

Abstract
Background and Purpose
PDGF‐BB is an angiogenic factor involved in cardiovascular diseases. Here, we investigated the possible effects of activation of the nicotinic ACh receptor α7 subtype (α7nAChR) on PDGF‐BB‐induced proliferation and migration in vascular smooth muscle cells (VSMCs).
Experimental Approach
PNU‐282987, a selective α7nAChR pharmacological agonist, was used to activate α7nAChR. The influences of α7nAChR activation on PDGF‐BB‐induced proliferation and migration, as well as the phosphorylation of focal adhesion kinase (FAK)/Src, a pro‐migration signalling pathway, were determined in VSMCs. A variety of biochemical assays were applied to explore the underlying molecular mechanisms.
Key Results
PDGF‐BB induced pronounced migration and proliferation of VSMCs. Activation of α7nAChRs by PNU‐282987 blocked PDGF‐BB‐induced migration but not proliferation in wild‐type (WT) VSMCs, whereas this effect was absent in α7nAChR‐knockout VSMCs. Accordingly, PNU‐282987 attenuated PDGF‐BB‐induced phosphorylation of FAKTyr397 and SrcTyr416 in WT VSMCs. Mechanistically, PNU‐282987 suppressed PDGF‐BB‐induced oxidative stress, as demonstrated by the alterations in ROS, H2O2 content, superoxide anion and total antioxidant activity. A sirtuin 3 (SIRT3) inhibitor 3‐(1H‐1,2,3‐triazol‐4‐yl) pyridine or shRNA‐mediated SIRT3 knockdown abolished the inhibitory effect of PNU‐282987. PNU‐282987 did not modulate SIRT3 protein expression but enhanced mitochondrial SIRT3 deacetylase activity. In line with this action, PNU‐282987 enhanced the deacetylation of mitochondrial FoxO3. Lastly, PNU‐282987 corrected the PDGF‐BB‐induced mitochondrial dysfunction by increasing mitochondrial citrate synthase activity, ATP content and nicotinamide adenine dinucleotide pool.
Conclusions
Pharmacological activation of α7nAChRs inhibits PDGF‐BB‐induced VSMC migration by activating the mitochondrial deacetylase SIRT3, implying an important role for α7nAChRs in mitochondria biology and PDGF‐related diseases.
Linked Articles
This article is part of a themed section on Mitochondrial Pharmacology: Featured Mechanisms and Approaches for Therapy Translation. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.22/issuetoc
Abbreviations
- 3‐TYP
3‐(1H‐1,2,3‐triazol‐4‐yl) pyridine
- CS
citrate synthase
- FAK
focal adhesion kinase
- FoxO3
forkhead box protein O3
- MLA
methyllycaconitine
- SIRT3
sirtuin 3
- T‐AOC
total‐antioxidant capacity
- VSMCs
vascular smooth muscle cells
- WT
wild‐type
- α‐SMA
α‐smooth muscle actin
- α7nAChR
α7 nicotinic ACh receptor
Introduction
PDGFs are a group of endogenous growth factors regulating cell growth and division. Among all PDGF ligands, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5039 is the most pluripotent form of PDGF (Hu and Huang, 2015). PDGF‐BB has mitogenic and angiogenic effects on many types of cells, including myocardial cells, endothelial cells, fibroblasts, osteogenic cells and tenocytes (Hu and Huang, 2015). Due to its potent ability to promote wound healing, PDGF‐BB has been approved by the US Food and Drug Administration for use in diabetic foot ulcers as a gel formulation. However, every coin has two sides. PDGF‐BB is also critically involved in the onset and development of several cardiovascular diseases such as atherosclerosis and restenosis due to its overwhelming stimulation of the growth of vascular smooth muscle cells (VSMCs) (Raines, 2004; Liu et al., 2017). In addition, PDGF‐BB induces pericytes to migrate and thereby promotes tumour metastasis by triggering neovascularization and lymphangiogenesis (Cao et al., 2004).
Vascular smooth muscle cells (VSMCs) and pericytes are periendothelial vascular mural cells. These two types of cell are considered to be of the same lineage, sharing common precursor and cellular markers in many organs (Armulik et al., 2011). Notably, PDGF‐BB activates the proliferation and migration of VSMCs and thereby induces VSMCs to convert to a different phenotype. VSMCs always exhibit a contractile phenotype with little proliferation/migration and extracellular matrix production but transfer to a synthetic phenotype after vascular injury, which is believed to be essential for the pathogenesis of atherosclerosis and neointimal hyperplasia driven by PDGF‐BB (Nguyen et al., 2013). A better understanding of the endogenous factors that regulate PDGF‐BB‐induced VSMCs' proliferation and migration might help to find new pharmacological targets for cardiovascular diseases, or even for malignancy.
http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 (nAChRs) are a group of cholinergic ligand‐gated ion channels that respond to the neurotransmitter http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=294 as well as chemical compounds such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2585. The α7 nAChR subtype (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=468) is composed of five α7 subunits. This receptor has been implicated in the regulation of cognitive processes and shows promise as a therapeutic target for treatment of Alzheimer's disease and schizophrenia (Bertrand et al., 2015; Echeverria et al., 2016). Recent investigations discovered that α7nAChR is also widely expressed in cells of peripheral immune system and is a key mediator of the so‐called cholinergic anti‐inflammatory pathway. The cholinergic anti‐inflammatory pathway is a novel physiological mechanism by which the brain limits peripheral inflammation at various locations in the body through stimulating the vagus nerve to release ACh and then activating α7nAChRs expressed in cells of immune system such as macrophages (de Jonge et al., 2005).
We have previously demonstrated that the α7nAChR participates in cardiovascular pathophysiological processes. Activation of α7nAChRs protected against damage caused by oxidative stress by reducing vascular peroxidase‐1 in a JNK signalling‐dependent manner in endothelial cells (Li et al., 2014). Moreover, α7nAChR activation reduced the chronic inflammation in hypertension (Li et al., 2011; Chen et al., 2012) and sepsis (Liu et al., 2009). We also provided the first evidence that the α7nAChR is expressed in VSMCs and plays a crucial role in VSMC senescence induced by angiotensin II (Li et al., 2016b) and inhibits neointimal hyperplasia by suppressing inflammation/oxidative stress (Li et al., 2018a). However, whether the α7nAChR is involved in the PDGF‐BB‐induced VSMCS proliferation and migration has not been elucidated. In the present study, we investigated the hypothesis that α7nAChR activation modulates the PDGF‐BB‐induced VSMCS phenotype switch, and further explored the underlying molecular mechanisms of this effect.
Methods
Animals
Wild‐type (WT) C57BL/6 mice were purchased from Sino‐British SIPPR/BK Lab Animal Ltd (Shanghai, China). The α7nAChR knockout (KO) mice were purchased from Jackson laboratory (C57BL/6 background, Stock number: 003232) as described in our previous studies (Li et al., 2011, 2016b). The α7nAChR KO mouse strain used in this study was backcrossed to C57BL/6 mice for at least six generations. Only male mice were used in this study. The mice were bred and housed (three to four mice per cage) in temperature‐controlled cages (~23°C) under a 12/12 h light/dark cycle with free access to water and standard chow in the Tongji University Animal Core. Animals were used in accordance with the Tongji University institutional guidelines for animal care and the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health. Experiments were approved by Ethics Committee on Animal Experiments of Tongji University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010). Every effort was taken to minimize the number of animals used and their suffering. The total number of mice used in the whole study was 32.
Randomization and blinding
Animals were randomized for treatment. Data collection and evaluation of all experiments were performed blindly of the group identity. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018).
Cell culture and treatment
Primary mouse VSMCs were isolated from WT and α7nAChR‐KO mice and cultured as described in our previous studies (Li et al., 2011, 2016b). At 4–5 weeks of age, the mice were killed with an overdose of pentobarbital sodium (100 mg·kg−1, i.p.) before the aorta was excised. Then, the aortae were transferred to serum‐free medium, cleaned of adventitia under a dissection stereoscope and placed in an enzymatic solution (1 mg·mL−1 collagenase and 0.25 mg·mL−1 elastase) for 1 h. After that, the partially digested tissue with enzyme solution was transferred into a 15 mL conical tube and gently triturated with a flame‐polished glass pipette. The mixture was centrifuged at 800× g for 10 min, and the supernatant was discarded. The dissociated cells were resuspended in 2–3 mL of culture medium (DMEM with 10% FBS) and incubated in 5% CO2 plus 95% O2 (Zhang et al., 2018). For one experiment, three to four mice were killed to isolate the aortic VSMCs. Experiments were performed using cells between passages 3 and 8.
Migration assays
VSMC migration was determined using two assays: the transwell migration assay and scratch‐wound migration assay. Transwell migration was performed as described as previously (Ward and Spiers, 2017). Briefly, this experiment was performed in polycarbonate transwell inserts (5 mm pore, Corning). The cultured VSMCs (1 × 105) were seeded in the upper compartment and were cultured at 37°C for 12 h. Then, the transwell was placed into a 24‐well plate containing Endothelial basal medium‐2 (EBM‐2) (Lonza, Basel, Switzerland) for 24 h. In the lower chamber, PDGF‐BB (Sigma‐Aldrich, St. Louis, MO, USA; 20 ng·mL−1) or PDGF‐BB (20 ng·mL−1) + http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3988 (Sigma‐Aldrich; 10 μM) were added. Migrated VSMCs were counted over three randomly selected high‐power fields in lower chambers. For the scratch‐wound migration assay, VSMCs were cultured on six‐well plates, and the confluent cells were growth‐arrested by additions of 0.1% FBS. The cells were scraped using sterilized 10 μL pipette tips and washed with 0.1% serum media and stimulated with PDGF‐BB (20 ng·mL−1) (Du et al., 2013). For the signalling pathway study, the following chemical inhibitors were used: S3I‐201 (100 μM) (Li et al., 2018c), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5282 (1 μM) (Pathria et al., 2016), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6004 (30 μM) (Liao et al., 2018), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=346 (50 nM) (Prins et al., 2017) and 3‐TYP (20 nM) (Shumin et al., 2018). All these inhibitors were purchased from Selleck Chemicals (Houston, TX, USA). At 24 h post wound, the number of migrated VSMCs into the denuded area was quantified with a computerized digital image analysis system from Leica (Wetzlar, Germany).
Cell viability and proliferation assay
Cell viability was measured using the CCK‐8 assay (Cell Counting Kit‐8, Beyotime, China) according to the manufacturer's instruction as described previously (Song et al., 2017). Briefly, VSMCs were seeded into 96‐well plates at an initial density of 1 × 104 cells per well. After incubation with different factors [PDGF‐BB, 20 ng·mL−1; PNU‐282987, 10 μM; http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4005 (MLA, Sigma‐Aldrich), 100 nM], 10 μL of kit reagent was added and incubated for an additional 2 h. Cell viability was obtained by scanning with a microplate reader (Thermo) at 450 nm. Cell proliferation was measured by the Click‐iT EdU Microplate Assay (#C10214, Invitrogen™ Thermo Fisher Scientific, Invitrogen, Carlsbad, CA) according to the manufacturer's instructions as described in our previous report (Li et al., 2016b).
Mitochondria isolation
Mitochondria were isolated using a commercial kit from Biovision (Mountain View, CA). VSMCs were homogenized in ice‐cold media (pH 7.4) containing 0.25 M sucrose, 1 mM EDTA and 5 mM Tris–HCl and protease inhibitors. The following experimental procedures were conducted according to the manufacturer's instructions. The final mitochondrial pellet was suspended in media containing 0.25 M sucrose, 1 mM EDTA and 5 mM Tris–HCl at a concentration of 50 mg·mL−1 (Gan et al., 2018).
Knockdown with siRNA
The plasmid containing shRNA targeting http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=848#2709 (SIRT3) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The transfection of shRNA‐plasmid was performed using Lipofectamine® LTX Reagent (Invitrogen) according to the manufacturer's instructions. Briefly, VSMCs were allowed to attach overnight. When the cells reached about 70% confluence, they were washed with PBS twice and then added into a prepared shRNA‐lipid complex. After incubation in shRNA‐lipid complex for 6 h, the medium was removed. The empty plasmid was used as a control vector for transfection. At day 3 after transfection, the efficiency of SIRT3 knockdown was checked by immunoblotting. The function analyses were performed on day 3 after transfection.
Measurement of oxidative stress
Intracellular ROS was determined by examining the immunofluorescence of 2,7‐dichlorodihydrofluorescein diacetate probe (DCFH‐DA, Invitrogen, Carlsbad, CA). VSMCs were seeded into six‐well plates (1 × 106 cells per well) in DMEM and grown to 70% confluence. After overnight serum deprivation, VSMCs were pretreated with different concentrations of PDGF‐BB (20, 50 and 200 ng·mL−1) or PDGF‐BB (200 ng·mL−1) + PNU‐282987 (10 μM) for 24 h. After treatment, the cells were washed with fresh serum‐free DMEM and incubated with 10 μM of DCFH‐DA at 37°C for 1 h. Images were captured by a confocal laser scanning microscope (Olympus Fluoview FV1000, Tokyo, Japan). The fluorescence intensity was analysed using Image‐J software (National Institutes of Health, USA). At least 10 images per group were included and analysed. The levels of H2O2, superoxide anion and total‐antioxidant capacity (T‐AOC) in VSMCs were measured using commercial kits from Beyotime Institute of Biotechnology (Haimen, China) and performed according to the manufacturer's instructions (Huang et al., 2017).
Immunofluorescence
The purity of primary mouse VSMCs was determined using immunofluorescence. Immunofluorescence was performed as described previously (Li et al., 2014, 2016a). VSMCs were seeded at low density in confocal dishes approximately 24 h prior to imaging to ensure proper cell attachment. The cells were fixed with 4% paraformaldehyde (Lv et al., 2017) and incubated with primary antibody against α‐smooth muscle actin (α‐SMA) overnight at 4°C and followed by Alexa‐488 labelled secondary antibody for 1 h. DAPI was used to stain nuclei. Images were captured by a confocal laser scanning microscope (Olympus Fluoview FV1000, Tokyo, Japan).
Immunoblotting
Immunoblotting was performed as described previously (Li et al., 2015; Wei et al., 2017; Yang et al., 2018). Protein was extracted from cells with RIPA buffer supplemented with a protease/phosphatase inhibitor cocktail (Pierce Technology, Rockford, IL, USA). Nuclear protein was further extracted using Nuclear Extraction Kit (#ab113474, Abcam, Cambridge, MA, USA). After centrifugation at 12 000× g for 10 min, the supernatant was collected and boiled for 5 min in SDS‐PAGE sample buffer and run on 10% SDS‐PAGE. The proteins were electrotransferred to nitrocellulose membranes, probed with primary antibody and incubated with Infrared‐Dyes‐conjugated secondary antibodies (Li‐Cor, Lincoln, NE). The images were obtained with Odyssey Infrared Fluorescence Imaging System (Li‐Cor). The following antibodies were used: anti‐phosphorylated‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2180 (p‐FAKTyr397, #8556, Cell Signaling Technology, 1:2000 dilution), anti‐total‐FAK (FAK, sc‐557, Santa Cruz Biotechnology, 1:2000 dilution), anti‐phosphorylated‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2206 Tyr416 (p‐SrcTyr416, #2113, Cell Signaling Technology, 1:2000 dilution), anti‐total‐Src (#2110, Cell Signaling Technology, 1:2000 dilution, 1:3000 dilution), anti‐FoxO3 (sc‐9808, Santa Cruz Biotechnology, 1:2000 dilution), anti‐SIRT3 (#07–1596, Upstate, 1:1000 dilution), anti‐acetylated‐lysine antibody (#9441, Cell Signaling Technology, 1:2000 dilution), anti‐MTCO1 (#ab14705, Abcam, 1:1000 dilution), anti‐succinate dehydrogenase complex subunit A (SDHA, #ab14715, Abcam, 1:1500 dilution), COX IV (sc‐69360, Santa Cruz Biotechnology, 1:1000 dilution) and anti‐GAPDH (sc‐0411, Santa Cruz Biotechnology, 1:2000 dilution). All immunoblotting experiments were repeated at least three times.
SIRT3 activity
The mitochondrial samples were incubated for 10 min at 30°C to allow for NAD+ degradation and incubated for 10 additional minutes with DTT 2 μM. SIRT3 in the tissues lysed by RIPA buffer was enriched by immunoprecipitation using a specific anti‐SIRT3 antibody (#sc‐365175, Santa Cruz, CA) and was then subjected to a deacetylation assay using a commercial SIRT3 Fluorimetric Kit (Biovision, Mountain View, CA).
Acetylation assay
Primary VSMCs were lysed in RIPA buffer with protease inhibitor cocktail. The crude lysates were cleared of insoluble debris by centrifugation at 1200× g. Immunoprecipitating antibody (Anti‐FoxO3, 3 μg) or normal IgG (negative control, Santa Cruz, #sc‐2762) was added and incubated on a rotator at 4°C overnight. The 20 μL G/A agarose beads (Santa Cruz, #sc‐2003) were added into the 200 μL lysates and incubated for 4 h with gentle agitation. The beads were washed three times with the lysis buffer and boiled with 10 μL 2× sample buffer (Fermentas, #R1011). The beads were removed by centrifugation (5 min at 1200× g). The supernatant was collected and used for immunoblotting with anti‐acetylated‐lysine antibody.
NAD+ concentration, citrate synthase activity and ATP content
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2451 + levels were determined with a commercial NAD+ quantification kit (BioVision, Mountain View, CA, USA). Briefly, cells were lysed using RIPA buffer, and the lysates were centrifuged at 14 000× g for 15 min at 4°C. The supernatant was used for subsequent procedures according to the manufacturer's instructions. Citrate synthase (CS) activity was measured by colorimetric assay kits (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China). In the kit, the enzyme is captured within the wells of the microplate, and activity is determined by recording colour development of 2‐nitro‐5‐thiobenzoate, which is generated from 5,5′‐dithiobis‐2‐nitrobenzoic acid present in the reaction of citrate synthesis. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 content was determined by colorimetric assay kits (Abcam). This kit utilized the phosphorylation of glycerol to generate a product that is easily quantified by a colorimetric assay (ODmax = 570 nm).
Statistical analysis
Data were analysed with GraphPad Prism‐5 statistic software (La Jolla, CA). All values are presented as the mean ± SEM and analysed by ANOVA followed by Tukey's post hoc test when the F statistic was significant. P < 0.05 was considered statistically significant. At least three independent experiments were performed in duplicate with all the assays.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Results
Activation of α7nAChRs inhibits PDGF‐BB‐induced VSMC migration
We cultured primary mouse VSMCs and confirmed their characteristics by immunofluorescence staining of α‐SMA (Figure 1A). Then, we tested the effects of α7nAChR activation on PDGF‐BB‐induced migration in these cells. The scratch‐wound migration assay showed that PDGF‐BB significantly enhanced migration in both WT normal and α7nAChR−/− VSMCs (Figure 1B). In WT VSMCs, the prominent PDGF‐BB‐induced migration was abolished by PNU‐282987, a selective agonist of α7nAChRs (Figure 1B). PNU‐282987 failed to inhibit the PDGF‐BB‐induced migration in α7nAChR−/− VSMCs (Figure 1B). Administration of the α7nAChR blocker MLA at a selective concentration (100 nM) totally prevented the effect of PNU‐282987 in WT VSMCs but not in α7nAChR−/− VSMCs (Figure 1B). We also performed a transwell migration assay to confirm these results (Figure 1C). Activation of α7nAChRs by PNU‐282987 also attenuated migration in WT VSMCs but not in α7nAChR−/− VSMCs in the transwell assay (Figure 1C). MLA also abolished the inhibitory action of PNU‐282987 on migration in WT VSMCs but not in α7nAChR−/− VSMCs (Figure 1C). PNU‐282987 itself did not show an effect on VSMC migration (low dose: 10 μM; high dose: 100 μM; Figure 1D). These results indicate that pharmacological activation of α7nAChRs inhibits PDGF‐BB‐induced migration in VSMCs.
Figure 1.

Activation of α7nAChRs inhibits PDGF‐BB‐induced VSMC migration. (A) Immunofluorescence staining of α‐SMA in primary mouse VSMCs. The cells were isolated from mouse aorta tissue and cultured. α‐SMA is a specific marker for VSMCs. DAPI is a probe used to stain nuclei. Scale bar, 50 μm. (B) Scratch‐wound assay showing primary VSMCs isolated from WT and α7nAChR−/− mice cultured on six‐well plates and scraped, followed by stimulation with PDGF‐BB (20 ng·mL−1) or PDGF‐BB (20 ng·mL−1) + PNU‐282987 (10 μM) for 24 h. MLA (100 nM) was used to block α7nAChRs. The VSMCs migrated into the denuded area were quantified to reflect the migration of VSMCs. The white dot lines indicate the scraped sign, and the red dot lines indicate the border of migrated cells. *P < 0.05 versus blank, #P < 0.05 versus PDGF‐BB. n = 8 per group. NS, no significance. (C) Transwell migration assay showing migration of WT and α7nAChR−/− primary VSMCs. MLA (100 nM) was used to block α7nAChRs. *P < 0.05 versus blank, #P < 0.05 versus PDGF‐BB. n = 8 per group. NS, no significance. (D) Scratch‐wound assay showing that the α7nAChR agonist PNU‐282987 (two concentrations: 10 and 100 μM) itself did not alter VSMC migration. NS, no significance. PNU, PNU‐282987. Two‐way ANOVA with a post hoc Tukey's test was used for all the statistical analyses.
Activation of α7nAChRs does not affect PDGF‐BB‐induced VSMC proliferation
Next, we applied the cell viability assay and Edu incorporation assay to determine the action of the α7nAChR agonist on PDGF‐BB‐induced proliferation of VSMCs. PDGF‐BB treatment for 48 and 72 h significantly enhanced the viability of VSMCs (Figure 2A). PNU‐282987 had no effect on PDGF‐BB‐induced proliferation at two time points (Figure 2A). In the Edu incorporation assay, we confirmed that PNU‐282987 failed to inhibit the proliferation of VSMCs induced by PDGF‐BB (Figure 2B). When PDGF‐BB was not present, activation of α7nAChRs by PNU‐282987 or inhibition of α7nAChRs by MLA also had no obvious effect on VSMC proliferation (Figure 2C,D). These results suggest that α7nAChR activation does not affect VSMC proliferation whether or not PDGF‐BB is present.
Figure 2.

Activation of α7nAChRs does not affect VSMC proliferation. (A) Cell viability assay showing the proliferation of WT and α7nAChR−/− VSMCs under PDGF‐BB (20 ng·mL−1) and PNU‐282987 (10 μM) treatment for 48 and 72 h. *P < 0.05 versus blank. NS, no significance. n = 8 per group. (B) Edu proliferative assay showing the proliferation of WT and α7nAChR−/− VSMCs under PDGF‐BB (20 ng·mL−1) and PNU (10 μM) treatment for 48 and 72 h. *P < 0.05 versus blank. NS, no significance. n = 8 per group. (C) Cell viability assay showing that there is no direct effect of the α7nAChR agonist PNU‐282987 or α7nAChR antagonist MLA (100 nM) on VSMC proliferation. NS, no significance. n = 8 per group. (D) Edu proliferative assay showing that there is no direct effect of α7nAChR agonist PNU‐282987 or α7nAChR antagonist MLA (100 nM) on VSMC proliferation. NS, no significance. n = 8 per group. PNU, PNU‐282987. Two‐way ANOVA with a post hoc Tukey's test was used for all the statistical analyses.
Activation of α7nAChRs suppresses PDGF‐BB‐induced oxidative stress in VSMCs
PDGF‐BB induces redox processes and oxidative stress in VSMCs, which critically contribute to PDGF‐BB‐induced VSMC migration and chemotaxis (ten Freyhaus et al., 2006). Thus, we evaluated the influence of α7nAChR activation on PDGF‐BB‐induced oxidative stress. DCFH fluorescent staining showed that PDGF‐BB treatment induced obvious ROS production in VSMCs in a dose‐dependent manner (Figure 3A). When PNU‐282987 was added into the medium to activate α7nAChRs, the ROS production in VSMCs was significantly reduced (Figure 3A). Administration of the α7nAChR agonist MLA abolished the inhibitory action of PNU‐282987 (Figure 3A). In line with this result, α7nAChR activation by PNU‐282987 reduced the concentrations of H2O2 (Figure 3B) and superoxide anion (Figure 3C) and enhanced the total antioxidant activity (Figure 3D). However, all these changes induced by PNU‐282987 were blocked by MLA treatment (Figure 3B–D).
Figure 3.

Activation of α7nAChRs suppresses PDGF‐BB‐induced oxidative stress. (A) ROS production was determined by DCFH‐DA staining in primary mouse VSMCs treated with difference doses of PDGF‐BB (20, 50 and 200 ng·mL−1) or PDGF‐BB (200 ng·mL−1) + PNU‐282987 (10 μM) for 72 h. MLA (100 nM) was used to block α7nAChRs. *P < 0.05. n = 8 per group. (B–D) The levels of H2O2 (B), superoxide anion (C) and total T‐AOC activity (D) in primary mouse VSMCs treated with difference doses of PDGF‐BB (20, 50 and 200 ng·mL−1) or PDGF‐BB (200 ng·mL−1) + PNU‐282987 (10 μM) for 72 h. MLA (100 nM) was used to block α7nAChRs. *P < 0.05. n = 8 per group. One‐way ANOVA with a post hoc Tukey's test was used for all the statistical analyses.
Activation of α7nAChRs attenuated PDGF‐BB‐induced activation of FAK‐Src signalling pathway in VSMCs
FAK‐Src signalling is a key signalling pathway involved in PDGF‐BB‐induced VSMC migration but not proliferation (Son et al., 2014). Consequently, we measured the influence of α7nAChR activation on FAK‐Src signalling. Our immunoblotting analysis showed that PDGF‐BB treatment (20, 50 and 200 ng·mL−1) significantly enhanced the level of phosphorylated FAK (Figure 4A) and Src (Figure 4B) in VSMCs in a dose‐dependent manner, suggesting that the FAK‐Src signalling pathway was activated by the PDGF‐BB. Treatment with PNU‐282987 significantly inhibited PDGF‐BB‐induced phosphorylation of FAK (Figure 4A) and Src (Figure 4B), suggesting that α7nAChR activation blocks PDGF‐BB‐induced activation of the FAK‐Src signalling pathway in VSMCs.
Figure 4.
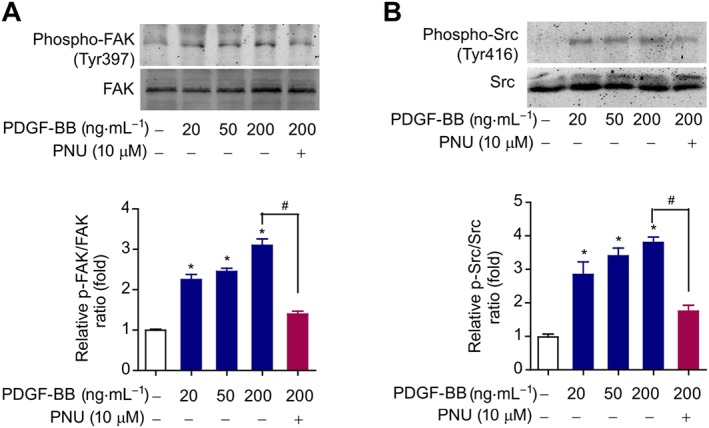
Activation of α7nAChRs inhibits the stimulation of the FAK‐Src signalling pathway induced by PDGF‐BB. (A) Immunoblotting analysis of FAK phosphorylation at Tyr397 site in primary mouse VSMCs treated with PDGF‐BB (20, 50 and 200 ng·mL−1) or PDGF‐BB (200 ng·mL−1) + PNU‐282987 (10 μM) for 72 h. *P < 0.05 versus blank, #P < 0.05 PDGF‐BB + PNU‐282985 versus PDGF‐BB. n = 6 per group. (B) Immunoblotting analysis of Src phosphorylation at Tyr416 site in primary mouse VSMCs treated with PDGF‐BB (20, 50 and 200 ng·mL−1) or PDGF‐BB (200 ng·mL−1) + PNU‐282987 (10 μM) for 72 h. *P < 0.05 versus blank, #P < 0.05 PDGF‐BB + PNU‐282985 versus PDGF‐BB. n = 6 per group. One‐way ANOVA with a post hoc Tukey's test was used for all the statistical analyses.
SIRT3 is involved in the inhibitory effect of α7nAChR on PDGF‐BB‐induced VSMC migration
We further explored the molecular mechanisms underlying the inhibitory action of α7nAChR on VSMC migration by screening several important intracellular signalling factors involved in cell migration: JAK2‐STAT3, MEK‐ERK1/2, PI3K‐Akt, PKC and SIRT3 pathways. S3I‐201, U0126, LY294002, staurosporine and 3‐TYP were used to block these signalling pathways or proteins respectively. PNU‐282987 still inhibited the PDGF‐BB‐induced VSMCs migration in the presence of S3I‐201, U0126, LY294002 and staurosporine (Figure 5A,B). However, PNU‐282987 no longer exhibited an inhibitory action on PDGF‐BB‐induced migration in the presence of the SIRT3 chemical inhibitor 3‐TYP (the last column, Figure 5A,B). To confirm this effect, we used shRNA to deplete SIRT3 in VSMCs. Transfection of shRNA targeting SIRT3 in VSMCs successfully reduced SIRT3 protein level by ~70% (Figure 5C). As expected, PNU‐282987 inhibited migration in control VSMCs (control vector transfected) but not in shRNA‐SIRT3 transfected VSMCs (Figure 5D). These data suggest that α7nAChR inhibits PDGF‐BB‐induced VSMC migration in a SIRT3‐dependent manner.
Figure 5.

SIRT3 is critical for the inhibitory action of α7nAChRs on PDGF‐BB‐induced VSMC migration. (A, B) Representative images (A) and quantitative analysis (B) in scratch‐wound assay showing VSMC migration under different signalling inhibitors. Only 3‐TYP, a specific chemical inhibitor of SIRT3, abolished the inhibitory action of α7nAChR activation on PDGF‐BB‐induced primary mouse VSMC migration. S3I‐201: a specific inhibitor of JAK2‐STAT3 signalling pathway. U0126: a specific inhibitor of MEK‐ERK1/2 signalling pathway. LY294002: a specific inhibitor of PI3K‐Akt signalling pathway. Staurosporine: a specific inhibitor of PKC signalling pathway. The following concentrations were used in the experiments: PDGF‐BB (20 ng·mL−1), PNU‐282987 (10 μm), S3I‐201 (100 μM), U0126 (1 μM); LY294002 (30 μM), staurosporine (50 nM) and 3‐TYP (1 μM). *P < 0.05 versus PDGF‐BB, #P < 0.05 versus PDGF‐BB + PNU. n = 8 per group. NS, no significance. (C) Immunoblotting was applied to determine the effect of shRNA‐mediated knockdown of SIRT3 in primary mouse VSMCs. *P < 0.05 versus control vector. n = 6 per group. (D) Representative images and quantitative analysis of the scratch‐wound assay showing that the inhibitory action of α7nAChR activation on PDGF‐BB‐induced VSMC migration is absent in SIRT3‐depleted VSMCs, which were transfected with specific shRNA targeting SIRT3. *P < 0.05 versus PDGF‐BB, #P < 0.05 versus PDGF‐BB + PNU. n = 8 per group. PNU, PNU‐282987. One‐way ANOVA with a post hoc Tukey's test was used for all the statistical analyses.
Activation of α7nAChR enshances mitochondrial SIRT3 deacetylase activity in VSMCs
SIRT3 is a mitochondria‐localized NAD+‐dependent deacetylase that is critically involved in cell migration (Dong et al., 2016; Ren et al., 2017), due to its ability to orchestrate intracellular metabolic responses and exert anti‐oxidative effects (Hirschey et al., 2010). Thus, we determined the mitochondrial expression and deacetylase activity of SIRT3 to further delineate the potential role of mitochondrial SIRT3 in the inhibitory action of α7nAChR activation on VSMC migration. We found that PDGF‐BB treatment reduced mitochondrial SIRT3 protein levels, while the α7nAChR agonist PNU‐282987 did not affect this effect (Figure 6A). Interestingly, PNU‐282987 partly prevented the decrease in mitochondrial SIRT3 deacetylase activity induced by PDGF‐BB (Figure 6B). To confirm this, we further measured the acetylation of FoxO3, a downstream target of SIRT3. Indeed, PNU‐282987 significantly decreased the acetylation of FoxO3 (Figure 6C), suggesting that SIRT3‐mediated FoxO3 deacetylation was enhanced. In addition, SIRT3‐mediated deacetylation of FoxO3 has been shown to increase the stability of FoxO3 protein (Tseng et al., 2014). In line with this, mitochondrial Foxo‐3 protein levels in VSMCs were significantly increased by PNU‐282987 (Figure 6D). All these results demonstrate that activation of α7nAChRs enhances mitochondrial SIRT3 deacetylase activity in PDGF‐BB‐treated VSMCs.
Figure 6.

Activation of α7nAChRs promotes mitochondrial SIRT3 deacetylase activity in PDGF‐BB‐treated VSMCs. (A) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on SIRT3 protein level after 72 h treatment in primary mouse VSMCs. *P < 0.05 versus control. NS, no significance. n = 5 per group. (B) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on SIRT3 deacetylase activity after 48 and 72 h treatment in mitochondrial extracts isolated from VSMCs. *P < 0.05 versus control, #P < 0.05 versus PDGF‐BB. n = 5 per group. (C) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on acetylation of FoxO3 in mitochondrial extracts. *P < 0.05 versus PDGF‐BB. n = 5 per group. (D) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on protein level of FoxO3 in mitochondrial extracts. *P < 0.05 versus control, #P < 0.05 versus PDGF‐BB. n = 5 per group. One‐way ANOVA with a post hoc Tukey's test was used for all the statistical analyses.
Activation of α7nAChRs corrects mitochondrial dysfunction in VSMCs
Due to the importance of SIRT3 in the regulation of mitochondrial function, we also assessed the influence of α7nAChR activation on mitochondrial function in VSMCs. PDGF‐BB treatment for 48 or 72 h reduced mitochondrial CS activity by 35 and 40% respectively in VSMCs (Figure 7A). The α7nAChR agonist PNU‐282987 partly attenuated this effect (Figure 7A). Similar results were observed on mitochondrial ATP content (Figure 7B) and mitochondrial NAD+ concentration (Figure 7C). Additionally, we applied immunoblotting to measure protein expression of MTCO1 and SDHA, two mitochondrial‐encoded proteins. PNU‐282987 treatment significantly increased MTCO1 and SDHA protein expression (Figure 7D). These data indicate that activation of the α7nAChR corrects mitochondrial dysfunction in VSMCs.
Figure 7.

Activation of α7nAChRs corrects mitochondrial dysfunction in PDGF‐BB‐treated VSMCs. (A) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on CS activity in mitochondrial extracts. (B) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on ATP content in mitochondrial extracts. (C) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on NAD+ concentration in mitochondrial extracts. (D) Influence of PDGF‐BB and α7nAChR agonist PNU‐282987 on protein expression of MTCO1 and SDHA in mitochondrial extracts. *P < 0.05 versus control, #P < 0.05 versus PDGF‐BB. n = 5 per group. One‐way ANOVA with a post hoc Tukey's test was used for all the statistical analyses.
Discussion
In the present study, we provide strong evidence that the α7nAChR inhibits PDGF‐BB‐induced VSMC migration but not proliferation in a SIRT3‐dependent manner (Figure 8). The major findings of our study include: (i) pharmacological activation of α7nAChRs using a selective agonist PNU‐282987 reduced PDGF‐BB‐induced VSMC migration but not proliferation; (ii) the α7nAChR agonist attenuated the activation and oxidative stress in VSMCs induced by PDGF‐BB via the FAK‐Src signalling pathway; (iii) a chemical inhibitor of SIRT3 or shRNA‐mediated knockdown of SIRT3 abolished the inhibitory effects of the α7nAChR agonist on PDGF‐BB‐induced VSMC migration; and (iv) α7nAChR activation increased mitochondrial NAD+ levels and thus enhanced mitochondrial SIRT3 deacetylase activity. These results are consistent with the main suggestions in our previous studies showing that the α7nAChR protects VSMCs from senescence induced by angiotensin II (Li et al., 2016b) and inhibits neointimal hyperplasia by suppressing inflammation/oxidative stress in a mouse model of aortic injury (Li et al., 2018a).
Figure 8.

A proposed working model for the effects of α7nAChR activation on VSMC migration induced by PDGF‐BB.
The first important finding in our study was that α7nAChR activation only inhibits PDGF‐BB‐induced VSMC migration but not proliferation, which suggests that PDGF‐BB may trigger migration and proliferation in VMSCs through distinct intracellular signalling pathways. Activation of α7nAChRs is likely to only inhibit the PDGF‐BB‐induced changes in the migration‐related signalling pathway in VSMCs. Previously, a lot of chemical compounds such as atorvastatin and apamin displayed inhibitory effects on both PDGF‐BB‐induced VSMC migration and proliferation (Chen et al., 2015; Kim et al., 2015; Mair et al., 2015). These dual actions on VSMC migration and proliferation were always mediated by mitogenic signalling pathways such as ERK1/2 and Akt (Chen et al., 2015; Kim et al., 2015). Also, some chemical agents only inhibit PDGF‐BB‐induced VSMC migration but not proliferation. For example, Freyhaus et al. reported that the NADPH oxidase inhibitor VAS2870 effectively suppressed PDGF‐BB‐mediated ROS generation and inhibited PDGF‐dependent VSMC migration, while not affecting VSMC proliferation (ten Freyhaus et al., 2006). Hence, we studied the effects on the FAK‐Src signalling pathway, a key intracellular cascade for cell migration but not proliferation (Sieg et al., 2000). As expected, PDGF‐BB triggered the phosphorylation of FAK at the Tyr397 site and Src at the Tyr416 site; however, α7nAChR activation substantially suppressed these effects, supporting the finding that α7nAChR activation only inhibits PDGF‐BB‐induced VSMC migration but not proliferation. There are a large number of modulators of α7nAChRs, including full agonists, partial agonists, antagonists and coagonists. For example, AR‐R17779 and WAY‐317538 are full agonists whereas GTS‐21, SSR‐180711 and bradanicline are partial agonists (Yang et al., 2017). Partial agonists are complex because they can increase the activity of α7nAChRs submaximally on their own but limit the ability of ACh to activate α7nAChRs. Currently, there are no data to show whether PNU‐282987 is a full or partial agonist. Thus, it should be noted that it is necessary to clearly distinguish the full effects of PNU‐282987 on migration from its partial effects on the FAK‐Src signal and ROS production (Figure 8). We speculated that the final action of PNU‐282987 on VMSC migration may be orchestrated and determined by the complex influences of PNU‐282987 on VSMCs.
Activation of α7nAChRs in the cholinergic anti‐inflammatory pathway was reported to protect against neurodegeneration or ischaemic stroke by inhibiting oxidative stress (Navarro et al., 2015). The α7nAChR agonist PNU‐282987 reduced the neurotoxicity and the pathological phenotypes caused by LPS by inducing HO‐1 expression (Navarro et al., 2015). We previously demonstrated that α7nAChR activation inhibited inflammatory reactions in cardiovascular cells (Li et al., 2014). So it may not be surprising that α7nAChR activation alleviated oxidative stress induced by PDGF‐BB in VSMCS. In the present study, we observed that the α7nAChR agonist PNU‐282987 significantly suppressed PDGF‐BB‐induced ROS production, H2O2, superoxide anion and the decline of T‐AOC. These results corroborate the potent antioxidant activity of α7nAChRs in other type of cells, which was shown by our recent fatty liver animal study (Li et al., 2018b). However, the mechanism of the cholinergic anti‐inflammatory pathway and the role of α7AChRs are still a matter of debate (Matteoli et al., 2014). The mechanism of action of the cholinergic anti‐inflammatory pathway seems to differ in the context of the disease. In fact, there is increasing evidence suggesting that the effect on α7AChRs is indirect rather than directly from the vagus nerve‐derived ACh (Cailotto et al., 2014; Matteoli et al., 2014). α7AChR activation can even have an opposite effect of that of nicotine (Snoek et al., 2010). Furthermore, other ACh receptors might also be involved in the cholinergic anti‐inflammatory pathway, like α4β2AChRs (van der Zanden et al., 2009).
The most important finding in this study is that α7nAChR activation exerts an inhibitory effect on PDGF‐BB‐induced migration mediated via activation of mitochondrial SIRT3 deacetylase activity. The mammalian sirtuins are a family of seven complex NAD+‐dependent class III histone deacetylases that are increasingly thought to underlie many biological processes. Unlike traditional histone deacetylases, sirtuins not only deacetylate as histones but also deacetylate quantities of substrates ranging from transcription factors to metabolic enzymes. SIRT3 is a mitochondrial‐localized sirtuin subtype and modulates mitochondrial integrity (Hirschey et al., 2010). Importantly, SIRT3 negatively regulates cell migration (Dong et al., 2016; Ren et al., 2017) due to its potent counteraction on intracellular metabolic responses and oxidative stress (Hirschey et al., 2010). In our work, only SIRT3 inhibitor 3‐TYR blocked the effect of PNU‐282987. This phenotype prompted us to postulate that SIRT3 may be the effector mediating the inhibitory action of α7nAChRs on PDGF‐BB signalling. As expected, we found that the α7nAChR activator PNU‐282987 indeed prevented the PDGF‐BB‐induced impairment of mitochondrial SIRT3 deacetylation activity. Since SIRT3 suppresses oxidative stress, the alterations in mitochondrial SIRT3 activity in VSMCs would lead to the changes in oxidative stress in these cells. In support of these postulates, activation of α7nAChRs by PNU‐282987 also enhanced FoxO3 deacetylation in a mitochondrial extract of VSMCs. These phenotypes are apparently in line with a previous study showing that SIRT3‐mediated deacetylation of FoxO3 promotes FoxO3 stabilization (Tseng et al., 2014).
We further questioned how α7nAChR activation affected mitochondrial SIRT3 deacetylation activity. To this end, we determined the protein expression of SIRT3 but failed to find any change in PNU‐282987‐treated VSMCs. By contrast, our results showed that the mitochondrial NAD+ concentration was increased by PNU‐282987. SIRT3 deacetylation activity would be totally compromised if NAD+ is depleted (Onyango et al., 2002). In fact, all sirtuins require NAD+ as a cofactor for their catalytic function. Hence, we inferred that the promoting effect of α7nAChR activation on NAD+ concentration may be the key reason for the increase in mitochondrial SIRT3 activity. It should be noted that SIRT3, SIRT4 and SIRT5 are all mitochondrial sirtuins (Qiu et al., 2010). Interestingly, because NADH (the reduced form of NAD+) cannot pass through the mitochondrial membranes, there are separate NAD+ pools in the mitochondria and the cytoplasm/nucleus (Yang et al., 2007). Nuclear and cytoplasmic NAD+ concentration are greatly depleted under genotoxic stress, whereas the mitochondrial NAD+ concentration still remains at physiological levels (Yang et al., 2007). This elegant intracellular regulation of the NAD+ pool allows different sirtuins to exert distinct functions based on their subcellular localization. Recently, a comprehensive study of the mitochondrial SIRT3 interaction network has elucidated the mechanisms controlling SIRT3 activity and candidate targets not previously associated with SIRT3 (Yang et al., 2016). The improvements in CS activity, ATP content and protein expression of MTCO1/SDHA induced by of α7nAChR activation support the beneficial action of α7nAChR activation on mitochondrial function, and suggest that SIRT3 is a key mediator of the inhibitory action of α7nAChR on PDGF‐BB‐induced migration in VSMCs.
The use of an α7nAChR agonist might have clinical value for the treatment of vascular injury. Due to the inhibitory action of α7nAChR agonists on VSMC migration, they can be used for therapy in atherosclerosis and restenosis. Several previous investigations have shown the therapeutic value of an α7nAChR agonist in an atherosclerotic mice model. Al‐Sharea et al. recently reported that another selective α7nAChR agonist GTS‐21 caused a reduction in atherosclerosis in the aortic arch and proximal aorta in ApoE−/− atherosclerotic mice fed a Western‐type diet (Al‐Sharea et al., 2017). In support of this finding, a group from Japan reported that stimulation of α7nAchRs by AR‐R17779 suppresses atherosclerosis and aortic aneurysm formation in ApoE−/− atherosclerotic mice (Hashimoto et al., 2014). Watanabe et al. also showed that administration of AR‐R17779 prevented enlargement of the abdominal aorta in association with reduced inflammation and extracellular matrix disruption (Watanabe et al., 2016). Our results in this study demonstrate a functional effect of α7nAChRs on PDGF‐BB‐induced VSMC proliferation and migration in vitro. These data may provide some clues about the development of treatment for PDGF‐BB‐associated diseases. However, most of the investigations, including ours, were performed in animal or laboratory cells, suggesting the necessity of more studies in humans.
In summary, our findings reveal that pharmacological activation of α7nAChRs attenuates migration in PDGF‐BB‐treated VSMCs via a mechanism dependent on mitochondrial SIRT3. Our results imply that the α7nAChR plays a critical role in mitochondrial biology and may be a promising pharmacological target of PDGF signalling for the prevention or treatment of human diseases.
Author contributions
D.J.L. and P.W. conceived the study, designed the experiments, analysed data and wrote the manuscript. H.W. reviewed and edited it and provided intellectual contributions. J.T., F.Y.Z. M.G. and Y.H.L. performed the experiments, and. P.W., D.J.L. and H.W. acquired funding.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by grants from the National Science Foundation of China (Nos 81473208, 81773719, 81673485, 81300081 and 81671304), National Key R&D Program of China (No. 2018YFA0108301), Shanghai Municipal Health and Family Planning Commission Foundation (No. 2016ZB0306) and Yantai University Key Laboratory of Molecular Pharmacology and Drug Evaluation of Ministry of Education (P201603).
Li D.‐J., Tong J., Zeng F.‐Y., Guo M., Li Y.‐H., Wang H., and Wang P. (2019) Nicotinic ACh receptor α7 inhibits PDGF‐induced migration of vascular smooth muscle cells by activating mitochondrial deacetylase sirtuin 3. British Journal of Pharmacology, 176, 4388–4401. 10.1111/bph.14506.
Contributor Information
Hongbo Wang, Email: hongbowang@ytu.edu.cn.
Pei Wang, Email: peiwang@tongji.edu.cn.
References
- Al‐Sharea A, Lee MKS, Whillas A, Flynn MC, Chin‐Dusting J, Murphy AJ (2017). Nicotinic acetylcholine receptor alpha 7 stimulation dampens splenic myelopoiesis and inhibits atherogenesis in Apoe(−/−) mice. Atherosclerosis 265: 47–53. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C (2011). Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21: 193–215. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Lee CH, Flood D, Marger F, Donnelly‐Roberts D (2015). Therapeutic potential of alpha7 nicotinic acetylcholine receptors. Pharmacol Rev 67: 1025–1073. [DOI] [PubMed] [Google Scholar]
- Cailotto C, Gomez‐Pinilla PJ, Costes LM, van der Vliet J, Di Giovangiulio M, Nemethova A et al (2014). Neuro‐anatomical evidence indicating indirect modulation of macrophages by vagal efferents in the intestine but not in the spleen. PLoS One 9: e87785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Bjorndahl MA, Religa P, Clasper S, Garvin S, Galter D et al (2004). PDGF‐BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell 6: 333–345. [DOI] [PubMed] [Google Scholar]
- Chen JK, Zhao T, Ni M, Li DJ, Tao X, Shen FM (2012). Downregulation of alpha7 nicotinic acetylcholine receptor in two‐kidney one‐clip hypertensive rats. BMC Cardiovasc Disord 12: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Liu B, Kong D, Li S, Li C, Wang H et al (2015). Atorvastatin calcium inhibits phenotypic modulation of PDGF‐BB‐induced VSMCs via down‐regulation the Akt signaling pathway. PLoS One 10: e0122577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, van der Zanden EP, The FO , Bijlsma MF, van Westerloo DJ, Bennink RJ et al (2005). Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2‐STAT3 signaling pathway. Nat Immunol 6: 844–851. [DOI] [PubMed] [Google Scholar]
- Dong XC, Jing LM, Wang WX, Gao YX (2016). Down‐regulation of SIRT3 promotes ovarian carcinoma metastasis. Biochem Biophys Res Commun 475: 245–250. [DOI] [PubMed] [Google Scholar]
- Du G, Zhu H, Yu P, Wang H, He J, Ye L et al (2013). SMND‐309 promotes angiogenesis in human umbilical vein endothelial cells through activating erythropoietin receptor/STAT3/VEGF pathways. Eur J Pharmacol 700: 173–180. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Yarkov A, Aliev G (2016). Positive modulators of the alpha7 nicotinic receptor against neuroinflammation and cognitive impairment in Alzheimer's disease. Prog Neurobiol 144: 142–157. [DOI] [PubMed] [Google Scholar]
- Gan L, Wang Z, Si J, Zhou R, Sun C, Liu Y et al (2018). Protective effect of mitochondrial‐targeted antioxidant MitoQ against iron ion (56) Fe radiation induced brain injury in mice. Toxicol Appl Pharmacol 341: 1–7. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46 (D1): D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Ichiki T, Watanabe A, Hurt‐Camejo E, Michaelsson E, Ikeda J et al (2014). Stimulation of alpha7 nicotinic acetylcholine receptor by AR‐R17779 suppresses atherosclerosis and aortic aneurysm formation in apolipoprotein E‐deficient mice. Vascul Pharmacol 61: 49–55. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB et al (2010). SIRT3 regulates mitochondrial fatty‐acid oxidation by reversible enzyme deacetylation. Nature 464: 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Huang Y (2015). Targeting the platelet‐derived growth factor signalling in cardiovascular disease. Clin Exp Pharmacol Physiol 42: 1221–1224. [DOI] [PubMed] [Google Scholar]
- Huang F, Ni M, Zhang JM, Li DJ, Shen FM (2017). TRPM8 downregulation by angiotensin II in vascular smooth muscle cells is involved in hypertension. Mol Med Rep 15: 1900–1908. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kim KH, Lee WR, An HJ, Lee SJ, Han SM et al (2015). Apamin inhibits PDGF‐BB‐induced vascular smooth muscle cell proliferation and migration through suppressions of activated Akt and Erk signaling pathway. Vascul Pharmacol 70: 8–14. [DOI] [PubMed] [Google Scholar]
- Li D‐J, Fu H, Tong J, Li Y‐H, Qu L‐F, Wang P et al (2018a). Cholinergic anti‐inflammatory pathway inhibits neointimal hyperplasia by suppressing inflammation and oxidative stress. Redox Biol 15 (Supplement C): 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DJ, Evans RG, Yang ZW, Song SW, Wang P, Ma XJ et al (2011). Dysfunction of the cholinergic anti‐inflammatory pathway mediates organ damage in hypertension. Hypertension 57: 298–307. [DOI] [PubMed] [Google Scholar]
- Li DJ, Fu H, Zhao T, Ni M, Shen FM (2016a). Exercise‐stimulated FGF23 promotes exercise performance via controlling the excess reactive oxygen species production and enhancing mitochondrial function in skeletal muscle. Metabolism 65: 747–756. [DOI] [PubMed] [Google Scholar]
- Li DJ, Huang F, Lu WJ, Jiang GJ, Deng YP, Shen FM (2015). Metformin promotes irisin release from murine skeletal muscle independently of AMP‐activated protein kinase activation. Acta Physiol (Oxf) 213: 711–721. [DOI] [PubMed] [Google Scholar]
- Li DJ, Huang F, Ni M, Fu H, Zhang LS, Shen FM (2016b). Alpha7 nicotinic acetylcholine receptor relieves angiotensin II‐induced senescence in vascular smooth muscle cells by raising nicotinamide adenine dinucleotide‐dependent SIRT1 Activity. Arterioscler Thromb Vasc Biol 36: 1566–1576. [DOI] [PubMed] [Google Scholar]
- Li DJ, Liu J, Hua X, Fu H, Huang F, Fei YB et al (2018b). Nicotinic acetylcholine receptor alpha7 subunit improves energy homeostasis and inhibits inflammation in nonalcoholic fatty liver disease. Metabolism 79: 52–63. [DOI] [PubMed] [Google Scholar]
- Li DJ, Zhao T, Xin RJ, Wang YY, Fei YB, Shen FM (2014). Activation of alpha7 nicotinic acetylcholine receptor protects against oxidant stress damage through reducing vascular peroxidase‐1 in a JNK signaling‐dependent manner in endothelial cells. Cell Physiol Biochem 33: 468–478. [DOI] [PubMed] [Google Scholar]
- Li LX, Zhou JX, Calvet JP, Godwin AK, Jensen RA, Li X (2018c). Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis 9: 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R, Yan F, Zeng Z, Wang H, Qiu K, Xu J et al (2018). Insulin‐like growth factor‐1 activates PI3K/Akt signalling to protect human retinal pigment epithelial cells from amiodarone‐induced oxidative injury. Br J Pharmacol 175: 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Shen FM, Le YY, Kong Y, Liu X, Cai GJ et al (2009). Antishock effect of anisodamine involves a novel pathway for activating alpha7 nicotinic acetylcholine receptor. Crit Care Med 37: 634–641. [DOI] [PubMed] [Google Scholar]
- Liu R, Heiss EH, Schachner D, Jiang B, Liu W, Breuss JM et al (2017). Xanthohumol blocks proliferation and migration of vascular smooth muscle cells in vitro and reduces neointima formation in vivo. J Nat Prod 80: 2146–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv G, Sun D, Zhang J, Xie X, Wu X, Fang W et al (2017). Lx2‐32c, a novel semi‐synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm Sin B 7: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair CE, Liu R, Atanasov AG, Wimmer L, Nemetz‐Fiedler D, Sider N et al (2015). Piperine congeners as inhibitors of vascular smooth muscle cell proliferation. Planta Med 81: 1065–1074. [DOI] [PubMed] [Google Scholar]
- Matteoli G, Gomez‐Pinilla PJ, Nemethova A, Di Giovangiulio M, Cailotto C, van Bree SH et al (2014). A distinct vagal anti‐inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut 63: 938–948. [DOI] [PubMed] [Google Scholar]
- Navarro E, Buendia I, Parada E, Leon R, Jansen‐Duerr P, Pircher H et al (2015). Alpha7 nicotinic receptor activation protects against oxidative stress via heme‐oxygenase I induction. Biochem Pharmacol 97: 473–481. [DOI] [PubMed] [Google Scholar]
- Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G et al (2013). Smooth muscle cell plasticity: fact or fiction? Circ Res 112: 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP (2002). SIRT3, a human SIR2 homologue, is an NAD‐dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A 99: 13653–13658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Garg B, Garg K, Wagner C, Wagner SN (2016). Dual c‐Jun N‐terminal kinase‐cyclin D1 and extracellular signal‐related kinase‐c‐Jun disjunction in human melanoma. Br J Dermatol 175: 1221–1231. [DOI] [PubMed] [Google Scholar]
- Prins KW, Tian L, Wu D, Thenappan T, Metzger JM, Archer SL (2017). Colchicine depolymerizes microtubules, increases junctophilin‐2, and improves right ventricular function in experimental pulmonary arterial hypertension. J Am Heart Assoc 6: e006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown KV, Moran Y, Chen D (2010). Sirtuin regulation in calorie restriction. Biochim Biophys Acta 1804: 1576–1583. [DOI] [PubMed] [Google Scholar]
- Raines EW (2004). PDGF and cardiovascular disease. Cytokine Growth Factor Rev 15: 237–254. [DOI] [PubMed] [Google Scholar]
- Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y et al (2017). MCU‐dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36: 5897–5909. [DOI] [PubMed] [Google Scholar]
- Shumin C, Wei X, Yunfeng L, Jiangshui L, Youguang G, Zhongqing C et al (2018). Genipin alleviates vascular hyperpermeability following hemorrhagic shock by up‐regulation of SIRT3/autophagy. Cell Death Discov 4: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH et al (2000). FAK integrates growth‐factor and integrin signals to promote cell migration. Nat Cell Biol 2: 249–256. [DOI] [PubMed] [Google Scholar]
- Snoek SA, Verstege MI, van der Zanden EP, Deeks N, Bulmer DC, Skynner M et al (2010). Selective alpha7 nicotinic acetylcholine receptor agonists worsen disease in experimental colitis. Br J Pharmacol 160: 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JE, Lee E, Jung SK, Kim JE, Oak MH, Lee KW et al (2014). Anthocyanidins, novel FAK inhibitors, attenuate PDGF‐BB‐induced aortic smooth muscle cell migration and neointima formation. Cardiovasc Res 101: 503–512. [DOI] [PubMed] [Google Scholar]
- Song Q, Liu L, Yu J, Zhang J, Xu M, Sun L et al (2017). Dihydromyricetin attenuated Ang II induced cardiac fibroblasts proliferation related to inhibitory of oxidative stress. Eur J Pharmacol 807: 159–167. [DOI] [PubMed] [Google Scholar]
- ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M et al (2006). Novel Nox inhibitor VAS2870 attenuates PDGF‐dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res 71: 331–341. [DOI] [PubMed] [Google Scholar]
- Tseng AH, Wu LH, Shieh SS, Wang DL (2014). SIRT3 interactions with FOXO3 acetylation, phosphorylation and ubiquitinylation mediate endothelial cell responses to hypoxia. Biochem J 464: 157–168. [DOI] [PubMed] [Google Scholar]
- van der Zanden EP, Snoek SA, Heinsbroek SE, Stanisor OI, Verseijden C, Boeckxstaens GE et al (2009). Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor alpha4beta2. Gastroenterology 137: 1029–1039. [DOI] [PubMed] [Google Scholar]
- Ward MP, Spiers JP (2017). Protein phosphatase 2A regulation of markers of extracellular matrix remodelling in hepatocellular carcinoma cells: functional consequences for tumour invasion. Br J Pharmacol 174: 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Ichiki T, Kojima H, Takahara Y, Hurt‐Camejo E, Michaelsson E et al (2016). Suppression of abdominal aortic aneurysm formation by AR‐R17779, an agonist for the alpha7 nicotinic acetylcholine receptor. Atherosclerosis 244: 113–120. [DOI] [PubMed] [Google Scholar]
- Wei CC, Kong YY, Hua X, Li GQ, Zheng SL, Cheng MH et al (2017). NAD replenishment with nicotinamide mononucleotide protects blood‐brain barrier integrity and attenuates delayed tissue plasminogen activator‐induced haemorrhagic transformation after cerebral ischaemia. Br J Pharmacol 174: 3823–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ et al (2007). Nutrient‐sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130: 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Xiao T, Sun Q, Wang K (2017). The current agonists and positive allosteric modulators of alpha7 nAChR for CNS indications in clinical trials. Acta Pharm Sin B 7: 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S et al (2016). Mitochondrial sirtuin network reveals dynamic SIRT3‐dependent deacetylation in response to membrane depolarization. Cell 167: 985–1000 e1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Guan D, Lei L, Lu J, Liu JQ, Yang G et al (2018). H6, a novel hederagenin derivative, reverses multidrug resistance in vitro and in vivo. Toxicol Appl Pharmacol 341: 98–105. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Qi D, Wang X, Gao Z, Li P, Liu W et al (2018). Protective effect of Salvianolic acid A on ischaemia‐reperfusion acute kidney injury in rats through protecting against peritubular capillary endothelium damages. Phytother Res 32: 103–114. [DOI] [PubMed] [Google Scholar]