

Abstract
Uridine-5’-diphosphate (UDP)-glucose is reported as one of the most versatile building blocks within the metabolism of pro- and eukaryotes. The activated sugar moiety is formed by the enzyme UDP-glucose pyrophosphorylase (GalU). Two homologous enzymes (designated as RoGalU1 and RoGalU2) are encoded by most Rhodococcus strains, known for their capability to degrade numerous compounds, but also to synthesize natural products such as trehalose comprising biosurfactants. To evaluate their functionality respective genes of a trehalose biosurfactant producing model organism—Rhodococcus opacus 1CP—were cloned and expressed, proteins produced (yield up to 47 mg per L broth) and initially biochemically characterized. In the case of RoGalU2, the Vmax was determined to be 177 U mg−1 (uridine-5’-triphosphate (UTP)) and Km to be 0.51 mM (UTP), respectively. Like other GalUs this enzyme seems to be rather specific for the substrates UTP and glucose 1-phosphate, as it accepts only dTTP and galactose 1-phoshate in addition, but both with solely 2% residual activity. In comparison to other bacterial GalU enzymes the RoGalU2 was found to be somewhat higher in activity (factor 1.8) even at elevated temperatures. However, RoGalU1 was not obtained in an active form thus it remains enigmatic if this enzyme participates in metabolism.
Keywords: glycosylation; UDP-glucose pyrophosphorylase; UDP-glucose; nucleotide donors; Rhodococcus, Actinobacteria, gene redundancy; Leloir glycosyltransferases; activated sugar; UTP
1. Introduction
Uridine-5’-diphosphate (UDP)-glucose is a key metabolite in most organisms and thus used in a variety of reactions of the sugar and starch metabolism, sugar interconversions, amino and nucleotide sugar metabolism, biosynthesis of antibiotics and cell envelope components, and as precursor for different primary and secondary metabolites [1,2,3]. Being an interesting and valuable compound, UDP-glucose attracted more and more attention for biotechnological applications. As an example, in glycosylation reactions, their production and application in fine chemical scale could be shown [1,2,3]. It was presented that whole cell catalysis by sucrose synthase (SuSy) with UDP as precursor led to large-scale production of 100 gUDP-glucose/L with a yield of 86% [3]. Another report with 0.1 g/L of free SuSy could achieve 144 gUDP-glucose/L with a comparable conversion rate of 85% based on the precursor UDP [1]. The enzymatic production of various sugar-nucleotides can be realized via several UDP-glucose synthesizing enzymes (Scheme 1). Glycosyltransferases, like sucrose synthase, are the most common used representatives [1,4,5,6,7]. Nucleotidyl transferases, like UDP-glucose pyrophosphorylase have been known for a long time but were seldom employed as biocatalysts to produce UDP-glucose, maybe due to low specific activities. Those enzymes are more often studied for their metabolic role.
Scheme 1.

Reactions in which UDP-glucose is consumed or formed. UDP: Uridine-5’-diphosphate, UMP: Uridine-5’-monophosphate, UTP: Uridine-5’-triphosphate, NAD+/NADH: Nicotinamide adenine dinucleotide in the oxidized and reduced form.
UDP-glucose pyrophosphorylases (GalU or UGPase; EC 2.7.7.9) catalyze the reversible reaction of glucose 1-phosphate and UTP into UDP-glucose and inorganic pyrophosphate (PPi). Enzymes of the GalU family are ubiquitous and can be found among the tree of life [8]. With respect to bacteria mostly proteobacterial GalUs have been studied. Like many other nucleotidyl transferases, also GalU requires divalent cations to promote the reaction (Scheme 2). In most cases magnesium ions are employed, and so far only magnesium chloride has been investigated [9,10,11,12,13,14,15,16]. The reaction mechanism follows a sequential bi-bi-mechanism starting with the binding of UTP to the active site, in presence of a magnesium ion [17], followed by the binding of glucose 1-phosphate. The octahedral coordination sphere of the magnesium positions the substrates in the right way and enables the nucleophilic attack of glucose 1-phosphate on UTP [18]. A lysine, an aspartate and several water molecules within the active site help to stabilize the position of the substrates and cofactor for the proper nucleophilic attack of the phosphoryl oxygen of glucose 1-phosphate towards the α-phosphor atom of UTP [17]. Finally, PPi is released from the GalU/Mg2+/UDP-glucose complex [17].
Scheme 2.

Proposed reaction mechanism of UDP-glucose pyrophosphorylase, adapted from Kim et al. [17].
GalU enzymes have been shown to play crucial roles in galactose fermentation [19]. Other metabolic pathways in which this enzyme family is active are similar like the ones UDP-glucose itself plays an important role. It produces UDP-glucose as precursor for sucrose, glucan and amylose in the sugar and starch metabolism. Furthermore, it plays a role in galactose metabolism and glucuronate interconversion, where galactose 1-phosphate is over all converted into UDP-glucuronate with the help of a hexose 1-phosphate uridylyl transferase and a UDP-glucose 6-dehydrogenase. In amino sugar and nucleotide sugar metabolism, GalU is active in interconverting different UDP-sugars via the intermediate UDP-glucose. Additionally, it can be found in glycolipid metabolism and the synthesis of antibiotics. Respectively, many GalU enzymes from several organisms have been investigated to date, e.g., GalUs of plants [20,21,22,23,24], of mammals [25,26,27], including human [28], parasites [29], and many different bacteria [9,10,12,13,30,31,32,33], only to name a few groups. Fields of study included the determination of structures [17,30,34], biochemical characterization [14,31,35,36] and biological function [15,33,37].
During those studies, it became clear that many organisms also carry a gene encoding for a GalF, a homologous and putative regulatory protein towards GalU. In E. coli the function of the GalF protein was determined by Ebrecht and co-workers [19] to putatively regulate or even interact with GalU while maintaining a low UDP-glucose pyrophosphorylase activity (0.004% of EcGalU). It seems to be an evolutionary artefact, which was observed for several other Enterobacteria, as exemplarily different species and strains of Klebsiella, Yersinia, Pectobacterium, Shigella, Escherichia, Pantoea, Salmonella, Serratia, and some others [19]. Both proteins, GalU and GalF, seem to have evolved independently from a common ancestor, but only GalU kept the initial enzymatic role. Apparently, GalF underwent several mutations, which led to a much lower activity compared to GalU. By means of mutagenesis of identified key residues, the activity of GalF was restored and comparable to GalU and even a physiological complementation of a GalU-knockout strain was possible [19]. GalUs are known to have two important conserved motifs: The N-terminal one, which is involved in the binding of the uracil ring with the sequence G-X-G-T-R-X-L-P-X-T-K (X stands for any amino acid) [8,38] and a V-E-K-P-motif with an important lysine for glucose 1-phosphate binding [38,39,40]. The GalF of E. coli was shown to have the same V-E-K-P-motif, but instead of G-L-G-T-R, GalF has G-L-G-M-H residues in the N-terminal motif [19]. By means of mutagenesis it could be shown that the alteration of T-R into M-H was possibly the reason for the reduced UDP-glucose pyrophosphorylase activity of E. coli GalF. But, the actual function of GalF among Enterobacteria still remains unclear. If gene redundancy and physiological flexibility here between E. coli GalU and GalF is of relevance is not yet clarified. Furthermore, it remains enigmatic why this presence of homologous GalU/GalF-proteins has so far only been reported for Enterobacteria among the bacteria. However, in the genomes of Rhodococcus species, gene redundancy plays a crucial role [20,22,27,41,42,43]. Interestingly, most Rhodococcus species also carry two galU genes which we will highlight herein for the first time. Thus, R. opacus 1CP encodes for two isoforms of the GalU enzyme—RoGalU1 and RoGalU2, which have not been characterized yet. In some cases, it could be shown that enzymatic isoforms derived from rhodococci have the same or only a slightly different function, so that it was assumed that the coding isogenes have evolved independently from a common ancestor [44]. However, the functionality of respective proteins has not been investigated so far. Furthermore, to the best of our knowledge studies on UDP-glucose pyrophosphorylases in Rhodocccus species have not been carried out. Herein we describe the redundant GalU enzymes of R. opacus 1CP whereas one was found to be active as recombinant enzyme and might participate in several metabolic pathways, which is discussed in detail. Phylogenetic analysis of the amino acid sequences of GalU proteins of several rhodococci, Enterobacteria, and Actinobacteria helps to further understand the role of RoGalU1 and RoGalU2. This will also give more information on gene redundancy in Actino- and Enterobacteria in general.
2. Results
2.1. Genome Mining and Phylogenetic Analyses
Via a genome mining approach two genes encoding for two UDP-glucose pyrophosphorylases (GalUs) were identified in Rhodococcus opacus 1CP. The respective genes and amino acid sequences used for codon usage optimization were derived from the NCBI protein accession numbers ANS26426 (RoGalU1, theoretical size: 33.2 kDa, GenBank accession number of the codon usage optimized nucleotide sequence: MN617759) and ANS26629 (RoGalU2, theoretical size: 33.9 kDa, GenBank accession number of the codon usage optimized nucleotide sequence: MN617760). They have a sequence identity of more than 70% on amino acid level to each other when compared over full length.
Having a look at the genomic environment of both genes, various genes were found flanking RogalU1, which were involved in the sugar- and nucleotide metabolism, e.g., a mannose 6-phosphate isomerase, a D-glycero-D-manno-heptose 1-phosphate guanosyltransferase or a undecaprenyl-phosphate galactose phosphotransferase, whereas RogalU2 is flanked by cation channels, heat shock proteins or putative regulatory proteins.
Analyzing the amino acid sequences of the GalUs originating from R. opacus 1CP, both contain the same motifs for uracil binding, G-X-G-T-R-F-L-P (start: amino acid 17 in EcGalU sequence; 18 and 21 in RoGalU1 and RoGalU2, respectively). Equally, both display the V-E-K-P motif (start: amino acid 200 in EcGalU sequence; 199 and 202 in RoGalU1 and RoGalU2, respectively) with the important lysine (K202 in EcGalU) for binding of glucose 1-phosphate (see Figure 1; the subsequent numbering of amino acids is according to EcGalU; [19]). Interestingly, only the GalF of E. coli shows an alteration from T20-R21 to M-H in the first motif, which was shown before to be the reason for the reduced UDP-glucose pyrophosphorylase activity of E. coli GalF. Large parts of RoGalU1 and RoGalU2 are identical and seem to be conserved. Several of those conserved amino acids can be found in EcGalU and/or EcGalF, respectively. Furthermore, all four sequences share the same residues for the hydrogen bonding of the uracil ring (A16-G17 and Q109), and at least the Rhodococcus GalUs and EcGalU have the same amino acids for the phosphoryl binding of UDP-glucose (G179, K202 and G-A-G-D, start: amino acid: 234). The overall alignment indicates that the structure of all four proteins should be highly similar, as the secondary elements presented by α-helical and β-sheet areas of EcGalU are present in RoGalU1 and RoGalU2, as well.
Figure 1.

Alignment of RoGalU1 and RoGalU2 with the GalU and GalF amino acid sequences of E. coli K-12 [19], executed with the MUSCLE algorithm [45,46] in the program MEGA X [47] for multiple sequence alignment with default settings and imaged with ESPript 3.0 [48]. Conserved amino acids are color-coded. Red box with white letters: strictly identical amino acids, yellow box with black bold letters: similar amino acids, which are conserved among at least two sequences, black squiggles with α or η signs: α-helical structures, black arrows with β sign: β-sheet structures, TT: strict β-turns, TTT: strict α-turns, grey stars: residues with alternate conformations.
The alignment (Figure 1; part of the complete alignment with all 48 amino acid sequences) that underlies the phylogenetic tree (Figure 2) shows remarkable sequence similarities and again huge conserved areas between the GalU sequences of different actinobacterial species and especially strains of Rhodococcus, Mycobacterium, Corynebacterium, and Gordonia. As they are close relatives, it is not surprising that those sequences cluster together. It is striking that there are two sequence sections around the G-X-G-T-R-F-L-P (start: amino acid 17) motif and the V-E-K-P (start: amino acid 200) motif, which are highly conserved among all chosen sequences. Besides those and the highly conserved motifs for binding of the substrates and products, there are also three more conserved motifs: One G-L-G-H sequence (start: amino acid 114) before and two sequences between the V-E-K-P motif and the C-terminus, namely G-R-Y-L-L (start: amino acid 216) and Q-L-T-D-A-I (start: amino acid 240) within all analyzed sequences. In addition, those seem to be highly conserved, not only among Actinobacteria, but also Enterobacteria. Furthermore, the sequences contain single conserved amino acids, like G53, F72, E79, D137, Y178, S210, G261, D265, and G267 (Figure 1).
Figure 2.

Phylogenetic tree of RoGalU1 and RoGalU2 with similar characterized or related UDP-glucose pyrophosphorylase sequences. The evolutionary history was inferred by using the Maximum Likelihood method and JTT matrix-based model [49]. The tree with the highest log likelihood (−13775.30) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measuring the number of substitutions per site. This analysis involved 48 amino acid sequences. There was a total of 375 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [47].
The above described alignment was used and completed by numerous GalU protein sequences in order to generate a distance tree for a phylogenetic analysis (Figure 2). The first big branch of the tree contains all actinobacterial proteins, which are very close relatives. Within those, also Rhodococcus enzymes form one big cluster. Interestingly, the enzymes from the same strains do not cluster together, but form two different smaller clusters. The Rhodococcus strains containing only one GalU protein are mapped within the branch of our tested and active RoGalU2. The second big branch of the tree contains Actinobacteria in the upper part, Firmicutes, and Proteobacteria in the lower part.
2.2. Recombinant Expression of RogalU1 in E. coli, Purification, and Renaturation
For characterization purposes in more detail RogalU1 was cloned into the expression vector pET16bP. After transformation of chemically competent E. coli BL21(DE3) pLysS with pET16bP-RogalU1 overexpression and respective protein production and purification were carried out as described in the material and methods section. The SDS-PAGE in Figure 3a shows the expected molecular weight of RoGalU1 of 33 kDa, but most of the produced enzyme is present as insoluble protein in the cells. The formation of those inclusion bodies was determined from the greyish to white color of the pellet after crude cell extract preparation. Hence, it was tried to renature the misfolded protein with the Thermo Scientific Pierce Protein Refolding Kit according to the manufacturer’s instruction. Unfortunately, it was not possible to obtain RoGalU1 in an active and soluble form. Also, protein within the inclusion bodies only had a marginal residual enzyme activity. This was determined to be <0.001 U mg−1 by means of the standard test which is described in the materials and methods section (4.6). In this work, we determined throughout enzyme activity with respect to product formation (1 U corresponds to the formation of 1 µmol UDP-glucose per minute). In order to turn inclusion bodies into native protein, the expression was altered with respect to various media and cultivation conditions. All efforts failed and thus this enzyme was not characterized further.
Figure 3.

SDS-PAGE gels of RoGalU1 (a) and RoGalU2 (b). Arrows indicate the expected molecular weight of 33 kDa for RoGalU1 and 34 kDa for RoGalU2. SP: soluble proteins, IB: inclusion bodies, PP: purified protein, M: Marker.
2.3. Recombinant Expression in E. coli, Purification and Production of Active RoGalU2
The gene RogalU2 was treated as described in the materials and methods and similarly as the RogalU1 described above. In this case, it was possible to produce about 47 mg of pure, soluble protein with an expected molecular weight of 34 kDa (see Figure 3b) from 25 g wet biomass. The maximum enzyme activity observed was 3.6 U mg−1 in the direction of UDP-glucose formation, using the standard reaction conditions. In addition, different media for gene expression were tested (LB, TB, or NB), but no significant difference in terms of protein yield or activity was found. TB medium was the fastest medium for cell growth and led to most biomass, therefore using TB auto induction medium for later large-scale production was an obvious choice.
In order to validate the oligomeric state of the enzyme, a size exclusion chromatography under non-denaturizing conditions was performed in duplicates with two different concentrations of the protein (0.2 and 2.0 mg mL−1), each. The elution profiles were similar in those cases (not shown). The respective calibration was also measured twice and revealed the occurrence of this protein mostly in a hexameric state (188 kDa elution volume vs. subunit size of 34 kDa; Figure 3 and Figure 4). Thus, we can state that under these conditions the protein was present mostly in a single hydrodynamic state and therefore protein characterization can be performed.
Figure 4.

Size exclusion chromatography of RoGalU2 (a) and the corresponding calibration measurements (b). The calibration curve was prepared with the following proteins of known molecular weight (given in brackets): Ribonuclease (13.7 kDa), Carbonic Anhydrase (29 kDa), Conalbumin (75 kDa), Aldolase (158 kDa) and Ferritin (440 kDa). All elutions were performed in a buffer containing 50 mM Hepes, pH 7.0 and 1 mM magnesium chloride. See methods Section 4.5. The protein concentration was either 0.2 or 2.0 mg mL−1 whereas the latter is presented herein.
2.4. Determining Optimum Reaction Conditions for RoGalU2
In order to collect proper kinetic data for the enzyme activity of RoGalU2 in the direction of UDP-glucose formation, it was necessary to find optimum reaction conditions. In addition, the product detection method and fitting procedure had to be established. UDP-glucose formation by RoGalU2 was determined by HPLC as described in materials and methods section. A typical result and a respective rate determination is presented in Figure 5.
Figure 5.

Enzymatic conversion of UTP and glucose 1-phosphate into UDP-glucose and PPi. In (a) the HPLC chromatograms of a time course from 0 to 7 min are presented and the determined concentration of UDP-glucose was used to calculate a product formation rate as shown in (b). For this analysis the standard enzyme assay and determination methods as described in materials and methods section have been used.
Firstly, the reaction temperature was varied from 4 °C to 87 °C. There was an activity plateau determined between 40 °C and 45 °C, with a maximum observed enzyme activity of 6.7 U mg−1 (Figure 6a). To monitor the stability of RoGalU2 at different temperatures, the enzyme was incubated for 30 min at temperatures ranging from 0 °C to 70 °C. The activity assay was then carried out at 45 °C. RoGalU2 shows at least 80% of activity when incubated for 30 min between 19 °C and 45 °C (see Figure 6b). Because of the higher activity that could be achieved at 42 °C versus 45 °C, it was decided to perform all subsequent experiments at 42 °C.
Figure 6.

Temperature-dependent activity (a) and stability (b) of purified RoGalU2 enzyme was measured in triplicates. Activity was measured with pre-incubation of the assay mixture (2 mM UTP, 2 mM glucose 1-phosphate, 4 mM MgCl2, 50 mM Hepes buffer, pH 7.0, 30 µg RoGalU2, 1 mL reaction volume) at indicated temperatures. The enzyme was stored on ice and directly used for measurements. For stability measurements the enzyme was incubated shaking for 30 min at indicated temperatures and directly used for activity assay at 45 °C. Means and standard deviations are shown. 100% relative RoGalU2 activity correspond to 6.7 U mg−1 ((a), activity) and 5.0 U mg−1 ((b), stability), respectively.
To verify the dependence of RoGalU2 on magnesium the concentration of magnesium chloride and magnesium sulfate present in the standard reaction setup at 42 °C was varied between 0–10 mM. For magnesium salt concentrations below 1 mM, the standard buffer of the enzyme had to be exchanged by a buffer without magnesium chloride freshly prior to respective experiments since RoGalU2 is not stable in a buffer without magnesium. The activity was also measured after every buffer exchange. Figure 7 shows the absolute dependence of the activity on the concentration of magnesium ions. There is no activity and thus UDP-glucose formation measurable without magnesium salt. The highest activity was determined to be 5.2 U mg−1 with magnesium chloride and 9.5 U mg−1 with magnesium sulfate, in the presence of only 1 mM of the respective salt. When increasing the magnesium concentration, the RoGalU2 activity decreases significantly, falling below 60% with 10 mM magnesium salt. Thus, it can be concluded that magnesium is a crucial cofactor for the enzyme as presented in Scheme 2, respectively.
Figure 7.

Relative RoGalU2 enzyme activity depending on the MgCl2 and MgSO4 concentration between 0 mM and 10 mM. Assay mixture contained 2 mM UTP, 2 mM glucose 1-phosphate, 0–10 mM MgCl2 or MgSO4, 50 mM Hepes buffer, pH 7.0, 30 µg RoGalU2. Reaction was carried out at 42 °C in 1 mL reaction volume. Means with standard deviations of triplicate measurements are shown. 100% relative RoGalU2 activity correspond to 5.2 U mg−1.
The activity of RoGalU2 with respect to magnesium salts was most similar at a concentration of 4 mM. Thus, further divalent cations were tested as metal chloride salts at 4 mM each like manganese, nickel, cobalt, calcium, and zinc. In addition, trivalent metal cations like iron and aluminum were tested. No activity was observed with the trivalent metal chlorides. For all other divalent chlorides, the relative activity was below 20%. Thus, a magnesium dependency was clearly determined.
To test whether masking of the magnesium by means of EDTA results in a reduced enzyme activity, we applied 1 mM EDTA to the reaction solution and could not observe any activity. Therefore, the standard assay was used but at a lower magnesium chloride concentration, which was set to 1 mM as well. We also tested the reaction in presence of acetonitrile as solvent and received the same result. This was of importance since acetonitrile was used to stop the reactions prior to HPLC analysis as well as to adjust samples towards elution conditions. Thus, it was proven that acetonitrile was a proper reagent to stop the reaction.
Different buffers like Bis-Tris, MOPS, sodium phosphate and imidazole were also tested to see whether the enzymatic activity can be further increased. The pH of the reaction solution was also varied ranging from 6.6–9.3 in total and split into the following ranges of different buffers: 6.6–7.4 (Imidazole), 7.4–8.4 (Hepes), and 7.6–9.3 (Tris-HCl). Figure 8 shows a graph with the comparison of RoGalU2 activities measured in those reaction solutions.
Figure 8.

RoGalU2 enzyme activity depending on the buffer (a) and pH (b) of the reaction solution. (a) Assay mixtures contained 1.5 mM UTP, 250 mM glucose 1-phosphate, 1 mM MgCl2, 50 mM buffer, pH 7.4, 1.7 µg RoGalU2. Reaction was carried out at 42 °C in 1 mL volume. (b) Assay mixtures contained 1.5 mM UTP, 250 mM glucose 1-phosphate, 1 mM MgCl2, 50 mM Imidazole, pH 6.6–7.4 (black triangle) or 50 mM Hepes, pH 7.4–8.4 (black circle) or 50 mM Tris-HCl, pH 7.6–9.3 (white circle), 1.7 µg RoGalU2. Reaction was carried out at 42 °C in 1 mL volume. Means with standard deviations are shown out of a minimum set of three independent measurements. 100% relative activities correspond to 126 U mg−1 (a) and 109 U mg−1 (b).
In none of the used buffers the relative RoGalU2 activity dropped below 60%. But it was possible to increase the relative activity up to 180% with Imidazole at pH 7.4. The pH profile even shows an increase up to 250% in activity when Imidazole or Tris-HCl are used at pH values of around 7.5. Interestingly, there is no significant difference when Hepes buffer is used at pH values between 7.4 and 8.3 or when Tris-HCl is used between pH 8.1 and 9.1. Here we only observed relative RoGalU2 activities around 80%.
The above described results allowed to formulate an improved enzyme assay with the following conditions: 1.5 mM UTP, 250 mM glucose 1-phosphate, 1 mM magnesium chloride, 50 mM Hepes, pH 7.4, 42 °C in 1 mL reaction volume. Even though the activity of RoGalU2 is higher with Imidazole pH 6.6–7.4 or Tris-HCl pH 7.6, we decided to continue using Hepes pH 7.4, because the standard deviation with Hepes buffer was found to be much smaller than with other buffers. This might be due to the weaker buffering properties of Tris-HCl or Imidazole in the applied range.
2.5. Enzyme Kinetics with RoGalU2
In order to collect proper kinetic data, the above described improved enzyme assay was employed. Thus, we could now determine the common kinetic constants Km, Vmax, kcat, and kcat/Km by means of varying the substrate concentrations. Already here we like to state the Km and thus kcat values have to be considered as apparent values as later indicated in Table 1. This is necessary as a saturation of the enzyme by both substrates cannot be secured as the later on presented data show. Pre-experiments defined 1.5 mM UTP and 250 mM of glucose 1-phosphate as most suitable fixed concentrations for the variation of the opposite substrate, respectively. As shown in Figure 9 UTP was varied between 10 µM and 5 mM with a fixed concentration of 250 mM glucose 1-phosphate. The data were fitted to the model of Yano and Koga [50] (Equation (2), see Methods section) for substrate inhibition, because above 1.5 mM UTP a strong substrate inhibition became obvious. Below this concentration, the RoGalU2-activity reaches a maximum observed activity of about 122 U mg−1. The calculated Vmax was determined to be 177 U mg−1 according to the inhibition fit of Yano and Koga [50].
Table 1.
Kinetic properties of RoGalU2.
| Varied Substrate | UTP 1 | Glucose 1-Phosphate 1 | |
|---|---|---|---|
| conc. [mM] | 0.01–5 | 0.05–10 | 0.05–500 |
| apparent Km [mM] | 0.51 | 0.9 | 150 |
| Vmax [U mg−1] 2 | 177 | 6.75 | - |
| Vmax,ob [U mg−1] 3 | 122 | 6.68 | 119 |
| apparent kcat [s−1] | 96.2 | 3.7 | 65 |
| KI [mM] | 2.61 | - | 225 |
| kcat/Km [µM−1 s−1] | 0.19 | 0.004 | 0.0004 |
1 Assay mixture contained substrates at varied concentrations given whereas the other substrate was set constant at 250 mM glucose 1-phosphate or 1.5 mM UTP, respectively, 1 mM MgCl2, 50 mM Hepes buffer, pH 7.4, 1.7 µg RoGalU2. Reaction was carried out at 42 °C in 1 mL volume. All experiments were performed at least in triplicates. Fits were according Michaelis–Menten or Yano and Koga [50]. 2 The specific activity was obtained from fitting the data. 3 The maximal observed activity was taken as a read out from the plot.
Figure 9.

RoGalU2 enzyme activity depending on the UTP concentration between 10 µM and 5 mM at a fixed glucose 1-phosphate concentration of 250 mM. Fit is calculated by the Model of Yano and Koga [50], for enzyme kinetics with substrate inhibition. The assay mixture contained 0.01–5 mM UTP, 250 mM glucose 1-phosphate, 1 mM MgCl2, 50 mM Hepes buffer, pH 7.4, 1.7 µg RoGalU2. Reaction was carried out at 42 °C in 1 mL volume. Mean values with standard deviations from triplicates are shown.
Figure 10 shows the graphs for substrate variation of glucose 1-phosphate between 50 µM and 500 mM with 1.5 mM UTP in each reaction. Figure 10a shows a Michaelis–Menten-like kinetic at concentrations between 50 µM and 10 mM. This is common for many GalU enzymes as discussed later. But with a closer look at the data, it became clear that the model does not describe the values in a proper way. In contrast to common Michaelis–Menten kinetics, the RoGalU2 activity still increased, beyond the concentration of 10 mM glucose 1-phosphate applied. Therefore, the collection of data for higher glucose 1-phosphate concentrations was necessary and showed a kinetic behavior that was different from general models that describe enzyme kinetics (Figure 10b), with a maximum observed activity of about 119 U mg−1. No model allowed to fit the obtained data properly. Further, we repeated the experiments with other batches of protein and the results were similar. To achieve a maximum formation rate of UDP-glucose, a high concentration of glucose 1-phosphate was mandatory, resulting in those data.
Figure 10.

RoGalU2 enzyme activity depending on the glucose 1-phosphate concentration between 50 µM and 500 mM at a fixed UTP concentration of 1.5 mM. Data for glucose 1-phosphate concentrations between 0 and 10 mM were fit to the kinetic model of Michaelis–Menten (a). The data obtained over the total glucose 1-phosphate variation are presented in (b), whereas no fit was able to describe the data. Assay mixture contained 0.05–500 mM glucose 1-phosphate, 1.5 mM UTP, 1 mM MgCl2, 50 mM Hepes buffer, pH 7.4, 1.7 µg RoGalU2. Reaction was carried out at 42 °C in 1 mL volume. Means with standard deviations are shown from a minimum of triplicates.
The kinetic properties of RoGalU2 are summarized in Table 1. Further, to describe the substrate scope of RoGalU2, we used UTP, ATP, GTP, CTP, and dTTP as nucleotides and glucose 1-phosphate, galactose 1-phosphate, ribose 5-phosphate and glucose as sugars in different combinations to each other. Only with sugar 1-phosphates and UTP or dTTP, product formation was detectable. Nevertheless, the relative activity of the reaction of dTTP and glucose 1-phosphate could not be evaluated due to unavailability of a product standard. A low relative RoGalU2 activity of 2% was observable when galactose 1-phosphate and UTP were tested (100% relative RoGalU2 activity corresponded to 8.1 U mg−1). With other nucleotides or sugars no activity or product formation was observable.
3. Discussion
UDP-glucose pyrophosphorylases are important enzymes for the general metabolism of many organisms as highlighted in the introduction. However, among the bacterial representatives mostly GalU enzymes from Proteobacteria have been studied. To a lesser extent, GalUs of Actinobacteria were described, as for example some information are available for the ones originating of Mycobacterium or Streptomyces species [10,13,51]. Here we wanted to add knowledge and studied the enzyme family from rhodococci, which is interesting from a phylogenetic as well as mechanistic point of view. In a model organism, Rhodococcus opacus 1CP, relevant for trehalose biosurfactant production or aromatic compound degradation [52,53], two isogenes encoding for UDP-glucose pyrophosphorylases were identified and described for the first time herein.
A phylogenetic tree based on an alignment of 48 protein sequences of partially characterized UDP-glucose pyrophosphorylases was generated (Figure 2). The tree is separated into two groups; proteobacterial and actinobacterial representatives. It should be stressed that some protein sequences have been annotated as GalU and others as GalF enzymes or regulatory proteins, respectively, but in most cases this has not been experimentally verified.
The first group contains GalU- or GalF-designated protein sequences of partially characterized UDP-glucose pyrophosphorylases or related sequences from Actinobacteria, the other contains GalUs and GalFs from Proteobacteria. The branch of actinobacterial enzymes is separated again into two subgroups; Corynebacteriales and other orders of Actinobacteria. Within the branch of corynebacterial enzymes different Rhodococcus species cluster together. Gene products derived from soil or wastewater populating rhodococci (which encode for two GalU enzymes) cluster into two subbranches, respectively. Within those, GalUs of the zoonotic pathogen or animal infecting Rhodococcus species, such as R. coprophilus and R. hoagie (equii), as well as those Rhodococcus strains that only comprise a single GalU gene form one cluster. Herein, the protein RoGalU2 from our model strain is localized. An exception is the plant pathogenic R. fascians, which also encodes for only one GalU protein. It seems more related to a pathogenic Nocardia then to other rhodococci enzymes. From those Rhodococcus species carrying two GalUs, a second branch is formed in which the protein RoGalU1 clusters. Another branch within corynebacterial representatives is formed by GalUs of the genera Gordonia, Mycobacterium and Corynebacterium. And another more distant group of the actinobacterial GalUs contains enzymes of the genera Streptomyces, Kineococcus and Arthrobacter. Interestingly, Arthrobacter also has strains carrying two isoforms of GalU proteins, but they form a group far from the Rhodococcus proteins. A similar behavior is present in the group of proteobacterial GalUs including some annotated but also experimentally verified GalFs. Here the enzymes are arranged according to their phylogenetic relation as expected. However, E. coli and Erwinia amylovora each encode two related proteins which form distinct branches.
As the branch of proteins named GalU2 also contains other actinobacterial GalUs only having one GalU isoform, it is possible that GalU2 is the older protein with the initial function in the genus Rhodococcus. Thus, GalU1 might have evolved from GalU2 and gained the function of a UDP-glucose pyrophosphorylase. But the genetic environment of the respective coding genes does not support this hypothesis. The genetic environment of RogalU1 is more related to the sugar metabolism with sugar-phospho transferases and sugar-phospho isomerases, whereas the genetic environment of RogalU2 comprises many putative and for the sugar or amino metabolism unspecific genes. It consists of different genes coding for cation and phosphate channels, transmembrane proteins, conductance and mechanosensitive channels, acting as an osmotic release valve in response to osmotic stress, as well as ATP converting enzymes and heat shock proteins. Interestingly, the gene cluster of RogalU1 is only available in R. jostii RHA1, whereas the gene cluster surrounding RogalU2 is present in many other Actinobacteria, like N. farcinica, M. smegmatis, M. tuberculosis, C. glutamicum, among others. Therefore, the RogalU2 cluster seems to be the more common one for Actinobacteria.
Interestingly, RoGalU2 has 99% amino acid sequence identity to the GalU regulator GalF of R. opacus PD630. However, this GalF designation for strain PD630 is based only on a bioinformatic annotation and no other galU gene is present in the genome of R. opacus PD630. Thus, this protein must also be an UDP-glucose pyrophosphorylase and not a regulatory element. RoGalU1 only has 83% sequence identity to this mentioned protein of R. opacus PD630. Thus, it seems likely that the protein more related to our RoGalU2 is of importance for rhodococci, which will need to be verified by further studies and representatives. GalF of E. coli only has a slight residual UDP-glucose pyrophosphorylase activity. E. coli GalF showed a drastically increased activity and substrate specificity for glucose 1-phosphate when exchanging the methionine in position 15 into a threonine and the histidine in position 16 into an arginine (both within the G-X-G-T-R-F-P-L motif of GalUs), which are the corresponding residues in E. coli GalU [19]. In the sequences of RoGalU1 and RoGalU2, there is no alteration in the G-X-G-T-R-F-L-P motif. Thus, a regulatory role among the Rhodococcus proteins is excluded at this stage.
The underlying alignment of the distance tree shows that GalU enzymes in general are very conserved among bacteria, and that the degree of conservation within Actinobacteria is even higher. The first 100 amino acids are almost fully conserved with only some exceptions. As the two largest conserved areas of all shown sequences are around the motifs for substrate and product binding, it is obvious that the reaction mechanism is very likely the same in all bacterial GalU enzymes. It is striking that there are also four more motifs and several single amino acids which are highly conserved among bacterial GalU sequences. The tyrosine of the G-R-Y-L-L motif and the leucine in the Q-L-T-D-A motif are also mentioned by Aragão et al. to form a hydrophobic cap to the base of the sugar ring in the active site [9]. It could be possible that the other motifs and amino acids are not only important for the structure formation, like establishing the subunit interaction, but also for the construction of the active site, the binding of substrate and product or even the binding of magnesium ions as cofactors. They could help to stabilize the nucleophilic attack of glucose 1-phosphate to UTP or to bring the active site in the right conformation when the substrates are bound.
Hence, both genes RogalU1 and RogalU2 were successfully cloned into expression systems and protein production for a subsequent biochemical characterization was studied. It was not possible to produce active and soluble RoGalU1. In addition, the inclusion bodies obtained had only a low residual enzyme activity. Neither the optimization of medium or expression conditions nor the renaturation of wrongly folded protein lead to soluble and active RoGalU1. Inclusion bodies can be avoided or reduced by different methods. Reduction of the expression temperature to 14–16 °C or even lower, reduction of the expression time or ITPG concentration could possibly increase the amount of soluble active protein [54]. Also, co-expression of the gene of interest with chaperones, are reported to help folding the protein in the right way [55]. The production of a fusion protein with mCherry or other suitable proteins can increase the protein solubility as well [2]. The sole production of inclusion bodies of a GalU from M. tuberculosis could be overcome by using an expression system which was more related to the donor organism, so gene expression was achieved in competent M. smegmatis cells and the mycobacterial specific expression vector pMIP12 for improved gene expression [51].
RoGalU2 was successfully overproduced and purified in reasonable amounts compared to literature, but with a higher yield of insoluble recombinant RoGalU2, as well [13]. SDS-PAGE revealed the expected molecular weight of about 34 kDa. Within bacteria this molecular weight corresponds to that of other GalUs [12,13,56]. The oligomeric state of the protein was determined to be hexameric by size-exclusion chromatography. But the other known bacterial GalUs were shown to be dimers [10,31] or tetramers [8,9,12,17,30]. Only Lai et al. reported the GalU of M. tuberculosis strain H37Rv to be dimeric and hexameric in solution [13]. As the latter one is closely related to our target RoGalU2 this fits well to our findings (Figure 2). But we did observe a hexameric state under applied conditions, which can be studied in more detail by altering the buffer or presence of divalent ions through the size exclusion chromatography. Respectively, the presence of a protein in a single hydrodynamic state allowed us to study the biochemical properties in more detail.
RoGalU2 with a Vmax of 177 U mg−1 (UTP) and 119 U mg−1 (G1P), respectively, has a high activity compared to other bacterial GalUs. Many other bacterial GalUs show activities between 0.1 U mg−1 for M. tuberculosis GalU [13] and 90 U mg−1 for Xantomonas campestris [31]. Similar or slightly higher activities for bacterial GalUs are known as well, like 270 U mg−1 for Streptomyces coelicolor GalU [10] and 340 U mg−1 for E. coli GalU [19]. Higher activities are only known from eukaryotes [24,57,58]. RoGalU2 also showed a high apparent turnover frequency (app. kcat) with 96 s−1. But this value is very heterogeneous among bacteria, showing lower, similar and higher turnover frequencies [13,19,30]. An overview on bacterial GalU enzymes is provided in Table 2.
Table 2.
Kinetic properties of UDP-glucose pyrophosphorylases used for the phylogenetic analysis. Units are given in brackets in the table head, unless otherwise indicated.
| Organism and Protein | Vmax [U mg−1] | Km [mM] | kcat [s−1] | c [mM] | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| UTP | G1P | UTP | G1P | UTP | G1P | UTP | G1P | ||
| R. opacus 1CP GalU2 | 177 | 6.751 1192 |
0.51 | 0.91 1502 |
96.2 | 3.71 652 |
0.01–5 | 0.05–500 | This study |
| E. coli K-12 GalU | 340 | 0.17 | 0.035 | 187 | 2 | 2 | [19] | ||
| E. coli K-12 GalF | 0.015 | 0.36 | 0.52 | 0.008 | 2 | 2 | [19] | ||
| M. tuberculosis H37Rv GalU | 2.5–2.73 9.8 U4 |
5.8 U4 | 0.13 0.0124 |
0.133 0.0454 |
93.444 | 55.034 | 0.001–0.014 | 0.008–0.054 | [51,13] |
| E. amylovora CFBP 1430 GalU | 14.4 | 32.5 | 0.027 | 0.007 | 7.9 | 17.2 | 0.006–0.1 | 0.003–0.1 | [30] |
| S. coelicolor A3 (2) GalU | 270 | >10 | 0.06 | 149 | ≤20 | n.s. | [10] | ||
| B. bifidum PRL2010 GalU | 13 | 0.042 | 0.098 | 12 | 0–2 | 0–2 | [59] | ||
| X. capestris pv. campestris str. 8004 GalU | 60–90 | 0.21 | 0.06 | 40 | n.s. | 0-8 | [31] | ||
| X. axonopodis pv. citris str. 306 GalU | 60–90 | 0.11 | n.s. | 29 | n.s. | n.s. | [31] | ||
| S.thermophilus LY03 GalU | 0.2 | n.s. | n.s. | 0.11 | 1.25 | 1 | [32] | ||
The maximum activity was observed between 40 °C and 45 °C and the protein was stable up to 45 °C. The enzymatic activity drastically decreased after an incubation for 30 min at higher temperatures. Other GalUs have temperature optima around only 37 °C [60,61]. Only nucleotidyltransfrases of thermophilic bacteria and Archaea have a higher temperature optimum [62,63] and a half-life of 30 min at 95 °C [36]. However, RoGalU2 originates from a mesophilic Actinobacterium, R. opacus 1CP, which grows best at 30 °C. Thus, this temperature optimum was not expected for a metabolically relevant enzyme. Only two other enzymes of this strain showed a higher temperature activity or stability so far; a flavin-dependent monooxygenase [64] and membrane-linked isomerase [65].
The use of magnesium sulfate with respect to UDP-glucose pyrophosphorylases was not described in literature before. Here we show the definite dependency on magnesium ions and an activity increase to 180% when using sulfate as anion instead of chloride. The total electronegativity of sulfate is higher than of chloride, which could be advantageous for charge neutralization and coordination of the phosphoryl oxygen. This could then help binding UTP and implementing the nucleophilic attack, which is the base for the proposed reaction mechanism (Scheme 2) [17,18]. Another possibility is that the sulfate variant of magnesium salt forms a more stable complex with UTP than the chloride. As Kleczkowski already reported, those complexes could be the actual substrates for GalUs and that free UTP inhibits the reaction when present in too high concentrations [66]. This is in agreement to our kinetic study in which substrate inhibition was determined at higher UTP concentrations (Figure 9). Furthermore, this is supported by the results obtained by the variation of magnesium salt concentration. There the activity of RoGalU2 decreased rapidly when the concentration of the magnesium salt was higher than 1–2 mM. It is likely that the activity increases when both, the cofactor and UTP, are fed in equimolar concentrations. But an experimental set up will need to verify this hypothesis. Other groups did not report the occurrence of substrate inhibition. Here, we describe a substrate inhibition for both, glucose 1-phosphate and UTP. The only report showing a similar behavior of a GalU enzyme at increasing glucose 1-phosphate concentrations is for the enzyme from potato by Gupta and colleagues [57]. But it has to be noted that the tested glucose 1-phosphate concentration range was quite small (0.05–1 mM) and the activity still increased at 1 mM [57].
GalUs show activities with the same divalent metal ions that were tested with RoGalU2 and also the same degree of inhibition by EDTA [12,36,63,67]. Thus, a clear preference for Mg2+ is demonstrated for all GalU enzymes.
The literature reported pH range tolerated by GalUs is huge, between pH 5.5 and pH 10.0, but the optimum is often found around pH 7.5 [13,36,63,67]. RoGalU2 behaves expectedly with a pH optimum of around 7.5, depending on the buffer. However, it maintains a comparable high activity up to a pH of about 9 and thus behaves as many GalU enzymes.
RoGalU2 accepts UTP and dTTP as nucleotides, as well as glucose 1-phosphate and galactose 1-phosphate as sugar phosphates. Substrate promiscuity is known for some GalUs, whereas some are very specific [39]. Indeed, it has been reported that GalUs from S. coelicolor, Salmonella enterica, Sphingomonas elodea, and E. coli are able to accept both UTP and dTTP as substrates [10,40,68,69], whereas for example the GalU from Helicobacter pylori is specific only for UDP [17]. It has been shown for H. pylori that configuration of the active site prevents a thymine from binding due to steric clashes with a methionine residue (M105) [17]. Superposition of a RoGalU2 homology model, produced using SWISS-MODEL [70,71], with the crystal structure from H. pylori in complex with UDP-glucose (pdbID 3JUK, [17]) shows that RoGalU2 does not have a methionine but a proline residue in the designated position. This observation is similar to what has been reported for S. elodea and E. coli GalUs having a proline and an alanine, respectively, at that position and also being active towards dTTP [39]. Furthermore, RoGalU2 is also able to take galactose 1-phosphate as a substrate, but the enzyme is less active (only 2% residual activity), as was observed for S. elodea and E. coli GalUs [30]. It has been suggested that this less favorable binding of galactose 1-phosphate is due to the loss of the H-bond formed between the glucose 1-phosphate and the main-chain nitrogen atom of a glycine residue (Gly179 E. coli and Gly180 in RoGalU2 homology model) [39].
Having all this in mind and the optimal conditions for RoGalU2 experimentally verified, we could use this knowledge to obtain a maximum observed activity value for this UDP-glucose pyrophosphorylases in the direction of UDP-glucose formation (Figure 11). Under optimal conditions the activity of RoGalU2 was 270 U mg−1, which is an increase in activity of about 37%. Thus this enzyme is among the most active GalUs of bacteria and might be interesting to be studied for various biotechnological applications described recently [72].
Figure 11.
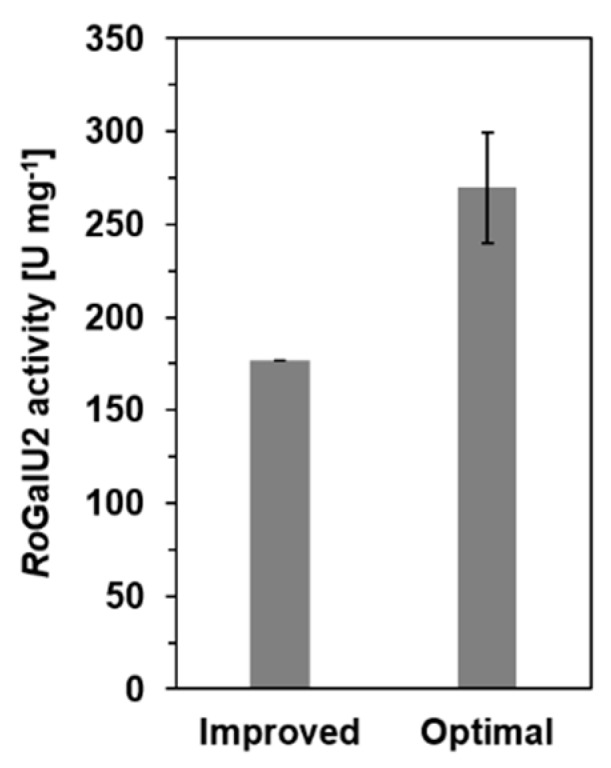
RoGalU2 activity of the improved activity assay compared to an assay with optimal reaction conditions. Reaction solution for improved test contained 1.5 mM UTP, 250 mM glucose 1-phosphate, 1 mM MgCl2, 50 mM Hepes, pH 7.4, 1.7 µg RoGalU2. Reaction was carried out at 42 °C in 1 mL scale. Reaction solution for optimal test contained 1.5 mM UTP, 250 mM glucose 1-P, 1 mM MgSO4, 50 mM Imidazol, pH 7.4, 1.7 µg RoGalU2. Reaction was carried out 43 °C in 1 mL scale. Means with standard deviations are shown (triplicates).
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Gene Synthesis
Protein sequences of the UDP-glucose pyrophosphorylases RoGalU1 and RoGalU2 of Rhodococcus opacus 1CP were taken from the NCBI accessions ANS26426 (RoGalU1) and ANS26629 (RoGalU2), respectively. The corresponding genes RogalU1 (914 bp) and RogalU2 (932 bp) used in this study were codon usage optimized to increase the expression level in E. coli and synthesized by Eurofins Genomics (Ebersberg, Germany) with flanking restriction sites of NdeI and NotI (GenBank accession numbers of the codon usage optimized nucleotide sequences of RogalU1: MN617759 and RogalU2: MN617760). Both genes were delivered in separate vectors (pEX-A2). They were cloned into the expression vector pET16bP (5740 bp) carrying a resistance against ampicillin, a DNA sequence that allowed the production of the GalU proteins with an N-terminal Histidine10-tag and an additional DNA sequence for the gene expression induction with isopropyl β-D-1-thiogalactopyranoside (IPTG) (see Table 3).
Table 3.
Strains, plasmids, and primers used in this study.
| Sample | Relevant Characteristics | Source, Reference |
|---|---|---|
| Strains | ||
| R. opacus 1 CP | Benzoate+, 4-hydroxybenzoate+, 3-chlorobenzoate+, phenol+, 4-chlorophenol+, 2,4-dichlorophenol+, 2-chlorophenol+, 3-methylphenol+, 4-methylphenol+, phthalate+, isophthalate+, n-alkanes+ (C10-C16), styrene+ | [73,64] |
| E. coli DH5α | fhuA2 Δ(argF-lacZ) U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | NEB 1 |
| E. coli BL21(DE3) pLysS | fhuA2 [lon] ompT gal (λ DE3) [dcm] ∆hsdS λ DE3 = λ sBamHIo ∆EcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5 | NEB 1 |
| Plasmids | ||
| pEX-A2 | multiple cloning site, Lac-Promoter, pUC origin, Ampr | Eurofins 2 |
| pET16bP | pET16b with additional multiple cloning site; allows production of recombinant proteins with N-terminal Histidine10-Tag and gene expression induction with IPTG | Wehmeyer 3 |
| pEX-A2-RogalU1 | pEX-A2 vector with recombinant UDP-glucose-pyrophosphorylase gene 1 of R. opacus 1 CP (RogalU1) 914 bp, NCBI protein accession: ANS26426 | This study |
| pEX-A2-RogalU2 | pEX-A2 vector with recombinant UDP-glucose-pyrophosphorylase gene 2 of R. opacus 1 CP (RogalU2) 932 bp, NCBI protein accession: ANS26629 | This study |
| pET16bP-RogalU1 | pET16bP vector with recombinant UDP-glucose-pyrophosphorylase gene 1 of R. opacus 1 CP (RogalU1) | This study |
| pET16bP-RogalU2 | pET16bP vector with recombinant UDP-glucose-pyrophosphorylase gene 2 of R. opacus 1 CP (RogalU2) | This study |
| Primer | ||
| pET16bP-fw | 5’ - CATCACAGCAGCGGCCATATCGAAG - 3’ | This study |
| pET16bP-rev | 5’ - CAGCTTCTTTTCGGGCTTTGTTAG - 3’ | This study |
1 New England Biolabs Inc. or 2 Eurofins Genomics as a commercial source. 3 personal communication by U. Wehmeyer.
4.2. Protein Production
Transformation of E. coli BL21(DE3) pLysS was carried out as recommended by New England Biolabs Inc. (Ipswich, Massachusetts, USA).
Protein production was realized in a 1 L scale in a Fernbach flask in standard LB medium (lysogenic broth: 10 g L−1 trypton, 5 g L−1 yeast extract, 10 g L−1 sodium chloride) with 100 mg L−1 ampicillin as well as 50 mg L−1 chloramphenicol. Expression cultures were inoculated 1:50 with an overnight pre-culture at 37 °C of E. coli BL21(DE3) pLysS-pET16bP-RogalU1 or E. coli BL21(DE3) pLysS-pET16bP-RogalU2 in the same medium used for the expression culture. The main culture was incubated for about 2 h at 37 °C until an OD600 of 0.2–0.3 could be observed. After cooling down to 20 °C, the gene expression and thus protein production started after induction with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at an OD600 of 0.3–0.4.
The large-scale protein production was performed in an analog manner in a 10 L Eppendorf bioreactor in TB autoinduction medium [74] with the same concentrations of antibiotics and inoculation culture as described above.
After 22 h the protein production was stopped in both cases by harvesting the cells at 4 °C and 5000× g for 30 min. After washing with 25 mM sodium phosphate buffer, pH 7.1, pelleted cells were frozen and stored at −80 °C in portions of about 25 g of wet biomass.
4.3. Purification of Recombinant Proteins
For purification 25 g of pelleted cells were thawed as fast as possible in a warm water bath and mixed gently with 25 mL of a buffer containing the following compounds: 25 mM sodium phosphate buffer, pH 7.1, 100 mM sodium chloride, 1 mM magnesium chloride, 240 U of DNase I and 20 mg of lysozyme. After incubation at 30 °C for 45 to 60 min the cells were sonicated 10 times for 30 s with an intensity of 70% (Bandelin Sonoplus HD 2070, MS 72) and centrifuged at 12,000× g at 4 °C for 20 min. Another two centrifugation steps with the supernatant followed at 4 °C, 50,000× g for 30 min, each. The clear supernatant was filtered through 0.45 µm and 0.2 µm filter, respectively, before purification with an Äkta Prime Plus FPLC system with 5 mL HisTrap HP nickel column (GE Healthcare) with a flow of 5 mL min−1. For equilibration of the affinity chromatography column a buffer containing 25 mM sodium phosphate buffer, pH 7.1, 300 mM sodium chloride and 25 mM imidazole was used. Protein loading was performed with equilibration buffer containing 25 mM imidazole. Unspecific proteins were washed off the column with equilibration buffer containing 40 mM imidazole. Protein purification was then performed by applying a linear gradient from 40–500 mM imidazole in this buffer in a course of total 10–20 mL elution, depending on the injection volume. The protein of interest was eluted at 500 mM imidazole.
Fractions that showed UDP-glucose pyrophosphorylase activity were pooled and precipitated with 80% saturated ammonium sulfate solution. The precipitated protein was dissolved and stored in a buffer of 50 mM Hepes, pH 7.0, 100 mM sodium chloride and 1 mM magnesium chloride. Protein aliquots were stored at –80 °C for long term storage or at 4 °C for short term storage and had concentrations of about 2–8 mg mL−1.
Here, it needs to be mentioned that RoGalU2 has a very low extinction coefficient due to the low amount of aromatic amino acids in the polypeptide chain and thus purifying by following the UV/VIS trace was somewhat difficult.
4.4. Renaturation of RoGalU1
Isolation of inclusion bodies and renaturation of the wrongly folded RoGalU1 was realized with the Thermo Scientific Pierce Protein Refolding Kit according to the manufacturer’s instruction, but without any EDTA in the buffers or solutions. The RoGalU1 concentrations for the renaturation were 1 mg mL−1 and 10 mg mL−1, respectively.
4.5. Protein Determination
Determination of the protein concentration was done as described before by Bradford [75] with a bovine serum albumin (BSA) standard. Identification of the proteins was performed by determination of the molecular weight with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and coomassie staining [76]. Furthermore, the oligomeric state of the proteins was determined by size exclusion chromatography using a 24 mL Superdex 200 10/300 GL column (GE Healthcare), the manufacturers protocol and a calibration standard mix containing Ferritin, Conalbumin, Carbonic Anhydrase, Ribonuclease, and Aprotinin. Dextran Blue was used to determine the void volume of the size exclusion column. The buffer that was used for this method contained 50 mM Hepes, pH 7.0 and 1 mM magnesium chloride and was the same as used for the most characterization experiments.
4.6. Enzyme Activity Assay
The specific enzyme activity of RoGalU1 and RoGalU2 was measured only in the direction of UDP-glucose formation in a reaction volume of 1 mL. The following assay composition was used in this work mostly and thus designated as standard test. The reaction solution for initial activity measurements contained 2 mM UTP, 2 mM glucose 1-phosphate, 4 mM magnesium chloride, and 50 mM Hepes, pH 7.0. Pre-incubation of the reaction samples was done for 15 min at 30 °C and the reaction was started by addition of 30 µg of enzyme, unless otherwise indicated. Samples of 100 µL were taken after defined time points to determine the initial reaction rates. Therefore, the reaction was stopped by adding 100 µL acetonitrile and vortexing in order to denature proteins. After centrifugation for 2 min at 20,000× g 100 µL of the clear supernatant were used for HPLC analysis.
In order to characterize GalU enzymes the above described standard assay was altered with respect to buffer, pH, temperature and various other additives during experimentation and to find optimal reaction conditions.
The enzyme activity is expressed as U mg−1. Respectively, 1 U corresponds to the formation of 1 µmol UDP-glucose per minute. This value is then referred to the amount of enzyme in the assay.
4.7. Product Determination by HPLC for Specific Enzyme Activity Evaluation
HPLC measurement was performed with a Thermo Scientific Dionex Ultimate 3000 with UV/VIS detector and Macherey-Nagel EC 150/4.6 Nucleoshell HILIC column with a particle size of 2.7 µm. For determination of UDP-glucose formation an isocratic chromatography program was used with 70% acetonitrile and 30% 134 mM ammonium acetate, pH 5.35 at a flow of 1.3 mL min−1 and an oven temperature of 30 °C. Detection of UDP-glucose was done at a wave length of 260 nm with an injection volume of 5 µL. Calibration was carried out with appropriate concentrations of a UDP-glucose standard under the same conditions.
For data evaluation and calculation of product formation the area of the UDP-glucose peak was used and referred to the time point of the enzyme assay to obtain an initial rate. Those rates were plotted according to the enzyme kinetic models for analysis.
The following two equations according to Michaelis–Menten (1) and Yano und Koga [50] (2);
| (1) |
| (2) |
have been used. Equation (1) was used for non-limiting conditions and 2 for the cases of substrate inhibition.
5. Conclusions
The biochemical characterization of UDP-glucose pyrophosphorylases has been reported for several organisms. Here, we described the first characterization of a UDP-glucose pyrophosphorylase from a Rhodococcus strain that contained two isogenes coding for GalUs. Based on phylogenetic analyses and activity data obtained it seems obvious that RoGalU2 represents the metabolically active enzyme, whereas the role of RoGalU1 remains enigmatic and needs to be investigated. Furthermore, the activity of RoGalU2 is higher than that of some other reported GalUs, but the use of conventional kinetic models is limited. Therefore, further biochemical investigation will be necessary. In addition, this is the first report of the use of magnesium sulfate as metal cofactor. The sulfate salt of magnesium was able to double the activity of RoGalU2. A maximum activity of RoGalU2 of about 270 U mg−1 was determined which renders it a candidate for further biocatalytic investigations.
Acknowledgments
We thank the collaborator of the ERA-IB project No-P for fruitful discussion and among those especially Katarzyna Szymańska, Andrzej Jarzębski, Daria Kowalczykiewicz and Marta Przypis from the Silesian University of Technology (Poland), Ulf Hanefeld, Peter-Leon Hagedoorn and Luuk Mestrom from the TU Delft (The Netherlands), as well as Arkadiusz Chruściel from MEXEO (Poland) and Rob Schoevaart from Chiral Vision (The Netherlands). In addition, we thank Daniel Eggerichs and Carolin Mügge for graphical support and constructively reviewing.
Author Contributions
Conceptualization, A.K., D.T. and I.B.; methodology, A.K., A.P. (Anett Partzsch) and A.P. (André Pollender); formal analysis, A.K. and A.P. (André Pollender); investigation, A.K. and A.P. (Anett Partzsch); data curation, A.K.; writing—original draft preparation, A.K. and D.T.; writing—review and editing, all authors.; visualization, A.K.; funding acquisition, D.T.
Funding
This research was funded by European Union and Saxonian Government, grant number 100263899 related to the ERA-IB project No-P, grant ERA-IB-15-110. The APC was partially funded by Ruhr-Universität Bochum.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- 1.Gutmann A., Nidetzky B. Unlocking the potential of Leloir glycosyltransferases for applied biocatalysis: Efficient synthesis of Uridine 5’-diphosphate-glucose by sucrose synthase. Adv. Synth. Catal. 2016;358:3600–3609. doi: 10.1002/adsc.201600754. [DOI] [Google Scholar]
- 2.Mestrom L., Marsden S.R., Dieters M., Achterberg P., Stolk L., Bento I., Hanefeld U., Hagedoorn P.-L. Artificial fusion of mCherry enhances trehalose transferase solubility and stability. Appl. Environ. Microbiol. 2019:85. doi: 10.1128/AEM.03084-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmölzer K., Lemmerer M., Gutmann A., Nidetzky B. Integrated process design for biocatalytic synthesis by a Leloir glycosyltransferase: UDP-glucose production with sucrose synthase. Biotechnol. Bioeng. 2017;114:924–928. doi: 10.1002/bit.26204. [DOI] [PubMed] [Google Scholar]
- 4.Bungaruang L., Gutmann A., Nidetzky B. Leloir glycosyltransferases and natural product glycosylation: Biocatalytic synthesis of the C-glucoside Nothofagin, a major antioxidant of redbush herbal tea. Adv. Synth. Catal. 2013;355:2757–2763. doi: 10.1002/adsc.201300251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai L., Li J., Yao P., Zhu Y., Men Y., Zeng Y., Yang J., Sun Y. Exploiting the aglycon promiscuity of glycosyltransferase Bs-YjiC from Bacillus subtilis and its application in synthesis of glycosides. J. Biotechnol. 2017;248:69–76. doi: 10.1016/j.jbiotec.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Diricks M., Gutmann A., Debacker S., Dewitte G., Nidetzky B., Desmet T. Sequence determinants of nucleotide binding in sucrose synthase: Improving the affinity of a bacterial sucrose synthase for UDP by introducing plant residues. Protein Eng. Des. Sel. 2017;30:141–148. doi: 10.1093/protein/gzw048. [DOI] [PubMed] [Google Scholar]
- 7.Schmölzer K., Gutmann A., Diricks M., Desmet T., Nidetzky B. Sucrose synthase: A unique glycosyltransferase for biocatalytic glycosylation process development. Biotechnol. Adv. 2016;34:88–111. doi: 10.1016/j.biotechadv.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Thoden J.B., Holden H.M. The molecular architecture of glucose-1-phosphate uridylyltransferase. Protein Sci. 2007;16:432–440. doi: 10.1110/ps.062626007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aragão D., Fialho A.M., Marques A.R., Mitchell E.P., Sa-Correia I., Frazao C. The complex of Sphingomonas elodea ATCC 31461 glucose-1-phosphate uridylyltransferase with glucose-1-phosphate reveals a novel quaternary structure, unique among nucleoside diphosphate-sugar pyrophosphorylase members. J. Bacteriol. 2007;189:4520–4528. doi: 10.1128/JB.00277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asención Diez M.D., Peirú S., Demonte A.M., Gramajo H., Iglesias A.A. Characterization of recombinant UDP- and ADP-glucose pyrophosphorylases and glycogen synthase to elucidate glucose-1-phosphate partitioning into oligo- and polysaccharides in Streptomyces coelicolor. J. Bacteriol. 2012;194:1485–1493. doi: 10.1128/JB.06377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ebrecht A.C., Asencion Diez M.D., Piattoni C.V., Guerrero S.A., Iglesias A.A. The UDP-glucose pyrophosphorylase from Giardia lamblia is redox regulated and exhibits promiscuity to use galactose-1-phosphate. Biochim. Biophys. Acta. 2015;1850:88–96. doi: 10.1016/j.bbagen.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Koo H.M., Yim S.-W., Lee C.-S., Pyun Y.R., Kim Y.S. Cloning, sequencing, and expression of UDP-glucose pyrophosphorylase gene from Acetobacter xylinum BRC5. Biosci. Biotechnol. Biochem. 2014;64:523–529. doi: 10.1271/bbb.64.523. [DOI] [PubMed] [Google Scholar]
- 13.Lai X., Wu J., Chen S., Zhang X., Wang H. Expression, purification, and characterization of a functionally active Mycobacterium tuberculosis UDP-glucose pyrophosphorylase. Protein Expr. Purif. 2008;61:50–56. doi: 10.1016/j.pep.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Meng M., Wilczynska M., Kleczkowski L.A. Molecular and kinetic characterization of two UDP-glucose pyrophosphorylases, products of distinct genes, from Arabidopsis. Biochim. Biophys. Acta. 2008;1784:967–972. doi: 10.1016/j.bbapap.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 15.Okazaki Y., Shimojima M., Sawada Y., Toyooka K., Narisawa T., Mochida K., Tanaka H., Matsuda F., Hirai A., Hirai M.Y., et al. A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell. 2009;21:892–909. doi: 10.1105/tpc.108.063925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roeben A., Plitzko J.M., Korner R., Bottcher U.M., Siegers K., Hayer-Hartl M., Bracher A. Structural basis for subunit assembly in UDP-glucose pyrophosphorylase from Saccharomyces cerevisiae. J. Mol. Biol. 2006;364:551–560. doi: 10.1016/j.jmb.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 17.Kim H., Choi J., Kim T., Lokanath N.K., Ha S.C., Suh S.W., Hwang H.-Y., Kim K.K. Structural basis for the reaction mechanism of UDP-glucose pyrophosphorylase. Mol. Cells. 2010;29:397–405. doi: 10.1007/s10059-010-0047-6. [DOI] [PubMed] [Google Scholar]
- 18.Thoden J.B., Holden H.M. Active site geometry of glucose-1-phosphate uridylyltransferase. Protein Sci. 2007;16:1379–1388. doi: 10.1110/ps.072864707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebrecht A.C., Orlof A.M., Sasoni N., Figueroa C.M., Iglesias A.A., Ballicora M.A. On the Ancestral UDP-Glucose Pyrophosphorylase Activity of GalF from Escherichia coli. Front. Microbiol. 2015;6:1253. doi: 10.3389/fmicb.2015.01253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balan D., Tokas J., Singal H.R. UDP-glucose pyrophosphorylase: Isolation, purification and characterization from developing thermotolerant wheat (Triticum aestivum) grains. Protein Expr. Purif. 2018;148:68–77. doi: 10.1016/j.pep.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Chen R., Zhao X., Shao Z., Zhu L., He G. Multiple isoforms of UDP-glucose pyrophosphorylase in rice. Physiol Plant. 2007;129:725–736. doi: 10.1111/j.1399-3054.2007.00865.x. [DOI] [Google Scholar]
- 22.Cotrim C.A., Soares J.S.M., Kobe B., Menossi M. Crystal structure and insights into the oligomeric state of UDP-glucose pyrophosphorylase from sugarcane. PLoS ONE. 2018;13:e0193667. doi: 10.1371/journal.pone.0193667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCoy J.G., Bitto E., Bingman C.A., Wesenberg G.E., Bannen R.M., Kondrashov D.A., Phillips G.N., Jr. Structure and dynamics of UDP-glucose pyrophosphorylase from Arabidopsis thaliana with bound UDP-glucose and UTP. J. Mol. Biol. 2007;366:830–841. doi: 10.1016/j.jmb.2006.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng M., Fitzek E., Gajowniczek A., Wilczynska M., Kleczkowski L.A. Domain-specific determinants of catalysis/substrate binding and the oligomerization status of barley UDP-glucose pyrophosphorylase. Biochim. Biophys. Acta. 2009;1794:1734–1742. doi: 10.1016/j.bbapap.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Dutta S.K. UDPglucose pyrophosphorylase from Ehrlich ascites carcinoma cell-purification and characterization. Indian J. Biochem. Biophys. 1985;22:203–207. [PubMed] [Google Scholar]
- 26.Granzow C., Kopun M., Zimmermann H.P. Role of nuclear glycogen synthase and cytoplasmic UDP glucose pyrophosphorylase in the biosynthesis of nuclear glycogen in HD33 Ehrlich-Lettré ascites tumor cells. J. Cell Biol. 1981;89:475–484. doi: 10.1083/jcb.89.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds T.H., Pak Y., Harris T.E., Manchester J., Barrett E.J., Lawrence J.C. Effects of insulin and transgenic overexpression of UDP-glucose pyrophosphorylase on UDP-glucose and glycogen accumulation in skeletal muscle fibers. J. Biol. Chem. 2005;280:5510–5515. doi: 10.1074/jbc.M413614200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Führing J.I., Cramer J.T., Schneider J., Baruch P., Gerardy-Schahn R., Fedorov R. A quaternary mechanism enables the complex biological functions of octameric human UDP-glucose pyrophosphorylase, a key enzyme in cell metabolism. Sci. Rep. 2015;5:9618. doi: 10.1038/srep09618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dickmanns A., Damerow S., Neumann P., Schulz E.-C., Lamerz A.-C., Routier F.H., Ficner R. Structural basis for the broad substrate range of the UDP-sugar pyrophosphorylase from Leishmania major. J. Mol. Biol. 2011;405:461–478. doi: 10.1016/j.jmb.2010.10.057. [DOI] [PubMed] [Google Scholar]
- 30.Benini S., Toccafondi M., Rejzek M., Musiani F., Wagstaff B.A., Wuerges J., Cianci M., Field R.A. Glucose-1-phosphate uridylyltransferase from Erwinia amylovora: Activity, structure and substrate specificity. Biochim. Biophys. Acta Proteins Proteom. 2017;1865:1348–1357. doi: 10.1016/j.bbapap.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 31.Bosco M.B., Machtey M., Iglesias A.A., Aleanzi M. UDPglucose pyrophosphorylase from Xanthomonas spp. Characterization of the enzyme kinetics, structure and inactivation related to oligomeric dissociation. Biochimie. 2009;91:204–213. doi: 10.1016/j.biochi.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Degeest B., de Vuyst L. Correlation of activities of the enzymes alpha-phosphoglucomutase, UDP-galactose 4-epimerase, and UDP-glucose pyrophosphorylase with exopolysaccharide biosynthesis by Streptococcus thermophilus LY03. Appl. Environ. Microbiol. 2000;66:3519–3527. doi: 10.1128/AEM.66.8.3519-3527.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Padilla L., Morbach S., Krämer R., Agosin E. Impact of heterologous expression of Escherichia coli UDP-glucose pyrophosphorylase on trehalose and glycogen synthesis in Corynebacterium glutamicum. Appl. Environ. Microbiol. 2004;70:3845–3854. doi: 10.1128/AEM.70.7.3845-3854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toccafondi M., Cianci M., Benini S. Expression, purification, crystallization and preliminary X-ray analysis of glucose-1-phosphate uridylyltransferase (GalU) from Erwinia amylovora. Acta Crystallogr. F Struct. Biol. Commun. 2014;70:1249–1251. doi: 10.1107/S2053230X14016458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawano Y., Sekine M., Ihara M. Identification and characterization of UDP-glucose pyrophosphorylase in cyanobacteria Anabaena sp. PCC 7120. J. Biosci. Bioeng. 2014;117:531–538. doi: 10.1016/j.jbiosc.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Kim J.S., Koh S., Shin H.J., Lee D.S., Lee S.Y. Biochemical characterization of a UDP-sugar pyrophosphorylase from Thermus caldophilus GK24. (Pt 1)Biotechnol. Appl. Biochem. 1999;29:11–17. [PubMed] [Google Scholar]
- 37.Holden H.M., Rayment I., Thoden J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem. 2003;278:43885–43888. doi: 10.1074/jbc.R300025200. [DOI] [PubMed] [Google Scholar]
- 38.Marques A.R., Ferreira P.B., Sá-Correia I., Fialho A.M. Characterization of the ugpG gene encoding a UDP-glucose pyrophosphorylase from the gellan gum producer Sphingomonas paucimobilis ATCC 31461. Mol. Genet. Genomics. 2003;268:816–824. doi: 10.1007/s00438-003-0805-7. [DOI] [PubMed] [Google Scholar]
- 39.Berbis M., Sanchez-Puelles J., Canada F., Jimenez-Barbero J. Structure and function of prokaryotic UDP-glucose pyrophosphorylase, a drug target candidate. CMC. 2015;22:1687–1697. doi: 10.2174/0929867322666150114151248. [DOI] [PubMed] [Google Scholar]
- 40.Silva E., Marques A.R., Fialho A.M., Granja A.T., Sá-Correia I. Proteins encoded by Sphingomonas elodea ATCC 31461 rmlA and ugpG genes, involved in gellan gum biosynthesis, exhibit both dTDP- and UDP-glucose pyrophosphorylase activities. Appl. Environ. Microbiol. 2005;71:4703–4712. doi: 10.1128/AEM.71.8.4703-4712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gröning J.A.D., Eulberg D., Tischler D., Kaschabek S.R., Schlömann M. Gene redundancy of two-component (chloro)phenol hydroxylases in Rhodococcus opacus 1CP. FEMS Microbiol. Lett. 2014;361:68–75. doi: 10.1111/1574-6968.12616. [DOI] [PubMed] [Google Scholar]
- 42.Letek M., González P., Macarthur I., Rodríguez H., Freeman T.C., Valero-Rello A., Blanco M., Buckley T., Cherevach I., Fahey R., et al. The genome of a pathogenic Rhodococcus: Cooptive virulence underpinned by key gene acquisitions. PLoS Genet. 2010;6:e1001145. doi: 10.1371/journal.pgen.1001145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizanur R.M., Pohl N.L. A thermostable promiscuous glucose-1-phosphate uridyltransferase from Helicobacter pylori for the synthesis of nucleotide sugars. J. Mol. Catal. B Enzymatic. 2008;50:13–19. doi: 10.1016/j.molcatb.2007.09.018. [DOI] [Google Scholar]
- 44.Tischler D., Gröning J.A.D., Kaschabek S.R., Schlömann M. One-component styrene monooxygenases: An evolutionary view on a rare class of flavoproteins. Appl. Biochem. Biotechnol. 2012;167:931–944. doi: 10.1007/s12010-012-9659-y. [DOI] [PubMed] [Google Scholar]
- 45.Edgar R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edgar R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320-4. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones D.T., Taylor W.R., Thornton J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 50.Yano T., Koga S. Dynamic behavior of the chemostat subject to substrate inhibition. Biotechnol. Bioeng. 1969;11:139–153. doi: 10.1002/bit.260110204. [DOI] [Google Scholar]
- 51.Asención Diez M.D., Demonte A.M., Syson K., Arias D.G., Gorelik A., Guerrero S.A., Bornemann S., Iglesias A.A. Allosteric regulation of the partitioning of glucose-1-phosphate between glycogen and trehalose biosynthesis in Mycobacterium tuberculosis. Biochim. Biophys. Acta. 2015;1850:13–21. doi: 10.1016/j.bbagen.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patil H.I., Pratap A.P. Production and quantitative analysis of trehalose lipid biosurfactants using high-performance liquid chromatography. J. Surfactants Deterg. 2018;21:553–564. doi: 10.1002/jsde.12158. [DOI] [Google Scholar]
- 53.Tischler D., Niescher S., Kaschabek S.R., Schlömann M. Trehalose phosphate synthases OtsA1 and OtsA2 of Rhodococcus opacus 1CP. FEMS Microbiol. Lett. 2013;342:113–122. doi: 10.1111/1574-6968.12096. [DOI] [PubMed] [Google Scholar]
- 54.Pacheco B., Crombet L., Loppnau P., Cossar D. A screening strategy for heterologous protein expression in Escherichia coli with the highest return of investment. Protein Expr. Purif. 2012;81:33–41. doi: 10.1016/j.pep.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 55.Strandberg L., Enfors S.O. Factors influencing inclusion body formation in the production of a fused protein in Escherichia coli. Appl. Environ. Microbiol. 1991;57:1669–1674. doi: 10.1128/aem.57.6.1669-1674.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aragão D., Marques A.R., Frazão C., Enguita F.J., Carrondo M.A., Fialho A.M., Sá-Correia I., Mitchell E.P. Cloning, expression, purification, crystallization and preliminary structure determination of glucose-1-phosphate uridylyltransferase (UgpG) from Sphingomonas elodea ATCC 31461 bound to glucose-1-phosphate. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006;62:930–934. doi: 10.1107/S1744309106030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gupta S.K., Sowokinos J.R., Hahn I.-S. Regulation of UDP-glucose pyrophosphorylase isozyme UGP5 associated with cold-sweetening resistance in potatoes. J. Plant Physiol. 2008;165:679–690. doi: 10.1016/j.jplph.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 58.Steiner T., Lamerz A.C., Hess P., Breithaupt C., Krapp S., Bourenkov G., Huber R., Gerardy-Schahn R., Jacob U. Open and closed structures of the UDP-glucose pyrophosphorylase from Leishmania major. J. Biol. Chem. 2007;282:13003–13010. doi: 10.1074/jbc.M609984200. [DOI] [PubMed] [Google Scholar]
- 59.De Bruyn F., Beauprez J., Maertens J., Soetaert W., de Mey M. Unraveling the Leloir pathway of Bifidobacterium bifidum: Significance of the uridylyltransferases. Appl. Environ. Microbiol. 2013;79:7028–7035. doi: 10.1128/AEM.02460-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Z., Fan H.-j., Lu C.-p. Molecular cloning and analysis of the UDP-glucose pyrophosphorylase in Streptococcus equi subsp. zooepidemicus. Mol. Biol. Rep. 2011;38:2751–2760. doi: 10.1007/s11033-010-0420-8. [DOI] [PubMed] [Google Scholar]
- 61.Weissborn A.C., Liu Q., Rumley M.K., Kennedy E.P. UTP: Alpha-D-glucose-1-phosphate uridylyltransferase of Escherichia coli: isolation and DNA sequence of the galU gene and purification of the enzyme. J. Bacteriol. 1994;176:2611–2618. doi: 10.1128/jb.176.9.2611-2618.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Honda Y., Zang Q., Shimizu Y., Dadashipour M., Zhang Z., Kawarabayasi Y. Increasing the thermostable sugar-1-phosphate nucleotidylyltransferase activities of the archaeal ST0452 protein through site saturation mutagenesis of the 97th amino acid position. Appl. Environ. Microbiol. 2017:83. doi: 10.1128/AEM.02291-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Z., Tsujimura M., Akutsu J.-i., Sasaki M., Tajima H., Kawarabayasi Y. Identification of an extremely thermostable enzyme with dual sugar-1-phosphate nucleotidylyltransferase activities from an acidothermophilic archaeon, Sulfolobus tokodaii strain 7. J. Biol. Chem. 2005;280:9698–9705. doi: 10.1074/jbc.M411211200. [DOI] [PubMed] [Google Scholar]
- 64.Tischler D., Eulberg D., Lakner S., Kaschabek S.R., van Berkel W.J.H., Schlömann M. Identification of a novel self-sufficient styrene monooxygenase from Rhodococcus opacus 1CP. J. Bacteriol. 2009;191:4996–5009. doi: 10.1128/JB.00307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oelschlägel M., Gröning J.A.D., Tischler D., Kaschabek S.R., Schlömann M. Styrene oxide isomerase of Rhodococcus opacus 1CP, a highly stable and considerably active enzyme. Appl. Environ. Microbiol. 2012;78:4330–4337. doi: 10.1128/AEM.07641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kleczkowski L.A. Glucose activation and metabolism through UDP-glucose pyrophosphorylase in plants. Phytochemistry. 1994;37:1507–1515. doi: 10.1016/S0031-9422(00)89568-0. [DOI] [Google Scholar]
- 67.Li Q., Huang Y.-Y., Conway L.P., He M., Wei S., Huang K., Duan X.-C., Flitsch S.L., Voglmeir J. Discovery and biochemical characterization of a thermostable glucose-1-phosphate nucleotidyltransferase from Thermodesulfatator indicus. Protein Pept. Lett. 2017;24:729–734. doi: 10.2174/0929866524666170724110408. [DOI] [PubMed] [Google Scholar]
- 68.Bernstein R.L., Robbind P.W. Control aspects of Uridine 5’-diphosphate glucose and Thymidine 5’-diphosphate glucose synthesis by microbial enzymes. J. Biol. Chem. 1965;240:391–397. [PubMed] [Google Scholar]
- 69.Lindquist L., Kaiser R., Reeves P.R., Lindberg A.A. Purification, characterization and HPLC assay of Salmonella glucose-1-phosphate thymidylyltransferase from the cloned rfbA gene. Eur. J. Biochem. 1993;211:763–770. doi: 10.1111/j.1432-1033.1993.tb17607.x. [DOI] [PubMed] [Google Scholar]
- 70.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W293–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumpf A., Tischler D. Structural investigations of GalU enzymes from Actinobacteria, Crystals. Unpublished work.
- 72.Mestrom L., Przypis M., Kowalczykiewicz D., Pollender A., Kumpf A., Marsden S.R., Bento I., Jarzębski A.B., Szymańska K., Chruściel A., et al. Leloir glycosyltransferases in applied biocatalysis: A multidisciplinary approach. Int. J. Mol. Sci. 2019;20:5263. doi: 10.3390/ijms20215263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moiseeva O.V., Solyanikova I.P., Kaschabek S.R., Gröning J., Thiel M., Golovleva L.A., Schlömann M. A new modified ortho cleavage pathway of 3-chlorocatechol degradation by Rhodococcus opacus 1CP: Genetic and biochemical evidence. J. Bacteriol. 2002;184:5282–5292. doi: 10.1128/JB.184.19.5282-5292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Studier F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 75.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 76.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]