

Abstract
Gemcitabine (Gem) is a standard first-line treatment for pancreatic cancer (PC). However, its chemotherapeutic efficacy is hampered by various limitations such as short half-life, metabolic inactivation, and lack of tumor localizing. We previously synthesized a lipophilic Gem derivative (Gem formyl hexadecyl ester, GemC16) that exhibited improved antitumor activity in vitro. In this study, a target ligand N,N-dimethyl-1,3-propanediamine was conjugated to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[hydroxyl succinimidyl (polyethylene glycol-2000)] (DSPE-PEG-NHS) to form DSPE-PEG-2N. Then, pancreas-targeting liposomes (2N-LPs) were prepared using the film dispersion-ultrasonic method. GemC16-loaded 2N-LPs displayed near-spherical shapes with an average size distribution of 157.2 nm (polydispersity index (PDI) = 0.201). The encapsulation efficiency of GemC16 was up to 97.3% with a loading capacity of 8.9%. In human PC cell line (BxPC-3) and rat pancreatic acinar cell line (AR42J), cellular uptake of 2N-LPs was significantly enhanced compared with that of unmodified PEG-LPs. 2N-LPs exhibited more potent in vitro cytotoxicity against BxPC-3 and AR42J cell lines than PEG-LPs. After systemic administration in mice, 2N-LPs remarkably increased drug distribution in the pancreas. In an orthotopic tumor mouse model of PC, GemC16-bearing liposomes were more effective in preventing tumor growth than free GemC16. Among these treatments, 2N-LPs showed the best curative effect. Together, 2N-LPs represent a promising nanocarrier to achieve pancreas-targeting drug delivery, and this work would provide new ideas for the chemotherapy of PC.
Keywords: pancreatic cancer, gemcitabine derivative, propanediamine, liposomes, targeted drug delivery
Introduction
Pancreatic cancer (PC) is a common digestive system malignant tumor, and almost 90% of PCs are pancreatic ductal adenocarcinoma (PDAC) [1]. According to the reported statistics, there were nearly 27.7 million new PC cases and 26.6 million death cases worldwide in 2008 [2]. Mortality and morbidity were basically the same. In recent years, the incidence of PC has increased yearly. PC is the fourth most common cause of cancer death in the United States and is projected to become the second leading cause of cancer-related deaths in 2030 [3, 4]. Due to the high incidence of metastatic disease at initial diagnosis, lack of effective therapies, and poor prognosis, the 5-year survival rate of PC is below 5% [5]. Surgery is the only method that can eradicate PC and is often combined with postoperative chemotherapy to reduce the recurrence rate [6]. Because of the insidious onset of this cancer, the majority of PC patients have no specific symptoms at an early stage [7–9]. Approximately 70%~80% of PC patients have been diagnosed as being in the advanced stages, thus losing the chance to be operated [10]. Therefore, improving the quality of life of patients and prolonging survival through nonsurgical treatments such as chemotherapy, radiotherapy, combined radiotherapy and chemotherapy, and immunotherapy have great significance [11–13].
Gemcitabine (2′,2′-difluoro-2′-deoxycytidine, Gem, Fig. 1a), a nucleoside analog, is a first-line chemotherapy for PC [14]. Gem itself has no antitumor efficacy until entering the cells and is finally phosphorylated in a stepwise manner to the active Gem diphosphate (dFdCDP) and gemcitabine triphosphate (dFdCTP) [15]. Nevertheless, Gem is extensively and rapidly metabolized in vivo by cytidine deaminase into the deaminated inactive metabolite 2′,2′-difluorodeoxyuridine (dFdU) and is then excreted in the urine [16]. Moreover, the small molecular weight of Gem promotes rapid renal clearance. Therefore, Gem has a very short plasma half-life (8–17 min) [17]. In addition, the efficacy of Gem is hindered by its indiscriminate targeting of both cancerous and noncancerous cells [18] and by poor cellular uptake due to the reliance on nucleoside transporters [19]. For all these reasons, even if the cytotoxic activity is potent, a dose of Gem must be up to 1 g/m2 to achieve the therapeutic drug level, which would further provoke side effects [20].
Fig. 1.
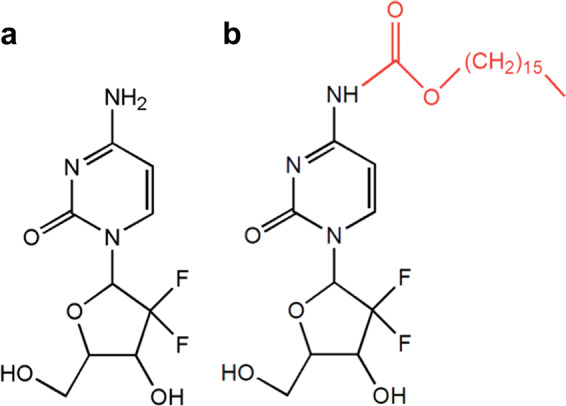
Chemical structures of a Gem and b GemC16
Various strategies have been reported to improve the bioavailability, metabolic stability, and efficacy of Gem. These strategies mainly include the use of Gem prodrugs/conjugates [21–23] and drug-loaded nanoformulations [20, 24–26]. Liposomes are nanosized lipid vesicles made from naturally occurring phospholipids, which consist of a hydrophilic core and a hydrophobic shell. Liposomes can protect drugs from rapid metabolic inactivation and improve pharmacokinetics and biodistribution [27]. Furthermore, the surface of liposomes could be structurally modified to prepare functionalized drug delivery systems [28]. However, Gem is a low-molecular-weight compound with good water solubility. Such molecules diffuse rapidly through the lipid bilayer, and drugs are difficult to be stably encapsulated in the core, thus limiting the shelf life and application of these liposomes [29–31].
In our previous research, a lipophilic Gem derivative, Gem formyl hexadecyl ester (GemC16), was designed and synthesized (Fig. 1b). Hexadecanoic acid, also known as palmitic acid, is a highly saturated fatty acid with good biocompatibility. Hexadecanoic acid was linked to Gem via a urethane bond [32]. With the introduction of a long carbon chain to Gem, lipophilicity was greatly increased. This strategy also provides chemical protection of the free amino group in the N4 position, which is susceptible to rapid hydrolysis and inactivation through cytidine deaminase presented in body fluids. In vitro cell proliferation and apoptosis analyses showed that GemC16 exhibited stronger antitumor activity than Gem and had potential for clinical application.
Lipophilic derivatives improve the lipophilicity of drugs and thereby facilitate drug entrapment in liposomes, which leads to further passive targeting to cancerous tissue via the enhanced permeability and retention effect (EPR) and improved efficacy [33, 34]. However, PDAC has been reported to be a dense solid tumor that cannot achieve effective EPR accumulation [35–37]. Hence, other mechanisms for GemC16 to target the pancreas, such as local drug delivery or active targeting using site-specific ligands, should be considered. N,N,Nʹ-trimethyl-Nʹ-(2-hydroxyl-3-methyl-5-[123I] iodobenzyl)-1,3-propanediamine (123I-labeled HMPDM), a pancreas-imaging agent, has a strong ability to cross the blood−pancreas barrier (BPB) [38, 39]. Our previous research has shown that the propanediamine moiety on 123I-labeled HMPDM might be responsible for pancreas-specific affinity, and the propylenediamine derivatives (e.g., N,N-dimethyl-1,2-ethanediamine) also displayed excellent pancreas-targeting ability [40, 41].
Based on the above research, we chose N,N-dimethyl-1,3-propanediamine, a small molecular compound with clear structure and good safety, as a targeting ligand to conjugate to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[hydroxyl succinimidyl (polyethylene glycol-2000)] (DSPE-PEG-NHS) to obtain DSPE-PEG-2N and to develop pancreas-targeted GemC16-loaded liposomes (2N-LPs). The study systemically investigated the development, characterization, targeting ability, and therapeutic efficacy of the 2N-modified liposomal formulation. All of these studies provide new ideas for the chemotherapy of PC and lay the foundation for studying novel pancreas-targeted drug delivery systems.
Materials And Methods
Materials and regents
Gem (purity > 98.0%), cetyl chloroformate (purity > 98.0%), 4-dimethylaminopyridine (DMAP), N,N-dimethyl-1,3-propanediamine and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD) were obtained from Best Reagent Co., Ltd. (Chengdu, China). Lipoid E80 was purchased from Lipoid Co., Ltd. (Ludwigshafen, Germany). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) and DSPE-PEG-NHS were obtained from Laysan Bio, Inc. (Hawaii, USA). Cholesterol was provided by Kelong Company (Chengdu, China). d-luciferin potassium salt and rabbit anti-Ki-67 polyclonal antibody were obtained from Abcam (Cambridge, UK). All other chemicals were of analytical or high-performance liquid chromatography grade and were purchased from commercial sources.
Animals and cell cultures
All animal experiments were performed in compliance with the guidelines for animal experiments of the Institutional Animal Care and Ethics Committee of Sichuan University (Chengdu, China). Female C57BL/6 mice (bodyweight: 20 ± 2 g) were supplied by the Experimental Animal Center of West China, Sichuan University. The mice were raised in a germ-free environment (temperature: 25 ± 2 °C, relative humidity: 50% ± 10%, dark−light cycle: 12 h) for at least 1 week to adapt to the housing conditions before the experiments. The animals were allowed free access to water and food.
BxPC-3 cells (human pancreatic adenocarcinoma cell line), AR42J cells (rat pancreatic acinar cell line), and Panc02-luc cells (luciferase-tagged murine pancreatic adenocarcinoma cell line) were obtained from ATCC (Virginia, USA). The BxPC-3, AR42J, and Panc02-luc cells were cultured in either RPMI-1640 (GIBCO, USA) or DMEM with high glucose (GIBCO, USA). Both RPMI-1640 and DMEM were supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. The cells were maintained in a 5% CO2 atmosphere at 37 °C, and the cell medium was changed every other day.
Synthesis of DSPE-PEG-2N
The synthetic route of DSPE-PEG-2N is shown in Scheme 1. Briefly, 40 mg of DSPE-PEG-NHS and 5 mg of DMAP were dissolved in 10 mL of dimethyl sulfoxide (DMSO). Then, 20 μL of N,N-dimethyl-1,3-propanediamine (700 mg/mL) was added, and the mixture was stirred gently at room temperature for 48 h. The resulting reaction solution was introduced into a dialysis membrane bag (molecular weight cutoff of 500 Da) and dialyzed against distilled water for 72 h. The water was changed every 4 h, and the solution after dialysis was freeze-dried. Finally, the DSPE-PEG-2N powder was stored at −20 °C and identified by nuclear magnetic resonance hydrogen spectrum (1H NMR).
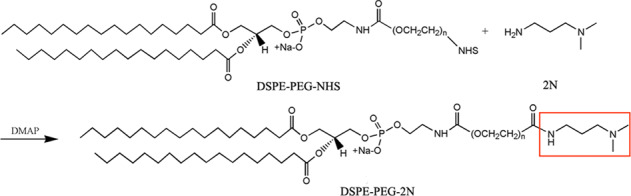
Scheme 1
Preparation of liposomes
GemC16 was synthesized and purified as described previously [32]. GemC16-loaded liposomes were prepared by the film dispersion-ultrasonic method. Briefly, E80 (12 mg), cholesterol (4 mg), DSPE-PEG (3.2 mg) and DSPE-PEG-2N (0.8 mg) were dissolved in 2 mL of chloroform at a mass ratio of 3:1:0.8:0.2. Next, 200 μL of ethanol-acetone (1:1, v/v) containing 2 mg of GemC16 was added, and the mixture was stirred for 10 min at room temperature. The final mixture was evaporated under reduced pressure in a rotary evaporator at 37 °C for 20 min to remove the organic solvent and form a uniform transparent film. Next, the film was hydrated with phosphate-buffered saline (PBS) (pH 7.4) at 37 °C and sonicated for 4 min at 80 W (3 s pulse and 8 s rest, ultrasonic cell crasher). Finally, 2N-LPs encapsulating GemC16 were successfully prepared. Furthermore, E80, cholesterol and DSPE-PEG at a mass ratio of 3:1:1 were used to prepare unmodified PEG-LPs. Additionally, blank PEG-LPs and 2N-LPs were prepared in the same way.
Characterization of liposomes
The particle size distribution and zeta potentials of PEG-LPs and 2N-LPs were determined by dynamic light scattering using a Malvern Zetasizer NanoZS90 system (Malvern, UK). All experiments were repeated three times. The morphologies of the PEG-LPs and 2N-LPs were observed using a transmission electron microscope (TEM) (H-600, Hitachi, Japan). A dispersion of liposomes was placed on a copper grid covered with nitrocellulose and negatively stained with 2% (w/v) phosphotungstic acid for 30 s. Then, the sample was dried and subjected to TEM observation.
The content of GemC16 encapsulated in the PEG-LPs and 2N-LPs was determined by a membrane ultrafiltration method. The encapsulation efficiency (EE) and drug-loading (DL) capacity were calculated according to the following equations [42]:
A high-performance liquid chromatography system equipped with an ultraviolet detector (Agilent Technologies, Santa Clara, CA, USA) was used to analyze the concentration of GemC16. The chromatographic separation was performed on an octadecyl silica (ODS) column (Kromasil, 150 mm × 4.6 mm, 5 μm), and the column temperature was maintained at 35 °C. The mobile phase consisted of (A) 0.3% (v/v) aqueous formic acid and (B) methanol (6/94, v/v). The flow rate was 1.0 mL/min, and the detection wavelength was set at 293 nm.
Cellular uptake study
DiD-labeled PEG-LPs and 2N-LPs were prepared as previously described in the sections on liposome preparation. BxPC-3 and AR42J cells in the logarithmic growth phase were seeded in six-well culture plates (5 × 105 cells/plate) and incubated for 24 h before the treatment. Then, the original medium was replaced with serum-free culture medium containing free DiD, DiD-labeled PEG-LPs and 2N-LPs (DiD concentration was 500 ng/mL). Each sample was analyzed in triplicate. After incubation at 37 °C for 1 and 2 h, the cells were washed three times with cold PBS. Thereafter, the cell suspensions were collected and centrifuged at 2000 r/min for 3 min. The supernatants were discarded, and the cells were washed two times with PBS. The cells were resuspended in 0.5 mL PBS after the final centrifugation. The fluorescence intensity of the cells was evaluated by a flow cytometer (Cytomics™ FC 500, Beckman Coulter, USA).
Cytotoxicity assay
The cytotoxicities of PEG-LPs and 2N-LPs were evaluated by the MTT assay. The BxPC-3 or AR42J cells were seeded in 96-well culture plates at a density of 1 × 104 cells/well and incubated in a 5% CO2 atmosphere at 37 °C for 24 h. Then, the cells were treated with different concentrations of PEG-LPs or 2N-LPs (dispersed in serum-free culture medium) for 4 h. Next, 20 μL of an MTT solution (5 mg/mL in PBS) was added into each well. The cells were incubated at 37 °C for another 4 h. Thereafter, the medium was discarded, and the cells were mixed with 150 μL DMSO. The absorbance at 570 nm was measured using a microplate reader (Thermo, Varioskan Flash). The cell viability (%) was determined according to the equation (Atest/Acontrol) × 100%. Cells treated with drug-free medium were considered 100% viable and were used as a control.
In vivo biodistribution study
The in vivo biodistribution of 2N-LPs was examined in C57BL/6 mice. Briefly, the mice were randomly divided into three groups (n = 3). Free DiD, DiD-labeled PEG-LPs and 2N-LPs were administered intravenously at an equivalent dose of 200 μg/kg DiD. The mice were sacrificed at 0.5 h after administration, and the major organs, including the heart, liver, spleen, lung, kidney, brain and pancreas, were collected. These tissues were carefully washed with cold physiological saline (0.9%, w/v) and dried with filter paper for further ex vivo imaging of DiD fluorescence using an in vivo imaging system (Quick View 3000, Bio-Real, Austria).
In vivo antitumor efficacy
A fluorescent orthotopic mouse model of PC was established as described previously [43, 44]. Briefly, each mouse was anesthetized with 1% sodium pentobarbital (50 mg/kg, i.p.) and subsequently underwent sterile laparotomy. Approximately 20 μL PBS containing Panc02-luc cells (1.25 × 108 cells/mL) was injected into the tail of the pancreas, keeping the needle inside for 30 s to avoid leakage. Then, the incision was sutured and observed until the mice woke up from anesthesia. After modeling for 2 weeks (day 0), the tumor-bearing mice were randomly divided into five groups of five each as follows: the control group, Gem-treated group, GemC16-treated group, GemC16-loaded PEG-LPs-treated group and GemC16-loaded 2N-LPs-treated group. On days 0, 4, 8, 12, and 16, physiological saline, free Gem, GemC16, PEG-LPs, and 2N-LPs (5 mg/kg, equivalent to Gem) were injected into the mice intravenously. On day 20, a D-luciferin potassium salt solution was administered intraperitoneally to tumor-bearing mice at a dose of 150 mg/kg. The tumor could be imaged under an in vivo imaging system 10 min later.
After collecting in vivo fluorescence data, the animals were sacrificed, and Panc02-luc-bearing pancreases were excised for follow-up analysis. The pancreases were fixed overnight in 4% buffered paraformaldehyde, sectioned and stained for histopathology and immunohistochemistry analysis [45].
Statistical analysis
All data are represented as the mean ± standard deviation (SD). Statistical analysis was performed by two-tailed Student’s t test. The difference was considered to be statistically significant at P value < 0.05.
Results
Synthesis of DSPE-PEG-2N
DSPE-PEG-2N was synthesized by conjugating the active ester of DSPE-PEG-NHS and the primary amine group of N,N-dimethyl-1,3-propanediamine (Scheme 1). The product was verified using 1H NMR (Fig. 2). The presence of the peak of a new amide hydrogen atom (δ 8.45) suggested that a new amide bond formed, proving the successful synthesis of DSPE-PEG-2N (Fig. 2b).
Fig. 2.

The 1H NMR spectroscopy of a DSPE-PEG and b DSPE-PEG-2N
Preparation and characterization of 2N-LPs
The liposomes were prepared according to the film dispersion-ultrasonic technique. N,N-dimethyl-1,3-propanediamine was stably assembled on the surface of 2N-LPs to establish a pancreas-targeted drug delivery system (Fig. 3a). The particle sizes of blank PEG-LPs and 2N-LPs were 124.7 ± 8.2 nm and 132.2 ± 7.5 nm, and the zeta potentials were −16.6 and −12.1 mV, respectively (Table 1). The low PDI indicated that these liposomes had a uniform size. After the encapsulation of GemC16 into liposomes, the particle sizes of PEG-LPs (150.7 ± 8.8 nm) and 2N-LPs (157.2 ± 10.3 nm) were slightly increased, while the potentials remained unchanged. The TEM images show that the PEG-LPs and 2N-LPs displayed a near-spherical morphology (Fig. 3b).
Fig. 3.
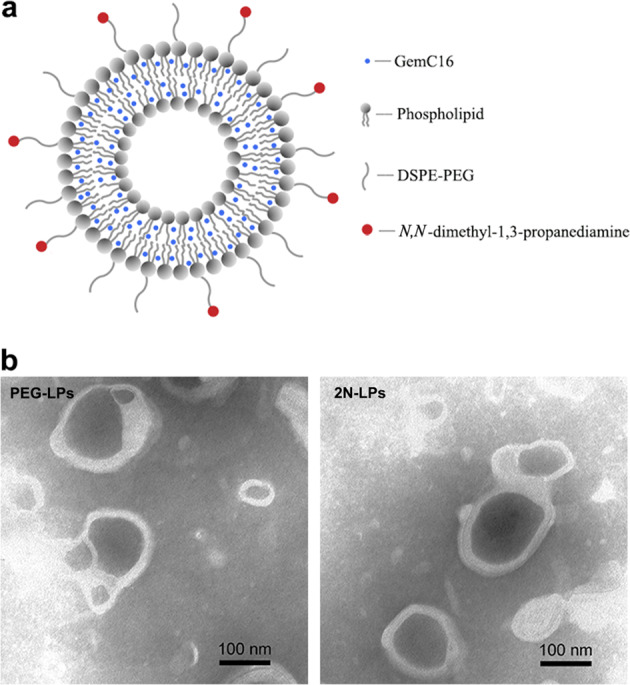
a The structural scheme of GemC16-loaded 2N-LPs. b Representative TEM images of PEG-LPs and 2N-LPs
Table 1.
Characterization of liposomes (n = 3, mean ± SD)
| Entry | Size (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| Blank PEG-LPs | 124.7 ± 8.2 | 0.152 ± 0.10 | –16.6 ± 1.6 |
| Blank 2N-LPs | 132.2 ± 7.5 | 0.186 ± 0.08 | –12.1 ± 1.3 |
| GemC16-loaded PEG-LPs | 150.7 ± 8.8 | 0.185 ± 0.09 | –16.2 ± 1.4 |
| GemC16-loaded 2N-LPs | 157.2 ± 10.3 | 0.201 ± 0.08 | –13.0 ± 1.2 |
PDI polydispersity index
In this work, the entrapment efficiency was studied by the ultrafiltration method, and an ultrafiltration membrane with an interception molecular weight of 3 kDa was used. Free GemC16 could pass through the ultrafiltration membrane, whereas drugs encapsulated into liposomes could not. The encapsulation efficiency of GemC16 in PEG-LPs (97.6%) and 2N-LPs (97.3%) was consistently above 97%, and the average loading capacity for both was determined to be 8.9% ± 0.1%. The results illustrated that GemC16 was well incorporated into the liposomes.
Cellular uptake study
To investigate cellular internalization, DiD-labeled PEG-LPs and 2N-LPs were incubated with BxPC-3 and AR42J cells. As shown in Fig. 4, the fluorescence intensity of BxPC-3 cells treated with 2N-LPs was approximately 1.7 times higher than those treated with PEG-LPs at 1 h and was approximately 2.1 times higher at 2 h. The results were similar for the AR42J cells. Both BxPC-3 and AR42J cells displayed significantly more efficient cellular uptake of 2N-LPs than that of PEG-LPs, suggesting superior cellular delivery efficiency by N,N-dimethyl-1,3-propanediamine.
Fig. 4.

Cellular uptake of DiD-labeled PEG-LPs and 2N-LPs after incubation with BxPC-3 and AR42J cells for a 1 h and b 2 h. ***P < 0.001 versus PEG-LPs group
In vitro cytotoxicity
We next evaluated the in vitro cytotoxicity of PEG-LPs and 2N-LPs in the BxPC-3 and AR42J cell lines, respectively, by using a standard MTT assay. As shown in Fig. 5, the cells were treated with serial concentrations of drugs, and the relative cell viability displayed concentration-dependent profiles for GemC16-loaded PEG-LPs and 2N-LPs. For cells under investigation, once the drug concentration exceeded 30 μmol/L, 2N-LPs showed higher cytotoxicity compared with that of PEG-LPs after 4 h of treatment.
Fig. 5.

In vitro cytotoxicity of 2N-LPs in a BxPC-3 and b AR42J cells as determined by standard MTT assay. *P < 0.05, ***P < 0.001 versus PEG-LPs group
In vivo biodistribution study
To evaluate the pancreas-targeting efficiency of 2N-LPs, the tissue distribution of DiD, DiD-labeled PEG-LPs and 2N-LPs was studied. Ex vivo fluorescence images of the main organs (heart, liver, spleen, lung, kidney, brain and pancreas) at 0.5 h after administration were obtained (Fig. 6a). For the free DiD group, no fluorescence signal was detected in each organ. PEG-LPs and 2N-LPs were distributed in various organs at different concentrations. Compared to that of PEG-LPs, the intensity of 2N-LPs in the pancreas was higher (Fig. 6b). This indicated that the proposed propanediamine derivative had good BPB-penetration efficiency and significantly improved pancreas-specific accumulation of drugs.
Fig. 6.

a Ex vivo DiD fluorescence images showing the drug biodistribution of free DiD, DiD-labeled PEG-LPs and 2N-LPs in mice at 0.5 h post injection. b Fluorescence intensity of DiD in the pancreas at 0.5 h after intravenous administration of DiD-labeled PEG-LPs and 2N-LPs. *P < 0.05 versus PEG-LPs group
In vivo antitumor efficacy
The in vivo antitumor efficacy of GemC16-loaded 2N-LPs was evaluated in a fluorescent orthotopic mouse model of PC [44]. The tumor volume and the bioluminescence signal increased with time. The experiment was performed by intravenously injecting physiological saline, free Gem, GemC16, GemC16-loaded PEG-LPs and 2N-LPs into tumor-bearing C57BL/6 mice. The detection of a fluorescence signal 2 weeks after implantation visually confirmed the successful implantation and growth of PC tumors. All of the test drugs exhibited varying degrees of antitumor activity compared to that of the control group (physiological saline group), and the fluorescence intensity of the tumor was sequentially decreased in the order of grouping (Gem < GemC16 < PEG-LPs < 2N-LPs) (Fig. 7a, b). The in vivo antitumor activity of GemC16 was stronger than that of Gem, which was in accordance with the in vitro results [32]. Compared with that of free GemC16, intravenous injection of PEG-LPs and 2N-LPs was more efficient in inhibiting tumor growth.
Fig. 7.

In vivo antitumor activity of Gem, GemC16, PEG-LPs and 2N-LPs in a fluorescent orthotopic mouse model of PC. Data represent mean ± SD. a Fluorescence images of orthotopic pancreatic tumors (n = 5). b Fluorescence intensity of orthotopic pancreatic tumors (n = 5). c H&E staining and Ki-67 antigen staining of Panc02-luc-bearing pancreases (n = 3)
Furthermore, hematoxylin and eosin (H&E) staining and Ki-67 antigen staining assays of Panc02-luc-bearing pancreases were investigated. For H&E staining (Fig. 7c), histopathological examination revealed obvious pancreatic tissue damage in the control group and Gem- and GemC16-treated groups, manifested as acinar cell necrosis, acinar epithelial cell nuclear pyknosis, and cell breakage or disappearance. Moreover, the intercellular space was enlarged. The PEG-LPs treatment resulted in less pancreatic cell damage. Nevertheless, the boundary of the acinar epithelial cells was not clear in the PEG-LPs-treated group, and the shape of some pancreatic acinar cells was changed. In contrast, 2N-LPs showed a better protective effect against tumor-bearing pancreas. The acinar cells in the 2N-LPs-treated group were structurally intact and regularly arranged. In addition, the interlobular stroma had a normal morphology.
Antigen Ki-67, a nuclear protein, is widely used as a cell proliferation marker [45]. Remarkably, the PEG-LPs- and 2N-LPs-treated groups showed lower Ki-67-positive rates (22.4% and 19.3%, respectively) than those of the control group (52.3%) and the Gem- (40.6%) and GemC16-treated groups (31.8%, Fig. 7c). The above results indicated that embedding GemC16 in liposomes significantly improved its efficacy, and the therapeutic effect of 2N-LPs was slightly better than that of PEG-LPs.
Discussion
Gem is a clinically approved (United States Food and Drug Administration: US-FDA) first-line therapy for PC as a single agent. A short half-life, rapid metabolic inactivation, and lack of tumor localization are the major deficiencies of Gem therapy [30]. Nanoengineered systems such as micelles [46], nanoparticles [47] and liposomes [48] have been proven to elevate the in vivo efficacy of cytotoxic agents via improving pharmacokinetic properties and biodistribution profiles and targeting drugs to cancerous tissues. Liposomes, nanosized artificial vesicles with a lipid-bilayer boundary similar to cell membranes, are widely used in tumor-targeting delivery of both lipophilic and hydrophilic drugs [49]. Highly lipophilic drugs are stably entrapped within the lipid bilayer of liposomes [50], while highly hydrophilic drugs such as Gem are located exclusively in the internal aqueous compartment, which rapidly diffuses through the bilayer [51]. Transforming hydrophilic drugs into lipophilic prodrugs or conjugates is a common approach to improve drug incorporation into liposomes [52, 53].
The N4 position of Gem is usually modified with lipophilic chains to improve its physical and chemical properties and biological characteristics [25]. This method also prevents Gem from being quickly metabolized to its deaminated inactive metabolite dFdU, which enhances the in vivo stability of the drug. GemC16, a novel lipophilic Gem derivative, was designed and synthesized in our previous study. Hexadecanoic acid was linked to the 4-amino group of Gem via a urethane bond, and the lipophilicity of Gem was increased. There is no doubt that GemC16 greatly improved the encapsulation efficiency and physical stability of liposomes. Importantly, it has been proved that GemC16 was more potent in inhibiting the proliferation of BxPC-3, AR42J and A549 (human lung adenocarcinoma cell line) cells than Gem. For the BxPC-3 cells, GemC16 induced cell cycle arrest and apoptosis more effectively than Gem. The above results indicated that GemC16 had much stronger antitumor activity than Gem and possessed potential application in clinical practice.
In previous works, small propanediamine derivatives significantly enhanced pancreas drug accumulation in rats [40, 41]. An organic cation transport protein mediates the transport of drugs across the cell membrane and contributes to pancreas-specific accumulation [40]. Herein, we chose N,N-dimethyl-1,3-propanediamine, a linear propanediamine derivative, to modify the liposomes and develop a novel pancreas-targeted drug delivery system of GemC16 for PC therapy. N,N-dimethyl-1,3-propanediamine was conjugated to DSPE-PEG-NHS to obtain DSPE-PEG-2N. Then, DSPE-PEG-2N was incorporated into the liposome surface to target 2N-LPs to the pancreas. The particle size of the liposomes was not significantly changed after modification with a small molecular compound, N,N-dimethyl-1,3-propanediamine, while the potential was slightly increased due to the presence of a tertiary amino group in N,N-dimethyl-1,3-propanediamine (Table 1). Modification of Gem with a long-chain hydrocarbon significantly promoted the encapsulation of drugs, and the encapsulation efficiency of GemC16 in both 2N-LPs and unmodified PEG-LPs was above 97%.
The BxPC-3 human PC cell line and the AR42J rat pancreatic acinar cell line were used to investigate the cellular uptake of 2N-LPs. AR42J is the only currently available cell line that maintains many characteristics of normal pancreatic acinar cells. AR42J has been extensively used as an in vitro model to study the exocrine pancreas [54]. The intracellular delivery efficiency of 2N-LPs in BxPC-3 and AR42J cells was prominently superior to that of PEG-LPs. The excellent cellular delivery efficiency indicated the possibility of outstanding pancreas-targeting ability of 2N-LPs in vivo. The tissue distribution studies in C57BL/6 mice confirmed our assumption. The tissue accumulation of PEG-LPs in the pancreas was remarkably increased after 2N modification (Fig. 6).
Cell lines or tumor fragments are usually implanted subcutaneously to establish heterotopic models, while pancreatic tumors grow in the organ of origin [55]. In contrast to heterotopic models, orthotopic models mimic the real microenvironment of tumors, providing an accurate prediction of therapeutic efficacy in response to drugs [56]. The orthotopic xenograft model established here derives tumors from the murine pancreatic adenocarcinoma cell line, Panc02-luc, which is genetically engineered to express luciferase [44]. This model enables imaging and monitoring of tumor growth in a noninvasive way. As a result, the antitumor efficacy of GemC16-loaded liposomes against PC was much stronger than that of free GemC16, and that of 2N-LPs was slightly better than that of PEG-LPs (Fig. 7). PEGylated liposomes are able to inhibit uptake by the reticuloendothelial system and increase the blood circulation time of drugs [57]. Therefore, PEG-LPs could also significantly improve the in vivo behavior of GemC16.
In summary, a novel pancreas-targeted drug delivery system was designed and constructed using N,N-dimethyl-1,3-propanediamine as a targeting ligand. A new carrier material (DSPE-PEG-2N) was synthesized, and PEG-LPs and 2N-LPs were successfully prepared by the film dispersion-ultrasonic method. Liposomes inhibited tumor growth more efficiently than did free GemC16, and the systemic delivery of 2N-LPs carrying GemC16 showed a better therapeutic effect in the orthotopic PC mouse model than that of PEG-LPs. Taken together, propylenediamine derivative-modified drug delivery systems represent a potential strategy for pancreas-targeted delivery of chemotherapy drugs.
Acknowledgements
We acknowledge the financial support of the Science & Technology Department of Sichuan Province (No. 2018SZ0012, No. 2019YJ0372), the Collaborative Innovation Center of Sichuan for Elderly Care and Health (No. YLZBZ1808), and Chengdu Medical College (No. CYZ17-07).
Author contributions
ZRZ and YCZ conceived and designed the study. PWL performed the experiments and generated and analyzed the data. SL and LYX helped with the animal and cell studies, and SL and LW helped with the synthesis studies. BLT assisted in data analysis. YCZ wrote the original manuscript, and ZRZ helped with manuscript editing.
Competing interests
The authors declare no competing interests.
References
- 1.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–17. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 4.Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–45. doi: 10.2147/DDDT.S60328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tran B, Whiteman DC, Webb PM, Fritschi L, Fawcett J, Risch HA, et al. Association between ultraviolet radiation, skin sun sensitivity and risk of pancreatic cancer. Cancer Epidemiol. 2013;37:886–92. doi: 10.1016/j.canep.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 6.Bockhorn M, Uzunoglu FG, Adham M, Imrie C, Milicevic M, Sandberg AA, et al. Borderline resectable pancreatic cancer: a consensus statement by the International Study Group of Pancreatic Surgery (ISGPS) Surgery. 2014;155:977–88. doi: 10.1016/j.surg.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Ahlgren JD. Chemotherapy for pancreatic carcinoma. Cancer. 1996;78:654–63. doi: 10.1002/(SICI)1097-0142(19960801)78:3<654::AID-CNCR46>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 8.Bria E, Carlini P, Gelibter A, Ruggeri E, Ceribelli A, Pino M, et al. Current status of targeted agents in advanced pancreatic cancer (APC): a pooled analysis of 2,361 patients (pts) enrolled in six phase III trials. J Clin Oncol. 2006;24:4126. [Google Scholar]
- 9.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–72. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 10.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–20. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corsini MM, Miller RC, Haddock MG, Donohue JH, Farnell MB, Nagorney DM, et al. Adjuvant radiotherapy and chemotherapy for pancreatic carcinoma: the Mayo Clinic experience (1975-2005) J Clin Oncol. 2008;26:3511–6. doi: 10.1200/JCO.2007.15.8782. [DOI] [PubMed] [Google Scholar]
- 12.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–10. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 13.Amedei A, Niccolai E, Prisco D. Pancreatic cancer: role of the immune system in cancer progression and vaccine-based immunotherapy. Hum Vaccin Immunother. 2014;10:3354–68. doi: 10.4161/hv.34392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown K, Dixey M, Weymouth-Wilson A, Linclau B. The synthesis of gemcitabine. Carbohydr Res. 2014;387:59–73. doi: 10.1016/j.carres.2014.01.024. [DOI] [PubMed] [Google Scholar]
- 15.Baraniak J, Pietkiewicz A, Kaczmarek R, Radzikowska E, Kulik K, Krolewska K, et al. N-Acyl-phosphoramidates as potential novel form of gemcitabine prodrugs. Bioorg Med Chem. 2014;22:2133–40. doi: 10.1016/j.bmc.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 16.Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- 17.Cattel L, Airoldi M, Delprino L, Passera R, Milla P, Pedani F. Pharmacokinetic evaluation of gemcitabine and 2’,2’-difluorodeoxycytidine-5’-triphosphate after prolonged infusion in patients affected by different solid tumors. Ann Oncol. 2006;17(Suppl 5):142–7. doi: 10.1093/annonc/mdj970. [DOI] [PubMed] [Google Scholar]
- 18.Song W, Tang Z, Li M, Lv S, Sun H, Deng M, et al. Polypeptide-based combination of paclitaxel and cisplatin for enhanced chemotherapy efficacy and reduced side-effects. Acta Biomater. 2014;10:1392–402. doi: 10.1016/j.actbio.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 19.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–57. [PubMed] [Google Scholar]
- 20.Oluwasanmi A, Al-Shakarchi W, Manzur A, Aldebasi MH, Elsini RS, Albusair MK, et al. Diels Alder-mediated release of gemcitabine from hybrid nanoparticles for enhanced pancreatic cancer therapy. J Control Release. 2017;266:355–64. doi: 10.1016/j.jconrel.2017.09.027. [DOI] [PubMed] [Google Scholar]
- 21.Pratt SE, Durland-Busbice S, Shepard RL, Donoho GP, Starling JJ, Wickremsinhe ER, et al. Efficacy of low-dose oral metronomic dosing of the prodrug of gemcitabine, LY2334737, in human tumor xenografts. Mol Cancer Ther. 2013;12:481–90. doi: 10.1158/1535-7163.MCT-12-0654. [DOI] [PubMed] [Google Scholar]
- 22.Han H, Jin Q, Wang Y, Chen Y, Ji J. The rational design of a gemcitabine prodrug with AIE-based intracellular light-up characteristics for selective suppression of pancreatic cancer cells. Chem Commun. 2015;51:17435–8. doi: 10.1039/c5cc06654a. [DOI] [PubMed] [Google Scholar]
- 23.Abu-Fayyad A, Nazzal S. Gemcitabine-vitamin E conjugates: synthesis, characterization, entrapment into nanoemulsions, and in-vitro deamination and antitumor activity. Int J Pharm. 2017;528:463–70. doi: 10.1016/j.ijpharm.2017.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mura S, Bui DT, Couvreur P, Nicolas J. Lipid prodrug nanocarriers in cancer therapy. J Control Release. 2015;208:25–41. doi: 10.1016/j.jconrel.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 25.May JP, Undzys E, Roy A, Li SD. Synthesis of a gemcitabine prodrug for remote loading into liposomes and improved therapeutic effect. Bioconjug Chem. 2016;27:226–37. doi: 10.1021/acs.bioconjchem.5b00619. [DOI] [PubMed] [Google Scholar]
- 26.Nesbitt H, Sheng Y, Kamila S, Logan K, Thomas K, Callan B, et al. Gemcitabine loaded microbubbles for targeted chemo-sonodynamic therapy of pancreatic cancer. J Control Release. 2018;279:8–16. doi: 10.1016/j.jconrel.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 27.Cattel L, Ceruti M, Dosio F. From conventional to stealth liposomes: a new frontier in cancer chemotherapy. Tumori. 2003;89:237–49. doi: 10.1177/030089160308900302. [DOI] [PubMed] [Google Scholar]
- 28.Mu LM, Ju RJ, Liu R, Bu YZ, Zhang JY, Li XQ, et al. Dual-functional drug liposomes in treatment of resistant cancers. Adv Drug Deliv Rev. 2017;115:46–56. doi: 10.1016/j.addr.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Patel KR, Baldeschwieler JD. Treatment of intravenously implanted Lewis lung carcinoma with liposome-encapsulated cytosine arabinoside and non-specific immunotherapy. Int J Cancer. 1984;34:415–20. doi: 10.1002/ijc.2910340320. [DOI] [PubMed] [Google Scholar]
- 30.Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100:331–46. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Crosasso P, Brusa P, Dosio F, Arpicco S, Pacchioni D, Schuber F, et al. Antitumoral activity of liposomes and immunoliposomes containing 5-fluorouridine prodrugs. J Pharm Sci. 1997;86:832–9. doi: 10.1021/js9604467. [DOI] [PubMed] [Google Scholar]
- 32.Li PW, Guo L, Deng L, Wang L, Luo S, Du ZW, et al. Study on the synthesis and in vitro anti-tumor activity of Gemcitabine derivative. West China. J Pharm Sci. 2018;3:122–5. [Google Scholar]
- 33.Sloat BR, Sandoval MA, Li D, Chung WG, Lansakara PD, Proteau PJ, et al. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm. 2011;409:278–88. doi: 10.1016/j.ijpharm.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ngoune R, Peters A, von Elverfeldt D, Winkler K, Putz G. Accumulating nanoparticles by EPR: a route of no return. J Control Release. 2016;238:58–70. doi: 10.1016/j.jconrel.2016.07.028. [DOI] [PubMed] [Google Scholar]
- 35.Hosoki T. Dynamic CT of pancreatic tumors. AJR Am J Roentgenol. 1983;140:959–65. doi: 10.2214/ajr.140.5.959. [DOI] [PubMed] [Google Scholar]
- 36.Sofuni A, Iijima H, Moriyasu F, Nakayama D, Shimizu M, Nakamura K, et al. Differential diagnosis of pancreatic tumors using ultrasound contrast imaging. J Gastroenterol. 2005;40:518–25. doi: 10.1007/s00535-005-1578-z. [DOI] [PubMed] [Google Scholar]
- 37.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–20. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto K, Som P, Srivastava SC, Meinken GE, Brill AB. Pancreas accumulation of radioiodinated HIPDM in mice and rats. J Nucl Med. 1985;26:765–9. [PubMed] [Google Scholar]
- 39.Yamamoto K, Shibata T, Saji H, Kubo S, Aoki E, Fujita T, et al. Human pancreas scintigraphy using iodine-123-labeled HIPDM and SPECT. J Nucl Med. 1990;31:1015–9. [PubMed] [Google Scholar]
- 40.Li J, Zhang J, Fu Y, Sun X, Gong T, Jiang J, et al. Dual pancreas- and lung-targeting therapy for local and systemic complications of acute pancreatitis mediated by a phenolic propanediamine moiety. J Control Release. 2015;212:19–29. doi: 10.1016/j.jconrel.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 41.Luo S, Li P, Li S, Du Z, Hu X, Fu Y, et al. N,N-dimethyl tertiary amino group mediated dual pancreas- and lung-targeting therapy against acute pancreatitis. Mol Pharm. 2017;14:1771–81. doi: 10.1021/acs.molpharmaceut.7b00028. [DOI] [PubMed] [Google Scholar]
- 42.Li M, Tang Z, Lv S, Song W, Hong H, Jing X, et al. Cisplatin crosslinked pH-sensitive nanoparticles for efficient delivery of doxorubicin. Biomaterials. 2014;35:3851–64. doi: 10.1016/j.biomaterials.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 43.Moreno JA, Sanchez A, Hoffman RM, Nur S, Lambros MP. Fluorescent orthotopic mouse model of pancreatic cancer. J Vis Exp. 2016;115:1–5. doi: 10.3791/54337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoki H, Aoki M, Katsuta E, Ramanathan R, Idowu MO, Spiegel S, et al. Host sphingosine kinase 1 worsens pancreatic cancer peritoneal carcinomatosis. J Surg Res. 2016;205:510–7. doi: 10.1016/j.jss.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou M, Li J, Li C, Guo L, Wang X, He Q, et al. Tertiary amine mediated targeted therapy against metastatic lung cancer. J Control Release. 2016;241:81–93. doi: 10.1016/j.jconrel.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Fan W, Dai X, Katragadda U, McKinley D, Teng Q, et al. Enhanced tumor delivery of gemcitabine via PEG-DSPE/TPGS mixed micelles. Mol Pharm. 2014;11:1140–50. doi: 10.1021/mp4005904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulhari H, Pooja D, Kota R, Reddy TS, Tabor RF, Shukla R, et al. Cyclic RGDfK peptide functionalized polymeric nanocarriers for targeting gemcitabine to ovarian cancer cells. Mol Pharm. 2016;13:1491–500. doi: 10.1021/acs.molpharmaceut.5b00935. [DOI] [PubMed] [Google Scholar]
- 48.Ji T, Lang J, Wang J, Cai R, Zhang Y, Qi F, et al. Designing liposomes to suppress extracellular matrix expression to enhance drug penetration and pancreatic tumor therapy. ACS Nano. 2017;11:8668–78. doi: 10.1021/acsnano.7b01026. [DOI] [PubMed] [Google Scholar]
- 49.Deshpande S, Spoelstra WK, van Doorn M, Kerssemakers J, Dekker C. Mechanical division of cell-sized liposomes. ACS Nano. 2018;12:2560–8. doi: 10.1021/acsnano.7b08411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gulati M, Grover M, Singh S, Singh M. Lipophilic drug derivatives in liposomes. Int J Pharm. 1998;165:129–68. [Google Scholar]
- 51.Moog R, Burger AM, Brandl M, Schuler J, Schubert R, Unger C, et al. Change in pharmacokinetic and pharmacodynamic behavior of gemcitabine in human tumor xenografts upon entrapment in vesicular phospholipid gels. Cancer Chemother Pharmacol. 2002;49:356–66. doi: 10.1007/s00280-002-0428-4. [DOI] [PubMed] [Google Scholar]
- 52.Tokunaga Y, Iwasa T, Fujisaki J, Sawai S, Kagayama A. Liposomal sustained-release delivery systems for intravenous injection. IV. Antitumor activity of newly synthesized lipophilic 1-beta-D-arabinofuranosylcytosine prodrug-bearing liposomes. Chem Pharm Bull. 1988;36:3574–83. doi: 10.1248/cpb.36.3574. [DOI] [PubMed] [Google Scholar]
- 53.Rubas W, Supersaxo A, Weder HG, Hartmann HR, Hengartner H, Schott H, et al. Treatment of murine L1210 lymphoid leukemia and melanoma B16 with lipophilic cytosine arabinoside prodrugs incorporated into unilamellar liposomes. Int J Cancer. 1986;37:149–54. doi: 10.1002/ijc.2910370123. [DOI] [PubMed] [Google Scholar]
- 54.Sata N, Klonowski-Stumpe H, Han B, Haussinger D, Niederau C. Cytotoxicity of peroxynitrite in rat pancreatic acinar AR4-2J cells. Pancreas. 1997;15:278–84. doi: 10.1097/00006676-199710000-00010. [DOI] [PubMed] [Google Scholar]
- 55.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7:645–58. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 56.Arranz A, Ripoll J. Advances in optical imaging for pharmacological studies. Front Pharmacol. 2015;6:189. doi: 10.3389/fphar.2015.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen TM, Newman MS, Woodle MC, Mayhew E, Uster PS. Pharmacokinetics and anti-tumor activity of vincristine encapsulated in sterically stabilized liposomes. Int J Cancer. 1995;62:199–204. doi: 10.1002/ijc.2910620215. [DOI] [PubMed] [Google Scholar]