

Abstract
Interferon regulatory factor 3 (IRF3) has both transcriptional and nontranscriptional functions. Transcriptional activity is dependent on serine phosphorylation of IRF3, while transcription‐independent IRF3‐mediated apoptosis requires ubiquitination. IRF3 also binds to inhibitor of nuclear factor kappa B kinase (IKKβ) in the cytosol, restricting nuclear translocation of p65. IRF3‐deficient mice are highly sensitive to high‐fat diet (HFD)‐induced liver injury; however, it is not known if transcriptional and/or nontranscriptional activity of IRF3 confers protection. Using a mouse model only expressing nontranscriptional functions of IRF3 (Irf3 S1/S1), we tested the hypothesis that nontranscriptional activity of IRF3 protects mice from HFD‐induced liver injury. C57BL/6, Irf3 −/−, and Irf3 S1/S1 mice were fed an HFD for 12 weeks. In C57BL/6 mice, the HFD increased expression of interferon (IFN)‐dependent genes, despite a decrease in IRF3 protein in the liver. The HFD had no impact on IFN‐dependent gene expression Irf3 −/− or Irf3 S1/S1 mice, both lacking IRF3 transcriptional activity. Liver injury, apoptosis, and fibrosis were exacerbated in Irf3 −/− compared to C57BL/6 mice following the HFD; this increase was ameliorated in Irf3 S1/S1 mice. Similarly, expression of inflammatory cytokines as well as numbers of neutrophils and infiltrating monocytes was increased in Irf3 −/− mice compared to C57BL/6 and Irf3 S1/S1 mice. While the HFD increased the ubiquitination of IRF3, a response associated with IRF3‐mediated apoptosis, in Irf3 S1/S1 mice, protection from liver injury was not due to differences in apoptosis of hepatocytes or immune cells. Instead, protection from HFD‐induced liver injury in Irf3 S1/S1 mice was primarily associated with retardation of nuclear translocation of p65 and decreased expression of nuclear factor kappa B (NFκB)‐dependent inflammatory cytokines. Conclusion: Taken together, these data identify important contributions of the nontranscriptional function of IRF3, likely by reducing NFκB signaling, in dampening the hepatic inflammatory environment in response to an HFD.

Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CCL
chemokine (C‐C) motif ligand
- CD
clusters of differentiation
- CXCL
chemokine (C‐X‐C) motif ligand
- ER
endoplasmic reticulum
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- HFD
high‐fat diet
- IκBα
NF-kappa B alpha
- IFIT
interferon‐induced protein with tetratricopeptide repeats
- IFN
interferon
- IKK
inhibitor of nuclear factor kappa B kinase
- IRF3
interferon regulatory factor 3
- MCP‐1
monocyte chemoattractant protein 1
- mRNA
messenger RNA
- MyD88
myeloid differentiation primary‐response 88 protein
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NFκB
nuclear factor kappa B
- PA
palmitic acid
- phospho
phosphorylated
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- rRNA
ribosomal RNA
- SEAP
secreted alkaline phosphatase
- STING
stimulator of interferon genes
- TLR
toll‐like receptor
- TNFα
tumor necrosis alpha
- TRIF
TIR‐domain‐containing adapter‐inducing interferon β
Obesity is a strong risk factor for the development of metabolic syndrome and is associated with insulin resistance and type 2 diabetes as well as nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH).1 Activation of the innate immune system is a key contributor to the pathogenesis of liver disease in obesity. Overnutrition results in microbial dysbiosis and impairs the gut barrier, allowing pathogen‐associated molecular patterns (PAMPs) into the circulation. Further, lipotoxicity of hepatocytes is associated with the release of damage‐associated molecular patterns (DAMPs).2 Recognition of PAMPs and DAMPs by pattern recognition receptors, such as toll‐like receptors (TLRs), on immune cells activates inflammatory pathways critical to the development of NAFLD/NASH.3, 4 Mice deficient in TLR4 are partially protected from high‐fat diet (HFD)‐induced liver injury, insulin resistance, and inflammation,5, 6 and TLR3‐deficient mice are protected from insulin resistance and hepatic steatosis in response to HFD‐induced obesity.7 TLR signaling by the myeloid differentiation primary‐response protein (MyD88)‐dependent pathway activates multiple signaling pathways, including nuclear factor kappa B (NFκB), to increase the expression of inflammatory mediators.8 Signaling by the MyD88‐independent TIR domain‐containing adapter‐inducing interferon β (TRIF)‐dependent pathway activates additional transcriptional factors, including interferon regulatory factor 3 (IRF3), which modulates the expression of interferons (IFNs).9, 10
IRF3‐stimulated expression of type 1 IFNs plays a key role in the innate immune response against viruses.11, 12 While phosphorylation of IRF3 is required for IRF3‐mediated expression of antiviral genes,12 IRF3 also has phosphorylation/transcription‐independent activities, including an IRF3‐mediated pathway of apoptosis, termed the RIG‐I‐like receptors‐induced IRF3‐mediated pathway of apoptosis.13 In IRF3‐mediated apoptosis, ubiquitinated IRF3 complexes with BAX, translocates to the mitochondria, where cytochrome c is released, resulting in apoptosis.13 In addition to IRF3‐mediated apoptosis, IRF3 interacts with the kinase domain of inhibitor of NFκB kinase β subunit (IKKβ) in the cytoplasm; this interaction prevents phosphorylation of IKKβ, thus restricting the release of phosphorylated (phospho) p65 from the IKK complex and impairing NFκB‐dependent expression of inflammatory genes.14
While IRF3 is classically associated with protection from viral infection, recent data also implicate a complex role for IRF3 in metabolic liver diseases. For example, Irf3 −/− mice are protected from chronic ethanol‐induced liver injury15, 16 and fibrosis.17 In contrast, Irf3 −/− mice develop severe hepatic injury, enhanced hepatic steatosis, and insulin resistance in response to HFDs.14 Interestingly, increased HFD‐induced liver injury in the Irf3 −/− mice is not associated with increased adipose inflammation,18 suggesting a liver‐specific protective effect of IRF3 in response to challenge with HFDs.
Given the multiple functional activities of IRF3, it is important to distinguish the specific transcriptional and nontranscriptional roles of IRF3 in metabolic liver diseases. The recent development of a mouse expressing a mutant Irf3 gene that encodes an IRF3 protein lacking key phosphorylation sites (SS388/390AA) required for the transcriptional function of IRF3,13, 19 termed Irf3 S1/S1, has allowed investigators to distinguish the transcriptional from nontranscriptional contributions of IRF3 to metabolic liver disease. Using these Irf3 S1/S1 mice, we reported that a nontranscriptional function of IRF3 contributes to ethanol‐induced liver injury in mice; IRF3‐mediated apoptosis promotes a proinflammatory immune environment in the livers of mice in response to acute on chronic ethanol challenge.16 This response was associated with IRF3‐mediated apoptosis in restorative Ly6Clow monocytes, resulting in a shift toward proinflammatory Ly6Chi monocytes and exacerbated liver injury.16 Making use of Irf3 S1/S1 mice, here we have tested the hypothesis that nontranscriptional activity of IRF3 is also involved in the hepatic response to HFD‐induced obesity. We find, in contrast to the proinflammatory phenotype of the Irf3 S1/S1 mice in response to acute on chronic ethanol, Irf3 S1/S1 mice were protected from HFD‐induced liver injury. This protection was associated with decreased nuclear translocation of phospho‐p65 to the nucleus and reduced expression of NFκB‐dependent proinflammatory cytokine expression.
Materials and Methods
Animals and Care
All animals received humane care and all procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Breeding colonies of Irf3 −/− and Irf3 S1/S1 on a C57BL/6 background were maintained at the Cleveland Clinic.13, 19 Five‐week‐old male C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) or Charles River Laboratories (Wilmington, MA). Animals were housed in pairs in standard micro‐isolator cages and maintained on a 12‐hour:12‐hour light–dark cycle, weighed weekly, and food intake per cage measured weekly. Mice were weight matched and randomly distributed to chow or an HFD. Mice were fed an HFD containing 42% kcal from fat and 18.8 kJ/g (TD 88137; Teklad Mills, Madison, WI) or a standard diet (chow) containing 6% fat and 13 kJ/g (TD2918; Teklad Mills, Madison, WI).20 Three independent feeding trials were conducted to collect sufficient material for all the analyses included in this study.
At the end of the 12‐week HFD feeding protocol, mice were fasted for 6 hours and then euthanized. Portions of liver and epididymal adipose tissue were flash frozen in liquid nitrogen and stored at −80°C, preserved in RNAlater (Thermo Fisher Scientific, Grand Island, NY), and stored at −20°C until RNA isolation or fixed in 10% formalin or frozen in optimal cutting temperature compound (Sakura Finetek U.S.A., Inc., Torrance, CA) for histology. Blood was transferred to ethylene diamine tetraacetic acid‐containing tubes for the isolation of plasma. Plasma was then stored at −80°C.
RNA Isolation and Quantitative Real‐Time Polymerase Chain Reaction
RNA was isolated from liver stored in RNAlater using RNeasy Mini kits per the manufacturer’s instructions (Qiagen, Germantown, MD). Two micrograms of liver RNA was reverse transcribed and analyzed with PowerSYBR quantitative real‐time polymerase chain reaction (qRT‐PCR) kits (Applied Biosystems, Foster City, CA) on a QuantStudio5 RT‐PCR Machine (Applied Biosystems). Relative messenger RNA (mRNA) expression was determined using gene‐specific primers (see Supporting Table S1). Statistical analyses were performed on the ∆Ct values (average Ct of gene of interest – average Ct of 18S).21
Biochemical Assays, Immunohistochemistry, Western Blot Analysis, and Flow Cytometry
Detailed methods for biochemical assays, immunohistochemistry, western blot analysis, and flow cytometry can be found in the Supporting Materials.
Statistical Analysis
Values shown in all figures represent the means ± SEM. Analysis of variance was performed using the general linear models procedure (SAS, Carey, IN). Data were log transformed as necessary to obtain a normal distribution. Follow‐up comparisons were made by least square means testing. P < 0.05 was considered significant.
Results
HFD Feeding Activated IRF3‐Dependent Transcription in Liver
Expression of the IRF3‐dependent genes IFN‐induced protein with tetratricopeptide repeats (IFIT)1 and IFIT3 were increased by HFD feeding in C57BL/6 but not in Irf3 −/− and Irf3 S1/S1 mice, consistent with the lack of transcriptional activity of IRF3 in these genotypes (Fig. 1A). Increased expression of the IFIT genes in response to HFD feeding occurred despite a reduction in the expression of IRF3 protein in livers of C57BL/6 mice compared to chow‐fed mice (Fig. 1B), consistent with reported results.14 A similar reduction in IRF3‐immunoreactive protein was observed in the Irf3 S1/S1 mice (Fig. 1B). Reduced IRF3 protein expression was independent of changes in Irf3 mRNA (Fig. 1C). As expected, Irf3 −/− mice showed low expression of IRF3 mRNA and protein (Fig. 1B,C).
Figure 1.
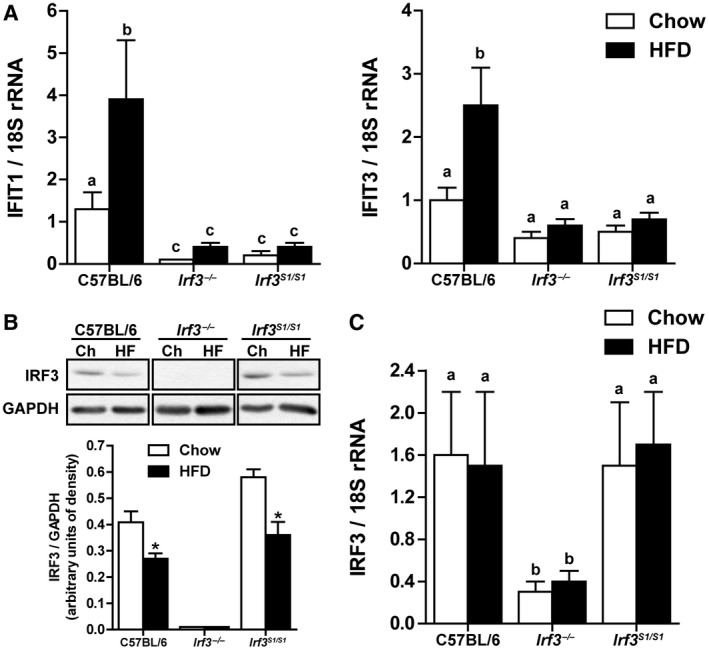
Hepatic IRF3 activation after HFD feeding. Male C57BL/6, Irf3 −/−, and Irf3 S1/S1 mice were fed an HFD or chow for 12 weeks. (A) Expression of IFIT1 and IFIT3 mRNA was analyzed by qRT‐PCR. Values were normalized to 18S and are shown as fold increase over the chow‐fed C57BL/6 control group, n = 12 per genotype. (B) Expression of IRF3 was assessed by western blot and normalized to GAPDH, n = 4 per genotype. (C) Expression of IRF3 mRNA was analyzed by qRT‐PCR. Values were normalized to 18S and are shown as fold increase over the chow‐fed C57BL/6 control group, n = 12 per genotype. Values represent means ± SEM. Values with different alphabetical superscripts are significantly different, P < 0.05. Abbreviations: Ch, chow; HF, high fat.
Restoration of Nontranscriptional Activity of IRF3 to Irf3‐Deficient Mice Reduced HFD‐Induced Liver Injury and Inflammation but Not Inflammatory Responses in Adipose Tissue
All genotypes gained more weight on the HFD compared to the chow diet. Despite similar or very small differences in food intake among genotypes (Supporting Fig. S1A,C), the final body weight of Irf3 −/− and Irf3 S1/S1 mice was 10% lower than C57BL/6 mice after HFD feeding (Supporting Fig. S1B,D). Kumari et al.18 also reported a slightly lower weight gain in Irf3 −/− mice on the HFD compared to wild‐type mice that was associated with an increase in energy expenditure in the Irf3 −/− mice compared to controls. As expected, HFD feeding impaired glucose tolerance; this response was independent of genotype (Supporting Fig. S1E‐G).
HFD feeding increased the liver injury markers alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in C57BL/6 mice; these responses were exacerbated in Irf3 −/− mice (Fig. 2A,B). In contrast, restoration of the nontranscriptional functions of IRF3 in the Irf3 S1/S1 mice reduced plasma ALT and AST compared to Irf3 −/− and C57BL/6 mice (Fig. 2A,B). Liver triglyceride content and histologic analysis of the livers by hematoxylin and eosin staining revealed an accumulation of lipid droplets in hepatocytes of C57BL/6 mice after HFD feeding (Fig. 2 C,D). However, hepatic triglyceride accumulation was lower in Irf3 −/− mice compared to C57BL/6 mice (Fig. 2C,D) and even further reduced in Irf3 S1/S1 mice after HFD feeding (Fig. 2C,D). These differences in lipid accumulation between genotypes were not due to differences in the ability of hepatocytes to accumulate lipid because the accumulation of lipid by primary cultures of hepatocytes exposed to palmitic acid (PA) was independent of genotype (Supporting Fig. S2). Expression of inflammatory cytokines and chemokines is a key pathophysiologic characteristic of HFD‐induced liver injury. Indeed, HFD feeding increased the expression of multiple inflammatory mediators in the liver, including tumor necrosis factor alpha (TNFα), interleukin‐1β (IL‐1β), and monocyte chemoattractant protein 1 (MCP‐1)/chemokine (C‐C) motif ligand 2 (CCL2) in C57BL/6 mice (Fig. 2E‐G). These responses were further increased in Irf3 −/− mice, but restoration of the nontranscriptional activity of IRF3 in the Irf3 S1/S1 mice ameliorated inflammation induced by HFD feeding (Fig. 2E‐G). Similarly, the appearance of inflammatory foci was increased in Irf3 −/− mice compared to C57BL/6 or Irf3 S1/S1 mice (see insets to Fig. 2D). Taken together, these data suggest that, while the transcriptional activity of IRF3 has a role in HFD‐induced hepatosteatosis, the nontranscriptional activity of IRF3 serves to protect mice from HFD‐induced inflammation and hepatocellular injury.
Figure 2.
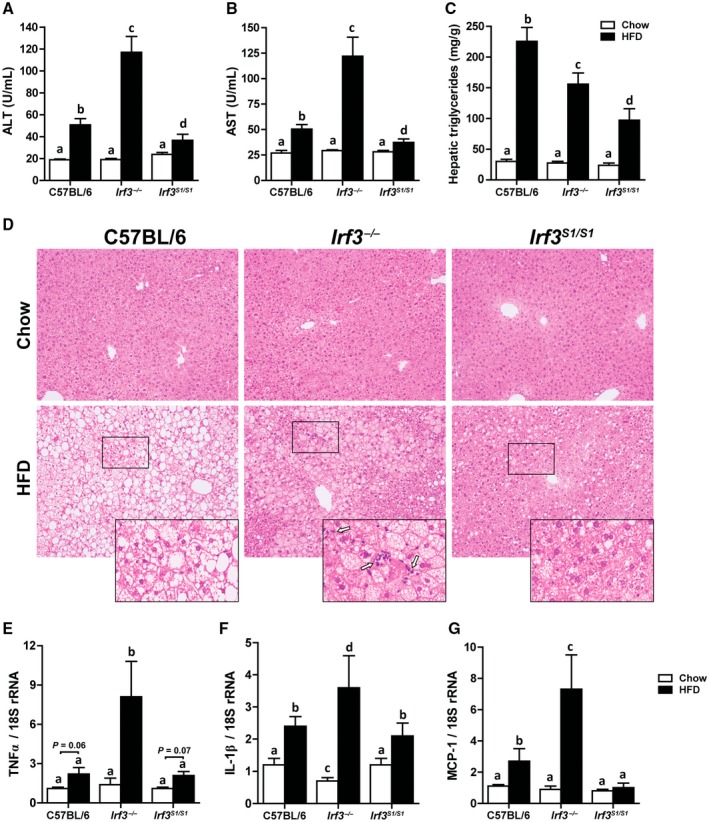
Irf3 S1/S1 mice reduced HFD‐induced liver injury, steatosis, and markers of inflammation. Male C57BL/6, Irf3 −/−, and Irf3 S1/S1 mice were fed an HFD or chow for 12 weeks. (A,B) ALT and AST activities were measured in plasma, n = 12 per genotype. (C) Hepatic triglyceride content was measured in whole‐liver homogenates, n = 12 per genotype. (D) Paraffin‐embedded liver sections were stained with hematoxylin and eosin. Images were acquired using 10× and 40× objectives, n = 4 per genotype. Expressions of (E) TNFα, (F) IL‐1β, and (G) MCP‐1 mRNA were analyzed by qRT‐PCR. Values were normalized to 18S and are shown as fold increase over the chow‐fed C57BL/6 control group, n = 12 per genotype. Values represent means ± SEM. Values with different alphabetical superscripts are significantly different, P < 0.05.
Adipose inflammation is another hallmark of the metabolic response to HFD feeding. Interestingly, in contrast to the impact of the Irf3 genotype on inflammatory responses in the liver, HFD feeding increased the expression of TNFα, MCP‐1, and collagen 6 in adipose tissue independently of genotype (Supporting Fig. S3A‐C). Similarly, crown‐like structures were increased in adipose tissue in response to HFD feeding in all genotypes (Supporting Fig. S3D). These data indicate that the impact of HFD feeding on adipose tissue inflammation was independent of Irf3 genotype.
Nontranscriptional Activity of IRF3 Reduced HFD‐Induced hepatocyte Apoptosis and Fibrosis
Hepatocyte apoptosis is considered a key driver of HFD‐induced liver injury.22 Accumulation of M30, a caspase cleavage product of cytokeratin‐18, is a specific marker of caspase activation and apoptosis in hepatocytes. HFD feeding increased M30 accumulation in C57BL/6 mice; this response was exacerbated in Irf3 −/− mice (Fig. 3A,B). However, when the nontranscriptional activity of IRF3 was restored in the Irf3 S1/S1 mice, the number of M30‐positive hepatocytes was equal to that of wild‐type mice (Fig. 3A,B). Inflammation and cellular death are also critical drivers of fibrosis in liver.23 Consistent with the increased inflammation and apoptosis in Irf3 −/− mice compared to C57BL/6 and Irf3 S1/S1 mice, both sirius red staining and expression of collagen 1 α 1 (Col1α1) mRNA were elevated in Irf3 −/− mice compared to C57BL/6 and Irf3 S1/S1 mice (Fig. 3C‐E). These data suggest that the nontranscriptional function of IRF3 ameliorated hepatocellular apoptosis and fibrosis.
Figure 3.
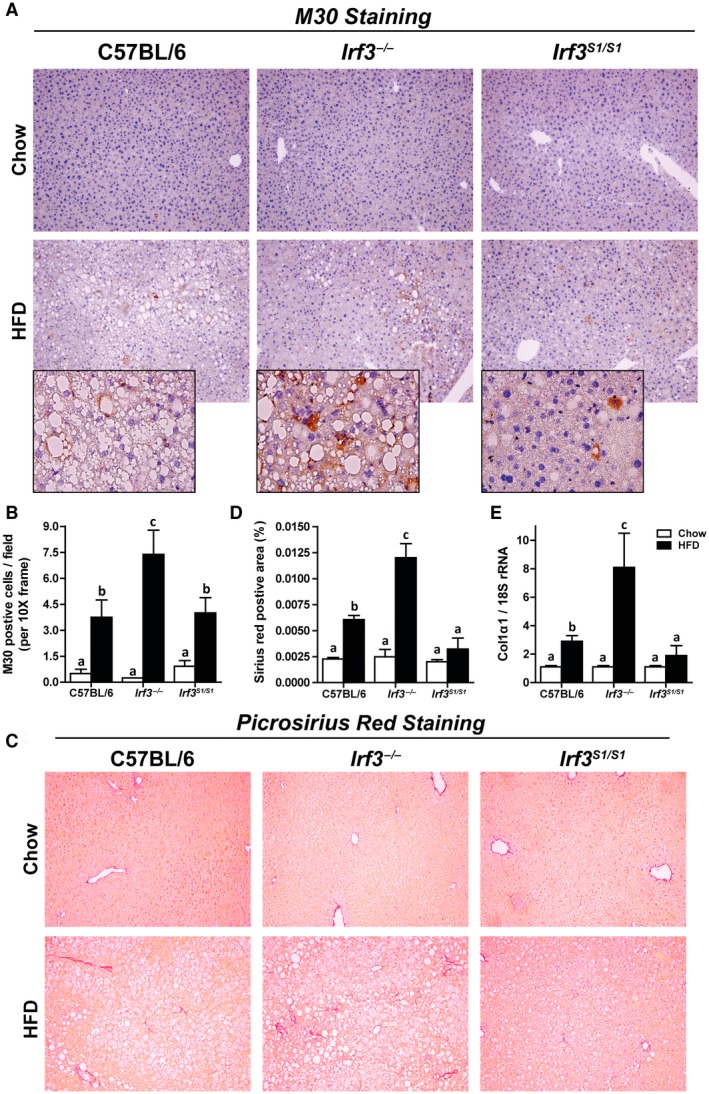
Enhanced markers of hepatocellular death and fibrosis in Irf3 −/− mice compared with wild‐type and Irf3 S1/S1 mice after HFD feeding. Male C57BL/6, Irf3 −/−, and Irf3 S1/S1 mice were fed an HFD or chow for 12 weeks. (A,B) Paraffin‐embedded liver sections were stained for M30, a caspase‐dependent cleavage product of cytokeratin 18. Images were acquired using 10× and 40× objectives, and the positive area was quantified, n = 4 per genotype. (C,D) Paraffin‐embedded liver sections were stained with sirius red. Images were acquired using a 40× objective, and the positive area was quantified, n = 4 per genotype. (E) Expression of Col1α1 mRNA was analyzed by qRT‐PCR. Values were normalized to 18S and are shown as fold increase over the chow‐fed C57BL/6 control group, n = 8 per genotype. Values represent means ± SEM. Values with different alphabetical superscripts are significantly different, P < 0.05. Abbreviation: Col1α1, collagen 1 α 1.
Restoration of Nontranscriptional Activity of IRF3 to Irf3‐Deficient Mice Reduced HFD‐Induced Accumulation of Immune Cells to the Liver
We next asked whether increased expression of inflammatory mediators in the liver of Irf3 −/− in response to HFD feeding was associated with increased accumulation of immune cells in the liver.24, 25 HFD feeding increased expression of clusters of differentiation (CD)45 mRNA in the liver (Fig. 4A) as well as the numbers of CD45‐positive cells assessed by flow cytometric analysis (Fig. 4B) and immunohistochemistry (Fig. 4C) in Irf3 −/− mice compared to C57BL/6 and Irf3 S1/S1 mice. Many CD45‐positive cells were found within clusters/inflammatory foci in Irf3 −/− mice but not C57BL/6 or Irf3 S1/S1 mice (Fig. 4C).
Figure 4.
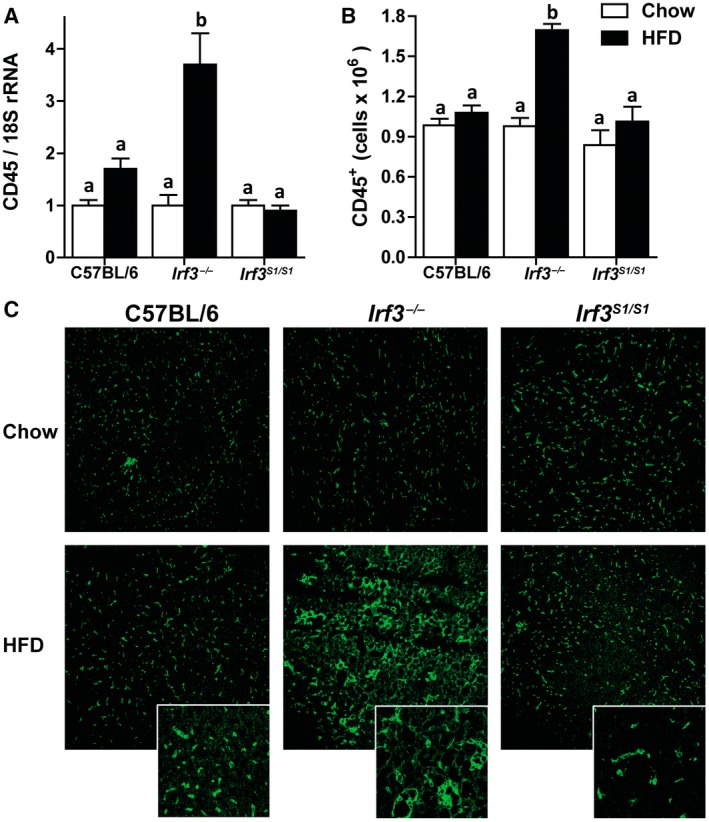
HFD enhanced CD45+ infiltration in Irf3 −/− but not in wild‐type and Irf3 S1/S1 livers. Male C57BL/6, Irf3 −/−, and Irf3 S1/S1 mice were fed an HFD or chow for 12 weeks. (A) Expression of CD45 mRNA was analyzed by qRT‐PCR. Values were normalized to 18S and are shown as fold increase over the chow‐fed C57BL/6 control group, n = 8 per genotype. (B) Hepatic CD45+ leukocytes were gated in isolated liver NPCs by flow cytometry, and total number of cells were quantified in isolated liver NPCs, n = 4 per genotype. (C) Paraffin‐embedded liver sections were stained for CD45. Images were acquired using a 20× objective, n = 4 per genotype. Values represent means ± SEM. Values with different alphabetical superscripts are significantly different, P < 0.05. Abbreviation: NPC, nonparenchymal cell.
Given the increase in CD45+ cells in Irf3 −/− livers, we then further studied the subpopulations of hepatic leukocytes using the gating strategy illustrated in Supporting Fig. S4. HFD feeding increased the accumulation of hepatic leukocytes to a greater extent in Irf3 −/− mice compared to C57BL/6 or Irf3 S1/S1 mice. Similar responses to diet and genotype were observed for the number of neutrophils (Ly6G+) (Fig. 5A), infiltrated monocytes (CD11b+) (Fig. 5B), inflammatory monocytes (Ly6Chigh) (Fig. 5C,D), and restorative monocytes (Ly6Clow) (Fig. 5C,E). In contrast, the number of Kupffer cells (F4/80+) in the liver was independent of diet or genotype (Fig. 5C,F). Taken together, these data indicate that the nontranscriptional activity of IRF3 restricted the accumulation of neutrophils, inflammatory monocytes, and restorative monocytes in the liver after HFD feeding.
Figure 5.
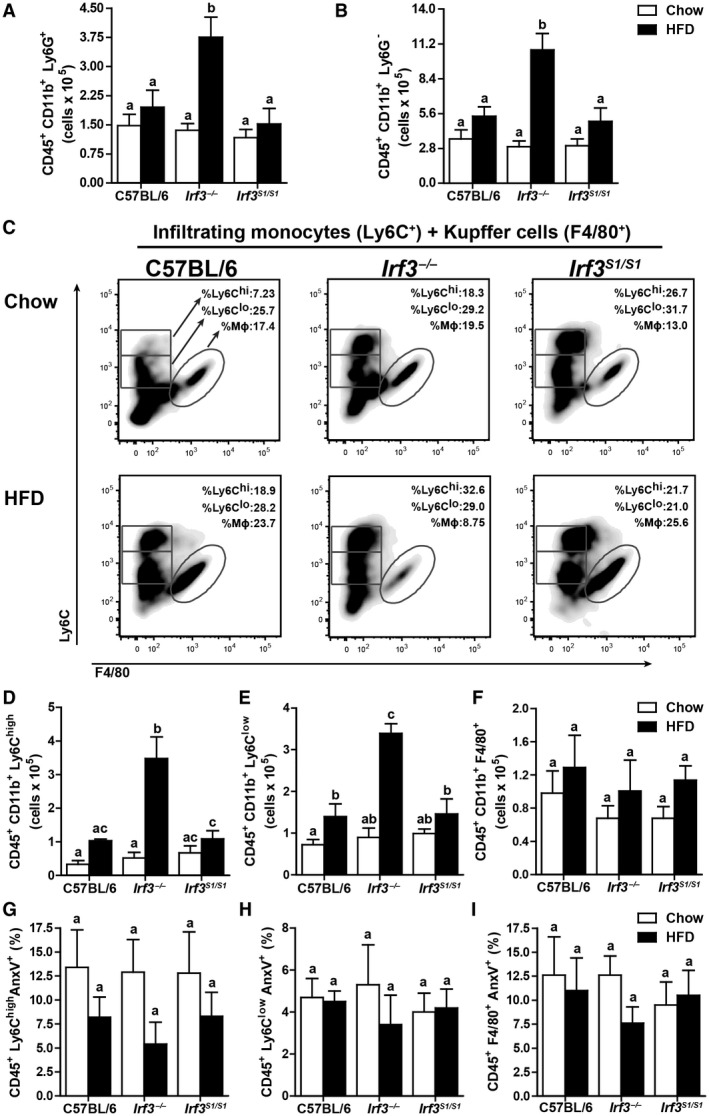
Infiltrated immune cell profile after HFD feeding in C57BL/6, Irf3 −/− , and Irf3 S1/S1 mice. Hepatic leukocytes were examined by flow cytometry from male C57BL/6, Irf3 −/−, and Irf3 S1/S1 mice following an HFD or chow diet for 12 weeks. CD45+ CD11b+ Ly6G+ were gated for (A) infiltrating neutrophils (CD45+ CD11b+ Ly6G+) and (B) infiltrating monocytes (CD45+ CD11b+ Ly6G–). (C‐E) Infiltrated monocytes were gated for (C,D) inflammatory monocytes (CD45+ CD11b+ Ly6Chigh), (C,E) restorative monocytes (CD45+ CD11b+ Ly6Clow), and (C,F) Kupffer cells (CD45+ CD11b+ F4/80+), n = 4 per genotype. (G‐I) Annexin V+ staining was used to assess apoptosis in isolated liver immune cells by flow cytometry for (G) inflammatory monocytes (Ly6Chigh), (H) restorative monocytes (Ly6Clow), and (I) Kupffer cells (F4/80+), n = 4 per genotype. Values represent means ± SEM. Values with different alphabetical superscripts are significantly different, P < 0.05. Abbreviation: AnxV, annexin V.
Reduced Accumulation of Leukocytes in the Liver of Irf3 S1/S1 Mice Was Not Due to Increased Apoptosis of Immune Cells
We recently reported the nontranscriptional activity of IRF3‐induced apoptosis of a specific population of hepatic immune cells in a murine model of acute on chronic ethanol‐induced liver injury.16 IRF3‐mediated apoptosis specifically in Ly6Clow restorative monocytes resulted in a more proinflammatory environment in the liver and exacerbated injury. Increased injury was associated with increased ubiquitination of IRF3 after ethanol feeding.16 In contrast to acute on chronic ethanol‐induced liver injury, HFD feeding did not increase annexin V‐positive Ly6Chigh, Ly6Clow, or F4/80+ cells in the liver (Fig. 5G‐I) or increase the ubiquitination of IRF3 (Supporting Fig. S5). Taken together, these data indicate that, in contrast to ethanol‐induced liver injury,16 ubiquitin‐dependent IRF3‐mediated apoptosis of innate immune cells in the liver does not contribute to HFD‐induced liver inflammation or injury.
Nontranscriptional Function of IRF3 Constrains the Translocation of p65‐NFκB to the Nucleus in Response to HFD Feeding
NFκB is a master regulator of inflammation in NAFLD.26 Following phosphorylation, p65 dissociates from the NFκB complex, translocates to the nucleus, and activates a proinflammatory transcriptional program. In a murine model of HFD‐induced liver injury, IRF3 was reported to restrict the movement of the p65 subunit of NFκB to the nucleus through an interaction with IKKβ.18 However, it is not known if this is dependent on the transcriptional or nontranscriptional activity of IRF3. If the nontranscriptional function of IRF3 were involved in this pathway, then the IRF3 S1 mutant protein would be expected to interact with NFκB subunits. RAW264.7 macrophages were transfected with empty vector, wild‐type Irf3, or Irf3 S1 mutant plasmids, and the interaction between p65 and IRF3 protein was assessed by immunoprecipitation (Fig. 6A) and confocal microscopy (Fig. 6B). Both wild‐type IRF3 and IRF3 S1 associated with p65, indicating that phosphorylation of IRF3 is not necessary for interaction with p65 (Fig. 6A,B). When RAW264.7 Blue cells, expressing secreted alkaline phosphatase (SEAP) under the control of NFκB, were challenged with Poly (I:C), SEAP activity increased in cells transfected with empty vector (Fig. 6C). However, in cells expressing either wild‐type IRF3 or IRF3 S1, SEAP activity was reduced (Fig. 6C), further confirming that both wild‐type IRF3 and IRF3 S1 can restrict the activity of NFκB.
Figure 6.
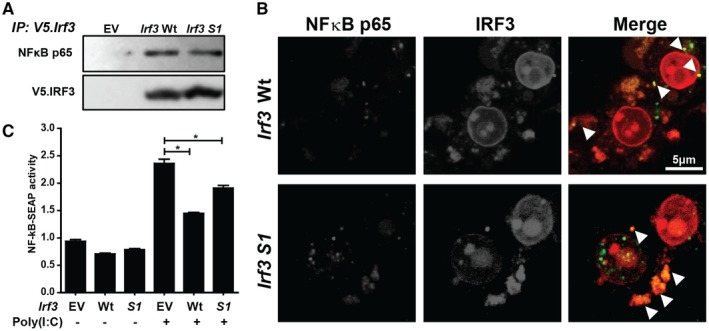
Interaction between IRF3 and NFκB in RAW264.7 macrophages. (A,B) RAW264.7 cells were transfected with EV, V5‐tagged Irf3 Wt, or Irf3 S1. After 48 hours, cells were (A) lysed, V5.Irf3 immunoprecipitated, and the interaction between IRF3 and the p65 subunit of NFκB analyzed by immunoblot or (B) fixed and immunostained with anti‐V5 and anti‐p65 antibodies for analysis by confocal microscopy. Arrows indicate the sites of colocalization. Images are representative of at least 20 fields from three independent experiments. (C) RAW‐Blue cells were transfected with V5‐tagged empty vector, Irf3 Wt, or Irf3 S1. After 24 hours, the cells were challenged with 50 ng/mL Poly (I:C) and NFκB‐driven SEAP activity measured in culture media 24 hours later using Quanti‐Blue reagent. Values were normalized to activity in cells transfected with EV and not treated with Poly (I:C). Values represent means ± SEM, n = 3. *P < 0.05. Abbreviations: EV, empty vector; IP, immunoprecipitated; Wt, wild type.
In C57BL/6 mice, HFD feeding activated NFκB signaling in the liver. Western blot analysis revealed that phosphorylation of IKKα/β and p65 were increased and the quantity of inhibitor of NF‐kappa B alpha (IκBα) decreased in HFD‐fed mice compared to chow‐fed mice. Phospho‐p65 was further increased in Irf3 −/− but not Irf3 S1/S1 mice compared to C57BL/6 (Fig. 7A). Immunohistochemical analysis also showed that HFD feeding increased the phosphorylation of p65 in the liver (Fig. 8A; Supporting Fig. S4), increasing the localization of p65 in the nuclei of both hepatocytes and nonparenchymal cells in the liver (Fig. 8A). If IRF3 restricts the movement of NFκB to the nucleus and subsequently reduces NFκB‐mediated inflammatory responses, then the HFD‐induced movement of p65 to the nucleus should be increased in Irf3 −/− mice and limited in Irf3 S1/S1 mice. Indeed, the accumulation of total phospho‐p65 assessed in 10× images (Supporting Fig. S6) as well as phospho‐p65 in hepatocyte and nonparenchymal cell nuclei visualized in 40× images (Fig. 7B) was more abundant in Irf3 −/− compared to Irf3 S1/S1 mice in response to HFD feeding. Similarly, more phospho‐p65 was found in nuclear extracts from Irf3 −/− mice compared to C57BL/6 or Irf3 S1/S1 mice (Fig. 7C). Elevated phospho‐p65 in the nucleus in Irf3 −/− mice was associated with increased expression of mRNA for a number of NFκB‐mediated inflammatory cytokines (Fig. 2E,F) as well as chemokines, including CCL5, (C‐X‐C) motif ligand (CXCL) 10, CXCL2, and CCL3 (Fig. 7D). These responses were attenuated in the Irf3 S1/S1 mice expressing only the nontranscriptional activities of IRF3.
Figure 7.
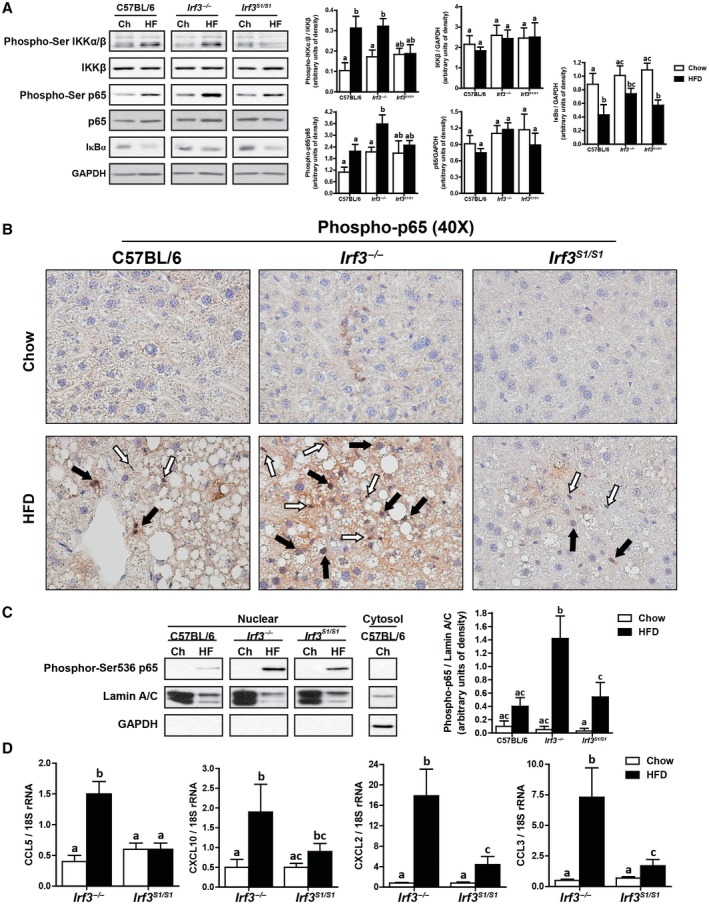
HFD feeding increased the translocation to the nucleus of p65 and expression of NFκB‐related genes in Irf3 −/− mice. Male C57BL/6, Irf3 −/−, and Irf3 S1/S1 mice were fed an HFD or chow for 12 weeks. (A) Expressions of phospho‐IKKα∕β, IKKβ, phospho‐p65, p65, and IκBα were assessed by western blot and normalized to GAPDH, n = 4 per genotype. (B) Formalin‐fixed paraffin‐embedded sections of liver were deparaffinized, and localization of phospho‐p65 was assessed by immunohistochemistry. Images were acquired at 40×. White arrows indicate nonparenchymal cells, and black arrows indicate hepatocytes. Images are representative of triplicate images from eight mice per treatment group. Values represent means ± SEM. (C) Nuclear fractions were isolated from livers, and equal amounts of protein were assessed by western blot and probed against phosphor‐Ser 536 p65‐NFκB. Lamin A/C was used as a nuclei marker, and GAPDH was used as a cytosolic marker. (D) Expressions of CCL5, CXCL10, CXCL2, and CCL3 were analyzed by qRT‐PCR. Values were normalized to 18S and are shown as fold increase over the chow‐fed C57BL/6 control group, n = 12 per genotype. Values with different superscripts are significantly different from each other, P < 0.05. Abbreviations: Ch, chow; HF, high fat.
Figure 8.
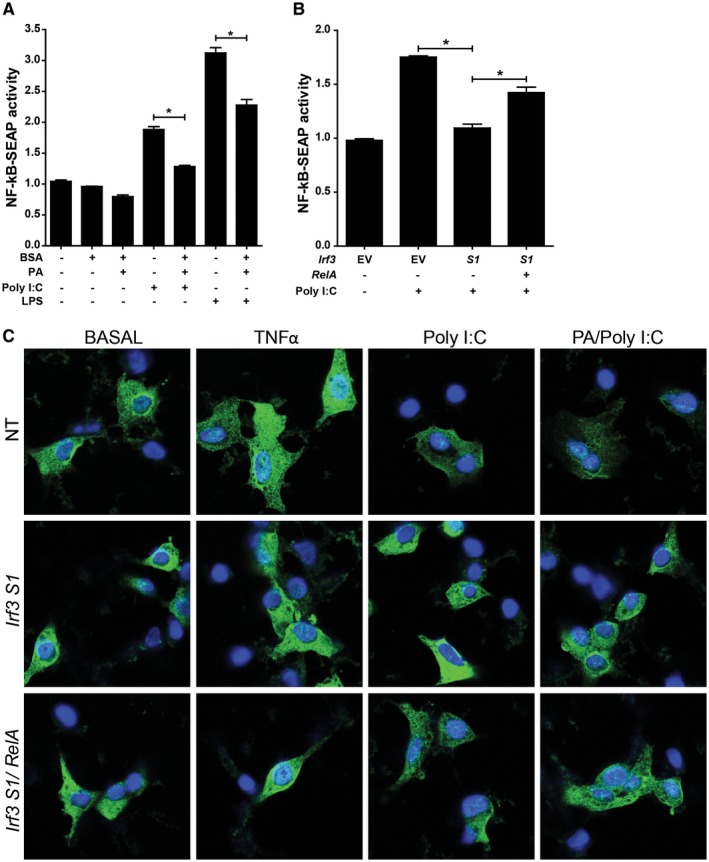
Overexpression of the RelA subunit of NFκB partially alleviates S1 IRF3‐mediated restriction of NFκB activity in RAW264.7 macrophages and AML12 hepatocytes. (A) RAW264.7 cells were challenged with 100 µM PA complexed to BSA or BSA (vehicle control) with or without 50 ng/mL Poly (I:C) or 1 µg/mL LPS. After 24 hours, NFκB‐driven SEAP activity was measured in the media. (B) RAW‐Blue cells were transfected with V5‐tagged empty vector or Irf3 S1 with or without an overexpression plasmid for the RelA subunit of NFκB. After 24 hours, the cells were challenged with 50 ng/mL Poly (I:C) and NFκB‐driven SEAP activity measured in culture media 24 hours later using Quanti‐Blue reagent. Values were normalized to activity in cells transfected with empty vector and not treated with Poly (I:C). Values represent means ± SEM, n = 3. *P < 0.05. (C) AML12 hepatocytes were transfected or not with V5‐tagged Irf3 S1 with or without an overexpression plasmid for the RelA subunit of NFκB. After 24 hours, the cells were challenged with 50 ng/mL TNFα, 500 µM PA, and 50 ng/mL Poly (I:C). Subcellular localization of p65 was assessed by confocal microscopy. Images are representative of triplicate images from three independent experiments. Abbreviations: BSA, bovine serum albumin; EV, empty vector; LPS, lipopolysaccharide; NT, no treatment.
While the reduction in inflammatory cytokines (Fig. 2) and chemokines (Fig. 7) in Irf3 S1/S1 mice is consistent with decreased NFκB signaling in immune cells, studies in whole liver cannot distinguish the specific cell types involved in NFκB activity. Therefore, we made use of cultured RAW264.7 macrophage‐like cells and AML12 hepatocytes to better understand cell‐specific responses in the liver. PA alone did not stimulate NFκB activity in RAW 264.7 (Fig. 8A) or AML12 cells (Fig. 8C), consistent with reported results.27 Poly I:C (Figs. 6C and 8A) and lipopolysaccharide (Fig. 8A) activated NFκB signaling in RAW264.7 Blue cells. Similarly, TNFα and Poly I:C increased the localization of p65 in the nucleus of AML12 cells (Fig. 8C). When S1 IRF3 was overexpressed in RAW264.7 (Fig. 8B) or AML12 cells (Fig. 8C), NFκB activity (Figs. 6C and 8B) and nuclear localization (Fig. 8C) were decreased. Importantly, when the p65 (RelA) subunit of NFκB was overexpressed, there was a partial restoration of NFκB activity (Fig. 8B) and nuclear localization (Fig. 8C).
Discussion
Development of NAFLD/NASH is driven by activation of innate immune and inflammatory responses. However, the mechanisms for the activation of these inflammatory responses in response to HFDs and obesity are not completely understood. NFκB‐dependent expression of inflammatory cytokines and chemokines is a well‐characterized pathway critical to HFD‐induced liver injury.26 There is also a growing appreciation that the IRF family of transcription factors also contributes to immune responses to a variety of metabolic stresses.28 For example, IRF3 has recently been implicated as a protective factor in HFD‐induced liver injury that is associated with an interaction between IRF3 and the NFκB complex in the cytosol.14 Here, using a knockin mouse that only expresses the nontranscriptional activity of IRF3, we identified that it is the nontranscriptional activity of IRF3 that mediates this interaction between NFκB and IRF3 signaling in response to HFD feeding. We find that IRF3, independent of its phosphorylation‐dependent transcriptional activity, serves an important anti‐inflammatory role in HFD‐induced liver injury by decreasing the translocation of p65 to the nucleus and decreasing the expression of inflammatory cytokines and chemokines. These data highlight the complex interactions between the NFκB‐ and IRF3‐mediated inflammatory responses, with IRF3 serving to dampen the proinflammatory effects of NFκB in the context of HFD‐induced liver injury.
HFD feeding increased the transcriptional activity of IRF3, evidenced by enhanced expression of the antiviral IFIT genes. However, expression of the IFIT genes was not required for the protective impact of IRF3 on HFD‐induced inflammation or hepatocyte injury because Irf3 S1/S1 mice, lacking IRF3‐mediated transcriptional activity, were protected from HFD‐induced increases in ALT/AST, inflammation, apoptosis, and fibrosis. Only HFD‐induced steatosis was modestly protected by the transcriptional function of IRF3. Increased expression of IFIT genes in response to HFD feeding in wild‐type mice occurred despite decreases in total IRF3 protein expression (Fig. 1B).14
Although these data indicate that HFD feeding potently activates IRF3, the pathways of activation are not well understood. While several receptors/effector molecules upstream of IRF3 activation have also been implicated in murine models of NAFLD/NASH, there are differential contributions to liver injury among activation pathways. For example, TLR4‐deficient mice, lacking both MyD88 and TRIF‐dependent signaling, are protected from HFD or fructose‐induced liver injury.29, 30 In contrast, mice deficient in TLR3, which signals only through TRIF, are protected from steatosis7 but not from inflammation/ hepatocyte injury.31 MyD88−/− mice are also protected from both steatosis and inflammation.32 In contrast, TRIF, a key intermediate in both TLR3 and TLR4 signaling pathways leading to the activation of IRF3, differentially increases hepatic steatosis while preventing inflammation and hepatocyte injury in the choline‐deficient amino acid‐defined (CDAA) mouse model of NASH.33 Although these studies were done in different models of NAFLD/NASH, including HFD feeding, CDAA, and methylcholine‐deficient diets, they can provide a general indication of the differential contributions among signaling pathways. Taken together with our results, these data suggest that there is a dichotomous contribution of MyD88 versus TRIF‐dependent signaling in modulating inflammation and hepatocyte injury, with MyD88 contributing to inflammation and injury and TRIF and IRF3 pathways serving anti‐inflammatory functions. IRF3 can also be activated by stimulator of interferon genes (STING). However, activation of STING promotes both inflammation and injury.34 Interestingly, in other model systems, STING is both activated by endoplasmic reticulum (ER) stress17 and induces ER stress.35 This relation to ER stress may contribute to the proinflammatory effects of this pathway in HFD‐induced liver injury.
HFD‐induced obesity and metabolic syndrome are associated with adipose tissue inflammation; adipose inflammation is typically a strong contributor to the development of HFD‐induced liver injury.36 Kumari et al.18 reported that IRF3 regulates expression of inflammatory cytokines in isolated adipocytes. Further, they found that Irf3 −/− mice were protected from HFD‐induced adipose inflammation and insulin resistance after 16 weeks of feeding. This protection was associated with an increase in browning of adipose tissue after long‐term HFD feeding.18 Interestingly, in our study using a 12‐week HFD‐feeding protocol, HFD‐induced glucose intolerance and adipose inflammation were independent of Irf3 genotype, suggesting that there may be time and/or disease stage‐dependent contributions of IRF3.
IRF3 may also differentially contribute to ethanol versus HFD‐induced liver injury. In contrast to the protective function of IRF3 in HFD‐induced liver injury, acute on chronic (Gao‐binge) ethanol‐induced liver injury was reduced in Irf3 −/− mice.16 In turn, when the nontranscriptional activity of IRF3 was restored in the Irf3 S1/S1 mice, injury was increased. Activation of NFκB was independent of Irf3 genotype in acute on chronic ethanol‐induced injury,16 unlike the interaction of the nontranscriptional activity of IRF3 with NFκB in HFD‐fed mice. Instead, increased ethanol‐induced injury in the Irf3 S1/S1 mice was associated with an IRF3‐mediated apoptosis of restorative Ly6Clow monocytes that shifted the innate immune environment in the liver to a more proinflammatory state.16 In the HFD‐feeding model, Irf3 genotype did not affect apoptosis of immune cells or hepatocytes. These differences in the impact of nontranscriptional activity of IRF3 between ethanol‐ and HFD‐induced injury were associated with differences in IRF3 ubiquitination; ethanol feeding increased ubiquitinated IRF3,16 while HFD feeding decreased ubiquitinated IRF3. In summary, the contributions of the nontranscriptional activity of IRF3 are very sensitive to the specific metabolic stressor and can have proinflammatory functions through modulation of immune cell apoptosis or anti‐inflammatory functions through restriction of NFκB activity.
Supporting information
Supported in part by the National Institutes of Health (grants P50 AA024333, R01AA027456, U01AA021890, and RO1AA023722 to L.E.N.; RO1AI073303 to G.S.; R21AA026017 to S.C.; and R21AA020941 and P30 DK097348 [pilot project] to S.R.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Khashab MA, Liangpunsakul S, Chalasani N. Nonalcoholic fatty liver disease as a component of the metabolic syndrome. Curr Gastroenterol Rep 2008;10:73‐80. [DOI] [PubMed] [Google Scholar]
- 2. Yu J, Marsh S, Hu J, Feng W, Wu C. The pathogenesis of nonalcoholic fatty liver disease: interplay between diet, gut microbiota, and genetic background. Gastroenterol Res Pract 2016;2016:2862173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 2015;33:257‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell 2013;152:673‐684. [DOI] [PubMed] [Google Scholar]
- 5. Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, et al. Hepatocyte Toll‐like receptor 4 regulates obesity‐induced inflammation and insulin resistance. Nat Commun 2014;5:3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid‐induced insulin resistance. J Clin Invest 2006;116:3015‐3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu LH, Huang CC, Adhikarakunnathu S, San Mateo LR, Duffy KE, Rafferty P, et al. Loss of toll‐like receptor 3 function improves glucose tolerance and reduces liver steatosis in obese mice. Metabolism 2012;61:1633‐1645. [DOI] [PubMed] [Google Scholar]
- 8. Gao D, Li W. Structures and recognition modes of toll‐like receptors. Proteins 2017;85:3‐9. [DOI] [PubMed] [Google Scholar]
- 9. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS‐TLR4 signaling to IRF‐3/7 and NF‐kappaB involves the toll adapters TRAM and TRIF. J Exp Med 2003;198:1043‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, et al. TRIF and IRF‐3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 2008;181:3049‐3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 2013;78:105‐116. [DOI] [PubMed] [Google Scholar]
- 12. Ysebrant de Lendonck L, Martinet V, Goriely S. Interferon regulatory factor 3 in adaptive immune responses. Cell Mol Life Sci 2014;71:3873‐3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, Sen GC. Ubiquitination of the transcription factor IRF‐3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 2016;44:1151‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang XA, Zhang R, She ZG, Zhang XF, Jiang DS, Wang T, et al. Interferon regulatory factor 3 constrains IKKbeta/NF‐kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 2014;59:870‐885. [DOI] [PubMed] [Google Scholar]
- 15. Petrasek J, Dolganiuc A, Csak T, Nath B, Hritz I, Kodys K, et al. Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology 2011;53:649‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanz‐Garcia C, Poulsen KL, Bellos D, Wang H, McMullen MR, Li X, et al. The non‐transcriptional activity of IRF3 modulates hepatic immune cell populations in acute on chronic ethanol administration in mice. J Hepatol 2019;70:974‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iracheta‐Vellve A, Petrasek J, Gyongyosi B, Satishchandran A, Lowe P, Kodys K, et al. Endoplasmic reticulum stress‐induced hepatocellular death pathways mediate liver injury and fibrosis via stimulator of interferon genes. J Biol Chem 2016;291:26794‐26805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest 2016;126:2839‐2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. The IRF‐3/Bax‐mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol 2011;85:3708‐3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roychowdhury S, McCullough RL, Sanz‐Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high‐fat diet‐induced liver injury. Hepatology 2016;64:1518‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim A, Paramanda S, Nagy L. miRNAs involved in M1/M2 hyperpolarization are clustered and coordinately expressed in alcoholic hepatitis. Front Immunol 2019;10:1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fierbinteanu‐Braticevici C, Sinescu C, Moldoveanu A, Petrisor A, Diaconu S, Cretoiu D, et al. Nonalcoholic fatty liver disease: one entity, multiple impacts on liver health. Cell Biol Toxicol 2017;33:5‐14. [DOI] [PubMed] [Google Scholar]
- 24. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011;29:415‐445. [DOI] [PubMed] [Google Scholar]
- 25. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of obesity‐induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol (Lausanne) 2013;4:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baker RG, Hayden MS, Ghosh S. NF‐kappaB, inflammation, and metabolic disease. Cell Metab 2011;13:11‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin J, Lu Z, Li Y, Cowart LA, Lopes‐Virella MF, Huang Y. Docosahexaenoic acid antagonizes the boosting effect of palmitic acid on LPS inflammatory signaling by inhibiting gene transcription and ceramide synthesis. PLoS ONE 2018;13:e0193343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao GN, Jiang DS, Li H. Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim Biophys Acta 2015;1852:365‐378. [DOI] [PubMed] [Google Scholar]
- 29. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll‐like receptor‐4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non‐alcoholic steatohepatitis. J Hepatol 2007;47:571‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll‐like receptor 4 is involved in the development of fructose‐induced hepatic steatosis in mice. Hepatology 2009;50:1094‐1104. [DOI] [PubMed] [Google Scholar]
- 31. Lee YS, Kim DY, Kim TJ, Kim SY, Jeong JM, Jeong WI, et al. Loss of toll‐like receptor 3 aggravates hepatic inflammation but ameliorates steatosis in mice. Biochem Biophys Res Commun 2018;497:957‐962. [DOI] [PubMed] [Google Scholar]
- 32. Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, et al. Deficiency in myeloid differentiation factor‐2 and toll‐like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol 2011;300:G433‐G441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang L, Miura K, Zhang B, Matsushita H, Yang YM, Liang S, et al. TRIF differentially regulates hepatic steatosis and inflammation/fibrosis in mice. Cell Mol Gastroenterol Hepatol 2017;3:469‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage‐mediated hepatic inflammation and fibrosis in mice. Gastroenterology 2018;155:1971‐1984.e1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING‐mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med 2019;216:867‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity‐induced insulin resistance. Biochim Biophys Acta 2014;1842:446‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials