

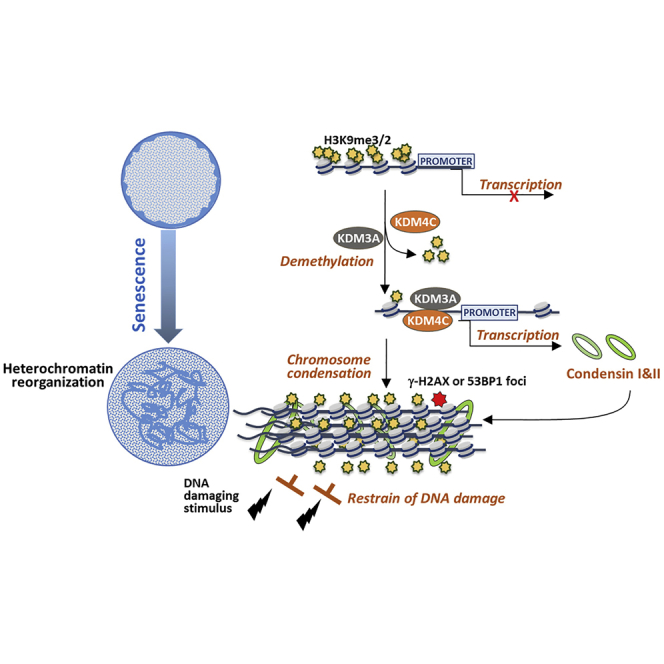
Summary
Epigenomic changes and stem cell deterioration are two hallmarks of aging. Accumulating evidence suggest that senescence of mesenchymal stromal cells (MSCs) perpetuates aging or age-related diseases. Here we report that two H3K9 demethylases, KDM3A and KDM4C, regulate heterochromatin reorganization via transcriptionally activating condensin components NCAPD2 and NCAPG2 during MSC senescence. Suppression of KDM3A or KDM4C by either genetic or biochemical approach leads to robust DNA damage response and aggravates cellular senescence, whereas overexpression of KDM3A/KDM4C or NCAPD2 promotes heterochromatin reorganization and blunts DNA damage response. Moreover, MSCs derived from Kdm3a−/− mice exhibit defective chromosome organization and exacerbated DNA damage response, which are associated with accelerated bone aging. Consistently, analysis of human bone marrow MSCs and transcriptome database reveals inverse correlation of KDM3A/KDM4C and/or NCAPD2/NCAPG2 with aging. Taken together, the present finding unveils that H3K9 demethylases function as a surveillance mechanism to restrain DNA damage accumulation in stem cells during aging.
Keywords: mesenchymal stromal cells, condensin, histone demethylase, DNA damage, bone aging, epigenetic regulation
Subject Areas: Molecular Mechanism of Gene Regulation, Cell Biology, Stem Cells Research
Graphical Abstract
Highlights
-
•
KDM3A and KDM4C restrain DNA damage response during MSC senescence
-
•
KDM3A and KDM4C promote heterochromatin reorganization via induction of condensin
-
•
Loss of Kdm3a exacerbates MSC senescence and bone aging in mice
-
•
Chronological aging of human MSCs is associated with reduced expression of KDM3A and KDM4C
Molecular Mechanism of Gene Regulation; Cell Biology; Stem Cells Research
Introduction
Aging is a complex multifactorial biological process manifested by a gradual decline of normal physiological functions in a time-dependent manner. The accumulation of senescent cells in aged tissues has been recognized as one of the most important common denominators of aging (Lopez-Otin et al., 2013). Mesenchymal stromal cells (MSCs) are extremely important adult stem cells for tissue homeostasis, regeneration, and repair. Despite their importance, it should be noted that MSCs are particularly sensitive to aging and age-related diseases, due to their central integration in the body (Baker et al., 2015). In fact, the regenerative capacity of bone marrow MSCs deteriorates after 30 years old (Caplan, 2005), and their deterioration has been recognized as an important hallmark of aging (Boyette and Tuan, 2014, Lopez-Otin et al., 2013). Reciprocally, MSC senescence contributes to the pathogenesis of age-related diseases, such as osteoporosis and osteoarthritis (Patel et al., 2016). Multiple signaling pathways leading to cellular senescence including p53/p21, p16/RB, and Akt/mTOR have been implicated in MSC senescence, which cause permanent cell cycle withdrawal and irreversible damage in very old MSCs (Gharibi et al., 2014, Lin et al., 2014, Wong et al., 2015, Yu and Kang, 2013). However, unlike a static endpoint, senescence reflects a series of progressive and phenotypically diverse cellular states acquired after the initial growth decline (Boyette and Tuan, 2014, Turinetto et al., 2016, Yu and Kang, 2013). A deeper understanding of the molecular mechanisms underlying the multi-step progression of MSC senescence and the link between MSC senescence and organism aging may lead to new therapeutic strategies for age-related diseases.
One striking characteristic of senescent cells is the large-scale alteration of genome architecture. For instance, in oncogene-induced senescence in fibroblasts, accumulation of constitutive heterochromatin and a dramatic rearrangement of heterochromatin into foci have been well documented (Hebbar et al., 2017, Narita et al., 2003). In contrast, a loss of heterochromatin has been described in replicative senescence and premature aging (progeroid) syndromes (Collinson et al., 2016, Goldman et al., 2004, Mani et al., 2017). The disparate nuclear phenotypes that accompany the different stress responses might be simply explained by the differences between acute and chronic senescence models (Chandra and Kirschner, 2016). Although the physiological function and molecular mechanism underlying the heterochromatin reorganization during senescence is still elusive, it should be noted that recent study demonstrated that MSCs derived from WRN null (−/−) embryonic stem cells displayed a pronounced senescence phenotype, which was attributed to disorganized heterochromatin (Zhang et al., 2015), raising a possibility that the unique and organized change of heterochromatin landscape functions as a protective mechanism against cellular senescence. Heterochromatin is characterized by typical post-translational modifications on histones, which have been postulated to be one of the mechanisms to facilitate the folding of heterochromatin into highly condensed structures (Benayoun et al., 2015, Chandra et al., 2015, Chandra and Narita, 2013, Ugarte et al., 2015, Zhu et al., 2013). These regions of compacted and transcriptionally repressive chromatin are critical for diverse aspects of nuclear biology, including the regulation of gene expression, the transcriptional silencing of genomic repeats, DNA repair, and the maintenance of genome stability (Bulut-Karslioglu et al., 2014). Methylation of histone H3 at lysine 9 has a well-recognized role in the establishment and maintenance of heterochromatin structure (Benayoun et al., 2015, Ugarte et al., 2015, Zhu et al., 2013). SUV39H1, the first-described H3K9 methyltransferase, has been associated with oncogene-induced senescence and pathological aging process (Braig et al., 2005, Peters et al., 2001); however, it is still unclear whether SUV39H1 regulates physiological aging. On the other hand, the steady state of methylation at H3 lysine 9 is dictated by the balance between addition and removal of methyl groups, which is achieved by a reciprocal action between lysine methyltransferases (KMTs) and histone demethylases (KDMs) (Munoz-Espin and Serrano, 2014). Indeed, several recent studies have provided experimental evidence that KDMs are involved in the longevity regulation in Caenorhabditis elegans (Johmura et al., 2016, Merkwirth et al., 2016, Sen et al., 2015), indicating the potential role of KDMs in cellular senescence and organism aging.
H3K9 demethylation is mainly catalyzed by Fe(II)- and α-ketoglutarate-dependent JmjC-domain-containing proteins, including the JMJD1/KDM3 family, JMJD2/KDM4 family, and PHF8/KDM7B (Mosammaparast and Shi, 2010). In this study, we identify two conserved H3K9 KDMs, KDM3A and KDM4C, that regulate heterochromatin reorganization to restrain DNA damage and progression of MSC senescence via transcriptionally activating condensin components NCAPD2 and NCAPG2. Decline in KDM3A and/or KDM4C leads to aggravated MSC senescence and bone aging in mice and is significantly correlated with chronological aging of MSCs in human. Our finding thus unveils a protective role of KDM-mediated heterochromatin reorganization in stem cells, which may provide potential targets for the diagnosis and intervention of age-related diseases.
Results
MSC Senescence Is Accompanied by Heterochromatin Reorganization
To experimentally assess MSC senescence in vitro, we established a replicative senescence model using either human bone marrow stromal cells (hBMSCs) or human umbilical cord-derived stromal cells (hUCMSCs). Late passage hMSCs presented typical phenotypes of cellular senescence, including increased β-galactosidase (SA-β-gal) activity, reduced colony forming ability, loss of proliferative potential, and upregulation of p21WAF1 (Figure S1A). To further evaluate the dynamic change of heterochromatin and H3K9 methylation accompanied by hMSC senescence, we focused on hUCMSCs, which sustains rapid multiplication until p16-p18 (Chen et al., 2015). A gradual aging process in hUCMSCs was demonstrated by progressively increased SA-β-gal–positive cells (Figure 1A). In consistence with the previous reports, the expression levels of H3K9 methylation and heterochromatin mark HP1-γ were dramatically reduced at the final stage of cellular senescence (p26), at which more than 85% of cells reached a senescence state (Figures 1A and 1B, p26). Of note, although the expression levels of H3K9 methylation were eventually lost, there was a mild induction of these repressive heterochromatin marks at the early stage of senescence (p10-p16) at which only small amount of cells exhibited senescent features (Figures 1B and S1B). The induction of heterochromatin marks was further validated by immunofluorescent staining using constitutive heterochromatin mark H3K9me3 and a centromere-specific heterochromatin mark CENPA (Figure 1C). In the young hUCMSCs (p6), heterochromatin was mainly localized underneath the nuclear membrane. On senescence entry, heterochromatin was markedly increased and reorganized into condensed heterochromatin foci (p16). Collectively, these data demonstrate that MSC senescence is accompanied by heterochromatin reorganization.
Figure 1.

MSC Senescence Is Accompanied by Heterochromatin Reorganization
Three hUCMSCs lines (hUC009, hUC011, hUC013) were used for serial passaging and characterized with various senescence markers.
(A)Morphology and β-Gal staining (scale bar = 100μm) in serial passage replicative senescence cell model with hUCMSCs. Quantification is shown at the right panel; data are presented as the mean ± SEM. ***p < 0.01 (t test, n = 3).
(B) Representative Western blot showing the expression levels of senescence marker genes and heterochromatin marks along with hUCMSCs aging; experiments were repeated three times.
(C) Representative images of immunofluorescence staining with H3K9me3 (green) and CENPA (red) in hUCMSCs at passage 6 and 16 (scale bar = 10μm). Experiments were repeated at least three times using three different hUCMSCs lines.
(D) Representative images of immunofluorescence staining of KDM3A or KDM4C and H3K9me2/3 in hUCMSCs at p7 and p16 (scale bar = 10μm). Experiments were repeated at least three times using three different hUCMSCs lines.
Identification of H3K9 Demethylases KDM3A and KDM4C that Are Potentially Involved in MSC Senescence
Post-translational modifications of histones such as methylation and acetylation are central in the regulation of heterochromatin structure. To investigate the potential roles of histone modification in heterochromatin reorganization and MSC senescence, we profiled the expression of histone modifying enzymes using both replicative senescence model (hBMSCs and hUCMSCs) and chronological aging model (rat primary young Vs old BMSCs) (Figure S1C). The screening results showed that the mRNA expression levels of KDM3A, KDM4C, and KDM5C were consistently upregulated, whereas SUV39H1 was consistently downregulated in the three models (Figures S2A–S2D). Of note, both KDM3A and KDM4C and SUV39H1 target on H3K9 methylation. We decided to focus on the two H3K9 KDMs, because the role of KDMs in stem cell senescence has not been characterized. The immunofluorescent staining and Western blot revealed that both KDM3A and KDM4C were readily upregulated at the early stage of senescence when heterochromatin is reorganized (Figures 1D and S2E).
To further investigate the potential role of KDM3A and KDM4C in heterochromatin reorganization and MSC senescence, we knocked down KDM3A or KDM4C using shRNA (Figure S3A) in the early passage hUCMSCs (p6). Our results showed that suppression of KDM3A or KDM4C induced a rapid onset of cellular senescence (p8) as demonstrated by increased β-gal activity, reduced colony formation, and increased expression of p21 and p15 in hUCMSCs (Figures 2A and 2B). Of particular interest, knockdown of KDM3A or KDM4C provoked a robust increase of γ-H2AX and 53BP1 foci, indicating an accumulation of DNA damage response (DDR) (Figure 2C). To answer the question of whether the effects of KDM3A and KDM4C on cellular senescence and DDR are related to their function as H3K9 demethylase, we used IOX1, a specific JmjC histone demethylase inhibitor, to treat hUCMSCs at p6-p8. In corroboration with the knockdown data, IOX1 treatment suppressed cell proliferation, increased β-gal activity, and enhanced DDR in a dose-dependent manner (Figures 2D–2F, S3B, and S3C). These results suggest that KDM3A and KDM4C protect MSCs from cellular senescence and DDR, the effect of which is related to their histone demethylase activity.
Figure 2.

Suppression of H3K9 Demethylases KDM3A and KDM4C Induces DNA Damage and Accelerates Cellular Senescence in hUCMSCs
Two different hUCMSCs lines (hUC009, hUC013) were used for knockdown or IOX experiments.
(A) Representative images of colony formation and β-Gal staining (scale bar = 100μm) in hUCMSCs treated with control shRNA or shRNAs targeting KDM3A or KDM4C. The cells were selected by ZsGreen positivity after lentiviral transduction and grew for two more passages. Quantification of β-Gal staining is shown at the right; data are presented as the mean ± SEM. ***p < 0.001 (t test: n = 3).
(B) RT-qPCR assay showing the expression levels of KDM3A, KDM4C, p15, and p21 after shRNA treatment; data are presented as the mean ± SEM. **p < 0.01; ***p < 0.001 (t test: n = 3).
(C) Representative images (scale bar = 5μm) and quantification of 53BP1, γ-H2AX foci in hUCMSCs transduced with lentiviral particles carrying shKDM3A-1, shKDM4C-1, or control shRNA. Quantification data are presented as mean ± SEM of values from three independent experiments with triplicate wells analyzed on 6–8 cells/field from five different fields. ***p < 0.001 (t test).
(D) MTT assay of hUCMSCs (p6) treated with different concentrations of IOX1 or DMSO for 72 h. Data are presented as the mean ± SEM of values from three independent experiments; **p < 0.01, IOX70μM compared with control; ##p < 0.01, IOX100μM compared to control group (t test).
(E) Quantification of β-Gal staining in hUCMSCs (p7) treated with different concentrations of IOX1 (20, 50, 70, and 100 μM). Data are presented as the mean ± SEM. *p < 0.05; **p < 0.01 (one-way ANOVA, n = 3).
(F) Quantification of 53BP1 and γ-H2AX foci in hUCMSCs (p6) treated with different concentration IOX1 (20, 50, 70, and 100 μM) and DMSO control. Quantification is shown at the right; mean ± SEM of values from three independent experiments with triplicate wells analyzed on 6–8 cells/field from five different fields. *p < 0.05; **p < 0.01 (one-way ANOVA).
KDM3A and KDM4C Regulate Condensin Complex Components NCAPD2 and NCAPG2 via Their Demethylase Activity
To elucidate the mechanism underlying the regulatory effect of KDM3A and KDM4C on MSC senescence, we analyzed the KDM3A- or KDM4C-dependent transcriptional program using RNA-seq in MSCs. Manipulation of KDM3A or KDM4C led to a consistent change in chromosome organization genes, in particular chromosome condensation genes (Figures S4A and S4B, GEO: GSE133098). Interestingly, among the chromosome condensation genes, we have identified that various components of the condensin complex I and II are consistently downregulated in KDM3A- or KDM4C-knockdown hUCMSCs or upregulated in KDM3A or KDM4C-overexpressing hUCMSCs (Figures 3A and S4B). We decided to focus on NCAPD2 and NCAPG2, the major components of condensin I and condensin II, respectively, which are unanimously regulated by KDM3A and KDM4C. Our real-time PCR and Western blot showed that siRNA knockdown of either KDM3A or KDM4C reduced the expression levels of both NCAPD2 and NCAPG2 (Figures 3B and S4C). Moreover, the binding of KDM3A and KDM4C on the promoter of NCAPD2 was significantly downregulated, whereas the occupancy of corresponding H3K9me3 and H3K9me2 was significantly upregulated in knockdown hUCMSCs (Figure 3C). In addition, inhibition of H3K9 demethylase activity by IOX1 reduced the expression levels of NCAPD2 and NCAPG2 (Figure 3D). IOX1 also reduced the expression level of Lamin B1 and increased the expression level of p53, supporting the senescence-inducing property of IOX1. The regulatory effect of IOX1 was attributed to the enzyme inhibitory function, as IOX1 increased the enrichment of H3K9me2 and H3K9me3 on NCAPD2 promoter (Figure 3E). Altogether, these data clearly indicate that KDM3A and KDM4C function as positive transcriptional regulators of condensin components via their H3K9 demethylase activity. As a first step to identify the potential mechanism underlying condensin-mediated effects, quantitative ChIP-PCR was used to determine the recruitment of NCAPD2 on repetitive DNA sequences, at which heterochromatin enriches. Our results showed that the binding of NCAPD2 on replicative DNA sequences was significantly reduced in KDM3A- or KDM4C-knockdown cells (Figure 3F), suggesting that KDM-mediated regulation of condensin might be critical for stabilizing the heterochromatin structure during senescence.
Figure 3.

KDM3A and KDM4C Transcriptionally Regulate Condensin Components NCAPD2 and NCAPG2 via Their Demethylase Activity
(A) Heatmap showing the global downregulation and upregulation of chromosome condensation genes in siKDM3A-/siKDM4C-knockdown (hUC009) or KDM3A-/KDM4C-overexpressing hUCMSCs (hUC013) compared with their relative control hUCMSCs.
(B) Representative Western blot showing that the expression levels of NCAPD2 and NCAPG2 are downregulated in KDM3A- or KDM4C-knockdown cells (hUC009). Data are presented as the mean ± SEM. **p < 0.01; ***p < 0.001 (t test).
(C) ChIP-qPCR showing the enrichment of H3K9me2, 3, KDM3A (siKDM3A-1+siKDM3A-3), or KDM4C (siKDM4C-1+siKDM4C-3) on NCAPD2 promoters relative to IgG enrichment after siRNA treatment. Data are presented as the mean ± SEM. *p < 0.05; **p < 0.01 (t test, n = 3).
(D) Representative Western blot showing the expression of senescence markers and NCAPD2 and NCAPG2 in hUCMSCs (p6-p8) treated with different concentration IOX1 (20, 50, 70, and 100μM). Experiments were repeated three times with two hUCMSCs lines (hUC009, hUC013). Data are presented as the mean ± SEM. *p < 0.05; **p < 0.01 (one-way ANOVA).
(E) ChIP-qPCR showing increased enrichment of H3K9me1, 2, and3 on the representative NCAPD2 promoter relative to IgG enrichment in 70μM IOX1-treated hUCMSCs. Data are presented as the mean ± SEM. **p < 0.01; ***p < 0.001 (t test: n = 3).
(F) ChIP-qPCR showing the enrichment of NCAPD2 on several repetitive DNA sequences loci in control- or KDM3A (siKDM3A-1+siKDM3A-3) or KDM4C (siKDM4C-1+siKDM4C-3) siRNA-treated hUCMSCs, data are presented as the mean ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001 (t test: n = 3).
KDM3A and KDM4C Induce Heterochromatin Reorganization to Restrain DNA Damage Response in Doxorubicin-Induced Senescence
The finding that suppression of KDM3A and KDM4C leads to increased DDR raises the possibility that KDM3A and KDM4C function as a protective mechanism against DNA damage during cellular senescence. To establish a direct link between KDM3A/KDM4C and DDR in senescent cells, we applied a DNA damage–induced acute senescence model in hUCMSCs using Doxorubicin (DOX). DOX treatment induced a gradual increase in KDM3A and KDM4C level peaked at 10–12 h, which thereafter decreased at 24 h and returned to virtually background level at 48 h. Of note, the kinetics of heterochromatin marks H3K9me3 and HP1γ, and condensin components NCAPD2 and NCAPG2 were consistent with the KDMs upon DOX treatment (Figure 4A), suggesting DOX treatment induces a heterochromatin reorganization process, which may be associated with KDM3A and KDM4C. In corroboration with the Western blot result, the immunofluorescent staining showed that heterochromatin mark gradually increased, which peaked at 10 h and decreased thereafter. Accordingly, 53BP1 and γ-H2AX foci were induced at 24 h, which stochastically distributed throughout the nucleus, and maximized around 48 h after DNA damage (Figures 4B and 4C). The sequential inductive effect on heterochromatin reorganization and DDR upon DOX treatment suggests a surveillance mechanism by heterochromatin rearrangement. In fact, knockdown of KDM3A and KDM4C or treatment with IOX1 markedly aggravated DOX-induced DDR and cellular senescence (Figures 4D, S5A, and S5B). By contrast, overexpression of KDM3A or KDM4C significantly reduced the number of DOX-induced 53BP1 and γ-H2AX foci and degree of cellular senescence as demonstrated by decreased β-gal activity and a dramatic downregulation of p21WAF1 (Figures 4E, S5C, and S5D). In addition, overexpression of condensin I complex component NCAPD2 alone significantly rescued DOX-induced DDR and cellular senescence, albeit at a lower level compared with KDM3A or KDM4C overexpression (Figures 4E, S5C, and S5D). The protective role of KDM3A and KDM4C is related to heterochromatin reorganization, as overexpression of KDM3A/KDM4C or NCAPD2 promoted heterochromatin reorganization as indicated by H3K9me2/3 staining (Figures S5C and S5D). The role of KDM3A/KDM4C and NCAPD2 in chromatin reorganization was further illustrated by Transmission Electron Microscope (TEM) analysis or immunofluorescent staining in KDM3A/KDM4C- or NCAPD2-overexpressing cells. As shown in Figure S6A, KDM3A or KDM4C overexpression induced chromosome condensation in the nuclear. Similarly, overexpression of NCAPD2 directly promoted heterochromatin reorganization as indicated by an increase of H3K9me3 positive foci (Figure S6B). Collectively, these results clearly demonstrate a protective role of KDM3A and KDM4C in DNA damage–induced senescence through induction of heterochromatin reorganization.
Figure 4.

Doxorubicin Induces Heterochromatin Reorganization and DNA Damage Response
Two hUCMSCs lines (hUC009, hUC013) were used for Doxorubicin-induced cellular senescence model.
(A) Early passage hUC-MSCs (p6-7) were treated with Doxorubicin for 48 h. Representative Western blot showing the expression levels of KDM3A, KDM4C, heterochromatin marks, NCAPD2, NCAPG2, and senescence marks at different time points (0–48 h) after Doxorubicin treatment.
(B) Representative immunofluorescence images of 53BP1 or γ-H2AX foci and H3K9me2/3 staining (scale bar = 5μm) in hUCMSCs treated with Doxorubicin for different time points.
(C) Quantification of 53BP1 or γ-H2AX and H3K9me2/3 immunofluorescence staining in hUCMSCs treated with Doxorubicin for different time points (CTCF, corrected total cell fluorescence). Data are presented as mean ± SEM of values from three different experiments with triplicate wells analyzed on 6–8 cells/field from five different fields; *p < 0.05; **p < 0.01, 53BP1, γ-H2AX compared with control group, #p < 0.05; ##p < 0.01, H3K9me2/3 compared with control group (Wilcoxon/Mann-Whitney test).
(D) Representative images and quantification of 53BP1 and γ-H2AX immunofluorescence staining (scale bar = 5μm) in Doxorubicin-treated hUCMSCs transfected with scrambled siRNAs or siRNA mixtures (siKDM3A-1 + siKDM4C-3). Data are presented as mean ± SEM of values from three different experiments with triplicate wells analyzed on 6–8 cells/field from five different fields; **p < 0.01; ***p < 0.001 (t test).
(E) Representative images and quantification of β-Gal staining (scale bar = 100μm) in control group or Doxorubicin-treated hUCMSCs transfected with Vector plasmid or KDM3A, KDM4C, or NCAPD2 plasmid.
Data are presented as the mean ± SEM. *p < 0.05; **p < 0.01 (t test, n = 3).
Dynamic Change of KDM3A/KDM4C and Condensin Complex Components during Bone Aging in Mice
Having established the role of KDM3A and KDM4C in MSC senescence in vitro, we asked whether the two H3K9 KDMs are related to heterochromatin reorganization of MSCs during aging in vivo. For this purpose, we established ovariectomized (OVX) rat model, which is a well-established model to study bone aging process. Successful OVX was validated at sacrifice, using uterus weight, bone CT, and morphometric indexes (Figures S7A–S7C). We then isolated rat bone marrow MSCs (rBMSCs) at different time points starting from 3 weeks to 12 weeks after OVX and determined the molecular changes and heterochromatin marks indicative of cellular senescence. The result illustrated a dramatic reduction of H3K9 methylation in rBMSCs collected at 9–12 weeks, at which the osteoporosis was utmost severe (Figure 5A). Of note, the kinetics of Kdm3a and Kdm4c expression peaked at both 6 weeks and 12 weeks (Figures 5A and 5B) recapitulating the expression dynamics in replicative senescence model (Figure S2E). Importantly, a gradual increase of senescence markers including p15, p19, and p27 was detected in rBMSCs along with disease development, indicating deterioration of rBMSCs along with bone aging process (Figure 5C). In addition, the expression levels of Ncapd2 and Ncapg2 reached the peak at 6 weeks, which was in line with Kdm3a and Kdm4c upregulation and decreased thereafter (Figure 5C), emphasizing the association between Kdm3a/Kdm4c and condensin complexes at the early stage of bone aging.
Figure 5.

Dynamic Change of KDM3A and KDM4C in OVX Rat Model
(A) Western blot analysis of Kdm3a, Kdm4c, and H3K9 methylation in BMSCs derived from OVX rat model (total n = 22; Sham, n = 4; OVX3w, n = 4; OVX6w, n = 4; OVX9w, n = 5; OVX12w, n = 5). The experiments were repeated for three times.
(B) The quantification analysis of Western blot results of Kdm3a, Kdm4c, and H3K9me3 show two peaks of Kdm3a and Kdm4c (n = 22). *p < 0.05, change of Kdm3a expression compared with sham; p < 0.05, change of Kdm4c expression compared with sham; #p < 0.05, change of H3K9me3 compared with sham (Wilcoxon/Mann-Whitney test).
(C) RT-qPCR assay of Ncapd2, Ncapg2, and several cell cycle inhibitor genes in BMSCs derived from OVX rat model.
Data are presented as mean ± SEM. *p < 0.05 (Wilcoxon/Mann-Whitney test, n = 22).
Deficiency of Kdm3a Leads to MSC Senescence and Bone Aging in Mice
The Kdm3a−/− mice have defects in sex determination, lipid metabolism, and spermatogenesis (Kuroki et al., 2013, Liu et al., 2010, Tateishi et al., 2009); however, the relationship between Kdm3a deficiency and aging has not been investigated. In this study, we took advantage of Kdm3a knockout mice to evaluate the effect of Kdm3a deficiency on bone aging (Figure S7D). We collected bone tissues from WT and KO mice at different ages and found that although no difference exists between WT and KO young mice (2 month), KO mice at 6 months old exhibited evident bone aging phenotype (Figure 6A). The expression levels of senescence markers p53, p21, and p16 were dramatically increased, whereas the expression of PCNA was significantly decreased in KO bones. In addition, senescence nuclear markers such as Lamin B1, H3K9me3, and H3K27me3 and condensin components were dramatically downregulated in KO bones, indicating the correlation between bone aging and heterochromatin destruction in vivo (Figures 6B and 6C). Of note, the expression of Suv39h1 was slightly increased in KO mice. To further correlate these changes with BMSCs, we isolated primary BMSCs from WT and KO mice. Our results showed that condensin components were significantly downregulated in KO BMSCs compared with WT BMSCs. In contrast, the expression levels of senescence genes such as p53, p21, and Gadd34 were significantly upregulated in KO BMSCs (Figure S7E). Moreover, when challenged with DOX, KO BMSCs elicited a much enhanced DDR as illustrated by increased expression of 53BP1 and p21 (Figure S7F). The specificity of the KO was validated by overexpressing Kdm3a in KO BMSCs, which showed that overexpression of Kdm3a completely rescued aggravated cellular senescence and increased DDR induced by DOX in KO cells (Figures S7G and S7H). In addition, overexpression of Ncapd2 alleviated the DOX-induced cellular senescence and DDR in KO BMSCs (Figures 6D and 6E). Altogether, these results strongly indicate that Kdm3a deficiency aggravates DNA damage and cellular senescence in BMSCs, which eventually contributes to the bone aging phenotype in mice.
Figure 6.

Deficiency of Kdm3a Leads to MSC Senescence and Bone Aging in Mice
(A) The representative three-dimensional reconstructed images of distal femur in a 6-month-old female Kdm3a−/− mice and WT mice by CT scanner. Quantification of morphometric parameters including bone volume fraction (BV/TV), trabecular number (Tb.N, 1/mm), and trabecular separation (Tb.Sp, mm) are shown at right. Data are presented as the mean ± SEM, **p < 0.01 (t test, n = 6).
(B) Western blot assay showing the expression of senescence markers p16, p21, p53, and heterochromatin markers H3K9me3 and H3K27me3 in bone tissues collected from 6-month-old Kdm3a−/− KO mice (n = 6) and WT mice (n = 6). Experiments were repeated three times, *p < 0.05; **p < 0.01 (Wilcoxon/Mann-Whitney test).
(C) RT-qPCR assay showing the reduction of condensin components in bone tissues collected from 6-month-old Kdm3a−/− KO mice. Data are presented as the mean ± SEM, *p < 0.05; **p < 0.01 (t test, n = 3).
(D) Representative images and quantification of β-Gal staining (scale bar = 100μm) show Ncapd2 rescues Doxorubicin-induced cellular senescence in KO BMSCs. Quantification data represents mean ± SEM of values from three independent experiments with three pairs of WT and KO mMSCs. *p < 0.05; **p < 0.01 (t test, n = 3).
(E) Representative images and quantification of γ-H2A.X and p21 immunofluorescence staining (scale bar = 5μm). Overexpression of ncapd2 in KO BMSCs completely rescues Doxorubicin-induced DNA damage and heterochromatin destruction. Quantification data are presented as mean ± SEM of values from three different experiments with three pairs of WT and KO mMSCs. In each experiment, cells were derived from triplicate wells and analyzed on 6–8 cells/field from five different fields. **p < 0.01; ***p < 0.001 (t test, n = 3).
The Expression Levels of KDMs and Condensin Components Are Inversely Correlated with Human Aging
Previous studies on gene expression analysis of young versus old human BMSCs have revealed large amounts of age-associated molecular changes related to MSC adhesion, cell cycle regulation, migration, and cytokine secretion (Bustos et al., 2014, Wilson et al., 2010). However, whether MSC aging is associated with a global change of genes involved in chromosome organization is unknown. In this study, we asked whether KDM3A/KDM4C and condensin components could be related to physiological aging in human stem cells. We first compared the expression levels of KDM3A/KDM4C, condensin components, and heterochromatin marks in primary BMSCs derived from nine young (13- to 32-year-old) and five old (52- to 61-year-old) individuals (Figure 7A). Our Western blot result showed a marked downregulation of KDM3A and KDM4C protein associated with a decrease in H3K9me3 and PCNA in BMSCs derived from old individuals. Importantly, the expression of NCAPG2 and NCAPD2 was dramatically decreased in aged BMSCs as well. It should be noted that the expression of SUV39H1 did not change in old BMSCs. To confirm the correlation of KDM3A/KDM4C and NCAPD2/NCAPG2 with aging, we examined publically available MSC gene expression datasets (GEO: GSE39540). GSE dataset analysis revealed that the expression levels of KDM3A, KDM4C, NCAPD2, and NCAPG2 were negatively correlated with human aging (Figure 7B). Taken together, these data clearly reveal that KDM3A and KDM4C are negatively correlated with stem cell aging in human.
Figure 7.

Expression Levels of KDM3A/KDM4C and NCAPD2/NCAPG2 Are Inversely Correlated with Human Aging
(A) Western blot assay showing the expression levels of KDM3A, KDM4C, heterochromatin marks, and condensin components are globally reduced in old hBMSCs compared with young hBMSCs. Human bone marrow MSCs from 14 healthy individuals (Male, Asian, age range from 13 to 61 years) were cultured for three passages and used for further analysis. Quantification of KDM3A and KDM4C is shown at right. Data are presented as the mean ± SEM, *p < 0.05 (Wilcoxon/Mann-Whitney test).
(B) Negative correlations between KDM3A, KDM4C, NCAPD2, and NCAPG2 (y axis) and age (x axis) in human MSCs samples (n = 60, age range from 19 to 84 years) (GEO: GSE39540).
Discussion
Although heterochromatin reorganization is observed during cellular senescence, the physiological function of this unique epigenetic change is still ill defined. In this study, we have unveiled that KDM3A and KDM4C regulate condensin-dependent heterochromatin reorganization to restrain DDR during MSC senescence. Deficiency of Kdm3a leads to accelerated MSC senescence and premature bone aging in mice. From a clinical perspective, reduced expression of KDM3A/KDM4C or condensin component genes NCAPD2 and NCAPG2 can be utilized as indicators for aging.
Significant chromatin structural changes occur during physiological aging and cellular senescence, which include global histone loss, alternation of epigenetic landscapes, loss of heterochromatic regions, and large-scale chromatin rearrangements(Corpet and Stucki, 2014, Dowen et al., 2013, Lopez-Otin et al., 2013, Murga et al., 2007). However, it is interesting to note that different senescence models may have disparate and unique chromatin changes. We have illustrated that MSC replicative senescence is accompanied by heterochromatin reorganization at the early stage and heterochromatin loss at the final stage. In particular, upon senescence entry, a spatial reorganization of heterochromatin structure is manifested by redistribution and condensation of heterochromatin from nuclear membrane to nucleoplasm (Figure 1C). The dynamic change of heterochromatin is also observed in DOX-induced senescence model, which displays a gradual heterochromatin reorganization followed by a rapid heterochromatin destruction after genotoxic treatment (Figures 4A and 4B). In line with the cell models, the stage-dependent change of heterochromatin has been recapitulated in OVX-induced bone aging model, which demonstrates a dynamic change of heterochromatin marks in primary BMSCs collected at different time points along with disease progression (Figures 5A and 5B). Altogether, it is clear that a stage-dependent heterochromatin reorganization process exists in conjunction with MSC senescence, misregulation of which could be detrimental as heterochromatin perturbation has been observed in progeria syndrome (Goldman et al., 2004, Scaffidi and Misteli, 2006, Shumaker et al., 2006, Zhang et al., 2015), and heterochromatin disorganization was proposed to underlie the pathogenesis of premature MSC aging in Werner syndrome (Zhang et al., 2015).
Interestingly, we have found that KDM3A and KDM4C are concomitantly upregulated along with heterochromation process at the early stage of replicative and DNA damage–induced senescence (Figures 1D and 4A–4C). Knockdown or suppression of KDM3A and KDM4C downregulates chromosome organization genes, whereas overexpression of KDM3A and KDM4C induces them (Figures 3 and S4). Condensin complex is of paramount importance for chromosome assembly and compaction during mitosis and meiosis (Hagstrom et al., 2002, Mishima et al., 2002). However, recent studies provide evidence that condensin complex might be involved in other biological processes. Previous studies have illustrated that condensins shape chromatin organization by its localization to topologically associating domain (TAD) boundaries in Drosophila, mouse, and human embryonic stem cells, indicating their regulatory role in chromatin high order organization in diverse interphase processes(Bauer et al., 2012). In addition, condensin complex binds to promoters and enhancers and participates in gene regulation in a variety of organisms (Ono et al., 2003, Ono et al., 2013, Tanaka et al., 2012). Despite of all these links, the potential role of condensin complex in the heterochromatin organization and cellular senescence had not been investigated until one recent study showed that condensin complex II subunit NCAPH2 was increased in oncogene-induced cancer cell lines (Yokoyama et al., 2015). In addition, overexpression of NCAPH2 was sufficient to induce the formation of SAHF, whereas knockdown of NCAPH2 reduced oncogene-induced cellular senescence in IMR90 fibroblasts (Yokoyama et al., 2015). This study raises unresolved fundamental questions of how condensin complex is induced upon senescence entry and what is the functional role of condensin complex during physiological aging. In the current study, we have unveiled that KDM3A and KDM4C induce the expression of both condensin I and II complex components at the early stage of MSC senescence via the H3K9 demethylase activity (Figures 3B–3E). Condensin complex I subunit NCAPD2 and condensin complex II subunit NCAPG2 promote heterochromatin condensation and reorganization, and overexpression of the condensin complex subunit rescues DOX-induced cellular senescence and heterochromatin destruction in Kdm3a KO MSCs (Figures 6D, 6E, and S6). In addition, suppression of KDMs alleviates the recruitment of condensin on the repetitive DNA sequences (Figure 3F). Positive correlation between KDM3A/KDM4C and condensin components has been demonstrated in Kdm3a KO mice and human stem cells, which are associated with aging (Figures 6C and 7). Altogether, these results suggest that condensin complex mediates the regulatory role of KDM3A and KDM4C in heterochromatin reorganization after the initial growth arrest during cellular senescence.
Activation of DDR is thought to enforce cellular senescence by imposing permanent checkpoints (d'Adda di Fagagna, 2008, d'Adda di Fagagna et al., 2003, Di Leonardo et al., 1994). Although DDR and chromatin reorganization have been both causally implicated in the establishment of cellular senescence, their relationship remains largely undefined (Liu et al., 2012, Mah et al., 2010, Sedelnikova et al., 2004). In our study, we observed that inhibition of KDM3A or KDM4C induced a striking DDR at the early stage of MSC senescence (Figure 2C). Because KDM3A and KDM4C is important for heterochromatin reorganization, we hypothesized that by hindering access of DNA damage sensors and associated DDR factors, heterochromatin can restrain DDR signaling in senescent stem cells, which functions as a protective machinery against senescence progression. To test this, we employed a genotoxic damage–induced cellular senescence model and clearly demonstrated that DOX induced a progressive heterochromatin reorganization process in advance of the activation of DDR (Figures 4A–4C). Suppression of KDM3A and KDM4C dramatically enhanced DOX-induced DDR signaling and cellular senescence (Figures 4D, S5A, and S5B), whereas overexpression of KDM3A/KDM4C or condensin component attenuated the DDR and senescence (Figures 4E and S5C), suggesting heterochromatin perturbation leads to an increase of DDR signaling in genotoxic damage–induced senescence. This assumption is further supported by the finding that Kdm3a KO mice that exhibit a defective condensin machinery are more sensitive to DOX-induced DDR (Figures S7G and S7H). Heterochromatin has been reported to pose a barrier to DDR signaling (d'Adda di Fagagna, 2008, d'Adda di Fagagna et al., 2003, Di Micco et al., 2011, Murga et al., 2007). In Drosophila, HP1 prevents DDR activation at chromosome ends (Fanti et al., 1998), and in mammals it modulates DDR activation (Ayoub et al., 2009, Kim and Haber, 2009). Our results are consistent with a scenario in which heterochromatinization plays important role in restraining DDR signaling pathways, which can be achieved by both confining the access of DNA damage sensors to DNA lesions and impairing local signal amplification, thus blocking the augmentation of DNA damage and preventing senescence progression (Di Micco et al., 2011). Given the established role of condensin complex in higher-order genome organization, our results strongly suggest that KDM3A and KDM4C restrain accumulation of DNA damage via condensin-mediated heterochromatin organization during senescence progression.
Another important finding from this study is that both KDM3A/KDM4C and condensin components are inversely correlated with stem cell aging in humans. We have found that the expression levels of KDM3A and KDM4C dramatically decrease in old BMSCs compared with young BMSCs, which is accompanied by a marked reduction of heterochromatin marks and NCAPD2 and NCAPG2, supporting the transcriptional regulatory role of KDM3A and KDM4C in the condensin components and other chromosome condensation genes (Figure 7). The global reduction of KDM3A, KDM4C, and core chromosome organization genes demonstrated in DOX-induced senescence model (Figure 4A) is in accordance with the marked reduction of these genes observed in aging human BMSCs. Given the established link between DNA damage and human aging, it is explicable that along with human aging, hBMSCs gradually lose KDM3A- and KDM4C-mediated heterochromatin organization machinery, which adversely makes these stem cells more susceptible to genotoxic damage. Of note, although previous study using a Werner syndrome stem cell model showed that SUV39H1 is critical for maintaining the normal heterochromatin organization, loss of which led to deregulated heterochromatin structure (Zhang et al., 2015), we did not observe any significant change of SUV39H1 in old BMSCs compared with young BMSCs (Figure 7A). Thus, it appears that distinctive epigenetic regulatory mechanisms may underlie chronological and pathological aging process.
In summary, our study has revealed a role of histone demethylases in modulating heterochromatin reorganization, which functions as a defensive mechanism against DNA damage accumulation in stem cell senescence. As chemically modifiable enzymes, KDM3A and KDM4C could be activated or deactivated to regulate stem cell senescence, thereby holding promising potentials as therapeutic targets for intervening geriatric diseases and stem cell–mediated regenerative medicine. Additionally, in view of the importance of chromosome organization in a wide variety of biological processes, the presently demonstrated critical role of H3K9 demethylases provides insights into the molecular basis underlying other physiological or pathological conditions, such as embryonic development and cancer.
Limitations of the Study
We demonstrate that a panel of condensin components is regulated by KDM3A and/or KDM4C in MSC replicative senescence model and Kdm3a KO mouse model. Although our data show that the regulatory effect is attributed to the demethylase activity of KDM3A and KDM4C, we cannot exclude the possibility that other histone modifiers or transcription factors are involved in the regulatory process. Further work would also be required to definitively identify the relevant demethylase targets. Moreover, the current understanding of the link between heterochromatin reorganization and DDR in physiological condition is still elusive. Although we found that deficiency of Kdm3a disrupts heterochromatin reorganization and aggravates DDR during MSC senescence, additional experiments are needed to elucidate the underlying protective mechanism.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgment
This work is supported by Hong Kong Food and Health Bureau (01120056, 03140496), and UGC/GRF, Hong Kong (14119516, 14165217, 14111519). The work is also supported by National Natural Science Foundation of China (NSFC No. 31771517), China and Guang Dong Province Science and Technology Grant (2017A050506043).
Author Contributions
Biao Huang: experimental design and preformation, collection and/or assembly of data, data analysis, interpretation, and manuscript writing.
Bin Wang: experimental preformation, collection and/or assembly of data, data analysis, and interpretation.
Wayne Yuk-Wai Lee, Kin Pong U, and Kam Tong Leung: isolation and collection of human bone marrow MSCs.
Liu Shi, Lai Ling Tsang, Jiacheng Lin, Hailong Liu, and Xican Li: conduction of in vivo experiment, collection and/or assembly of in vivo data, data analysis, and interpretation.
Zhenqing Liu: collection and/or assembly of in vitro data, data analysis, and interpretation.
Rui Chen: CHIP-seq analysis.
Baohua Liu: generation of progeria mice and sample provision.
Yechun Ruan: discussion and interpretation of the data.
Hsiao Chang Chan: provision of study material, data analysis, and interpretation.
Gang Li: provision of study material, financial support, and technical support.
Xiaohua Jiang: conception and experimental design, financial support, provision of study material, data analysis and interpretation, and manuscript writing.
Declaration of Interests
The authors declare no competing interests.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.041.
Supplemental Information
References
- Ayoub N., Jeyasekharan A.D., Venkitaraman A.R. Mobilization and recruitment of HP1: a bimodal response to DNA breakage. Cell Cycle. 2009;8:2945–2950. [PubMed] [Google Scholar]
- Baker N., Boyette L.B., Tuan R.S. Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone. 2015;70:37–47. doi: 10.1016/j.bone.2014.10.014. [DOI] [PubMed] [Google Scholar]
- Bauer C.R., Hartl T.A., Bosco G. Condensin II promotes the formation of chromosome territories by inducing axial compaction of polyploid interphase chromosomes. PLoS Genet. 2012;8:e1002873. doi: 10.1371/journal.pgen.1002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun B.A., Pollina E.A., Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015;16:593–610. doi: 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette L.B., Tuan R.S. Adult stem cells and diseases of aging. J. Clin. Med. 2014;3:88–134. doi: 10.3390/jcm3010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braig M., Lee S., Loddenkemper C., Rudolph C., Peters A.H., Schlegelberger B., Stein H., Dorken B., Jenuwein T., Schmitt C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- Bulut-Karslioglu A., De La Rosa-Velazquez I.A., Ramirez F., Barenboim M., Onishi-Seebacher M., Arand J., Galan C., Winter G.E., Engist B., Gerle B. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell. 2014;55:277–290. doi: 10.1016/j.molcel.2014.05.029. [DOI] [PubMed] [Google Scholar]
- Bustos M.L., Huleihel L., Kapetanaki M.G., Lino-Cardenas C.L., Mroz L., Ellis B.M., McVerry B.J., Richards T.J., Kaminski N., Cerdenes N. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am. J. Respir. Crit. Care Med. 2014;189:787–798. doi: 10.1164/rccm.201306-1043OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan A.I. Review: mesenchymal stem cells: cell-based reconstructive therapy in orthopedics. Tissue Eng. 2005;11:1198–1211. doi: 10.1089/ten.2005.11.1198. [DOI] [PubMed] [Google Scholar]
- Chandra T., Ewels P.A., Schoenfelder S., Furlan-Magaril M., Wingett S.W., Kirschner K., Thuret J.Y., Andrews S., Fraser P., Reik W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015;10:471–483. doi: 10.1016/j.celrep.2014.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T., Kirschner K. Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol. 2016;40:161–167. doi: 10.1016/j.ceb.2016.03.020. [DOI] [PubMed] [Google Scholar]
- Chandra T., Narita M. High-order chromatin structure and the epigenome in SAHFs. Nucleus. 2013;4:23–28. doi: 10.4161/nucl.23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.Y., Mou X.Z., Du X.C., Xiang C. Comparative analysis of biological characteristics of adult mesenchymal stem cells with different tissue origins. Asian Pac. J. Trop. Med. 2015;8:739–746. doi: 10.1016/j.apjtm.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Collinson A., Collier A.J., Morgan N.P., Sienerth A.R., Chandra T., Andrews S., Rugg-Gunn P.J. Deletion of the polycomb-group protein EZH2 leads to compromised self-renewal and differentiation defects in human embryonic stem cells. Cell Rep. 2016;17:2700–2714. doi: 10.1016/j.celrep.2016.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet A., Stucki M. Chromatin maintenance and dynamics in senescence: a spotlight on SAHF formation and the epigenome of senescent cells. Chromosoma. 2014;123:423–436. doi: 10.1007/s00412-014-0469-6. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer. 2008;8:512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F., Reaper P.M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N.P., Jackson S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Di Leonardo A., Linke S.P., Clarkin K., Wahl G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–2551. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- Di Micco R., Sulli G., Dobreva M., Liontos M., Botrugno O.A., Gargiulo G., dal Zuffo R., Matti V., d'Ario G., Montani E. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011;13:292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen J.M., Bilodeau S., Orlando D.A., Hubner M.R., Abraham B.J., Spector D.L., Young R.A. Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem Cell Reports. 2013;1:371–378. doi: 10.1016/j.stemcr.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanti L., Dorer D.R., Berloco M., Henikoff S., Pimpinelli S. Heterochromatin protein 1 binds transgene arrays. Chromosoma. 1998;107:286–292. doi: 10.1007/s004120050310. [DOI] [PubMed] [Google Scholar]
- Gharibi B., Farzadi S., Ghuman M., Hughes F.J. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells. 2014;32:2256–2266. doi: 10.1002/stem.1709. [DOI] [PubMed] [Google Scholar]
- Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B., Gruenbaum Y., Khuon S., Mendez M., Varga R. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom K.A., Holmes V.F., Cozzarelli N.R., Meyer B.J. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev. 2002;16:729–742. doi: 10.1101/gad.968302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbar M., Chandra T., Shukla A., Kadavigere R., Girisha K.M. Complexities in genotype-phenotype correlation and genetic counseling in collagen VI - related myopathy. Indian J. Pediatr. 2017;84:330–331. doi: 10.1007/s12098-016-2279-8. [DOI] [PubMed] [Google Scholar]
- Johmura Y., Sun J., Kitagawa K., Nakanishi K., Kuno T., Naiki-Ito A., Sawada Y., Miyamoto T., Okabe A., Aburatani H. SCF(Fbxo22)-KDM4A targets methylated p53 for degradation and regulates senescence. Nat. Commun. 2016;7:10574. doi: 10.1038/ncomms10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.A., Haber J.E. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc. Natl. Acad. Sci. U S A. 2009;106:1151–1156. doi: 10.1073/pnas.0812578106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki S., Matoba S., Akiyoshi M., Matsumura Y., Miyachi H., Mise N., Abe K., Ogura A., Wilhelm D., Koopman P. Epigenetic regulation of mouse sex determination by the histone demethylase Jmjd1a. Science. 2013;341:1106–1109. doi: 10.1126/science.1239864. [DOI] [PubMed] [Google Scholar]
- Lin S.P., Chiu F.Y., Wang Y., Yen M.L., Kao S.Y., Hung S.C. RB maintains quiescence and prevents premature senescence through upregulation of DNMT1 in mesenchymal stromal cells. Stem Cell Reports. 2014;3:975–986. doi: 10.1016/j.stemcr.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Yip R., Zhou Z. Chromatin remodeling, DNA damage repair and aging. Curr. Genomics. 2012;13:533–547. doi: 10.2174/138920212803251373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Zhou S., Liao L., Chen X., Meistrich M., Xu J. Jmjd1a demethylase-regulated histone modification is essential for cAMP-response element modulator-regulated gene expression and spermatogenesis. J. Biol. Chem. 2010;285:2758–2770. doi: 10.1074/jbc.M109.066845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah L.J., El-Osta A., Karagiannis T.C. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- Mani P., Keshavarz T., Chandra T.S., Kyazze G. Decolourisation of Acid orange 7 in a microbial fuel cell with a laccase-based biocathode: influence of mitigating pH changes in the cathode chamber. Enzyme Microb. Technol. 2017;96:170–176. doi: 10.1016/j.enzmictec.2016.10.012. [DOI] [PubMed] [Google Scholar]
- Merkwirth C., Jovaisaite V., Durieux J., Matilainen O., Jordan S.D., Quiros P.M., Steffen K.K., Williams E.G., Mouchiroud L., Tronnes S.U. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165:1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima M., Kaitna S., Glotzer M. Central spindle assembly and cytokinesis require a kinesin-like protein/RhoGAP complex with microtubule bundling activity. Dev. Cell. 2002;2:41–54. doi: 10.1016/s1534-5807(01)00110-1. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N., Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- Munoz-Espin D., Serrano M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- Murga M., Jaco I., Fan Y., Soria R., Martinez-Pastor B., Cuadrado M., Yang S.M., Blasco M.A., Skoultchi A.I., Fernandez-Capetillo O. Global chromatin compaction limits the strength of the DNA damage response. J. Cell Biol. 2007;178:1101–1108. doi: 10.1083/jcb.200704140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M., Nunez S., Heard E., Narita M., Lin A.W., Hearn S.A., Spector D.L., Hannon G.J., Lowe S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Ono T., Losada A., Hirano M., Myers M.P., Neuwald A.F., Hirano T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell. 2003;115:109–121. doi: 10.1016/s0092-8674(03)00724-4. [DOI] [PubMed] [Google Scholar]
- Ono T., Yamashita D., Hirano T. Condensin II initiates sister chromatid resolution during S phase. J. Cell Biol. 2013;200:429–441. doi: 10.1083/jcb.201208008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H., Mayl J., Chandra B., Pritchett C., Chandra T. Dermoid of the oral cavity: case report with histopathology correlation and review of literature. J. Radiol. Case Rep. 2016;10:19–27. doi: 10.3941/jrcr.v10i12.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A.H., O'Carroll D., Scherthan H., Mechtler K., Sauer S., Schofer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Scaffidi P., Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova O.A., Horikawa I., Zimonjic D.B., Popescu N.C., Bonner W.M., Barrett J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004;6:168–170. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- Sen P., Dang W., Donahue G., Dai J., Dorsey J., Cao X., Liu W., Cao K., Perry R., Lee J.Y. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015;29:1362–1376. doi: 10.1101/gad.263707.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker D.K., Dechat T., Kohlmaier A., Adam S.A., Bozovsky M.R., Erdos M.R., Eriksson M., Goldman A.E., Khuon S., Collins F.S. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. U S A. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A., Tanizawa H., Sriswasdi S., Iwasaki O., Chatterjee A.G., Speicher D.W., Levin H.L., Noguchi E., Noma K. Epigenetic regulation of condensin-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56 acetylation. Mol. Cell. 2012;48:532–546. doi: 10.1016/j.molcel.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K., Miura Y., Hayashi S., Takahashi M., Kurosaka M. DcR3 protects THP-1 macrophages from apoptosis by increasing integrin alpha4. Biochem. Biophys. Res. Commun. 2009;389:593–598. doi: 10.1016/j.bbrc.2009.09.027. [DOI] [PubMed] [Google Scholar]
- Turinetto V., Vitale E., Giachino C. Senescence in human mesenchymal stem cells: functional changes and implications in stem cell-based therapy. Int. J. Mol. Sci. 2016;17:1164–1182. doi: 10.3390/ijms17071164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte F., Sousae R., Cinquin B., Martin E.W., Krietsch J., Sanchez G., Inman M., Tsang H., Warr M., Passegue E. Progressive chromatin condensation and H3K9 methylation regulate the differentiation of embryonic and hematopoietic stem cells. Stem Cell Reports. 2015;5:728–740. doi: 10.1016/j.stemcr.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A., Shehadeh L.A., Yu H., Webster K.A. Age-related molecular genetic changes of murine bone marrow mesenchymal stem cells. BMC Genomics. 2010;11:229. doi: 10.1186/1471-2164-11-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong T.Y., Solis M.A., Chen Y.H., Huang L.L. Molecular mechanism of extrinsic factors affecting anti-aging of stem cells. World J. Stem Cells. 2015;7:512–520. doi: 10.4252/wjsc.v7.i2.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama Y., Zhu H., Zhang R., Noma K. A novel role for the condensin II complex in cellular senescence. Cell Cycle. 2015;14:2160–2170. doi: 10.1080/15384101.2015.1049778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K.R., Kang K.S. Aging-related genes in mesenchymal stem cells: a mini-review. Gerontology. 2013;59:557–563. doi: 10.1159/000353857. [DOI] [PubMed] [Google Scholar]
- Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348:1160–1163. doi: 10.1126/science.aaa1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Adli M., Zou J.Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P.L. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.