

Abstract
Autophagy is a process of cell self-renewal that is dependent on the degradation of the cytoplasmic proteins or organelles of lysosomes. Many diseases, such as metabolic diseases, cancer, neurodegenerative diseases, and lung diseases, have been confirmed to be associated with elevated or impaired levels of autophagy. At present, studies have found that autophagy participates in the regulation of chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis, pulmonary hypertension, acute lung injury, lung cancer, and other pulmonary diseases. Using recent literature on the signal transduction mechanisms of autophagy and the effects of autophagy signalling on lung diseases, this review intends to clarify the mechanisms of lung disease to guide the treatment of related diseases.
The reviews of this paper are available via the supplemental material section.
Keywords: acute lung injury, autophagy, idiopathic pulmonary fibrosis, infectious lung disease, lung cancer, pulmonary artery hypertension, pulmonary disease
Introduction
Autophagy, one type of cell death, has attracted increasing attention and discussion in recent years. Japanese molecular and cell biologist Yoshinori Ohsumi, who won the Nobel Prize in physiology or medicine, explained the basic principle behind the process of autophagy: autophagy is the key to removing ‘garbage’ in cells, preventing abnormal cell death, and maintaining normal cell functions. It is a mechanism that relies on lysosomes to degrade their own organelles or proteins for cell self-protection and self-renewal. An increasing number of studies have found that autophagy plays an important role in maintaining the stability of the intracellular environment, and participates in cellular mechanisms. Conversely, some studies have found that autophagy is involved in the pathogenesis of many diseases, such as cancer and inflammatory disorders. The process of autophagy requires the participation of dozens of autophagy-related genes. These core proteins mediate the process of autophagy through different protein complexes. However, how autophagy participates in the pathogenesis of different diseases is unclear. Disease pathogenesis may involve the regulation of autophagy-related genes, activation of related proteins, and positive and negative regulation. Once autophagy becomes dysfunctional, disease results.
Types of autophagy
Autophagy is an evolutionarily conserved, lysosomal-dependent, subcellular degradation pathway that can be divided into three types according to the different ways a substrate can enter the lysosome: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA).1,2 Research on autophagy focuses mainly on macroautophagy, and the mechanism of macroautophagy is also the most well defined. Macroautophagy is often defined as cell autophagy in a narrow sense, and includes initiation, extension, closure, and degradation processes. The critical event in autophagosome biogenesis is the nucleation of a small membrane sac, called a phagophore, in the cytoplasm. After nucleation, the phagophore elongates at both ends to form a cup-shaped structure that sequesters a portion of the cytoplasm. Once the two ends of the phagophore meet, the membranes seal to close the autophagosomes and sequester the cytoplasmic cargo. Autophagosomes can sequester cargo either nonselectively in a bulk fashion, or selectively by engaging specific cargo with the autophagic membranes.3,4 Microautophagy is a form of cargo delivery that bypasses vesicular intermediates and directly internalizes the cargo into lysosomes via direct invaginations in the lysosomal membrane. A similar process can also occur along the surface of late endosomes, leading to the formation of multivesicular bodies (MVBs). MVBs then fuse with lysosomes for cargo degradation. This latter form of autophagy is termed endosomal microautophagy.5,6 In contrast to capturing cargo with a vesicular intermediate, CMA delivers individual substrates directly to the lysosomal lumen. CMA has thus far been described only in mammalian cells.7,8
Although the three types of autophagy occur in different ways, they play important roles in the processes of cell responses to external stimuli and their removal of damaged substances. In the above processes, dozens of proteins are formed by autophagy-related genes (ATGs), whose products mediate autophagy by forming different protein complexes. (Figure 1).
Figure 1.

Three types of autophagy in mammalian cells. Macroautophagy relies on de novo formation of cytosolic double-membrane vesicles, autophagosomes, to sequester and transport cargo to the lysosome. Chaperone-mediated autophagy transports individual unfolded proteins directly across the lysosomal membrane. Microautophagy involves the direct uptake of cargo through invagination of the lysosomal membrane. All three types of autophagy lead to degradation of cargo and release of the breakdown products back into the cytosol for reuse by the cell.
Molecular biological mechanism of autophagy
In the early 1990s, Yoshinori Ohsumi’s team discovered the autophagy process in yeast, and identified most of the key genes involved in autophagy. After consulting among themselves, in 2003, different research groups combined the genes involved in autophagy into a category known as ATGs. At present, 40 key ATGs have been identified. The molecular core mechanism of autophagy is regulated by proteins encoded by approximately 18 core genes,9–11 and can be summarized as follows: the Autophagy-related protein 1/ Unc-51-like kinase 1 complex (Atg1/ULK1 complex), including Atg1, Atg13, Atg11, Atg17, Atg29 and Atg31, plays an important role in the initiation of autophagy; vesicles containing Atg9 and Atg2-Atg18 complexes are also involved in autophagy. Atg9-expressing vesicles can circulate in the bilayer membrane and cytoplasm, relying on the Atg17 or Atg11 complex to localize the vesicles to the pre-autophagosomal structure (PAS) and on the Atg2-Atg18 complex to leave the PAS; phosphatidylinositol 3-kinase (PI3K) complexes, including Vacuolar protein sorting-associated protein (Vps)34, Vps15, Atg6/Beclin-1, Atg14, and Atg38, bind to the membrane and catalyze the conversion of phosphatidylinositol (PI) to phosphatidylinositol-3-phosphate (PI3P), thereby recruiting proteins that bind to PI3P; two ubiquitin systems, one including Atg8/Autophagy marker Light Chain 3 (LC3), Atg4, Atg3, Atg7, and the other including Atg12, Atg7, Atg5, Atg10, and Atg16 have been described. Beclin-1 (Atg6) was first found to be an important regulatory factor in the process of autophagy, and the level of LC3 (Atg8) is directly proportional to the number of autophagy bubbles. These two proteins are the most commonly used autophagy markers.
In recent years, researchers have identified a new type of gene-dependent autophagy that is regulated by Na+, K+ ATPase, and nonapoptotic cell death, termed ‘autosis’, which can be induced by autophagy-inducing peptides (Tat-Beclin1), characterized by the disappearance of the endoplasmic reticulum and focal swelling of the nuclear space. Tat-Beclin1 increase levels of autophagy through a mechanism that is thought to involve disruption of Beclin1/ GAPR-1 binding in the Golgi complex.12
Autophagy and pulmonary disease
Autophagy and COPD
Chronic obstructive pulmonary disease (COPD) is a common, preventable, and treatable disease. COPD is caused by significant exposure to toxic particles or gases that cause airway or alveolar abnormalities, and typically presents with persistent respiratory symptoms and airflow limitations.13,14 Cigarette smoke extract (CSE) exposure is the most common risk factor for COPD.15,16 The mechanisms underlying the pathogenesis of COPD remain incompletely understood, but an increasing number of experiments have shown that autophagy may be involved in the pathogenesis of COPD.
In clinical studies, Chen and colleagues found elevations in the expression of general autophagy markers (LC3-II, microtubule-associated protein-1 light chain-3B, Atg4, Atg5/12, Atg7) in COPD lung tissue, decreased histone deacetylase (HDAC) activity as a result of CSE, resulting in increased binding of early growth response-1 (Egr-1) and E2F factors to the LC3B promoter on the autophagy gene, and increased LC3B expression. Inhibition of autophagy by LC3B-knockdown protected epithelial cells from CSE-induced apoptosis.17 Through comparing the autophagy levels in the peripheral blood mononuclear cells (PBMCs) of patients with COPD and healthy patients, Wu and colleagues found that the levels of autophagy (LC3II/I and beclin‑1 levels) in PBMCs in patients with COPD were increased, negatively correlated with the predicted FEV1, and positively correlated with the circulating levels of pro inflammatory cytokines (IL‑6, IL‑8, and TNF‑α).18 Furthermore, Puig-Vilanova’s working group showed that levels of autophagy markers, including ultrastructural autophagosome counts, were increased in the muscles of cachectic COPD patients.19 These studies suggest that autophagy is an important indicator for assessing the onset and the severity of COPD; the same conclusion has been confirmed in animal experiments. Zeng found that in the lung tissue of mice, the level of autophagy in the COPD model group was significantly higher than that in the control group, and microRNA-21 aggravated COPD by promoting autophagy and apoptosis in bronchial epithelial cells.20 Chen’s group, however, employed a model of emphysematous airspace enlargement in mice subjected to chronic (6 months) CSE exposure; increased autophagosome numbers and increased expression of autophagy proteins (LC3B) were observed in the lung tissue of mice that were chronically exposed to CSE compared with healthy mice, and LC3B−/− mice had significantly decreased levels of apoptosis in the lungs after CSE exposure, and displayed resistance to CSE-induced air space enlargement.21 Autophagy-deficient Becn1−/− mice were resistant to mucociliary clearance disruption in the airways after subchronic CSE exposure in vivo.22 In COPD, impaired airway clearance induced by ciliophagy-mediated cilia shortening may affect the removal of pathogens from airways, resulting in repeated respiratory infections.23 In addition, recent studies have demonstrated the critical role of autophagy in regulating inflammasome activation and the release of pro-inflammatory cytokines, such as IL-1β, which contribute to the initiation of hyperinflammatory responses in COPD.24–27 Moreover, Xiao-Xi and colleagues identified that CSE contributes to the pathogenesis of COPD (induced neutrophil death and elastase release), partly by inducing PAFR-dependent autophagic death in neutrophils.28 These findings suggest that autophagy serves different functions throughout the pathogenesis of COPD progression. Pathologically, COPD is characterized primarily by airway inflammation, alveolar destruction, and cell apoptosis, and the ultimate process of autosis is apoptosis. It remains to be determined, therefore, if the pathogenesis of COPD is a result of the autosis pathway. How autophagy participates in the pathogenesis of COPD remains unclear. Further studies will be necessary to improve the understanding of the role of autophagy in the pathogenesis of COPD (Figure 2).
Figure 2.

COPD and autophagy pathway. A typical macroautophagic process starts from initiation induced by autophagic activators followed by nucleation, elongation/closure, fusion, and finally degradation/recycling. CSE induces autophagy nucleation through upregulating the expression of microRNA-21. In addition, CSE decreases the activity of histone deacetylase (HDAC), resulting in the increase of LC3B expression by enhancing the binding of Egr-1 and E2F factors, thus inducing autophagy. Beclin-1 not only participates in autophagy nucleation, but is also an important gene in the process of autosis.
COPD, Chronic obstructive pulmonary disease; CSE, cigarette smoke extract; HDAC, histone deacetylase.
Autophagy and lung cancer
Studies have shown that autophagy is closely related to tumor cell movement, tumor cell invasion, tumor stem cell differentiation, and immune escape. On the one hand, autophagy can promote the survival of lung cancer cells, which is conducive to the occurrence and development of tumors. On the other hand, autophagy can also cause apoptosis or death in lung cancer cells through related channels, and inhibit tumor development.
Autophagy is involved in the occurrence and development of lung cancer
Xue and colleagues investigated the potential function and molecular mechanism of Inhibitor of Apoptosis Stimulating Protein of P53 (iASPP) in autophagy in human nonsmall cell lung cancer (NSCLC), and their data showed that iASPP could promote tumor growth by increasing autophagic flux.29 Autophagy provides the metabolic substrates for Ras-driven lung cancer cells to meet their energy needs and maintain their nucleic acids, thereby promoting their survival.30 A recent study demonstrated an oncosupportive role of BECN1 in the migration of NSCLC cells through regulation of the ubiquitination of vimentin. BECN1 overexpression increased cell migration, and knocking down BECN1 significantly reduced the migratory ability of NSCLC cells.31 Paradoxically, as a crucial component of cellular defence mechanisms, autophagy has a putative anticarcinogenic effect through the preservation of the mitochondria, the clearance of subcellular debris, the recycling of metabolic precursors, and the dampening of inflammation.32 A study of 66 patients with stage I/II NSCLC found that patients with high expression of autophagy protein LC3 had better prognoses than patients with low expression of LC3.33 Beclin-1 expression is inversely correlated with tumor size, and primary tumor stage in human lung adenocarcinoma, and is reduced in NSCLC tissue relative to that in normal tissue.34,35 Autophagy is induced by epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI), which can decrease the levels of EGFR, and shows a strong inhibitory effect on NSCLC both in vitro and in vivo.36 Through AMPK-mTOR and JNK signalling pathways, ginsenoside metabolite compound K (C-K) can promote autophagy-mediated apoptosis of NSCLC cells.37 Based on these results, the effect of autophagy may differ at different stages of tumor development.
Role of autophagy in lung cancer therapy
Chemotherapy often induces tumor cell autophagy, and inhibiting autophagy can enhance the sensitivity of lung cancer cells to chemotherapy, which provides a reasonable basis for the combined application of autophagy inhibitors and chemotherapy, provided that the chemotherapy-induced autophagy has a protective effect on tumor cells.38,39 Gemcitabine induces lung cancer cell autophagy, thus changing the sensitivity of lung cancer cells to chemotherapy; this gemcitabine-induced autophagy protects human lung cancer cells from apoptotic death.40 Autophagy promotes the resistance of lung adenocarcinoma cells to cisplatin through activated adenosine monophosphate-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) signalling pathways. Cisplatin in combination with autophagy inhibitors leads to increased apoptosis in lung cancer cells. Blocking autophagy can increase the sensitivity of lung cancer to europium, prevent tumor-related immune tolerance, increase immune cell infiltration in the tumor microenvironment, and inhibit lung cancer progression and metastasis.41 Autophagy exerts a protective role in camptothecin (CPT)-treated lung cancer cells, and the inhibition of autophagy enhances the levels of CPT-induced DNA damage in lung cancer cell lines.42 Combined with current offerings, most research shows that inhibiting autophagy enhances the sensitivity of lung cancer to chemotherapy drugs. Therefore, inhibiting autophagy may become a new strategy for the treatment of lung cancer (Figure 3).
Figure 3.
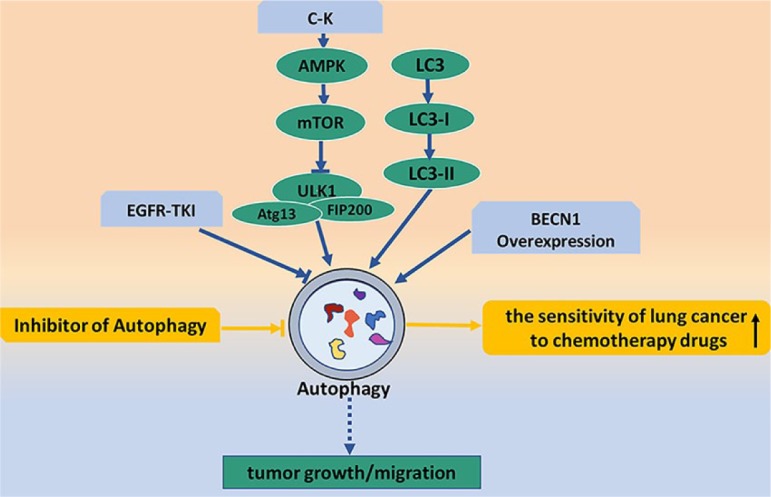
Lung cancer and autophagy pathway. The blue arrows show the relationships between autophagy and occurrence/development of lung cancer. C-K activates mTOR through AMPK signal pathway, inactivation of ULKI induced by mTOR increases autophagy and participates in the growth and migration of tumor cells. EGFR-TKI involves in the development of lung cancer by inhibiting autophagy levels. The yellow arrows show that inhibiting autophagy enhances the sensitivity of lung cancer to chemotherapy drugs.
AMPK, adenosine monophosphate-activated protein kinase; C-K, compound K; EGFR-TKI, epidermal growth factor receptor-tyrosine kinase inhibitor; mTOR mammalian target of rapamycin.
Autophagy in infectious lung disease
Autophagy is involved in host-pathogen interactions and plays a decisive role in the process of infection. Invading pathogens are transferred to lysosomes by autophagy, forming autophagic lysosomes, and destroying the pathogens. Autophagy makes an important contribution to host defences against various microbes, including bacteria, fungi, and viruses. At present, the antibacterial and antipathogenic functions of autophagy have been widely demonstrated.24
In bacterial studies, Wang and colleagues first demonstrated that invading Acinetobacter baumannii induced a complete, ubiquitin-mediated autophagic response that is dependent upon the septins SEPT2 and SEPT9 in mammalian cells, and that autophagy induced by A. baumannii was Beclin-1 dependent via the AMPK/ERK/mTOR pathway.43 Group A streptococcus (GAS) can continue to replicate and proliferate in macrophages by promoting autophagy escape through streptococcus haemolysin O.44 Neumann and colleagues demonstrated the induction of selective autophagy during Staphylococcus aureus infection as well as the escape of S. aureus from autophagosomes and the proliferation of S. aureus in the cytoplasm using live cell imaging. After invasion, S. aureus becomes ubiquitinated and recognized by receptor proteins, such as Sequestosome1/p62 (SQSTM1/p62), leading to phagophore recruitment. However, S. aureus evades phagophores and prevents further degradation through a mitogen-activated protein kinase 14/ p38α mitogen-activated protein kinase (MAPK14/p38α MAP kinase)-mediated blockade of autophagy.45 In addition, autophagy activation aids in the control of inflammation, contributing to a more efficient innate immune response against Mycobacterium tuberculosis. The activation of autophagy by isoniazid, an anti-tb drug, can inhibit the pro-inflammatory response induced by M. tuberculosis in macrophages.46 A recent study reported that autophagy activation participated in the pathophysiologic process of sepsis, and alleviated the cytokine [cytokine tumour necrosis factor (TNF), interleukin (IL)-6, IL-10, monocyte chemotactic protein (MCP)1, and high-mobility group box (HMGB)1] excessive release and lung injury in sepsis.47 At the same time, relevant studies have found that autophagy plays a protective role in fungal infection. Noncanonical fungal autophagy inhibits inflammation in response to Interferon-γ (IFN-γ) via Death-associated protein kinase 1(DAPK1).48 The absence of the autophagy-related Atg7 gene allows host defence mechanisms to easily kill Cryptococcus neoformans, and makes C. neoformans implantation in the host difficult.49 Moreover, studies have shown that autophagy plays an important role in the antiviral process. Infections with influenza viruses can promote the induction of autophagy and autophagosome formation, which is required for viral replication. During influenza A virus (IAV) infection, autophagy can activate extracellular vesicle-mediated protein secretion and contribute to the enhancement of virus infectivity by downregulating superoxide dismutase 1 expression in alveolar epithelial cells.50–53 Therefore, autophagy is considered a key player in infection progression (Figure 4).
Figure 4.

Infectious lung disease and autophagy pathway. Invading pathogens are transferred to lysosomes by autophagy, forming autophagic lysosomes and destroying the pathogens. Autophagy makes an important contribution to host defence against various microbes, including bacteria, fungi, and viruses.
Autophagy and idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and fatal form of fibrosing interstitial pneumonia of unknown cause. The typical clinical course includes dyspnoea, decreased exercise capacity, dry cough, and death 2.5–5 years after diagnosis.54 Fibroblast-to-myofibroblast differentiation is a crucial process in the development of IPF.55 Studies have shown that autophagy is involved in the pathogenesis of IPF, and that it also plays a role in promoting fibrosis. Autophagy promotes the profibrotic effects of Transforming Growth Factor β1( TGF-β1) in human lung fibroblasts.56 Akt1-mediated mitophagy contributes to alveolar macrophage apoptosis resistance and is required for pulmonary fibrosis development.57 In addition, the balance between cell autophagy and apoptosis is an important regulator of IPF. There is an interaction between apoptosis and autophagy; in the process of pulmonary fibrosis, the insufficient apoptosis of fibroblasts becomes an important pathogenic process. Lung tissue from IPF patients and human lung fibroblasts treated with TGF-β demonstrate increased cellular senescence and decreased autophagic activity characterized by decreased autophagy marker LC3B protein expression.58,59 However, there are more studies suggesting that the induction or enhancement of autophagy may have antipulmonary fibrosis effects. First, fibrosis is characterized by excessive extracellular matrix (ECM) protein deposition in the basement membrane and interstitial tissue of the injured epithelium, and the expansion of activated mesenchymal cells. Del Principe found that an autophagy deficiency can promote the deposition of ECM in lung fibroblasts, and accelerate the process of fibrosis.60,61 The latest report found that eEF2K might inhibit TGF-β1-induced normal lung fibroblast (NHLF) proliferation and differentiation and activate NHLF cell apoptosis and autophagy through p38 MAPK signalling, which might ameliorate lung fibroblast-to-myofibroblast differentiation.62 Reduced autophagy induces EMT of alveolar epithelial cells, and can contribute to fibrosis via aberrant epithelial–fibroblast crosstalk.63 Pirfenidone and nintedanib are the only two United States (US) Food and Drug Administration (FDA)-approved drugs to treat pulmonary fibrosis. Nintedanib plays an antifibrotic role by downregulating ECM secretion and inhibiting TGF-β signaling, and induces beclin-1-dependent, ATG7-independent autophagy.64,65 Moreover, Atg4b is an important factor regulating autophagy. When Atg4b is knocked out, damage to bronchial and alveolar epithelial cells is aggravated and apoptosis is increased.66 Using fibroblasts from IPF patients compared with those from healthy controls, Ricci and colleagues demonstrated that beclin-1 expression decreased, bcl-2 expression increased, autophagy was insufficient, and apoptosis was inhibited.67 These studies suggest that inhibition of mTOR, inhibition of TGF-β signalling, and enhancement of autophagy may be therapeutic strategies for IPF treatment (Figure 5).
Figure 5.

Antifibrotic effects of Nintedanib through the autophagy pathway. Nintedanib plays an antifibrotic role by inhibiting TGF-β signaling and downstream ECM secretion. Beclin-1-dependent and ATG7-independent autophagy is also involved in the process of Nintedanib antifibrotic action in the lung.
ECM, extracellular matrix; TGF-β, transforming growth factor β1.
Autophagy and pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a serious disease affecting the pulmonary vasculature that is characterized by the sustained elevation of pulmonary arterial pressure (>25 mmHg at rest).68 PAH is classified as a pulmonary-selective vascular remodelling disease in which vascular smooth muscle cells display a proliferative and antiapoptotic phenotype. Pulmonary artery smooth muscle cell and pulmonary vascular endothelial cell autophagy are involved in pulmonary vascular remodelling and play an important role in the pathogenesis of PAH.69
Autophagy has been reported to have an antiproliferation effect on pulmonary vascular cells.70,71 Oestradiol can directly inhibit the proliferation of vascular endothelial cells, improve hemodynamics, and inhibit the occurrence of pulmonary hypertension by enhancing autophagy, especially mitochondrial autophagy.72 By enhancing autophagy and apoptosis, carfilzomib can reverse pulmonary vascular remodelling and PAH.73
However, other studies have shown that autophagy can promote the development of pulmonary hypertension. In the lung tissues of PAH patients, mTOR expression is significantly downregulated. In mTOR overexpression, autophagy inhibition has been shown to be triggered by hypoxia resulting from blocked LC3B expression. Hypoxia also enhanced the proliferation of human pulmonary artery smooth muscle cells (HPASMCs), which was markedly abrogated by mTOR overexpression.74 Autophagy deficiency generated by the knockdown of the expression of the autophagy protein beclin-1 resulted in improved angiogenic functions in pulmonary artery endothelial cells from foetal lambs with persistent pulmonary hypertension.75 In HIV-associated PAH, autophagy accelerates the transformation of pulmonary vascular endothelial cells from an apoptotic to a hyperproliferative phenotype.76 Fatty acid synthase (FAS) expression and activity increases in hypoxic human pulmonary artery smooth muscle cells (HPASMCs). The works of Singh demonstrated that inhibiting FAS can increase HPASMC apoptosis and reduce autophagy in HPASMCs, thereby reducing pulmonary vascular remodelling and pulmonary endothelial dysfunction.77 The glucagon-like peptide-1 (GLP-1) receptor agonist can mitigate the proliferation of PASMCs by inhibiting the Atg-5/Atg-7/Beclin-1/LC3β-dependent pathways of autophagy in PAH.78 Finally, nuclear factor-kappaB (NF-κB)-induced autophagy contributes to the development of PAH in MCT-treated rats (Figure 6).79
Figure 6.

Pulmonary arterial hypertension and autophagy pathway. GLP-1 regulates the level of autophagy via atg-5/atg-7/beclin-1-dependent pathways, and further participates in pulmonary vascular remodeling. Hypoxia affects the nucleation and elongation of autophagy by changing the expression levels of NF-κB and LC3, involved in the formation of PAH.
GLP-1, glucagon-like peptide-1; NF-κB, nuclear factor-kappaB; PAH, pulmonary arterial hypertension.
Autophagy and acute lung injury
Acute lung injury (ALI) is a common clinical disorder that causes substantial health problems worldwide. The pathophysiology of ALI is characterized by complex mechanisms that involve cell inflammation, cytokines, and abnormal apoptosis with pulmonary cells, including pulmonary alveolar type II epithelial cells, pulmonary vascular endothelial cells, and alveolar macrophages.80,81 An excessive inflammatory response is the central feature of ALI, but the mechanism is still unclear, especially in regard to the role of autophagy.
Autophagy plays a protective role in acute lung injury
Autophagy plays a protective role in lipopolysaccharide (LPS)-induced ALI in mice by regulating the expression level of the chemokine CXCL16. The activation of MTOR in the epithelium promotes LPS-induced ALI, likely through the downregulation of autophagy, and the inhibition of autophagy by 3-MA exacerbates the cytotoxicity induced by LPS in A549 alveolar epithelial cells.82–84 The lungs of diabetic patients are more vulnerable to myocardial ischaemia reperfusion (IR), which involves impaired autophagy. Diabetic rats treated with autophagy inhibitor 3-methyladenine (3-MA) showed more serious ALI, with higher lung injury scores when myocardial IR occurs, in addition to impaired autophagy as indicated by reduced beclin-1 expression.85 Chlorine gas (Cl2) induces the ALI reaction process, and the upregulation of autophagy protects against Cl2-induced lung inflammation.86 High-mobility group box-1 (HMGB1) also induces an inflammatory response to aggravate ALI through the PI3K/AKT/mTOR pathway.87
Autophagy can aggravate acute lung injury
Studies have shown that autophagy can aggravate ALI. First, autophagy activation in the lungs during mechanical ventilation is dramatically attenuated in alveolar macrophage-depleted mice. Selective silencing of autophagy-related protein 5 in lung macrophages abolishes mechanical ventilation-induced nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome activation and lung inflammatory injury. Pharmacological inhibition of autophagy also significantly attenuates the inflammatory responses caused by lung hyperinflation.88 MiR-34a might suppress excessive autophagic activity (LC3 II/I and p62) in alveolar type II epithelial (AT-II) cells by targeting FoxO3 to reduce the damage of LPS-induced ALI.89 Moreover, autophagy in endothelial cells (ECs) could contribute to lung vascular injury. By regulating the expression of autophagy, visfatin activates the PI3K/AKT signalling pathway and reduces the autophagy level of ALI alveolar epithelial cells, resulting in a protective effect on lung tissue.90
In addition, studies have reported that the levels of autophagy detected at different stages of ALI differ. In the early stages (1 h and 2 h) of ALI, autophagy in the LPS group reached a peak at 2 h. As the ALI process progressed, apoptosis gradually increased in the lung tissue and reached its maximal level at later stages (6 h), while autophagy was time-dependently decreased.91 Therefore, the role of autophagy in ALI may be related to the pathophysiological process of ALI, and whether autophagy is over-activated; further research is needed to determine the relationships between autophagy and the molecular mechanisms of ALI (Figure 7).
Figure 7.

Acute lung injury and autophagy pathway. Several pharmacological and nutritional interventions as autophagy initiators/inhibitors affect the nucleation and elongation stages of autophagy. These autophagy promoters/inhibitors such as 3-MA, Cl2, MiR-34a, visfatin, virus, bacteria, and HMGB1 regulate the autophagy level by affecting the expression of beclin-1 and LC3 as well as LKB1/PI3K pathways, and lead to the release of chemokines, ROS, and other cytokines that contribute to ALI. The final effects of these interactions may vary, depending on the outcome of interactions between cell types.
ALI, acute lung injury; HMGB1, high-mobility group box-1; LC3, light chain 3; LKB1/PI3K, liver kinase B1/phosphatidylinositol 3-kinase; 3-MA, 3-methyladenine; ROS, reactive oxygen species.
Conclusion
In conclusion, autophagy is a cellular behavior that has been highly conserved throughout the course of evolution, and different types of cells have unique autophagy regulation mechanisms. Although preocular studies on autophagy are increasingly advanced in humans, they are still limited to the cellular level. The pathogenesis of autophagy in pulmonary disease remains unclear. Methods to exploit the positive effect, and avoid or reduce the ill effects, of autophagy still need further study. The transition from the laboratory to clinical practice remains far in the future.
Supplemental Material
Supplemental material, Author_response_v.1 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental Material
Supplemental material, Author_response_v.2 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental Material
Supplemental material, Reviewer_1_v.1 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental Material
Supplemental material, Reviewer_1_v.2 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental Material
Supplemental material, Reviewer_2_v.1 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Acknowledgments
The authors would like to thank Yao Ou-Yang for guidance.
Footnotes
Authors’ contributions: Shi-xia Liao contributed the idea and wrote the text, and Peng-peng Sun generated the figures. All authors read and approved the final manuscript.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and publication of this article: The present study was supported by the National Nature Science Foundation of China (grant no. 81460008).
Conflict of interest statement: The authors declare that there is no conflict of interest.
Ethics approval and consent to participate: Not applicable.
Patient consent for publication: Not applicable.
ORCID iD: Yao Ou-Yang  https://orcid.org/0000-0002-2649-6951
https://orcid.org/0000-0002-2649-6951
Availability of data and materials: Not applicable.
Supplemental material: The reviews of this paper are available via the supplemental material section.
Contributor Information
Shi-xia Liao, Department of Respiratory Medicine, Affiliated Hospital of ZunYi Medical College, Guizhou, China.
Peng-peng Sun, Department of Osteopathy, Affiliated Hospital of ZunYi Medical College, Guizhou, China.
Yan-hui Gu, Department of Respiratory Medicine, Affiliated Hospital of ZunYi Medical College, Guizhou, China.
Xi-min Rao, Department of Respiratory Medicine, Affiliated Hospital of ZunYi Medical College, Guizhou, China.
Lan-ying Zhang, Department of Respiratory Medicine, Affiliated Hospital of ZunYi Medical College, Guizhou, China.
Yao Ou-Yang, Department of Respiratory Medicine, Affiliated Hospital of ZunYi Medical College, 201 Daliang Road, Zunyi City, Guizhou 563003, P.R. China.
References
- 1. Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res 2014; 24: 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010; 221: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335: 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43: 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci 2012; 69: 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 2011; 7: 673–682. [DOI] [PubMed] [Google Scholar]
- 7. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014; 24: 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaushik S, Cuervo AM. Chaperones in autophagy. Pharmacol Res 2012; 66: 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klionsky DJ, Cregg JM, Dunn WA, Jr, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003; 5: 539–545. [DOI] [PubMed] [Google Scholar]
- 10. Harding TM, Morano KA, Scott SV, et al. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol 1995; 131: 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeshige K, Baba M, Tsuboi S, et al. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 1992; 119: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 2015; 22: 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dal-Ré RJ. Worldwide behavioral research on major global causes of mortality. Health Educ Behav 2011; 38: 433. [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez-Roisin R, Rabe KF, Vestbo J, et al. Global Initiative for Chronic Obstructive Lung Disease (GOLD) 20th Anniversary: a brief history of time. Eur Respir J 2017; 50: 1700671. [DOI] [PubMed] [Google Scholar]
- 15. Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176: 532–555. [DOI] [PubMed] [Google Scholar]
- 16. Strzelak A, Ratajczak A, Adamiec A, et al. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: a mechanistic review. Int J Environ Res Public Health 2018; 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 2008; 3: e3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Y, Xu B, He X, et al. Correlation between autophagy levels in peripheral blood mononuclear cells and clinical parameters in patients with chronic obstructive pulmonary disease. 2018; 17: 8003–8009. [DOI] [PubMed] [Google Scholar]
- 19. Puig-Vilanova E, Rodriguez DA, Lloreta J, et al. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic Biol Med 2015; 79: 91–108. [DOI] [PubMed] [Google Scholar]
- 20. Zeng Z, He S, Lu J, et al. MicroRNA-21 aggravates chronic obstructive pulmonary disease by promoting autophagy. Exp Lung Res 2018; 44: 89–97. [DOI] [PubMed] [Google Scholar]
- 21. Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. 2010; 107: 18880–18885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lam HC, Cloonan SM, Bhashyam AR, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest 2013; 123: 5212–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cloonan SM, Lam HC, Ryter SW, et al. Ciliophagy: the consumption of cilia components by autophagy. Autophagy 2014; 10: 532–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med 2017; 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol 2016; 138: 28–36. [DOI] [PubMed] [Google Scholar]
- 26. Ravindran R, Loebbermann J, Nakaya HI, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 2016; 531: 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shibutani ST, Saitoh T, Nowag H, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol 2015; 16: 1014–1024. [DOI] [PubMed] [Google Scholar]
- 28. Lv XX, Liu SS, Li K, et al. Cigarette smoke promotes COPD by activating platelet-activating factor receptor and inducing neutrophil autophagic death in mice. Oncotarget 2017; 8: 74720–74735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xue Y, Han H, Wu L, et al. iASPP facilitates tumor growth by promoting mTOR-dependent autophagy in human non-small-cell lung cancer. Cell Death Dis 2017; 8: e3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo JY, Teng X, Laddha SV, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 2016; 30: 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng Z, Xin H, Han T. BECN promotes the migration of NSCLC cells through regulating the ubiquitination of Vimentin. Cell Adh Migr 2019; 13: 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schläfli AM, Adams O, Galván JA, et al. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget 2016; 7: 39544–39555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu J, Lin Y, Yang H, et al. The expression of p33(ING1), p53, and autophagy-related gene Beclin1 in patients with non-small cell lung cancer. Tumour Biol 2011; 32: 1113–1121. [DOI] [PubMed] [Google Scholar]
- 35. Won KY, Kim GY, Lim SJ, et al. Decreased Beclin-1 expression is correlated with the growth of the primary tumor in patients with squamous cell carcinoma and adenocarcinoma of the lung. Hum Pathol 2012; 43: 62–68. [DOI] [PubMed] [Google Scholar]
- 36. Remon J, Planchard D. AZD9291 in EGFR-mutant advanced non-small-cell lung cancer patients. Future Oncol 2015; 11: 3069–3081. [DOI] [PubMed] [Google Scholar]
- 37. Li C, Dong Y, Wang L, et al. Ginsenoside metabolite compound K induces apoptosis and autophagy in non-small cell lung cancer cells via AMPK-mTOR and JNK pathways. Biochem Cell Biol 2019; 97: 406–414. [DOI] [PubMed] [Google Scholar]
- 38. Gewirtz DA. The challenge of developing autophagy inhibition as a therapeutic strategy. Cancer Res 2016; 76: 5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fan J, Zhang X, Wang S, et al. Regulating autophagy facilitated therapeutic efficacy of the sonic Hedgehog pathway inhibition on lung adenocarcinoma through GLI2 suppression and ROS production. Cell Death Dis 2019; 10: 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu HM, Shao LJ, Jiang ZF, et al. Gemcitabine-induced autophagy protects human lung cancer cells from apoptotic death. Lung 2016; 194: 1–8. [DOI] [PubMed] [Google Scholar]
- 41. Wu T, Wang MC, Jing L, et al. Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des Devel Ther 2015; 9: 6421–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiu YH, Hsu SH, Hsu HW, et al. Human nonsmall cell lung cancer cells can be sensitized to camptothecin by modulating autophagy. Int J Oncol 2018; 53: 1967–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang W, Zhang K, Shi X, et al. Critical role of bacterial isochorismatase in the autophagic process induced by Acinetobacter baumannii in mammalian cells. FASEB J 2016; 30: 3563–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O’Neill AM, Thurston TLM, Holden DW. Erratum for O’Neill et al., Cytosolic replication of Group A Streptococcus in human macrophages. mBio 2016; 7: e00020–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neumann Y, Bruns SA, Rohde M, et al. Intracellular Staphylococcus aureus eludes selective autophagy by activating a host cell kinase. 2016; 12: 2069–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim JJ, Lee HM, Shin DM, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2012; 11: 457–468. [DOI] [PubMed] [Google Scholar]
- 47. Zhao H, Chen H, Xiaoyin M, et al. Autophagy activation improves lung injury and inflammation in sepsis. Inflammation 2019; 42: 426–439. [DOI] [PubMed] [Google Scholar]
- 48. Oikonomou V, Moretti S, Renga G, et al. Noncanonical fungal autophagy inhibits inflammation in response to IFN-γ via DAPK1. Cell Host Microbe 2016; 20: 744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oliveira DL, Fonseca FL, Zamithmiranda D, et al. The putative autophagy regulator Atg7 affects the physiology and pathogenic mechanisms of Cryptococcus neoformans. Future Microbiol 2016; 11: 1405–1419. [DOI] [PubMed] [Google Scholar]
- 50. Zhang R, Chi X, Wang S, et al. The regulation of autophagy by influenza A virus. Biomed Res Int 2014; 2014: 498083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cypryk W, Lorey MB, Puustinen A, et al. Proteomic and bioinformatic characterization of extracellular vesicles released from human macrophages upon influenza A virus infection. J Proteome Res 2016; 16: 217. [DOI] [PubMed] [Google Scholar]
- 52. Gannage M, Dormann D, Albrecht R, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009; 6: 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jung KI, Pyo CW, Choi SY. Influenza A virus-induced autophagy contributes to enhancement of virus infectivity by SOD1 downregulation in alveolar epithelial cells. Biochem Biophys Res Commun 2018; 498: 960–966. [DOI] [PubMed] [Google Scholar]
- 54. Erratum: An Official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. an update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015; 192: 644. [DOI] [PubMed] [Google Scholar]
- 55. Sgalla G, Iovene B, Calvello M, et al. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res 2018; 19: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ghavami S, Yeganeh B, Zeki AA, et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta1 in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2018; 314: L493–L504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Larsoncasey JL, Deshane JS, Ryan AJ, et al. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 2016; 44: 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Araya J, Kojima J, Takasaka N, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2013; 304: L56–L69. [DOI] [PubMed] [Google Scholar]
- 59. Patel AS, Lin L, Geyer A, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One 2012; 3: e41394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Manni ML, Oury TD. Oxidative stress and pulmonary fibrosis. In: Laher I. (ed.) Systems biology of free radicals and antioxidants. Berlin, Heidelberg: Springer, 2014. [Google Scholar]
- 61. Del PD, Vona R, Giordani L, et al. Defective autophagy in fibroblasts may contribute to fibrogenesis in autoimmune processes. Curr Pharm Des 2011; 17: 3878–3887. [DOI] [PubMed] [Google Scholar]
- 62. Wang Y, Huang G, Wang Z, et al. Elongation factor-2 kinase acts downstream of p38 MAPK to regulate proliferation, apoptosis and autophagy in human lung fibroblasts. Exp Cell Res 2018; 363: 291–298. [DOI] [PubMed] [Google Scholar]
- 63. Hill C, Li J, Liu D, et al. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis 2019; 10: 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rangarajan S, Kurundkar A, Kurundkar D, et al. Novel mechanisms for the antifibrotic action of nintedanib. Am J Respir Cell Mol Biol 2016; 54: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Prashanth Goud M, Bale S, Pulivendala G, et al. Therapeutic effects of Nimbolide, an autophagy regulator, in ameliorating pulmonary fibrosis through attenuation of TGF-β1 driven epithelial-to-mesenchymal transition. Int Immunopharmacol 2019; 75: 105755. [DOI] [PubMed] [Google Scholar]
- 66. Sandra C, Mariana M, Iliana H, et al. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy 2015; 11: 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ricci A, Cherubini E, Scozzi D, et al. Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J Cell Physiol 2013; 228: 1516–1524. [DOI] [PubMed] [Google Scholar]
- 68. Hahn SS, Makaryus M, Talwar A, et al. A review of therapeutic agents for the management of pulmonary arterial hypertension. Ther Adv Respir Dis 2017; 11: 46–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004; 351: 1655–1665. [DOI] [PubMed] [Google Scholar]
- 70. Krymskaya VP, Snow J, Cesarone G, et al. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J 2011; 25: 1922–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang W, Liu J, Ma A, et al. mTORC1 is involved in hypoxia-induced pulmonary hypertension through the activation of Notch3. J Cell Physiol 2014; 229: 2117–2125. [DOI] [PubMed] [Google Scholar]
- 72. Lahm T, Frump AL, Albrecht ME, et al. 17beta-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2016; 311: L375–L388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang X, Ibrahim YF, Das D, et al. Carfilzomib reverses pulmonary arterial hypertension. Cardiovasc Res 2016; 110: 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li L, Wang X, Wang L, et al. Mammalian target of rapamycin overexpression antagonizes chronic hypoxia-triggered pulmonary arterial hypertension via the autophagic pathway. Int J Mol Med 2015; 36: 316–322. [DOI] [PubMed] [Google Scholar]
- 75. Teng RJ, Du J, Welak S, et al. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2012; 302: L651–L663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dalvi P, Sharma H, Chinnappan M, et al. Enhanced autophagy in pulmonary endothelial cells on exposure to HIV-Tat and morphine: role in HIV-related pulmonary arterial hypertension. Autophagy 2016; 12: 2420–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Singh N, Manhas A, Kaur G, et al. Inhibition of fatty acid synthase is protective in pulmonary hypertension. Br J Pharmacol 2016; 173: 2030–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wu YC, Wang WT, Lee SS, et al. Glucagon-like peptide-1 receptor agonist attenuates autophagy to ameliorate pulmonary arterial hypertension through Drp1/NOX- and Atg-5/Atg-7/Beclin-1/LC3beta pathways. Int J Mol Sci 2019; 20: E3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhai C, Shi W, Feng W, et al. Activation of AMPK prevents monocrotaline-induced pulmonary arterial hypertension by suppression of NF-κB-mediated autophagy activation. Life Sci 2018; 208: 87–95. [DOI] [PubMed] [Google Scholar]
- 80. Yang L, Zhang Z, Zhuo Y, et al. Resveratrol alleviates sepsis-induced acute lung injury by suppressing inflammation and apoptosis of alveolar macrophage cells. Am J Transl Res 2018; 10: 1961–1975. [PMC free article] [PubMed] [Google Scholar]
- 81. Patel VJ, Biswas Roy S, Mehta HJ, et al. Alternative and natural therapies for acute lung injury and acute respiratory distress syndrome. BioMed Res Int 2018; 2018: 2476824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zeng M, Sang W, Chen S, et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett 2017; 271: 26–37. [DOI] [PubMed] [Google Scholar]
- 83. Gao Y, Wang N, Li RH, et al. The role of autophagy and the chemokine (C-X-C Motif) ligand 16 during acute lung injury in mice. Med Sci Monit 2018; 24: 2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hu Y, Jian L, Mao YY, et al. Late-breaking abstract: activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Euro Res J 2016; 48: OA3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhan L, Zhang Y, Su W, et al. The roles of autophagy in acute lung injury induced by myocardial ischemia reperfusion in diabetic rats. J Diabetes Res 2018; 2018: 5047526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Carlisle M, Lam A, Svendsen ER, et al. Chlorine-induced cardiopulmonary injury. Ann N Y Acad Sci 2016; 1374: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Qu L, Chen C, Chen Y, et al. High-mobility group box 1 (HMGB1) and autophagy in acute lung injury (ALI): a review. Med Sci Monit 2019; 25: 1828–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang Y, Liu G, Dull RO, et al. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. American journal of physiology. Am J Physiol Lung Cell Mol Physiol 2014; 307: L173–L185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Song L, Zhou F, Cheng L, et al. MicroRNA-34a suppresses autophagy in alveolar type II epithelial cells in acute lung injury by inhibiting FoxO3 expression. Inflammation 2017; 40: 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wu XT, Ansari AR, Pang XX, et al. Visfatin plays a significant role in alleviating lipopolysaccharide-induced apoptosis and autophagy through PI3K/AKT signaling pathway during acute lung injury in mice. Arch Immunol Ther Exp (Warsz) 2019; 67: 249–261. [DOI] [PubMed] [Google Scholar]
- 91. Lin L, Zhang L, Yu L, et al. Time-dependent changes of autophagy and apoptosis in lipopolysaccharide-induced rat acute lung injury. Iran J Basic Med Sci 2016; 19: 632–637. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Author_response_v.1 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental material, Author_response_v.2 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental material, Reviewer_1_v.1 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental material, Reviewer_1_v.2 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease
Supplemental material, Reviewer_2_v.1 for Autophagy and pulmonary disease by Shi-xia Liao, Peng-peng Sun, Yan-hui Gu, Xi-min Rao, Lan-ying Zhang and Yao Ou-Yang in Therapeutic Advances in Respiratory Disease