

Abstract
A significantly higher proportion of patients with nonalcoholic steatohepatitis (NASH) who received obeticholic acid (OCA) had histological improvement relative to placebo in the FLINT (farnesoid X nuclear receptor ligand obeticholic acid for noncirrhotic, NASH treatment) trial. However, genetic predictors of response to OCA are unknown. We conducted a genome‐wide association study (GWAS) in FLINT participants to identify variants associated with NASH resolution and fibrosis improvement. Genotyping was performed using the Omni2.5 content GWAS chip. To avoid false positives introduced by population stratification, we focused our GWAS on white participants. Six regions on chromosomes 1, 4, 6, 7, 15, and 17 had multiple single nucleotide polymorphisms (SNPs) with suggestive association (P < 1 × ) with NASH resolution. A sentinel SNP, rs75508464, near CELA3B on chromosome 1 was associated with NASH resolution, improvement in the nonalcoholic fatty liver disease activity score, portal inflammation, and fibrosis. Among individuals carrying this allele, 83% achieved NASH resolution with OCA compared with only 33% with placebo. Eight regions on chromosomes 1, 2, 3, 11, 13, and 18 had multiple SNPs associated with fibrosis improvement; of these, rs12130403 near TDRD10 on chromosome 1 was also associated with improvement in NASH and portal inflammation, and rs4073431 near ANO3 on chromosome 11 was associated with NASH resolution and improvement in steatosis. Multiple SNPs on chromosome 11 had suggestive association with pruritus, with rs1379650 near ANO5 being the top SNP. Conclusion: We identified several variants that may be associated with histological improvement and pruritus in individuals with NASH receiving OCA. The rs75508464 variant near CELA3B may have the most significant effect on NASH resolution in those receiving OCA.
We conducted a genome wide association study (GWAS) in FLINT trial participants to identify variants associated with NASH resolution and fibrosis improvement. We identified several variants associated with histological improvement and pruritus in individuals with NASH receiving obeticholic acid (OCA). The rs75508464 variant near CELA3B had the most significant effect on NASH resolution in those receiving OCA.

Abbreviations
- ALT
alanine aminotransferase
- BMI
body mass index
- chr
chromosome
- GWAS
genome‐wide association study
- MAF
minor allele frequency
- NAFLD
nonalcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
nonalcoholic steatohepatitis
- NASH CRN
NASH Clinical Research Network
- OCA
obeticholic acid
- PC
principal component
- PCA
principal component analysis
- QC
quality control
- Q‐Q
quantile‐quantile
- SNP
single nucleotide polymorphism
Nonalcoholic steatohepatitis (NASH) is a subphenotype of nonalcoholic fatty liver disease (NAFLD) that is associated with hepatic inflammation, cell injury, and fibrosis.1 NASH is a progressive condition that can lead to cirrhosis, liver failure, and liver cancer. Thus, there is great interest in developing novel therapies for NASH in both adults and children.2 We published the results of a multicenter, randomized, placebo‐controlled trial of farnesoid X nuclear receptor ligand obeticholic acid for noncirrhotic, NASH treatment (FLINT).3 This study showed that obeticholic acid (OCA) administered daily for 72 weeks was associated with significant improvement in steatosis, necroinflammation, and fibrosis in patients with NASH. However, only a subgroup of patients receiving OCA achieved favorable changes in liver histology, and similarly adverse events such as pruritus were observed only in a subgroup of patients receiving OCA.
Pharmacogenomics is the study of the role of genetic variants in the response to a particular medication and development of adverse events with such medication.4 Previous studies in other liver diseases have shown that pharmacogenomics may be helpful in predicting treatment response or side effects to medications. Genetic variants in the IL28 gene were highly predictive of response to interferon therapy in patients with chronic hepatitis C.5 Variations in the HLA region are a well‐known risk factor for liver injury due to antibacterial agents such as flucloxacillin, amoxicillin‐clavulanate, or minocycline.6, 7, 8, 9 However, pharmacogenomics research within the NAFLD area is somewhat limited. One study showed that genetic variation in cytochrome P450 4F2 was not a significant predictor of response to vitamin E in nondiabetic patients with biopsy‐proven NASH.10 More recently, genetic variation in haptoglobin was found to be significantly associated with histological response to vitamin E.11 Studies showed the PNPLA3 148 M/M allele to be associated with poor response to omega‐3 polyunsaturated fatty acids.12, 13 Another recent study showed that variants in PNPLA3 and TM6SF2 modify the risk for hepatic steatosis caused by a novel glucagon receptor antagonist, LY2409021, in patients with type 2 diabetes.14
Recently, the clinical and laboratory factors associated with histological response to OCA in FLINT have been reported.15 These include treatment with OCA, baseline NAFLD activity score (NAS) > 5, baseline triglyceride level ≤154 mg/dL, baseline international normalized ratio ≤1, baseline aspartate aminotransferase level ≤49 U/L, and a decrease in alanine aminotransferase (ALT) level at week 24 by 17 U/L or more. Although we have previously identified several genetic variants associated with histological features of NAFLD,16 the genetic variants associated with NASH histological response to OCA are unknown.
We performed a GWAS of participants in the FLINT trial to identify genetic variants associated with histological improvement, specifically with NASH resolution and fibrosis improvement. As a secondary objective, we also conducted a GWAS to identify genetic variants associated with the development of pruritus among study participants.
Materials and Methods
Participants
The details of the FLINT study have been published previously.3 Briefly, FLINT was a randomized, placebo‐controlled clinical trial that enrolled 283 participants with biopsy‐proven noncirrhotic NASH. Almost half of the study participants had type 2 diabetes. Participants were randomized to receive OCA 25 mg orally daily or matching placebo for 72 weeks. Due to interim review of the study, only 200 participants underwent baseline and end‐of‐treatment (week 72) liver biopsies. We genotyped 244 patients who participated in FLINT with available DNA, of whom 198 (81%) were white.
Assessment of Liver Histology
The primary endpoint of FLINT was improvement in NAFLD histology defined as a reduction of NAS ≥ 2 points without worsening fibrosis. Liver histology was blindly and centrally assessed by the NASH Clinical Research Network (NASH CRN) Pathology Committee. Evaluation and scoring of baseline and end‐of‐treatment liver biopsies were done according the NASH CRN system.17 NASH resolution was defined as diagnostic classification of either not NAFLD, or NAFLD but not NASH, on week 72 biopsy. Fibrosis was assessed on a scale of 0‐4, and fibrosis improvement was defined as a decrease in fibrosis stage by at least one stage.
Genotyping
DNA was isolated from whole blood using standard procedures and transferred to the genotyping laboratory at the Los Angeles Biomedical Research Institute. Genotyping was performed using the Omni2.5 content GWAS chip. Genotypes were named using Illumina software (GenomeStudio 2.0). Allele naming included an extensive review of cluster plots, particularly for the exome content. Participants were removed for failed genotyping, unresolvable gender discrepancies, being outliers by principal component analyses, and by relatedness with . Successful genotyping rates were >98% per subject and >98% per SNP. The 198 white participants with 2,051,521 SNPs were further examined for quality control (QC) (Supporting Table S1). Cryptic relative checking found one pair of monozygotic twins, so one of them was randomly removed. Additional SNP‐level QC removed 613 SNPs due to missing rate >5% and another 32,941 SNPs due to Hardy‐Weinberg Equilibrium <10−6. In total, 197 white samples with 2,017,967 SNPs remained in the analysis data set.
Principal Component Analysis
To investigate potential population substructures in the sample, we carried out principal component analysis (PCA) using the GWAS data and the program smartPCA.18, 19 SNPs were filtered by minor allele frequency (MAF) < 0.05 and further pruned by linkage disequilibrium of 0.3, leaving 68,381 SNPs in the PCA. We found some modest population substructure in the study sample, and a significant percentage of the variance can be explained by the top three principal component (PCs). Hence, the top three PCs were included in the GWAS analysis (Supporting Fig. S1A,B).
Imputation
The genotype data were imputed to the Haplotype Reference Consortium population. Among the 2,017,967 SNPs that passed QC, 1,919,289 passed the pre‐imputation QC, which removed SNPs not in the reference panel, all A/T G/C SNPs with MAF > 40% in the reference data set, and all SNPs with an AF difference >0.2 between the reference and data set frequency file. In total, 197 samples with 1,906,129 SNPs on autosomes were then submitted to the Michigan Imputation Server. The server removed 50 SNPs due to invalid alleles, so 1,906,079 genotyped SNPs passed through and 37,225,499 SNPs were imputed. In total, there were 39,131,578 SNPs after imputation, with 5,417,859 SNPs having MAF > 0.05 and the rest 33,713,719 having MAF ≤ 0.05.
Genome‐Wide Association Analysis
Given most of the participants were white (81%), we focused our GWAS study on white participants only to avoid false positives introduced by population stratification. Common variant (MAF > 0.05) GWAS was then carried out for NASH resolution, a binary trait with 1 indicating NASH resolution and 0 indicating lack of NASH resolution. As NASH resolution is a binary trait, we used a logistic model as follows:
where denotes the resolution status for the ith subject; denotes the m covariates (e.g,. age, gender, baseline body mass index [BMI], percentage change in BMI, and top principal components of genetic variation); and denotes the genotypes for the p variants.
First, only the top three PCs were included as covariates:
Then, we included age and gender together with three PCs as covariates:
Next, we included baseline BMI together with three PCs, age, and gender as covariates:
Finally, because participants on OCA experienced an average weight loss of 2.3 kg versus none in those on placebo (P = 0.008), we included percentage change in BMI with all covariates in model 3:
The analysis was run by the Efficient and Parallelizable Association Container Toolbox (EPACTS) program at http://genome.sph.umich.edu/wiki/EPACTS, first on all 197 FLINT white participants whose samples passed QC, then on the OCA‐treated group (106 samples), and finally on the placebo‐treated group (91 samples). Of those, 141 white participants (76 in the OCA group and 65 in the placebo group) had paired liver biopsies at baseline and at end of treatment and were included for genotype‐histological response association analyses, whereas the pruritus GWAS was carried out in 197 white participants. Common variant (MAF > 0.05) GWAS was also carried out for fibrosis improvement, a binary trait with 1 indicating fibrosis improvement and 0 indicating lack of fibrosis improvement. The Wald test was applied to test the true value of the parameter based on the sample estimate. Based on the relatively small sample size, a P value of less than 10−4 was used to define top SNPs for further analysis. Additional analyses were conducted in those chromosomal regions where more than one SNP met the criterion for evaluation.
We used two criteria to select the SNPs and genetic regions for follow‐up. First, we focus on SNPs with greatest significance (smallest P value) and identified the genes to which the SNPs belonged. If the SNP was not rare (MAF > 0.05) and it is genotyped or imputed with high imputation quality, it is highly possible that the association is “real.”
Second, we focused on regions when having multiple SNPs with suggestive association (P < 1 × 10−4) within the same region. It is quite possible that SNPs within the same region represent the same genes that are likely to be significantly associated with histological resolution of NASH. Moreover, regions with multiple hits are more likely to have “real” association than single SNPs of the same significance.
Results
FLINT trial
The results of FLINT have been previously published.3 Briefly, the study primary outcome, histological improvement as indicated by reduction of NAS ≥ 2 points without worsening fibrosis, was observed in 45% of participants on OCA versus 21% of those on placebo (P = 0.0002). Resolution of definite NASH was achieved in 22% of patients on OCA versus 13% of those on placebo (P = 0.08). More patients on OCA versus placebo had significant improvement in fibrosis (35% versus 19%, P = 0.004), steatosis (61% versus 38%, P = 0.001), lobular inflammation (53% versus 35%, P = 0.006), and hepatocyte ballooning (46% versus 31%, P = 0.03). Pruritus was more commonly reported in patients on OCA versus placebo (23% versus 6%).
Genome‐Wide Association Analysis for NASH Resolution
Among the 141 white participants who were analyzed, NASH resolution was observed in 22.3% (17 of 76) in the OCA group versus 12.3% (8 of 65) in the placebo group. These rates are very similar to that observed in each group in the entire FLINT study. Genome‐wide association results are summarized in Manhattan plot and quantile‐quantile (Q‐Q) plots (Fig. 1). There were 120 SNPs that have suggestive association (P value < 1 × ) for the binary trait NASH resolution in the entire FLINT white group. These suggestive SNPs were distributed over 28 chromosomal loci on 16 different chromosomes. After adjusting for the top three PCAs plus age and gender (model 2), adding baseline BMI (model 3), and percentage change in BMI from baseline (model 4), the overall findings of associated chromosomal regions and sentinel SNPs were unchanged (Supporting Table S2). We therefore chose the first analysis model, which has the largest sample size.
Figure 1.
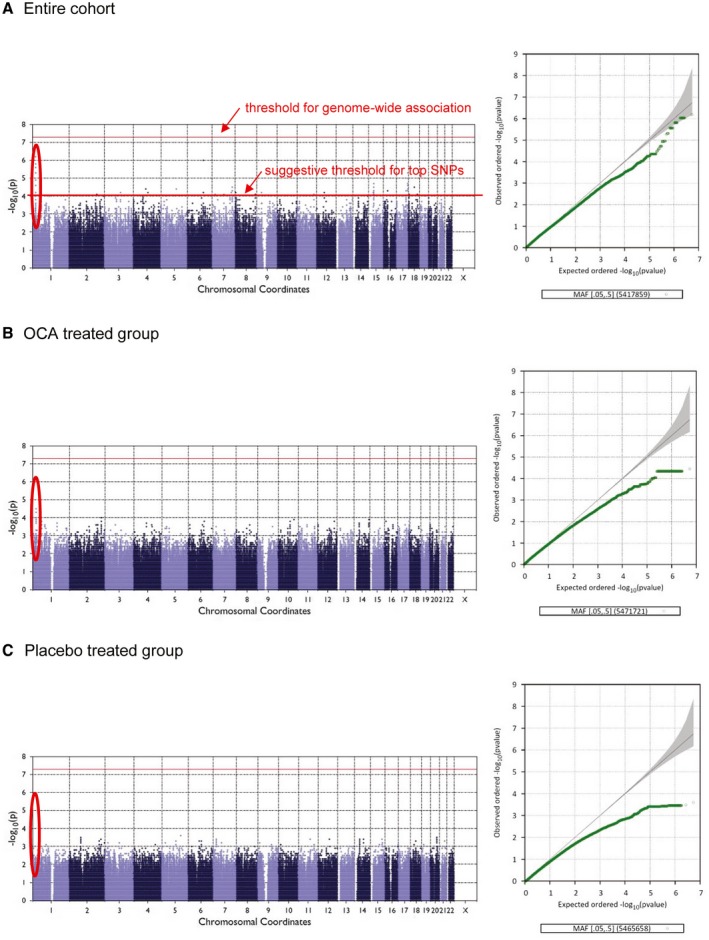
Manhattan and Q‐Q plots for NASH resolution in the entire cohort (A), in the OCA‐treated group (B), and in the placebo‐treated group (C).
Chromosomal Regions and SNPs Associated With NASH Resolution
Six chromosomal regions had multiple SNPs with suggestive association on chromosomes (chr) 1, 4, 6, 7, 15, and 17 (Supporting Fig. S2). Genes nearest to the sentinel SNPs include CELA3B (chr1), MAPK10 (chr4), FYN (chr6), CREB3L2 (chr7), TMCO5A (chr15), and RPTOR (chr17).
Next we assessed the interacting effects of each of the identified sentinel SNPs in these loci with that of treatment received, on NASH resolution. By comparing the effect in the OCA group versus placebo group, each sentinel SNP can have either a SNP effect only, or a SNP and treatment interaction effect (Fig. 2).
Figure 2.
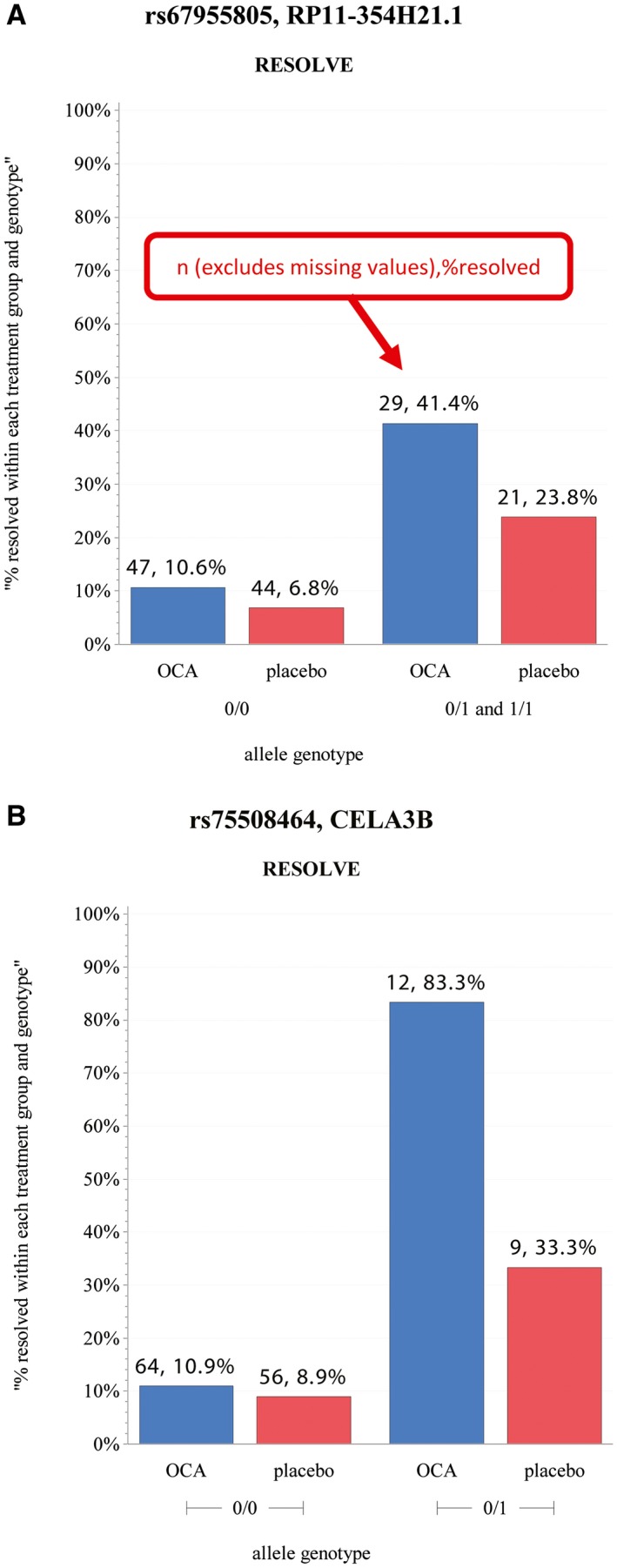
Illustrative examples of interacting sentinel SNP and treatment effects on NASH resolution. (A) SNP‐only effect (the addition of an effect allele, coded as 1, increases NASH resolution. Treatment and placebo have approximately the same rate of increase in resolution). (B) SNP and treatment interaction effect (the addition of an effect allele increases NASH resolution, and this increase is further heightened by treatment group, as the OCA group increases at a much larger rate than the placebo group).
Of the 28 identified sentinel SNPs, 15 demonstrated a SNP‐only effect, whereas 13 had a SNP and treatment interaction effect in model 1 (Table 1). The data are most supportive for the six regions with multiple associated SNPs. The effects of the top SNPs in these six regions on NASH resolution are highlighted in the genotype bar plots in Fig. 3.
Table 1.
Sentinel SNPs Associated With NASH Resolution Demonstrating Effect on Phenotype Regardless of Treatment Received (SNP‐Only Effect), or Those Interacting With Treatment to Affect the Phenotype (SNP and Treatment Interaction Effect)
| SNP‐Only Effect | SNP and Treatment Interaction Effect |
|---|---|
| rs61811017 | rs75508464 |
| rs67955805 | rs113171415 |
| rs2971355 | rs7677101 |
| rs7712188 | rs13148620 |
| rs118086828 | rs1859173 |
| rs1866671 | rs17133280 |
| rs71550675 | rs34105677 |
| rs78542190 | rs12593699 |
| rs34668456 | rs11649290 |
| rs35787382 | rs35871798 |
| rs75520616 | rs4969304 |
| rs11216486 | rs2202067 |
| rs73112049 | rs111832226 |
| rs2959341 | |
| rs36111385 |
Figure 3.
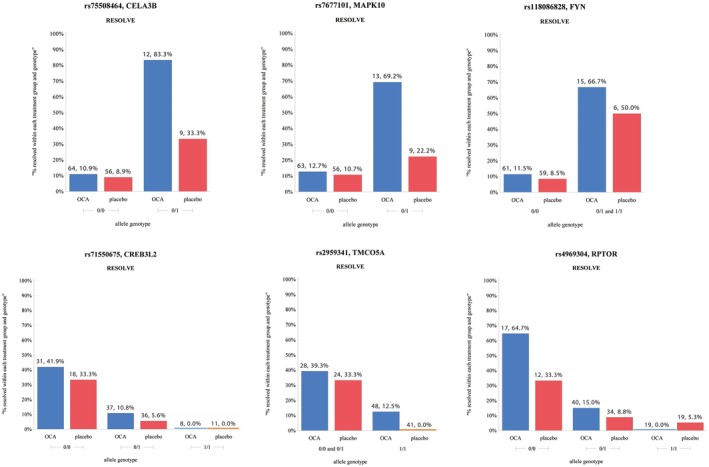
Genotype bar plot for top SNPs from model 1. (*Excludes missing data; 0/0 = homozygous reference; 0/1 = heterozygous; 1/1 = homozygous alternate; above each bar is the sample size for each group and relative frequency.)
We then assessed the effects of these six sentinel SNPs associated with NASH resolution on other NASH histological phenotypes, ALT, and pruritus (Table 2). Although SNPs in chromosomes 7, 15, and 17 were also associated with improvement in steatosis, the chromosome 1 sentinel SNP (rs75508464) near CELA3B was also associated with improvement in NAS, fibrosis, and portal inflammation. The association with improvement in fibrosis was still observed when the analysis was restricted to the OCA‐treated group. None of these sentinel SNPs was associated with ALT or development of pruritus.
Table 2.
Association of NASH Resolution–Associated SNPs With Improvement in Other Histological Phenotypes, ALT, and Pruritus
| rsid | CHROM | BEGIN | END | MARKER_ID | Resolve | Improve | Fibrosis | Steatosis | LI | PI | HB | ALT | Pruritus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs75508464 | 1 | 22288656 | 22288656 | 1:22288656_T/C_Intergenic | 6.4414E‐07 | 0.027994 | 0.0076437 | 0.076186 | 0.21147 | 0.0088855 | NA | 0.56131 | 0.33945 |
| rs7677101 | 4 | 86935417 | 86935417 | 4:86935417_T/A_Intergenic | 0.000044436 | 0.093126 | 0.27472 | 0.42503 | 0.89904 | 0.72379 | NA | 0.74975 | 0.68704 |
| rs118086828 | 6 | 112041632 | 112041632 | 6:112041632_T/C_Intron:FYN | 9.1433E‐07 | 0.018826 | 0.061389 | 0.056418 | 0.47975 | 0.7159 | NA | 0.833 | 0.56116 |
| rs71550675 | 7 | 137618322 | 137618322 | 7:137618322_G/A_Intron:CREB3L2 | 0.000097596 | 0.079524 | 0.21698 | 0.023096 | 0.71182 | 0.90641 | 0.080996 | 0.56733 | 0.69629 |
| rs2959341 | 15 | 38240315 | 38240315 | 15:38240315_G/C_Intron:TMCO5A | 0.000019486 | 0.075866 | 0.8993 | 0.036325 | 0.25038 | 0.10811 | 0.41474 | 0.57015 | 0.38786 |
| rs4969304 | 17 | 78892047 | 78892047 | 17:78892047_C/T_Intron:RPTOR | 5.4763E‐06 | 0.0079578 | 0.51532 | 0.01939 | 0.84053 | 0.053913 | 0.21721 | 0.72661 | 0.92022 |
Bold values are statistically significant.
Abbreviations: CHROM, chromosome; HB, hepatocyte ballooning; LI, lobular inflammation; and PI, portal inflammation.
Genome‐Wide Association Analysis for Fibrosis Improvement
Among the 141 white participants who were analyzed, fibrosis improvement was observed in 35.5% (27 of 76) in the OCA group versus 16.9% (11 of 65) in the placebo group. These rates are very similar to that observed in each group in the entire FLINT study. There were 252 SNPs with suggestive association (P value < 1 × in model 1 with the binary trait fibrosis improvement (yes or no) in the entire FLINT white group. These suggestive SNPs were distributed over 26 chromosomal loci on 16 different chromosomes (Fig. 4).
Figure 4.
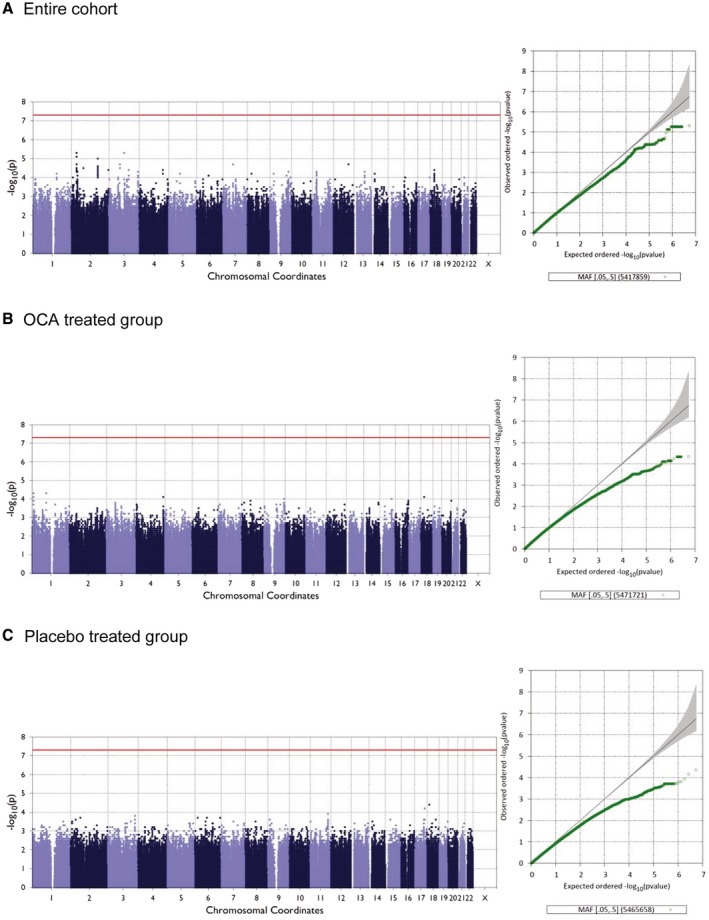
Manhattan and Q‐Q plots for fibrosis improvement in the entire cohort (A), in the OCA‐treated group (B), and in the placebo‐treated group (C).
After adjusting for the top three PCs plus age and gender (model 2), adding baseline BMI (model 3) and percentage change in BMI from baseline (model 4), the sentinel SNPs and chromosomal regions associated with fibrosis improvement remained at the same level of significance (Supporting Table S3).
Chromosomal Regions and SNPs Associated With Fibrosis Improvement
Of the 26 chromosomal regions that had SNPs with suggestive associations with fibrosis improvement, we focused on the eight regions with multiple associated SNPs on chromosomes 1, 2, 3, 11, 13, and 18 (Supporting Fig. S3). Genes nearest to the sentinel SNPs include TDRD10 (chr1), DYNC1I2 (chr2), TPRG1 (chr1), ANO3 (chr11), LINC00348 (chr13), and CDH2 (chr18).
Supporting Table S4 summarizes the top SNPs that showed suggestive evidence of association with fibrosis improvement. Four SNPs had primarily a SNP‐only effect on fibrosis improvement, while another four SNPs appeared to affect fibrosis improvement through interaction with treatment effect (Supporting Fig. S4).
We then assessed the effects of these SNPs associated with fibrosis improvement on other NASH histological phenotypes, ALT, and pruritus (Table 3). Several sentinel SNPs were also nominally associated with NASH resolution or improvement, but rs12130403 near TDRD10 on chromosome 1 and rs4073431 near ANO3 on chromosome 11 were also associated with improvement in portal inflammation and steatosis, respectively. None of these sentinel SNPs was associated with ALT or development of pruritus.
Table 3.
Association of Fibrosis Improvement Associated Sentinel SNPs With Improvement in Other Histological Phenotypes, ALT, and Pruritus
| rsid | CHROM | BEGIN | END | MARKER_ID | Fibrosis | Resolve | Improve | Steatosis | Lobular_inflammation | Portal_inflammation | Ballooning | ALT | Pruritus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs12130403 | 1 | 154549470 | 154549470 | 1:154549470_T/C_Utr3:CHRNB2 | 5.76E‐05 | 0.090209 | 0.0028111 | 0.1488 | 0.083912 | 0.024397 | 0.38314 | 0.80754 | 0.94369 |
| rs6718351 | 2 | 35337270 | 35337270 | 2:35337270_C/G_Intron:AC012593.1 | 5.4E‐06 | 0.13877 | 0.0032052 | 0.21698 | 0.8466 | 0.67615 | 0.12519 | 0.11843 | 0.4273 |
| rs61588295 | 2 | 172532728 | 172532728 | 2:172532728_C/T_Intergenic | 1.11E‐05 | 0.14913 | 0.041331 | 0.26493 | 0.74642 | 0.50986 | 0.67555 | 0.65066 | 0.54445 |
| rs73138351 | 3 | 99159241 | 99159241 | 3:99159241_A/G_Intergenic | 4.81E‐06 | 0.0021231 | 0.004839 | 0.12206 | 0.53096 | 0.78479 | 0.051509 | 0.81376 | 0.32564 |
| rs11917264 | 3 | 189150773 | 189150773 | 3:189150773_A/G_Intergenic | 5.47E‐05 | 0.37456 | 0.087554 | 0.18451 | 0.89726 | 0.62822 | 0.059201 | 0.21756 | 0.37259 |
| rs4073431 | 11 | 26171987 | 26171987 | 11:26171987_A/T_Intergenic | 4.79E‐05 | 0.046251 | 0.086436 | 0.027919 | 0.52158 | 0.85299 | 0.79978 | 0.12502 | 0.7981 |
| rs59879172 | 13 | 71864207 | 71864207 | 13:71864207_T/G_Intergenic | 5.04E‐05 | 0.044179 | 0.58848 | 0.066344 | 0.74708 | 0.99385 | 0.6243 | 0.79135 | 0.92511 |
| rs1910560 | 18 | 26169692 | 26169692 | 18:26169692_T/G_Intergenic | 4.02E‐05 | 0.038593 | 0.005606 | 0.51667 | 0.12457 | 0.53761 | 0.06709 | 0.60546 | 0.4725 |
Bold values are statistically significant.
Abbreviation: CHROM, chromosome.
Genome‐Wide Association Analysis for Pruritus
In the 197 white participants analyzed (106 on OCA, 91 on placebo), pruritus was commonly self‐reported during the FLINT trial occurring in 25% of participants receiving OCA versus 4% of those receiving placebo. In the genome‐wide association analysis to identify variants associated with pruritus in the entire cohort (Fig. 5), we noted multiple SNPs on chromosome 11 with suggestive association with pruritus. The top sentinel SNP was rs1379650 (P = 2.1 × 10−6). ANO5 was as the nearest upstream gene to this SNP.
Figure 5.
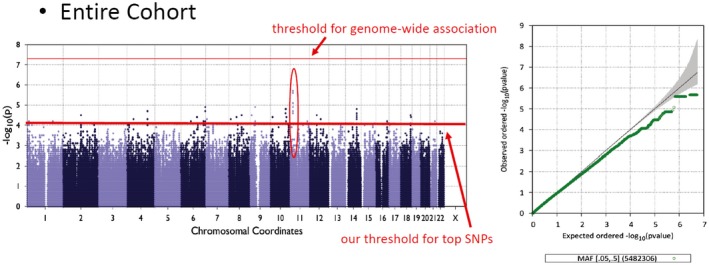
Manhattan and Q‐Q plots for pruritus.
This SNP demonstrates a SNP and treatment interaction effect, and appears to augment the OCA effect on pruritus (odds ratio [OR] of entire cohort = 6.94, OR of OCA = 7.36) (Supporting Fig. S5).
We subsequently compared the SNPs associated with NASH resolution to those associated with pruritus in the OCA group (Supporting Fig. S6). We noted no overlap between these SNPs, suggesting that if there is indeed genetic contribution to pruritus in this setting, the genetic mechanisms and regions contributing to development of pruritus on OCA are independent of those associated with NASH resolution.
Discussion
NAFLD is the most common liver disease in the United States.20 Liver failure and liver cancer due to NASH are on the rise, with NASH currently being the second most common indication for liver transplant listing in the United States.21 Therefore, there is an urgent need to develop effective therapies to halt or reverse the progression of NASH and prevent its complications. This has translated into a remarkable increase in clinical trials to test the potential therapeutic agents for NASH.22 However, as seen in major randomized clinical trials for NASH, only a subgroup of participants achieves the desired histological improvement. NASH resolution, when chosen as the primary endpoint, has been observed in 20%‐47% of participants in different trials.3, 23, 24 Understanding the host genetic factors that may influence response to a certain therapeutic agent may therefore allow a more effective and personalized delivery of therapy to patients with NASH who have a high chance of response. It may also spare patients with a low chance of response the exposure to a therapeutic agent that they may not respond to and also spare them the side effects of such agent.25, 26
In this study, we identified several genetic variants associated with NASH resolution and fibrosis improvement in participants in the FLINT trial. To our knowledge, except for variants in MAPK10, which were previously reported to be associated with liver enzymes levels in another GWAS,27 the other variants and nearby genes identified in this study had not been previously reported to be associated with NASH or with response of liver disease to therapeutic agents (Supporting Table S5).
Although five of the six sentinel SNPs associated with NASH resolution were also associated with improvement of one or two additional NAFLD histological phenotypes, rs75508464 near CELA3B on chromosome 1 was notably associated with improvement in NAS, fibrosis, and portal inflammation. The CELA3B encodes for elastase 3B, which is secreted from the pancreas and helps in digestion. The elastase 3B isoform can be measured in stool as a measure of pancreatic exocrine function.28 A variant in CELA3B has a possible protective effect on the risk of chronic alcoholic pancreatitis,29 and hypermethylation of the gene promoter may be associated with pancreatic cancer.30 However, no data are currently available on the role of CELAB3 in liver disease.
Similarly, while seven of the eight sentinel SNPs associated with fibrosis improvement were also nominally associated with NASH resolution or improvement, rs12130403 near TDRD10 on chromosome 1 and rs4073431 near ANO3 on chromosome 11 were also associated with improvement in portal inflammation and steatosis, respectively. Variants in TDRD10 and ANO3 have been linked to neurological diseases31, 32 but not yet to liver disease.
This study highlights the concept that host genetic variants may on their own affect the outcomes of intervention, positively or negatively, and regardless of treatment received (SNP‐only effect) (e.g., rs118086828, FYN, and rs71550675, CREB3L2 in Fig. 3.). This could partly explain improvements seen in recipients of placebo. Indeed, 15 of the 28 sentinel SNPs associated with NASH resolution had a SNP‐only effect.
In some patients, certain genetic variants not only increased the likelihood of response but also enhanced the effect of treatment (SNP and treatment effects). This is best highlighted in the 21 participants who were heterozygotes for the rs75508464 variant in CELA3B; these participants already had a higher NASH resolution rate compared with the null homozygotes and demonstrated further increase in response to OCA. As seen in Fig. 2B, 83% of those participants who received OCA achieved NASH resolution, compared with only 33% of those who received placebo.
Nearly a quarter of the participants in this analysis developed pruritus on OCA. We identified a sentinel SNP (rs1379650) near ANO5, which was associated with pruritus. ANO5 has been linked to thyroid cancer, gnathodiaphyseal dysplasia and myopathy,33, 34, 35 but not yet to liver disease. This variant also shows a SNP and treatment effect and increased the likelihood of pruritus in participants receiving OCA.
This pharmacogenomics study size was relatively small, but this was a consequence of the already established size of the original FLINT trial. Because this is a unique population with NASH exposed to OCA, validation in another similar cohort was not feasible at the time. The upcoming phase 3 randomized trial of OCA in NASH (NCT02548351) may offer an opportunity to validate these findings. We restricted our GWAS to white participants, who represented most of the cohort, to avoid false positives introduced by population stratification. The results therefore may not be applicable to patients of other races. Due to the modest study size, the significance threshold for examining associations was set at 10−4, which is lower than the traditional 10−8 for GWAS. Despite these limitations, this study has several strengths. It evaluated a unique population with NASH that received either OCA or placebo. The histological phenotypes were blindly and systematically well‐defined by the NASH CRN Pathology Committee. The observation that a number the genetic regions we identified had multiple associated SNPs, and that the sentinel SNPs in several of these regions were associated with improvement in multiple histological phenotypes, are also encouraging, and increase the likelihood of true significance and reduce the possibility of false discovery. Although these results could be viewed as tentative, they can trigger further research to confirm and explore the biological and clinical significance of identified sentinel SNPs.
In conclusion, we identified several genetic variants that may be associated with NASH resolution and fibrosis improvement in participants in the FLINT trial. The rs75508464 variant in CELA3B on chromosome 1 may have a significant effect on the rate of NASH resolution in those who received OCA. If confirmed, these variants may improve the effectiveness and precision of selecting NASH patients for OCA therapy by offering it to those with variants that increase the likelihood of NASH resolution. Once established, NASH patients with genetic variants associated with low likelihood of response to OCA and high likelihood of pruritus may be directed to receive different therapies.
Supporting information
Nonalcoholic Steatohepatitis Clinical Research Network
Cleveland Clinic Foundation, Cleveland, OH (Daniela Allende, M.D., Srinivasan Dasarathy, M.D., Arthur J. McCullough, M.D., Revathi Penumatsa, M.P.H., and Jaividhya Dasarathy, M.D.); Columbia University, New York, NY (Joel E. Lavine, M.D., Ph.D.); Duke University Medical Center, Durham, NC (Manal F. Abdelmalek, M.D., M.P.H., Mustafa Bashir, M.D., Stephanie Buie, Anna Mae Diehl, M.D., Cynthia Guy, M.D., Christopher Kigongo, M.B., Ch.B., Mariko Kopping, M.S., R.D., David Malik, and Dawn Piercy, M.S., F.N.P.); Indiana University School of Medicine, Indianapolis, IN (Linda Ragozzino, R.N., Kumar Sandrasegaran, M.D., and Raj Vuppalanchi, M.D.); Saint Louis University, St Louis, MO (Elizabeth M. Brunt, M.D. [2002‐2008], Theresa Cattoor, R.N., Danielle Carpenter, M.D., Janet Freebersyser, R.N., Debra King, R.N. [2004‐2015], Jinping Lai, M.D. [2015‐2016], Brent A. Neuschwander‐Tetri, M.D., Joan Siegner, R.N. [2004‐2015], Susan Stewart, R.N. [2004‐2015], Susan Torretta, and Kristina Wriston, R.N. [2015]); Swedish Medical Center, Seattle, WA (Maria Cardona Gonzalez, Jodie Davila, Manan Jhaveri, M.D., Nizar Mukhtar, M.D., Erik Ness, M.D., Michelle Poitevin, Brook Quist, and Sherilynn Soo); University of California San Diego, San Diego, CA (Brandon Ang, Cynthia Behling, M.D., Ph.D., Archana Bhatt, Michael S. Middleton, M.D., Ph.D., and Claude Sirlin, M.D.); University of California San Francisco, San Francisco, CA: Maheen F. Akhter, BS; Nathan M. Bass, M.D., Ph.D. [2002‐2011], Danielle Brandman, M.D., M.A.S., Ryan Gill, M.D., Ph.D., Bilal Hameed, M.D., Jacqueline Maher, M.D., Norah Terrault, M.D., M.P.H., and Ashley Ungermann, M.S.); University of Washington Medical Center, Seattle, WA (Matthew Yeh, M.D., Ph.D.); Virginia Commonwealth University, Richmond, VA (Sherry Boyett, R.N., B.S.N., Melissa J. Contos, M.D., Sherri Kirwin, Velimir A.C. Luketic, M.D., Puneet Puri, M.D. [2009‐2017], Arun J. Sanyal, M.D., Jolene Schlosser, R.N., B.S.N., Mohammad S. Siddiqui, M.D., and Leslie Yost‐Schomer, R.N.); Washington University, St. Louis, MO (Elizabeth M. Brunt, M.D. [2008‐2015], and Kathryn Fowler, M.D. [2012‐2015]); National Cancer Institute, Bethesda, MD (David E. Kleiner, M.D., Ph.D.); National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD (Edward C. Doo, M.D., Sherry Hall, M.S., Jay H. Hoofnagle, M.D., Patricia R. Robuck, Ph.D., M.P.H. [2002‐2011], Averell H. Sherker, M.D., and Rebecca Torrance, R.N., M.S.); and Data Coordinating Center, Johns Hopkins University, Bloomberg School of Public Health, Baltimore, MD (Patricia Belt, B.S., Jeanne M. Clark, M.D., M.P.H., John Dodge, Michele Donithan, M.H.S. [2002‐2017], Milana Isaacson, B.S., Mariana Lazo, M.D., Ph.D., Sc.M., Jill Meinert, Laura Miriel, B.S., Jacqueline Smith, A.A., Michael Smith, B.S., Alice Sternberg, Sc.M., James Tonascia, Ph.D., Mark L. Van Natta, M.H.S., Annette Wagoner, and Goro Yamada, Ph.D., M.H.S., M.H.S., M.M.S.).
Supported by the Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (U01DK061718, U01DK061728, U01DK061731, U01DK061732, U01DK061734, U01DK061737, U01DK061738, U01DK061730, and U01DK061713); the National Center for Advancing Translational Sciences (UL1TR000439, UL1TR000436, UL1TR000006, UL1TR000448, UL1TR000100, UL1TR000004, UL1TR000423, UL1TR000058, and UL1TR001881); and the NIDDK (DK063491 to the Southern California Diabetes Endocrinology Research Center). The FLINT trial was conducted by the NASH CRN and supported in part by a Collaborative Research and Development Agreement between NIDDK and Intercept Pharmaceuticals.
ClinicalTrials.gov: FLINT: NCT01265498.
Potential conflict of interest: Dr. Cummings consults for EiRx Theraputics and received grants from Novo‐Nordisk. Dr. Gawrieh consults for Transmedics and received grants from Cirius, Galmedm, and Zydus. Dr. Chalsani consults for Abbvie, Nosirt, Allergan, Madrigal, Siemens, Genetech, Coherus, and La Jolla, and received grants from Intecept, Exact, and Eli Lilly. Dr. Loomba serves as a consultant or advisory board member for Arrowhead Pharmaceuticals, AstraZeneca, Bird Rock Bio, Boehringer Ingelheim, Bristol‐Myer Squibb, Celgene, Cirius, CohBar, Conatus, Eli Lilly, Galmed, Gemphire, Gilead, Glympse bio, GNI, GRI Bio, Intercept, Ionis, Janssen, Merck, Metacrine, NGM Biopharmaceuticals, Novartis, Novo Nordisk, Pfizer, Prometheus, Sanofi, Siemens, and Viking Therapeutics. In addition, his institution has received grant support from Allergan, Boehringer‐Ingelheim, Bristol‐Myers Squibb, Cirius, Eli Lilly, Galectin Therapeutics, Galmed Pharmaceuticals, GE, Genfit, Gilead, Intercept, Grail, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, NuSirt, Pfizer, pHPharma, Prometheus, and Siemens. He is also co‐founder of Liponexus. Dr. Sreekumar owns stock in and is employed by Eli Lilly. Dr. Kowdley consults for, is on the speakers’ bureau for, and received grants from Intercept. Dr. Pillai owns stock in and is employed by Eli Lilly.
Contributor Information
Jerome I. Rotter, Email: jrotter@labiomed.org.
Naga Chalasani, Email: nchalasa@iu.edu, Email: jrotter@labiomed.org.
NASH Clinical Research Network:
Daniela Allende, Srinivasan Dasarathy, Arthur J McCullough, Revathi Penumatsa, Jaividhya Dasarathy, Joel E Lavine, Manal F Abdelmalek, Mustafa Bashir, Stephanie Buie, Anna Mae Diehl, Cynthia Guy, Christopher Kigongo, Mariko Kopping, David Malik, Dawn Piercy, Linda Ragozzino, Kumar Sandrasegaran, Raj Vuppalanchi, Elizabeth M Brunt, Theresa Cattoor, Danielle Carpenter, Janet Freebersyser, Debra King, Jinping Lai, Brent A Neuschwander, Joan Siegner, Susan Stewart, Susan Torretta, Kristina Wriston, Maria Cardona Gonzalez, Jodie Davila, Manan Jhaveri, Nizar Mukhtar, Erik Ness, Michelle Poitevin, Brook Quist, Sherilynn Soo, Brandon Ang, Cynthia Behling, Archana Bhatt, Michael S Middleton, Claude Sirlin, Maheen F Akhter, Nathan M Bass, Danielle Brandman, Ryan Gill, Bilal Hameed, Jacqueline Maher, Norah Terrault, Ashley Ungermann, Matthew Yeh, Sherry Boyett, Melissa J Contos, Sherri Kirwin, Velimir A.C. Luketic, Puneet Puri, Arun J Sanyal, Jolene Schlosser, Mohammad S Siddiqui, Leslie Yost‐Schomer, Elizabeth M Brunt, Kathryn Fowler, David E Kleiner, Edward C Doo, Sherry Hall, Jay H Hoofnagle, Patricia R Robuck, Averell H Sherker, Rebecca Torrance, Patricia Belt, Jeanne M Clark, John Dodge, Michele Donithan, Milana Isaacson, Mariana Lazo, Jill Meinert, Laura Miriel, Jacqueline Smith, Michael Smith, Alice Sternberg, James Tonascia, Mark L Natta, Annette Wagoner, and Goro Yamada
References
- 1. Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 2017;377:2063‐2072. [DOI] [PubMed] [Google Scholar]
- 2. Gawrieh S, Chalasani N. Pharmacotherapy for nonalcoholic fatty liver disease. Semin Liver Dis 2015;35:338‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weinshilboum RM, Wang L. Pharmacogenomics: precision medicine and drug response. Mayo Clinic Proc 2017;92:1711‐1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment‐induced viral clearance. Nature 2009;461:399‐401. [DOI] [PubMed] [Google Scholar]
- 6. Aithal GP, Grove JI. Genome‐wide association studies in drug‐induced liver injury: step change in understanding the pathogenesis. Semin Liver Dis 2015;35:421‐431. [DOI] [PubMed] [Google Scholar]
- 7. Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe'er I, Floratos A, et al. HLA‐B*5701 genotype is a major determinant of drug‐induced liver injury due to flucloxacillin. Nat Genet 2009;41:816‐819. [DOI] [PubMed] [Google Scholar]
- 8. Urban TJ, Nicoletti P, Chalasani N, Serrano J, Stolz A, Daly AK, et al. Minocycline hepatotoxicity: clinical characterization and identification of HLA‐B *35:02 as a risk factor. J Hepatol 2017;67:137‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, et al. Susceptibility to amoxicillin‐clavulanate‐induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 2011;141:338‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Athinarayanan S, Wei R, Zhang M, Bai S, Traber MG, Yates K, et al. Genetic polymorphism of cytochrome P450 4F2, vitamin E level and histological response in adults and children with nonalcoholic fatty liver disease who participated in PIVENS and TONIC clinical trials. PLoS ONE 2014;9:e95366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asgharpour A, Cazanave SC, Yates KP, Vincent R, Kohli R, Hameed B, et al. Haptoglobin genotype 2 allele identifies adults and children with nonalcoholic steatohepatitis (NASH) who improve liver histology and cardiometabolic risk factors with vitamin E treatment. Hepatology 2015;62:290a. [Google Scholar]
- 12. Nobili V, Bedogni G, Donati B, Alisi A, Valenti L. The I148M variant of PNPLA3 reduces the response to docosahexaenoic acid in children with non‐alcoholic fatty liver disease. J Med Food 2013;16:957‐960. [DOI] [PubMed] [Google Scholar]
- 13. Scorletti E, West AL, Bhatia L, Hoile SP, McCormick KG, Burdge GC, et al. Treating liver fat and serum triglyceride levels in NAFLD, effects of PNPLA3 and TM6SF2 genotypes: results from the WELCOME trial. J Hepatol 2015;63:1476‐1483. [DOI] [PubMed] [Google Scholar]
- 14. Guzman CB, Duvvuru S, Akkari A, Bhatnagar P, Battioui C, Foster W, et al. Coding variants in PNPLA3 and TM6SF2 are risk factors for hepatic steatosis and elevated serum alanine aminotransferases caused by a glucagon receptor antagonist. Hepatology Communications 2018;2:561‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loomba R, Sanyal AJ, Kowdley KV, Terrault N, Chalasani NP, Abdelmalek MF, et al. Factors associated with histologic response in adult patients with nonalcoholic steatohepatitis. Gastroenterology 2019;156:88‐95.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, et al. Genome‐wide association study identifies variants associated with histologic features of nonalcoholic fatty liver disease. Gastroenterology 2010;139:1567‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 18. Price A, Plenge R, Weinblatt M, Shadick N, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet 2006;38:904‐909. [DOI] [PubMed] [Google Scholar]
- 19. Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐Meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64(1):73‐84. [DOI] [PubMed] [Google Scholar]
- 21. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547‐555. [DOI] [PubMed] [Google Scholar]
- 22. Gawrieh S, Chalasani N. Emerging treatments for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Clin Liver Dis 2018;22:189‐199. [DOI] [PubMed] [Google Scholar]
- 23. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐alpha and ‐delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016;150:1147‐1159.e1145. [DOI] [PubMed] [Google Scholar]
- 24. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kawaguchi‐Suzuki M, Cusi K, Bril F, Gong Y, Langaee T, Frye RF. A genetic score associates with pioglitazone response in patients with non‐alcoholic steatohepatitis. Front Pharmacol 2018;9:752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lorbek G, Urlep Z, Rozman D. Pharmacogenomic and personalized approaches to tackle nonalcoholic fatty liver disease. Pharmacogenomics 2016;17:1273‐1288. [DOI] [PubMed] [Google Scholar]
- 27. Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, et al. Genome‐wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet 2011;43:1131‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toth AZ, Szabo A, Hegyi E, Hegyi P, Sahin‐Toth M. Detection of human elastase isoforms by the ScheBo Pancreatic Elastase 1 Test. Am J Physiol Gastrointest Liver Physiol 2017;312:G606‐G614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parniczky A, Hegyi E, Toth AZ, Szücs A, Szentesi Á, Vincze Á, et al. Genetic analysis of human chymotrypsin‐like elastases 3A and 3B (CELA3A and CELA3B) to assess the role of complex formation between proelastases and procarboxypeptidases in chronic pancreatitis. Int J Mol Sci 2016;17:2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao J, Zhu F, Lv S, Li Z, Ling Z, Gong Y, et al. Identification of pancreatic juice proteins as biomarkers of pancreatic cancer. Oncol Rep 2010;23:1683‐1692. [DOI] [PubMed] [Google Scholar]
- 31. Kauwe JS, Bailey MH, Ridge PG, Perry R, Wadsworth ME, Hoyt KL, et al. Genome‐wide association study of CSF levels of 59 alzheimer's disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet 2014;10:e1004758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Charlesworth G, Plagnol V, Holmstrom KM, Bras J, Sheerin UM, Preza E, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet 2012;91:1041‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang Z, Cai C, Han D, Gao Y, Li Q, Feng L, et al. Anoctamin5 regulates cell migration and invasion in thyroid cancer. Int J Oncol 2017;51:1311‐1319. [DOI] [PubMed] [Google Scholar]
- 34. Di Zanni E, Gradogna A, Scholz‐Starke J, Boccaccio A. Gain of function of TMEM16E/ANO5 scrambling activity caused by a mutation associated with gnathodiaphyseal dysplasia. Cell Mol Life Sci 2018;75:1657‐1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Papadopoulos C, LaforEt P, Nectoux J, et al. Hyperckemia and myalgia are common presentations of anoctamin‐5‐related myopathy in French patients. Muscle Nerve 2017;56:1096‐1100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials