures. Mice were killed at 3 and 7 days (TG/WT) or 14 days (DKO/WT, C57BL6) after BDL. Blood samples were collected from the inferior vena cava at the time of death. For C57BL6 studies, four mice at baseline and five mice after BDL were analyzed. For both DKO/WT and TG/WT at baseline, three mice per genotype were analyzed. Five WT and five DKO mice were analyzed 14 days after BDL, six WT and seven TG mice were analyzed 3 days after BDL, and eight WT and seven TG mice were analyzed 7 days after BDL.
Additional methods are available in the Supporting Materials.
Results
Mice Lacking LRP5/6 in Hepatocytes Have Equivalent Injury to WT Mice After BDL
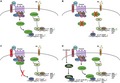
To address any potential role of Wnt signaling in either positive or negative regulation of the inhibitory β‐catenin/FXR complex, we subjected liver‐specific LRP5/6 double knockout mice and their WT counterparts to BDL and killed them after 14 days.22 Western blot (WB) confirmed maintenance of β‐catenin expression but loss of β‐catenin transcriptional target glutamine synthetase (GS) in DKO mice before and after BDL, demonstrating inhibited β‐catenin activation (Supporting Fig. S1A). Analysis of serum biochemistry showed no change in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) between WT and DKO mice after BDL; however, both alkaline phosphatase (ALP) and bilirubin were significantly decreased in DKO mice (Fig. 1A). To determine whether this resulted in a protective phenotype similar to β‐catenin KO mice, we next examined the histology of WT and DKO livers after BDL. We found no significant differences in the number of bile infarcts (Fig. 1B). Furthermore, fibrosis, as assessed by sirius red, was equivalent in both WT and DKO livers (Fig. 1C).
Figure 1.
LRP5/6 KO (DKO) mice have decreased biliary injury but equivalent parenchymal injury, fibrosis, inflammation, and ductular response after BDL. (A) Both ALP and bilirubin are significantly decreased in DKO livers at 14 days after BDL compared to WT livers, while AST and ALT are equivalent; **P < 0.01 versus WT+BDL (t test). (B) No change in the number of bile infarcts in WT and DKO mice after BDL (magnification ×100). (C) Sirius red staining shows equivalent fibrosis in DKO mice after BDL compared to WT mice (magnification ×100). (D) IHC shows fewer PCNA + proliferating hepatocytes in DKO mice after BDL compared to WT mice (magnification ×100); ****P < 0.0001 versus WT+BDL (t test). (E) IHC for panCK shows that ductular response is unchanged between DKO and WT mice after BDL (magnification ×100). (F) Inflammation is equivalent in DKO and WT mice after BDL, as assessed by CD45 IHC (magnification ×100). Horizontal lines in the graphs represent mean ± SEM. Abbreviations: H&E, hematoxylin and eosin; IU, international units; PCNA, proliferating cell nuclear antigen.
Histologically, DKO livers had significantly fewer proliferating hepatocytes than WT livers after BDL (Fig. 1D), which is in line with the described role of canonical Wnt signaling in regulating β‐catenin during liver regeneration.22 This did not translate to increased parenchymal injury in DKO livers, however, as markers of hepatic injury were not significantly altered (Fig. 1A). The extent of ductular response, as assessed by pan cytokeratin (panCK) immunohistochemistry (IHC), was similar in both WT and DKO mice, despite decreased markers of biliary injury (Fig. 1E). Furthermore, clusters of differentiation (CD)45 IHC showed a strong periductal inflammatory response that was equivalent in both WT and DKO mice (Fig. 1F). Taken together, these findings showed that DKO mice, unlike β‐catenin KO mice, are not protected from BDL‐induced cholestasis but instead phenocopy WT mice.
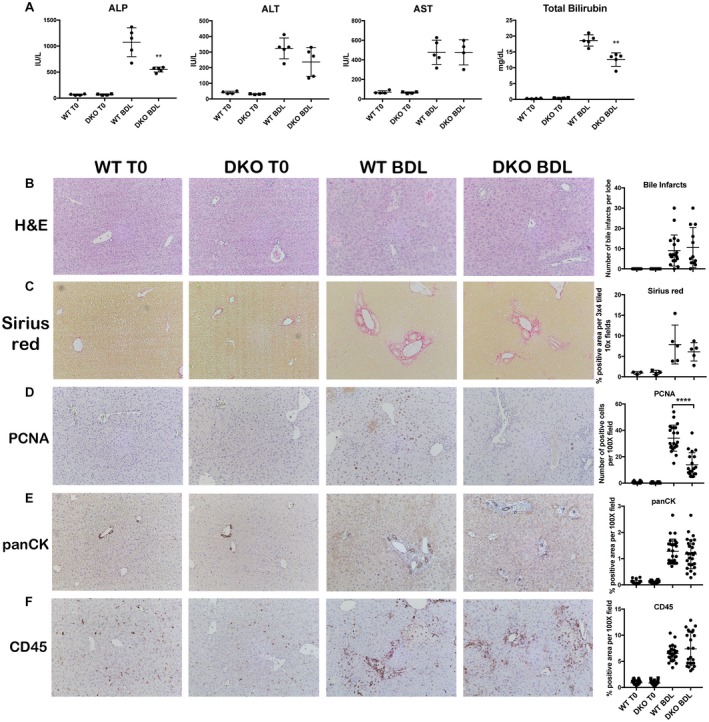
DKO MICE HAVE MODEST CHANGES IN SOME BA SYNTHESIS, UPTAKE, AND EXPORT GENES AFTER BDL COMPARED TO WT MICE
We next wanted to determine if comparable regulation of BA synthesis, metabolism, and transport after BDL contributes to the equivalent phenotype of DKO and WT mice. SHP, which is activated by FXR and suppresses BA synthesis through repression of Cyp7a1 expression,10 is unchanged in DKO mice before and after BDL. However, Cyp7a1, which catalyzes the rate‐limiting step in the classical BA biosynthesis pathway, is decreased in DKO mice at baseline, similar to β‐catenin KO mice,20 and is nonsignificantly decreased after BDL (Fig. 2A). Cyp27, a key enzyme in the alternative BA synthesis pathway, is likewise suppressed in DKO mice before and after BDL, while Cyp8b1 expression is unchanged (Fig. 2A). Interestingly, Cyp2b10 and Cyp3a11, which detoxify BA and are up‐regulated in β‐catenin KO mice after BDL,20 are not induced in DKO mice (Fig. 2B).
Figure 2.
Hepatic BAs in DKO mice are comparable to WT mice after BDL despite changes in BA metabolism genes. (A) Expression of SHP is unchanged between WT and DKO mice, while Cyp7a1 and Cyp27 are decreased in DKO mice before and after BDL. (B) Detoxification enzymes Cyp2b10 and Cyp3a11 are not induced in DKO mice after BDL. (C) Expression of uptake transporter OATP4 is decreased in DKO mice before and after BDL, and NTCP is decreased at baseline. (D) Expression of efflux transporter MRP2 is decreased in DKO mice after BDL, while BSEP is unaffected; additionally, MRP4 is significantly decreased in DKO mice compared to WT mice after BDL. (E) Total liver BAs are elevated in DKO mice at baseline but are equivalent to WT mice after BDL. (F) IP with FXR shows maintenance of the FXR/β‐catenin complex in DKO mice before and after BDL. For A‐E, *P < 0.05, **P < 0.01, and ***P < 0.001 versus WT (t test). Data in the graphs represent mean ± SEM. Abbreviations: IgG, immunoglobulin G; mRNA, messenger RNA; ns, not significant.
Sodium‐taurocholate cotransporting polypeptide (NTCP), a basolateral uptake transporter, was decreased in DKO compared to WT mice before BDL but equivalent after BDL, while organic anion transporting polypeptide 4 (OATP4), another uptake transporter, was decreased in DKO mice before and after BDL (Fig. 2C). Apical transporters bile salt export pump (BSEP) and multidrug resistance‐associated protein 2 (MRP2), which efflux bile into the biliary canaliculi, showed a differential regulation in DKO mice, with BSEP remaining equivalent to WT mice while MRP2 decreased significantly in DKO mice after BDL (Fig. 2D). Similarly, MRP4 but not MRP3, both of which efflux bile from the basolateral membrane of hepatocytes into the blood when biliary excretion is impaired,24 decreased in DKO mice after BDL (Fig. 2D). The organic solute transporter (Ost)α/β also participates in alternative basolateral BA export and is a target of FXR.25 Both subunits were unaffected by loss of Wnt signaling before or after BDL, although expression of Ostβ increased significantly after BDL, indicating FXR activation and increased efflux (Supporting Fig. S2A). Altogether, these findings suggest that DKO mice have decreased BA synthesis after BDL, similar to β‐catenin KO mice. However, loss of Wnt signaling did not further induce FXR activity, as demonstrated by the lack of SHP up‐regulation and equivalent efflux transporter expression.
Hepatic BA Levels are Equivalent in WT and DKO Mice and Coincide With Maintenance of the FXR/β‐Catenin Complex
To determine the net effect of these changes in BA metabolism genes, we analyzed total BA levels in livers of WT and DKO mice before and after BDL. At baseline, total BAs were mildly but significantly increased in DKO livers; however, after BDL, BAs were equivalent in both genotypes (Fig. 2E). Immunoprecipitation (IP) with FXR antibody was next performed to assess the dynamics of the FXR/β‐catenin complex. WB for β‐catenin showed comparable FXR/β‐catenin association in both WT and DKO livers before BDL (Fig. 2F); after BDL, FXR/β‐catenin association was maintained and may even have increased in DKO livers compared to both WT after BDL and DKO livers at baseline. Thus, the susceptibility of DKO mice to BDL‐induced cholestasis may be attributed to persistence of the FXR/β‐catenin complex, which results in equivalent BA accumulation and injury in WT and DKO livers.
Mice With β‐Catenin Overexpression in Hepatocytes Have Equivalent Injury to WT After BDL
We next examined the impact of β‐catenin overexpression on FXR/β‐catenin association, BA metabolism, and biliary injury. We have previously shown that while TG mice are able to successfully prevent excessive activation of β‐catenin at homeostasis, hepatocytes isolated from these TG mice have a growth advantage in culture. Additionally, the presence of excess β‐catenin provides a regenerative advantage to TG livers after partial hepatectomy.23 We subjected TG mice and age‐matched WT to BDL and killed mice after 7 days instead of 14 because we hypothesized that TG mice would have accelerated injury or mortality due to decreased FXR activation and increased BA accumulation. WB confirmed that expression of β‐catenin (and its surrogate activation target GS) were not significantly increased in TG mice before or after BDL. This is consistent with our published report showing that TG livers have adapted to and successfully monitored excess β‐catenin by 2 months of age (Supporting Fig. S1B).23 Analysis of serum biochemistry also showed no significant differences in either biliary (ALP, bilirubin) or hepatic (AST, ALT) injury in TG mice after BDL (Fig. 3A). Survival was unchanged between WT and TG mice over the duration of the experiment (Supporting Fig. S3A). Hematoxylin and eosin staining showed that the number of bile infarcts between WT and TG mice were equivalent (Fig. 3B); fibrosis after BDL was also unchanged between the two genotypes (Fig. 3C).
Figure 3.
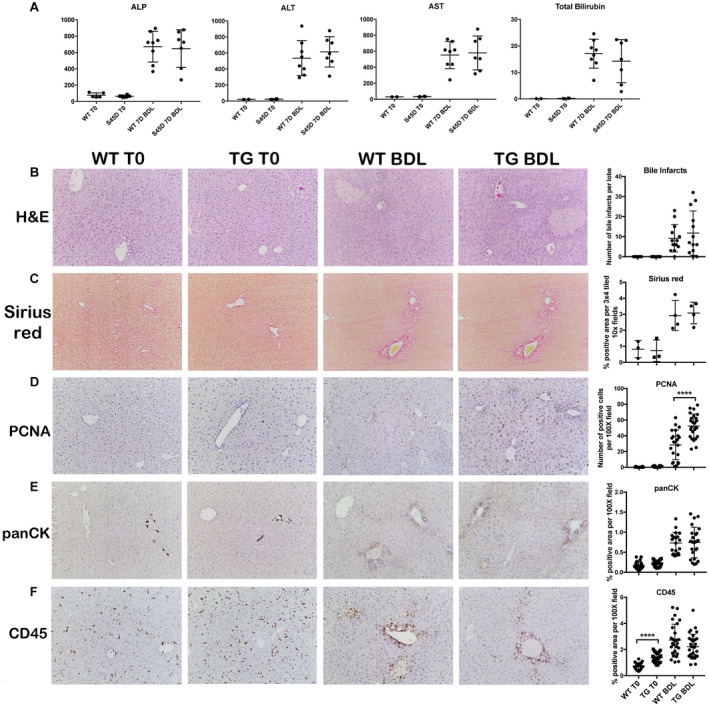
Overexpression of mutated nondegradable S45D β‐catenin does not increase injury, fibrosis, inflammation, or ductular response following BDL. (A) There is no difference in hepatic (AST, ALT) or biliary (ALP, bilirubin) injury between WT and TG mice after BDL. (B) No change in the number of bile infarcts in WT and TG mice after BDL (magnification ×100). (C) Sirius red staining shows equivalent fibrosis in TG mice after BDL compared to WT mice (magnification ×100). (D) IHC shows an increased number of PCNA+ proliferating hepatocytes in TG mice after BDL compared to WT mice (magnification ×100); ****P < 0.0001 versus WT+BDL (t test). (E) Ductular response is equivalent between TG and WT mice after BDL, as assessed by panCK IHC (magnification ×100). (F) Although CD45 staining is increased in TG compared to WT mice at baseline, there is no difference in inflammation between TG and WT mice after BDL (magnification ×100). Data in the graphs represent mean ± SEM. Abbreviations: H&E, hematoxylin and eosin; PCNA, proliferating cell nuclear antigen.
Unsurprisingly, TG livers had more proliferating hepatocytes after BDL owing to the referenced role of β‐catenin in inducing proliferation (Fig. 3D). However, the extent of ductular response and inflammation was similar in both WT and TG livers after 7 days of BDL (Fig. 3E,F), although inflammation was increased in TG livers at baseline. To ensure that we did not miss an earlier onset of injury that was compensated for at a later time point, we killed WT and TG mice after 3 days of BDL. TG mice did not have significantly higher levels of ALP, ALT, AST, or bilirubin at 3 days after BDL compared to WT mice, indicating that excess β‐catenin did not worsen injury after BDL (Supporting Fig. S3B).
BA Metabolism and FXR/β‐Catenin Association are Equivalent in TG and WT Mice After BDL
As with DKO mice, we next analyzed BA metabolism after BDL to determine the effect of β‐catenin overexpression on this process. Interestingly, SHP expression was maintained in TG mice at baseline levels after BDL compared to WT mice. Expression of BA synthesis genes Cyp7a1, Cyp27, and Cyp8b1 were comparable in TG compared to WT mice (Fig. 4A). There were also no significant changes in expression of uptake transporters NTCP and OATP4 or BA detoxification genes Cyp2b10 or Cypa11 between WT and TG mice after BDL (Fig. 4B,C). Likewise, efflux transporters, such as MRP4 and Ostα/β, were largely unchanged between WT and TG mice after BDL. The exception was MRP3, which was significantly decreased in TG compared to WT mice (Fig. 4D; Supporting Fig. S2B). Analysis of livers harvested 3 days after BDL also showed no significant differences between WT and TG mice in expression of SHP or Cyp7a1 (Supporting Fig. S3C), demonstrating that β‐catenin overexpression did not affect the synthesis and transport of BAs after BDL.
Figure 4.
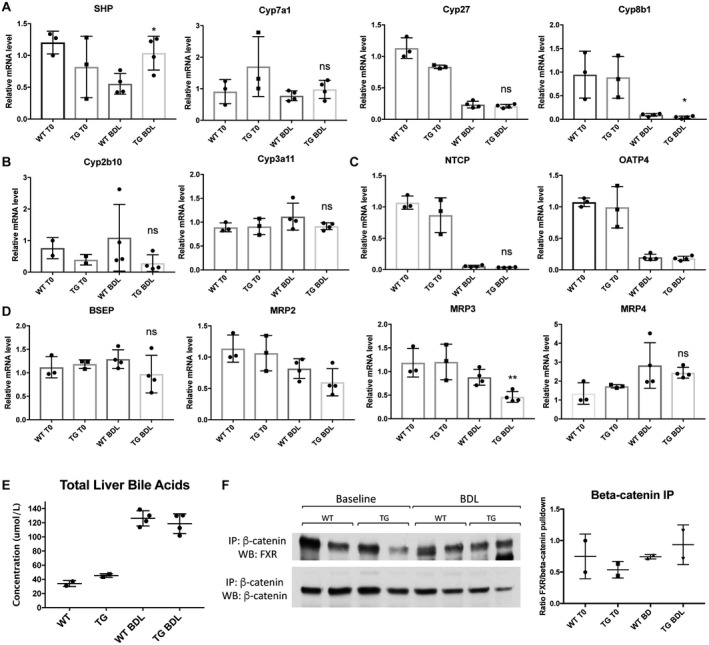
BA metabolism and FXR/β‐catenin association are comparable in TG and WT mice after BDL. (A) Expression of SHP is increased in TG compared to WT mice after BDL, while Cyp7a1 and Cyp27 are unchanged before and after BDL. (B) Detoxification P450 enzymes are equivalently expressed in WT and TG mice after BDL. (C) BA uptake is unaffected by overexpression of β‐catenin after BDL. (D) Expression of apical and basolateral BA exporters are not different between WT and TG mice in response to BDL, with the exception of MRP3, which is decreased in TG compared to WT mice. (E) Total liver BAs are equivalent in TG and WT mice after BDL. (F) IP with β‐catenin shows maintenance of the FXR/β‐catenin complex in TG mice before and after BDL. For A‐E, *P < 0.05 and **P < 0.01 versus WT (t test). Data in the graphs represent mean ± SEM. Abbreviations: mRNA, messenger RNA; ns, not significant.
As expected from the lack of change in BA metabolism genes, total liver BAs were similarly increased in WT and TG mice after 7 days of BDL (Fig. 4E). IP with β‐catenin antibody followed by WB for FXR showed an equivalent FXR/β‐catenin association in WT and TG mice at baseline and after BDL (Fig. 4F); similar results were obtained when FXR antibody was used for IP (data not shown). Thus, overexpression of S45D β‐catenin in hepatocytes did not adversely impact hepatic BA levels or alter the FXR/β‐catenin complex after BDL.
BDL Induces a Distinct Pattern of β‐Catenin Activation and Transcriptional Targets in Liver
Both DKO and TG mice phenocopied the WT after BDL, and maintenance of the FXR/β‐catenin complex appeared to be critical for the progression of injury. To further understand the role of Wnt/β‐catenin signaling in cholestasis, we next examined the activation of β‐catenin after BDL. WB showed that there was no net change in total β‐catenin protein levels after BDL (Fig. 5A); however, the distribution of β‐catenin changed significantly, as shown by IHC (Fig. 5B). At baseline, β‐catenin was strongly localized to the hepatocyte membrane. After BDL, much of this membranous β‐catenin was lost, and cytoplasmic stabilization was observed in periportal hepatocytes. Because of increased biliary mass, there was also notably more β‐catenin in the cholangiocyte compartment compared to baseline (Fig. 5B). These results suggest that BDL induced a differential expression of β‐catenin that may impact target gene expression and function.
Figure 5.
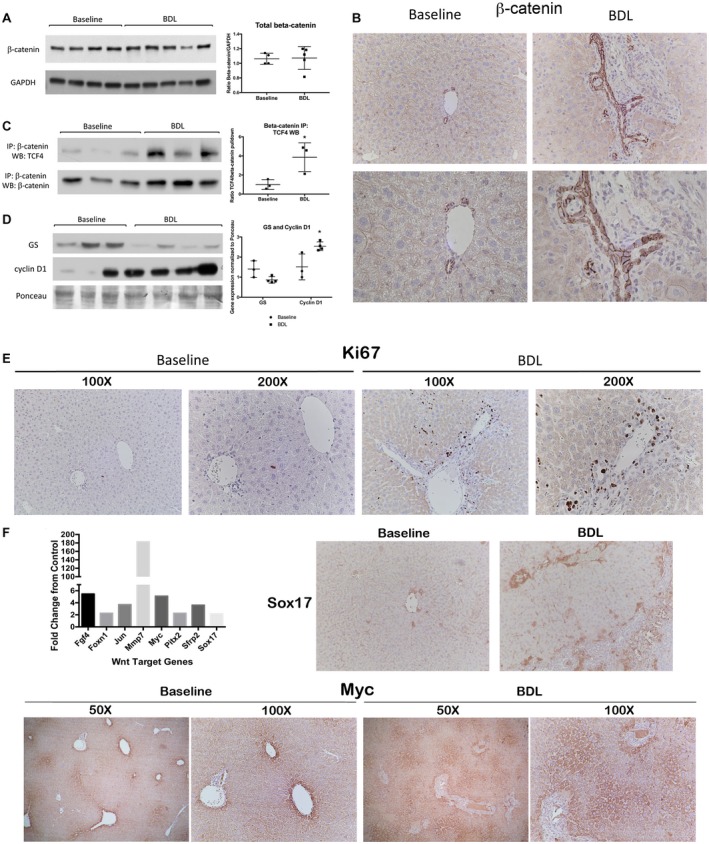
A distinct set of β‐catenin target genes is activated in liver after BDL. (A) WB and quantification show no change in total β‐catenin protein expression after BDL. (B) IHC shows that at baseline β‐catenin is primarily localized to the hepatocyte membrane, while after BDL, there is cytoplasmic staining in periportal hepatocytes and an increased proportion of total β‐catenin in the cholangiocyte compartment (top row images, magnification ×200; bottom row images, magnification ×400). (C) IP shows that TCF4/β‐catenin association increases after BDL in WT livers; *P < 0.05 versus baseline (t test). (D) WB shows an increase in cyclin D1 protein expression in WT livers after BDL, while expression of GS is decreased; *P < 0.05 versus baseline (t test). (E) Ki67 IHC demonstrates that BDL induces robust proliferation of cholangiocytes and nonparenchymal cells but not hepatocytes. (F) Left: There is increased expression of several Wnt/β‐catenin target genes in WT livers after BDL. Right: Sox17 staining is increased in the periportal region after BDL (magnification ×100). Bottom: Myc expression, which is normally restricted to the central vein, expands to the mid‐zonal and periportal regions after BDL, as assessed by IHC (magnification ×100). Data in A,C,D represent mean ± SEM. Abbreviations: Fgf4, fibroblast growth factor 4; Foxn1, forkhead box N1; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Jun, jun proto‐oncogene; Mmp7, matrix metallopeptidase 7; Pitx2, paired‐like homeodomain transcription factor 2; Sfrp2, secreted frizzled‐related protein 2.
We next examined the activation status of β‐catenin after BDL. IP studies demonstrated that BDL increased the T‐cell factor 4 (TCF4)/β‐catenin association, which coincided with β‐catenin stabilization in periportal hepatocytes (Fig. 5C). WB showed a significant decrease in GS expression after BDL but increased expression of cyclin D1 (Fig. 5D). However, the increase in cyclin D1 was likely due to robust proliferation of cholangiocytes and nonparenchymal cells because we observed very few proliferating hepatocytes by Ki67 staining (Fig. 5E). In order to gain more insight into the role of catenin activation during BDL, we used a polymerase chain reaction array to profile the expression of genes related to Wnt‐mediated signal transduction. Analysis revealed up‐regulation of genes, such as jun proto‐oncogene (Jun), matrix metallopeptidase 7 (Mmp7), myelocytomatosis oncogene (Myc), and sex‐determining region Y‐box 17 (Sox17), after BDL (Fig. 5F), several of which are not normally expressed in liver. At baseline, Sox17 was expressed at low abundance in hepatocytes near the central vein; however, IHC showed a marked increase in Sox17 expression after BDL, with staining appearing in periportal hepatocytes as well (Fig. 5F). Similarly, Myc, which was restricted primarily to the central vein at baseline, expanded into both the midzonal and periportal regions after BDL (Fig. 5F). Altogether, these data suggest that β‐catenin was preferentially activated in the periportal region after BDL where it induced expression of a unique set of transcriptional targets during cholestasis.
Discussion
In these studies, we sought to expand on previous findings that absence of β‐catenin protein and not its transcriptional activity regulates FXR during cholestasis. In our previous publication,20 we showed that β‐catenin small interfering RNA increased SHP activity secondary to a decreased FXR/β‐catenin association, while treatment with ICG‐001, a small molecule that antagonizes β‐catenin/TCF4 transcriptional activity without reducing protein levels of β‐catenin, does not induce SHP. To determine the relevance of these findings in the development and progression of cholestasis in vivo, we used LRP5/6 KO (DKO) mice, which have disrupted canonical Wnt signaling but intact β‐catenin.22 We showed that despite decreased biliary injury, DKO mice are as susceptible to BDL‐induced cholestasis as WT mice. Maintenance of the FXR/β‐catenin complex is associated with lack of SHP induction, equivalent liver BAs and bile infarcts, and similar accumulation of fibrosis and inflammation in DKO mice after BDL, supporting our hypothesis that the physical presence of β‐catenin prevents optimal FXR activation.
Notably, however, DKO mice had a significant decrease in serum markers of biliary injury after BDL compared to their WT counterparts as well as increased level of hepatic BAs at baseline, which parallels findings in β‐catenin KO mice.20 Compensatory down‐regulation of Cyp7a1 (seen in both β‐catenin transcriptionally deficient models) likely occurs in response to the retention of liver BAs. However, although hepatic BA levels are ~17.5‐fold higher in WT mice after BDL, they only increase ~9‐fold in DKO mice. Decreased expression of both Cyp7a1 and Cyp27, a direct target of Wnt/β‐catenin signaling,26 in DKO mice may account for this milder elevation in BAs after BDL and also explain the decreased biliary injury. However, suppression of BA synthesis alone is insufficient to protect DKO livers from other deleterious effects of BDL, such as bile infarcts, inflammation, fibrosis, and ductular response. Our studies in Wnt signaling‐deficient mice, therefore, confirm that most of the protective phenotype in β‐catenin KO mice can be attributed to the lack of a physical association of FXR and β‐catenin and not the lack of β‐catenin transcriptional activity.
We previously demonstrated that FXR activation can be altered by changing the amounts of FXR binding partners in the cell. Overexpression of retinoic X receptor (RXR)α increased FXR/RXRα heterodimer formation at the expense of FXR/β‐catenin association.20 Similarly, stable nondegradable S33Y β‐catenin increased the FXR/β‐catenin association and suppressed SHP activity compared to cells with WT β‐catenin.20 Surprisingly, however, mice TG for S45D β‐catenin, a nondegradable form of the protein,23 did not show accelerated injury or significant mortality at either 3 or 7 days after BDL but instead had comparable injury parameters to their WT counterparts at both time points. We found similar levels of hepatic BAs in both groups; this was likely due to the equivalent FXR/β‐catenin association in TG and WT mice both before and after BDL. One explanation for the discrepancy between in vitro (S33Y) and in vivo (S45D) findings is that mutation of serine 45 to aspartic acid could change the secondary structure of β‐catenin, disallowing its physical association with FXR. In this case, only endogenous β‐catenin, which is still present in TG livers, will bind to and inhibit FXR. Another explanation is that the different “pools” of β‐catenin in the cell are functionally distinct, with the pool bound to FXR unaffected by excess β‐catenin, which is sequestered at the hepatocyte membrane in association with E‐cadherin in TG livers.23 In the presence of an activating stimulus, such as BDL, membrane‐bound S45D β‐catenin preferentially translocates to the nucleus, contributing to the increased hepatocyte proliferation seen in TG mice after BDL rather than to the FXR/β‐catenin complex.
In support of this hypothesis, our data show that, overall, FXR activation during cholestasis is likely unaffected by active Wnt/β‐catenin signaling. The association of FXR with β‐catenin is not significantly altered after BDL (see Figs. 2F and 4F, WT lanes), nor does loss of Wnt signaling in LRP5/6 KO mice affect this complex. The β‐catenin/TCF4 association does increase after BDL, however, suggesting that transcriptionally active β‐catenin is distinct from and does not affect the pool of FXR‐bound β‐catenin. Instead, this complex is likely regulated by BAs as we have previously shown a transient dissociation of FXR/β‐catenin following stimulation both in vitro and in vivo with the BA agonist GW4064.20
The increased association of β‐catenin/TCF4 after BDL led us to investigate whether cholestasis causes transcriptional activation of β‐catenin. Surprisingly, expression of GS, a surrogate β‐catenin target gene, decreased after BDL. Loss of GS expression from hepatocytes could indicate a shift in β‐catenin activation to other zones of liver at the expense of pericentral targets like GS. Indeed, we identified a distinct set of β‐catenin target genes that showed increased expression in the periportal region after BDL, including Sox17 and Myc. β‐catenin‐dependent Sox17 expression regulates endoderm specification and also plays a role in maturation of gallbladder and bile duct epithelia during late organogenesis.27, 28, 29 Myc is a key factor in the reprogramming of fibroblasts to induced pluripotent stem cells.30, 31 Based on this, we believe that β‐catenin is activating an atypical set of target genes during cholestasis that may be involved in either hepatocyte to biliary reprogramming or cholangiocyte growth. We have previously described increased expression of Wnt7A during cholestasis that activates a biliary profile in periportal hepatocytes in a β‐catenin‐dependent manner,32 supporting this hypothesis. Transcriptional co‐activators that regulate target gene expression in a spatiotemporal and context‐dependent manner, as described by others, may be critical in regulating the switch between pericentral‐ and periportal‐specific β‐catenin‐driven gene expression.33, 34
In conclusion, we show that the FXR/β‐catenin complex is not under the control of Wnt signaling, which is instead activating target genes that may be involved in hepatocyte reprogramming after BDL. These data also support the hypothesis that the FXR/β‐catenin association, or lack thereof, is a key factor in determining susceptibility to cholestatic injury (Fig. 6). Our in‐depth mechanistic analysis provides additional insight into the role of β‐catenin signaling during cholestasis that is essential in designing inhibitors of this pathway for targeted therapeutics.
Figure 6.
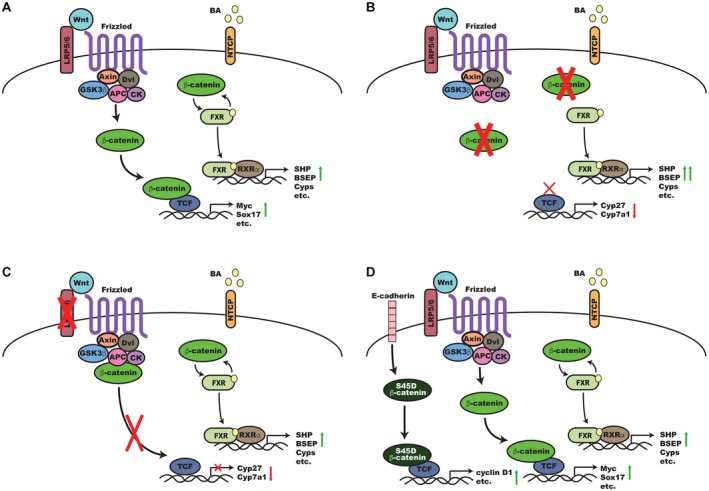
A proposed model for the regulation of β‐catenin and FXR activity during cholestasis. (A) In a normal liver after BDL, Wnts, such as Wnt7A, bind to coreceptors Frizzled and LRP5/6 KO, which triggers recruitment of Dvl and the β‐catenin destruction complex (Axin, APC, CK, andGSK3β) to the membrane. This results in hypophosphorylation of β‐catenin and its translocation to the nucleus where it binds to TCF to induce expression of target genes, such as Sox17 and Myc. Simultaneously, BAs accumulate and are taken up by NTCP; BAs bind to and activate FXR, which transiently dissociates from the inhibitory β‐catenin complex and, in partnership with RXRα, induces expression of target genes, such as BSEP and SHP. (B) In the absence of β‐catenin, transcriptional targets, such as Cyp27, are suppressed, while increased basal hepatic BAs also lead to compensatory suppression of Cyp7a1. Simultaneously, loss of the inhibitory β‐catenin/FXR complex results in increased activation of FXR, increased expression of downstream targets, such as SHP and BSEP, and optimal reduction and alteration of the hepatic BA pool during cholestasis. (C) In the absence of Wnt coreceptor LRP5/6, β‐catenin target genes are suppressed, but the continued presence of β‐catenin represses FXR and prevents optimal activation. (D) Mice overexpressing β‐catenin have similar kinetics of β‐catenin and FXR activation as normal mice except that S45D β‐catenin, which is usually sequestered at the hepatocyte membrane, is activated and turns on expression of additional β‐catenin targets, such as cyclin D1, that are involved in hepatocyte proliferation. Abbreviations: APC, adenomatous polyposis coli; Dvl, Dishevelled; GSK3, glycogen synthase kinase 3.
Supporting information
Supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (grant 1R01DK103775 to K.N.B.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Heuman DM, Mills AS, McCall J, Hylemon PB, Pandak WM, Vlahcevic ZR. Conjugates of ursodeoxycholate protect against cholestasis and hepatocellular necrosis caused by more hydrophobic bile salts. In vivo studies in the rat. Gastroenterology 1991;100:203‐211. [DOI] [PubMed] [Google Scholar]
- 2. Yerushalmi B, Dahl R, Devereaux MW, Gumpricht E, Sokol RJ. Bile acid‐induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 2001;33:616‐626. [DOI] [PubMed] [Google Scholar]
- 3. Slott PA, Liu MH, Tavoloni N. Origin, pattern, and mechanism of bile duct proliferation following biliary obstruction in the rat. Gastroenterology 1990;99:466‐477. [DOI] [PubMed] [Google Scholar]
- 4. Georgiev P, Jochum W, Heinrich S, Jang JH, Nocito A, Dahm F, et al. Characterization of time‐related changes after experimental bile duct ligation. Br J Surg 2008;95:646‐656. [DOI] [PubMed] [Google Scholar]
- 5. Johnstone JM, Lee EG. A quantitative assessment of the structural changes the rat's liver following obstruction of the common bile duct. Br J Exp Pathol 1976;57:85‐94. [PMC free article] [PubMed] [Google Scholar]
- 6. Miyoshi H, Rust C, Roberts PJ, Burgart LJ, Gores GJ. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999;117:669‐677. [DOI] [PubMed] [Google Scholar]
- 7. Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte‐specific inflammatory response. JCI Insight 2017;2:e90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alpini G, Glaser SS, Ueno Y, Rodgers R, Phinizy JL, Francis H, et al. Bile acid feeding induces cholangiocyte proliferation and secretion: evidence for bile acid‐regulated ductal secretion. Gastroenterology 1999;116:179‐186. [DOI] [PubMed] [Google Scholar]
- 9. Chiang JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol 2004;40:539‐551. [DOI] [PubMed] [Google Scholar]
- 10. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Mol Cell 2000;6:517‐526. [DOI] [PubMed] [Google Scholar]
- 11. Boyer JL. New perspectives for the treatment of cholestasis: lessons from basic science applied clinically. J Hepatol 2007;46:365‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fiorucci S, Clerici C, Antonelli E, Orlandi S, Goodwin B, Sadeghpour BM, et al. Protective effects of 6‐ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen‐induced cholestasis. J Pharmacol Exp Ther 2005;313:604‐612. [DOI] [PubMed] [Google Scholar]
- 13. Liu Y, Binz J, Numerick MJ, Dennis S, Luo G, Desai B, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra‐ and extrahepatic cholestasis. J Clin Invest 2003;112:1678‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monga SP. Role and regulation of beta‐catenin signaling during physiological liver growth. Gene Expr 2014;16:51‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Russell JO, Monga SP. Wnt/beta‐catenin signaling in liver development, homeostasis, and pathobiology. Annu Rev Pathol 2018;13:351‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monga SP. beta‐Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 2015;148:1294‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thompson MD, Monga SP. WNT/beta‐catenin signaling in liver health and disease. Hepatology 2007;45:1298‐1305. [DOI] [PubMed] [Google Scholar]
- 18. Behari J, Yeh TH, Krauland L, Otruba W, Cieply B, Hauth B, et al. Liver‐specific beta‐catenin knockout mice exhibit defective bile acid and cholesterol homeostasis and increased susceptibility to diet‐induced steatohepatitis. Am J Pathol 2010;176:744‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yeh TH, Krauland L, Singh V, Zou B, Devaraj P, Stolz DB, et al. Liver‐specific beta‐catenin knockout mice have bile canalicular abnormalities, bile secretory defect, and intrahepatic cholestasis. Hepatology 2010;52:1410‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson MD, Moghe A, Cornuet P, Marino R, Tian J, Wang P, et al. beta‐Catenin regulation of farnesoid X receptor signaling and bile acid metabolism during murine cholestasis. Hepatology 2018;67:955‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta‐catenin reveals its role in liver growth and regeneration. Gastroenterology 2006;131:1561‐1572. [DOI] [PubMed] [Google Scholar]
- 22. Yang J, Mowry LE, Nejak‐Bowen KN, Okabe H, Diegel CR, Lang RA, et al. beta‐Catenin signaling in murine liver zonation and regeneration: a Wnt‐Wnt situation!. Hepatology 2014;60:964‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nejak‐Bowen KN, Thompson MD, Singh S, Bowen WC Jr, Dar MJ, Khillan J, et al. Accelerated liver regeneration and hepatocarcinogenesis in mice overexpressing serine‐45 mutant beta‐catenin. Hepatology 2010;51:1603‐1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kock K, Brouwer KL. A perspective on efflux transport proteins in the liver. Clin Pharmacol Ther 2012;92:599‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL. Genome‐wide tissue‐specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 2010;51:1410‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gougelet A, Torre C, Veber P, Sartor C, Bachelot L, Denechaud PD, et al. T‐cell factor 4 and beta‐catenin chromatin occupancies pattern zonal liver metabolism in mice. Hepatology 2014;59:2344‐2357. [DOI] [PubMed] [Google Scholar]
- 27. Sinner D, Rankin S, Lee M, Zorn AM. Sox17 and beta‐catenin cooperate to regulate the transcription of endodermal genes. Development 2004;131:3069‐3080. [DOI] [PubMed] [Google Scholar]
- 28. Engert S, Burtscher I, Liao WP, Dulev S, Schotta G, Lickert H. Wnt/beta‐catenin signalling regulates Sox17 expression and is essential for organizer and endoderm formation in the mouse. Development 2013;140:3128‐3138. [DOI] [PubMed] [Google Scholar]
- 29. Uemura M, Ozawa A, Nagata T, Kurasawa K, Tsunekawa N, Nobuhisa I, et al. Sox17 haploinsufficiency results in perinatal biliary atresia and hepatitis in C57BL/6 background mice. Development 2013;140:639‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Okita K, Ichisaka T, Yamanaka S. Generation of germline‐competent induced pluripotent stem cells. Nature 2007;448:313‐317. [DOI] [PubMed] [Google Scholar]
- 31. Araki R, Hoki Y, Uda M, Nakamura M, Jincho Y, Tamura C, et al. Crucial role of c‐Myc in the generation of induced pluripotent stem cells. Stem Cells 2011;29:1362‐1370. [DOI] [PubMed] [Google Scholar]
- 32. Okabe H, Yang J, Sylakowski K, Yovchev M, Miyagawa Y, Nagarajan S, et al. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology 2016;64:1652‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta‐catenin in vertebrates. EMBO J 2000;19:1839‐1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta‐catenin‐mediated survivin gene expression. Oncogene 2005;24:3619‐3631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials