

Abstract
Purpose
Vascular endothelial cells demonstrate severe injury in sepsis, and a reduction in endothelial inflammation would be beneficial. Inter-α-Inhibitor (IαI) is a family of abundant plasma proteins with anti-inflammatory properties and has been investigated in human and animal sepsis with encouraging results. We hypothesized that IαI may protect endothelia from sepsis-related inflammation.
Methods
IαI-deficient or sufficient mice were treated with endotoxin or underwent complement-induced lung injury. VCAM-1 and ICAM-1 expression was measured in blood and lung as marker of endothelial activation. Human endothelia were exposed to activated complement C5a with or without IαI. Blood from human sepsis patients was examined for VCAM-1 and ICAM-1 and levels were correlated with blood levels of IαI.
Results
IαI-deficient mice showed increased endothelial activation in endotoxin/sepsis- and complement-induced lung injury models. In vitro, levels of endothelial pro-inflammatory cytokines and cell growth factors induced by activated complement C5a were significantly decreased in the presence of IαI. This effect was associated with decreased ERK and NFκB activation. IαI levels were inversely associated with VCAM-1 and ICAM-1 levels in a human sepsis cohort.
Conclusions
IαI ameliorates endothelial inflammation and may be beneficial as a treatment of sepsis.
Keywords: Sepsis, Complement, Inter-alpha-inhibitor, Endothelial injury, Lipopolysaccharide
Introduction
Sepsis, defined as the systemic inflammatory response to infection, remains a significant health problem worldwide. In the United States, septic shock causes > 200,000 deaths annually at a cost of > $15 billion [1]. Sepsis treatment remains largely supportive, and mortality is high at 30–50% [2], mostly from multi-organ failure (MOF) [3].
Sepsis causes endothelial injury, which induces inflammatory mediators, tissue factors, and coagulation pathways, leading to tissue hypoxia and MOF. Several mechanisms lead to endothelial injury in sepsis, for example, recent evidence suggests that oxidative stress and inflammation lead to damage in the endothelial glycocalyx, a gel-like layer covering the luminal surface of endothelial cells and shedding of glycocalyx components like heparin sulfate into the circulation [4]. This in turn leads to a disruption in the hemostatic function of endothelia and their ability to function effectively as a barrier [5]. The ultimate result is disruption of the microcirculation, leading to organ dysfunction which is a major hallmark of severe sepsis. Several upstream mediators promote endothelial injury, like bacterial endotoxins, reactive oxygen species, and inflammatory cytokines; however, activation of the complement cascade is a major contributor and resides at the apex of the pathophysiological processes leading to endothelial injury and organ failure [6].
Complement is a family of serum proteins acting via a catalytic cascade to amplify inflammation and induce opsonization and chemotaxis. Activated complement component 5 (C5a) is a central mediator of the complement cascade, and promotes a strong inflammatory response [7]. Blockade of C5a generation improves survival and decreases MOF in sepsis models [8].
Complement activation is central in endothelial injury [9]. C5a directly activates endothelial cells causing upregulation of cellular adhesion molecules E-selectin, vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1) [10]. These molecules assist inflammatory cell binding to activated endothelia and infiltration into the inflamed tissues [11–13].
Inter-alpha-inhibitor (IαI) is a family of plasma heavy- and light-chain anti-inflammatory proteins which have been investigated in sepsis with encouraging results (6). Two major forms of the protein family are found in circulation: Inter-α-inhibitor and Pre-α-inhibitor. Both proteins are collectively referred to as IαI in this report. IαI administration improves survival in sepsis models, e.g., after administration of lipopolysaccharide (LPS) [14–16]. Importantly, IαI administration in sepsis improves coagulation parameters which are markers of endothelial dysfunction [16]. We have shown that IαI inhibits complement activation and ameliorates lung injury in sepsis [17]. We now investigated the IαI effect on LPS- and C5a-induced endothelial activation. We show that IαI-deficient mice have increased markers of endothelial activation (ICAM-1, VCAM-1) in two sepsis models. We further show in vitro that IαI decreases ERK and NFκB signaling and the downstream expression of inflammatory cytokines after C5a exposure. Finally, we demonstrate an inverse correlation of IαI serum levels with VCAM-1 and ICAM-1 levels in human sepsis patients. In aggregate, our results suggest that IαI protects endothelial cells from inflammation by activated complement, thus being beneficial in sepsis.
Materials and Methods
Experimental Animals
IαI-deficient mice have been described previously [17]. Mice were backcrossed > 10 generations into the C57BL/6J mouse strain before used. Experimental animals were provided water and chow ad libitum and were kept in an enriched environment with a 12-h day-night cycle. All protocols were approved by the National Institute of Environmental Health Sciences Animal Care Committee.
Endothelial Cell Exposures
HUVEC cells were plated at 5000 cells/cm2 in 96-well dishes. Cells were incubated in a humidified incubator at 37 °C with 5% CO2 until they are 80% confluent in EGM (Lonza, Anaheim, CA) with 2% FBS. 24 h prior to exposure, cells were serum starved with basal EGM and incubated with either C5a (50 nM), C5a (50 nM), and IαI (50 nM), or media only. Supernatant was assessed using Bioplex (Biorad, Hercules, CA).
Western Blot Analysis
Lysates were collected after washing the cell layer twice with 1 ml ice-cold TBS in RIPA (Sigma-Aldrich, St Louis, MO) with HALT proteinase inhibitors (Thermo Scientific, Waltham, MA). Protein levels were quantified by BCA (Thermo Scientific). Western blot membranes were blocked with 4% Milk/TBS overnight and incubated for 4 h in TBS/0.5% BSA. Primary antibodies were ERK1/2 p44/42, Phospho-p44/42 ERK1/2 (Thr202/Tyr204), and NFKB p100/p52 (1:1000, Cell Signaling, Danvers, MA). Goat anti-Rabbit HRP secondary antibody was incubated at 1:10,000 and developed using Supersignal chemiluminescent substrate (Thermo-Fisher Scientific, Waltham, MA) and developed using BIOMax film (Kodak, Rochester, NY). Densitometry analysis was with ImageJ (National Institutes of Health, Bethesda, MD).
In Vivo Sepsis Model
LPS: Mice aged 6–8 weeks received i.p. LPS at 10 mg/kg (low dose), or 40 mg/kg (high dose) body weight and were sacrificed 24 h later by CO2.
C5a: Anti-BSA rabbit-fractionated antiserum (Sigma-Aldrich) was reconstituted in sterile-filtered ddH2O, diluted in LPS-free PBS to a concentration of 250 μg in 50 μl, and instilled retropharyngeally in isoflurane-anesthetized mice. Mice were then given 1 mg of LPS-free BSA (Equitech-Bio, Kerrville, TX) in 400 μl of LPS-free PBS into the retro-orbital sinus and sacrificed 4 h later by CO2.
For all mice, plasma was collected and lung tissue was snap frozen and stored at −80°C until used.
Quantitative RT-PCR
RNA was isolated from mouse lungs via the Trizol method. Expression of ICAM-1, VCAM-1, TNF-α, and E-selectin was examined (primers are listed in Table 1). Results were normalized to the ribosomal protein 18S.
Table 1.
Primer sequences for adhesion molecules and cytokines used in this study
| Gene | Forward | Reverse |
|---|---|---|
| ICAM-1 | GAG AGT GGA CCC AAC TGG AA | GCC ACA GTT CTC AAA GCA CA |
| VCAM-1 | CAA TGG GGT GGT AAG GAA | GTC ACA GCG CAC AGG TAA GA |
| TNFα | TGT TGC CTC CTC TTT TGC TT | TGG TCA CCA AAT CAG CGT TA |
| E-selectin | AGT CTA GCG CCT GGA TGA AA | CCA GCG AGG AGA ACA AAA AC |
| 18s | CGG CTA CCA CAT CCA AGG AA | GCT GGA ATT ACC GCG GCT |
Immunohistochemistry
Formalin-fixed mouse lungs were sectioned at 5 μm and stained with rabbit anti-IαI (Dako, Carpinteria, CA), or biotinylated HA binding protein (Seikagaku America, East Falmouth, MA).
Human Clinical Sepsis Study
Plasma samples were from a previous prospective multicenter phase III clinical trial of 266 patients with severe sepsis [18]. Specimens used for this study were exempt from IRB approval according to 45 CFR Part 46.1 01 (b) (4) because (a) this study used existing specimens collected for other research purposes or non-research activities; (b) subject identity and personal health information was de-identified and no clinical data were prospectively collected. Plasma samples were analyzed at admission (day 0) and day 5. Because some plasma samples had been used up, only 90 day-0 samples were analyzed for VCAM-1, 112 day-0 samples for ICAM-1, and 36 day-5 samples were analyzed for either. Investigators were blinded to plasma IαI level results. Duoset ELISA kits (R&D Systems, Minneapolis, MN) were used for sVCAM-1 and sICAM-1 measurements. Absorbance was measured at 490 nm/540 nm using Biotek Powerwave XS. Plasma IαI levels were measured by investigators who were unaware of sVCAM-1/sICAM-1 results. IαI levels were determined with a quantitative competitive ELISA using a single monoclonal antibody (MAb 69.26). Measurements were performed in triplicate.
Statistical Analysis and Regression Model
Analysis was performed using the GraphPad Prism 6 software (GraphPad Software, La Jolla, CA). ANOVA with Bonferroni post hoc test and Student’s t test was used and P < 0.05 was considered significant.
Results
Iαi Deficiency is Associated with Increased Markers of Endothelial Injury in Sepsis Models
E-selectin, VCAM-1, and ICAM-1 are well-described markers of endothelial activation in sepsis, promote leukocyte adhesion on activated endothelia, and have been associated with morbidity, MOF, and prognosis [19, 20]. We determined the effect of IαI deficiency on the expression of endothelial activation in two sepsis and organ injury models. Gene expression in lung tissue of IαI-deficient mice showed significant increase in VCAM-1, ICAM-1, and E-selectin (Fig. 1a) when compared to IαI-sufficient mice. Soluble VCAM-1 (sVCAM-1) is shed from activated endothelia, and n general is associated with severity of endothelial inflammation in septic patients [20]. Because measurement of VCAM-1 tissue expression is not easily feasible in septic patients, many investigators use sVCAM-1 as a surrogate [19]. Since we investigated the association of sVCAM-1 with serum IαI in sepsis patients (see below), we tested whether sVCAM-1 levels parallels tissue VCAM-1 expression in our mouse model. We found a significant increase in serum sVCAM-1 in IαI-deficient mice compared to sufficient mice, 24 h post intraperitoneal exposure to 10 mg/Kg or 40 mg/Kg LPS (Fig. 1b), confirming that both tissue expression and serum levels are increased in the absence of IαI. We also found significant upregulation of VCAM-1 and ICAM-1 in lung tissue of IαI-deficient mice compared to sufficient mice (Fig. 2a) in a second sepsis model, immune complex lung injury [17]. We then assessed serum cytokines and chemokines linked to endothelial inflammation in the context of sepsis [21–27]. We found significantly elevated levels of VEGF, PDGF, CXCL-9, M-CSF, IL-18, and GM-CSF in IαI-deficient mice relative to sufficient mice (Fig. 2b).
Fig. 1.
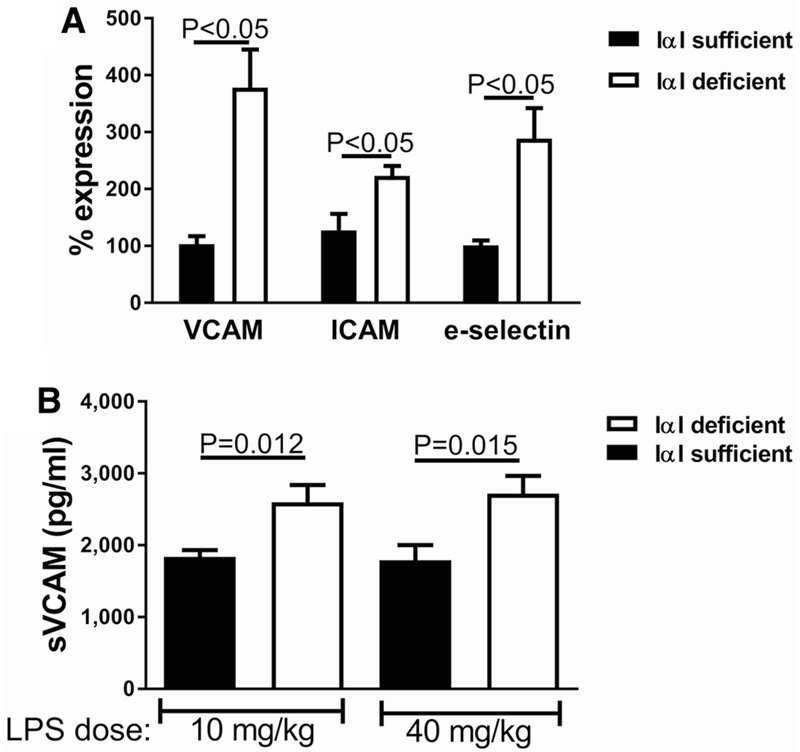
IαI-deficient mice dosed with LPS had increased expression of vascular adhesion molecules compared to IαI-sufficient mice. a Gene expression in lung tissue of VCAM-1, ICAM-1, and E-selectin was severalfold higher in IαI-deficient mice. b Increased levels of sVCAM in LPS-exposed IαI-deficient mice for high- and low-LPS dose
Fig. 2.
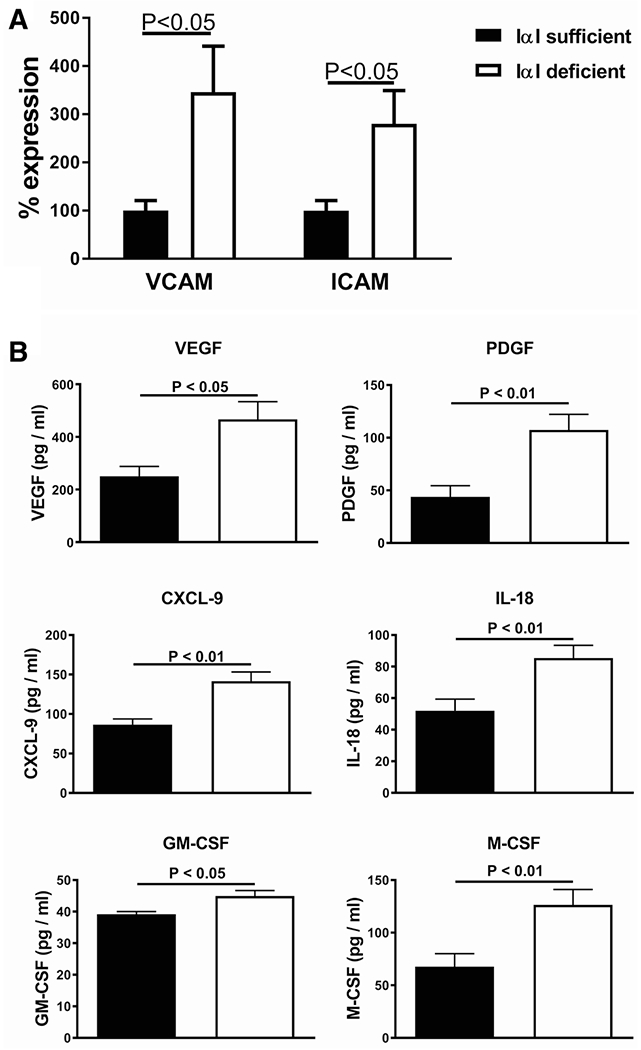
IαI-deficient mice in the C5a-induced lung injury model had increased endothelial injury and inflammation compared to IαI-sufficient mice. a Gene expression in lung tissue of VCAM-1 and ICAM-1 was 2- to 3-fold higher in IαI-deficient mice. b Endothelial growth factors and relevant cytokine levels in lung lavage fluid of exposed mice were higher in IαI-deficient mice compared to IαI-sufficient mice
Iαi Ameliorates Endothelial Inflammation Induced by C5a In Vitro
We then investigated the effect of IαI on endothelial inflammation induced by C5a, using an in vitro model of human umbilical vascular endothelial cells (HUVEC). We observed a strong upregulation of inflammatory cytokines after exposure of HUVEC to C5a. IαI significantly ameliorated C5a effects on IL-6, FGF, GM-CSF, MCP-1, PDGF, and IFN-γ. For some cytokines, e.g., IL-9, IαI did not have a protective effect on C5a-induced upregulation (Fig. 3a, b). Taken together our results suggest that, in addition to inhibiting complement activation [17], IαI also inhibits C5a-induced inflammation in endothelial cells.
Fig. 3.
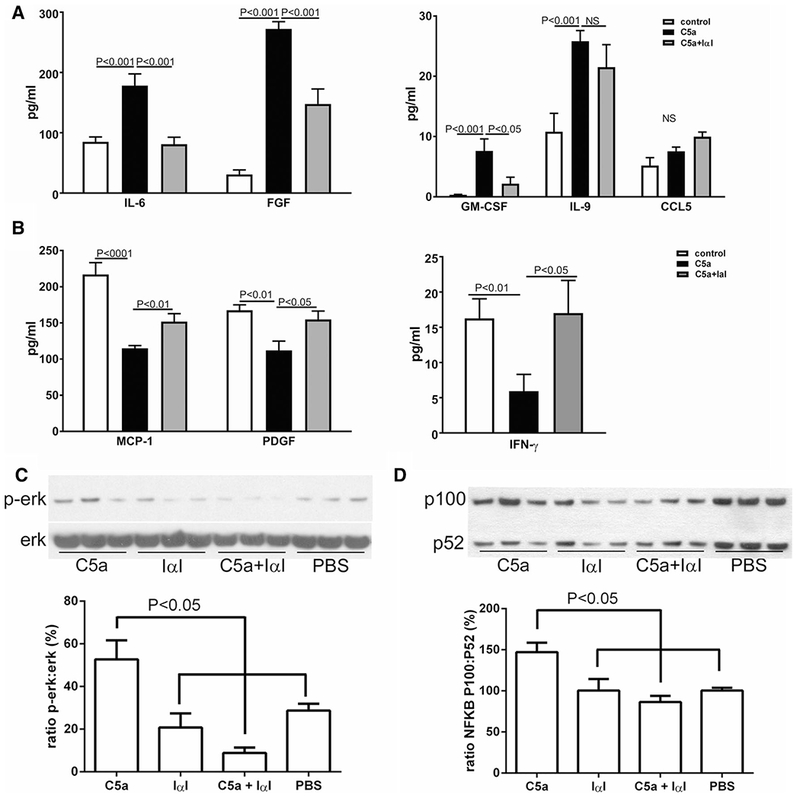
In vitro exposure of HUVEC to C5a demonstrated amelioration of the inflammatory response by IαI. a IαI reduced levels of IL-6, FGF-2 and GM-CSF that were increased by C5a. b IαI increased levels of MCP-1, PDGF, and IFN-γ that were reduced by C5a exposure. c IαI decreased activation of ERK and d IαI decreased activation of NFκB when dosed together with C5a. PBS=phosphate buffered saline vehicle
Iαi Ameliorates Nfkb And ERK Activation After Endothelial Exposure To C5a In Vitro
To further elucidate the mechanism of IαI protection against C5a-induced endothelial inflammation, we investigated the effect of IαI on signaling pathways which are known to be activated by C5a in endothelia, such as ERK [28] and NFκB [10], in the HUVEC model. We found that IαI significantly reduced ERK and NFκB activation after C5a exposure (Fig. 3c, d).
Plasma IαI Levels are Inversely Associated with sVCAM-1 and sICAM-1 Levels in Human Sepsis Patients
It has been previously shown that IαI plasma levels on admission are directly associated with survival and inversely correlated with organ failure scores and plasma IL-6 levels in a cohort of sepsis patients [18]. We utilized the same cohort to investigate whether there was an association between plasma IαI levels and plasma VCAM-1 and ICAM-1 levels, as markers of endothelial inflammation. We found a modest but significant inverse correlation between IαI levels and plasma VCAM-1 and ICAM-1 in sepsis patients on admission (day 0), but not at day 5 after admission (Fig. 4). This finding agreed with the experimental results in mouse sepsis models and in vitro endothelial injury models.
Fig. 4.
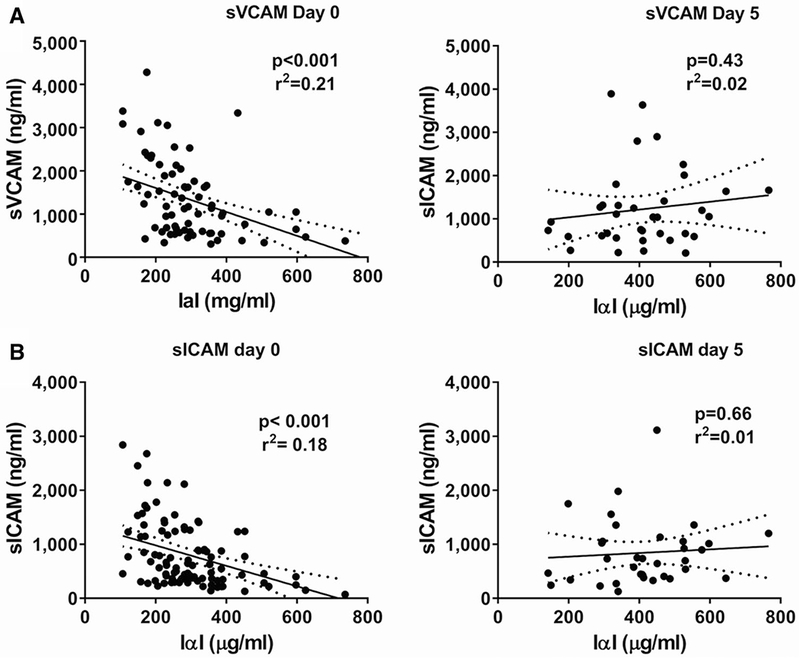
Inverse association between serum IαI and sVCAM a or sICAM b levels at day 0 (admission, left panels) but not at day 5 (right panels)
Discussion
It is increasingly recognized that sepsis therapy should not be limited to antimicrobial treatment. Instead, great health benefits may be realized by correcting the dysregulated immune response which underlies septic shock pathobiology [29]. Endothelial inflammation is a hallmark of sepsis [30–32] and leads to shock and MOF [33, 34]. Several cytokines produced by activated endothelia have effects on the endothelia themselves and immune cells that interact with them: GM-CSF promotes inflammatory cell adhesion to endothelia [35]; PDGF promotes endothelial integrity [36] and is associated with survival in sepsis [27]; IL-6 has a role in angiogenesis [37]; FGF-2 promotes endothelial migration through activation of ERK1/2 [38]; and MCP-1 expression by endothelia promotes the accumulation of monocytes in the injury site [39]. Endothelial activation induces expression of VCAM-1 and ICAM-1, which are involved in sepsis pathogenesis and are markers of end-organ failure, morbidity, and mortality in severe sepsis [20, 40]. Thus, VCAM-1 and ICAM-1 are representative markers of endothelial injury and activation in sepsis [19, 20]. We have now demonstrated that IαI suppresses VCAM-1 and ICAM-1 in two distinct models of sepsis lung injury (LPS and C5a). IαI also normalized in vitro the expression of relevant cytokines and endothelial factors. In addition, IαI is inversely associated with soluble VCAM-1 and ICAM-1 levels in patients admitted with sepsis. IαI is abundant in the circulation (normal levels of 0.2–0.6 mg/ml) and can be found in direct contact with vascular endothelia (Fig. 5). Thus, IαI is an excellent candidate modifier protein for endothelial inflammation and dysfunction in sepsis.
Fig. 5.
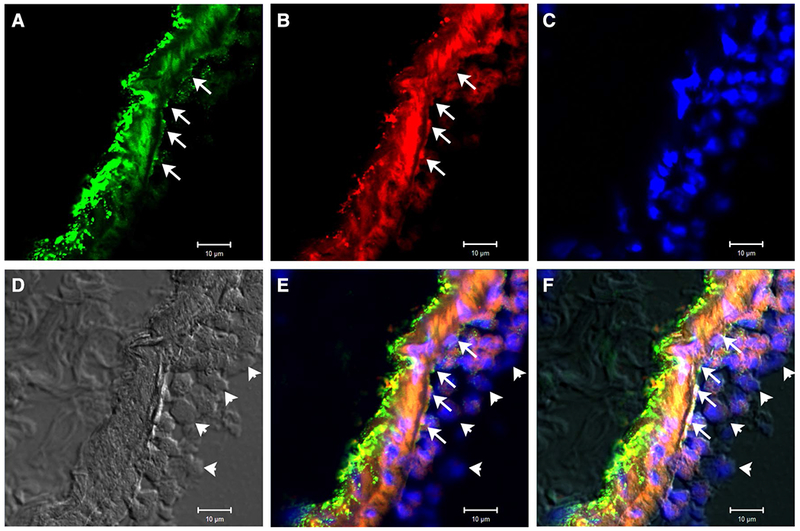
Mouse lung staining for hyaluronan (panel a, green), IαI (panel b, red), and DAPI (panel c, blue) after i.p. endotoxin exposure shows deposition of hyaluronan and IαI along the luminal endothelial surface of a venule (arrows). Panel d (transmitted light) is provided for structural reference. Panels e and f show merged images without and with transmitted light image, respectively. Note immune cells rolling along the endothelium which partially stain for IαI as well (arrowheads). Magnification: ×250
VCAM-1 and ICAM-1 upregulation in sepsis is mediated by inflammatory stimuli present in the serum of septic animals [41], including cytokines such as TNFα, IFN-γ, and IL-1β, glycosaminoglycans like short fragments of hyaluronan [42] and bacterial components such as LPS [43], which lead to the activation of NFκb in endothelial cells [42]. IαI is a potent anti-inflammatory protein and ameliorates sepsis-induced inflammation through several pathways. The IαI light-chain inhibits LPS-induced increase of cytoplasmic free Ca++ in immune and structural cells [44], and protects epithelia from injury after IL-1β and TNF-α exposure [45] through a Ca++ -inhibitory effect. Ca++ inhibition was seen in several cell types [44, 46, 47]. IαI also inhibits the activation of neutrophils after LPS exposure, by reducing ERK1/2 and p38 [48]. Furthermore, IαI has been shown to bind extracellular histones and neutralize histone-induced adverse effects in sepsis [49]. Finally, we recently described that IαI complexing with hyaluronan in septic animals leading to its rapid clearance and ameliorated sepsis mortality [50, 51]. This suggests that IαI is able to reduce the inflammatory cascade in injured endothelia through many pathways, ultimately leading to reduced VCAM-1 and ICAM-1 upregulation and secretion.
We investigated IαI in endothelial inflammation in vitro by using C5a exposure. We believe that this is a representative model since C5a is central to endothelial injury and MOF in sepsis [52–54]; furthermore, complement activation occurs throughout the course of systemic inflammation associated with sepsis [53]. A major pathway of C5a inflammation in endothelia is via ERK [28] and NFκB [10] activation. These pathways may be connected, since ERK leads to NFκB activation [55]. We demonstrate that IαI ameliorates both ERK and NFκB activation in endothelia. The IαI light-chain bikunin inhibits ERK activation in LPS-treated monocytes in vitro and LPS-dosed rats in vivo [56]; the effect may be at least partly due to inhibition of Ca++ influx [57], since bikunin inhibits LPS-induced Ca++ influx in neutrophils [48] and HUVEC [47]. Ca++ influx occurs after C5a binding to its receptor [58] and leads to the C5a-induced ERK activation [59]; thus, Ca++ influx inhibition is a plausible mechanism of IαI effects. The hypothesis that IαI effects target ERK is supported by the observation that C5a-induced IL-9 upregulation, which is not modulated by ERK [60], was not changed by IαI. IαI heavy chains may also contribute to its effect. Endothelial cells express several integrins [61] which IαI heavy chains can bind to [62]. Furthermore, IαI heavy chains are necessary for binding to hyaluronan, a crucial component of the endothelial glycocalyx [63]. IαI heavy chains may provide the necessary attachment to endothelial cells (see Fig. 5).
We established translational relevance of our findings by replicating the inverse association of IαI with VCAM-1 and ICAM-1 that we found in mice in a cohort of sepsis patients which in a previous work had demonstrated an inverse association of IαI plasma levels with mortality, MOF score, and serum IL-6 levels [18]. We found that IαI and VCAM-1/ICAM-1 levels are inversely associated in sepsis patients on admission (day 0), but not at day 5 after admission. On admission, patients have received very few treatment interventions. After 5 days, patients have received several potentially confounding interventions and possible complications, which may explain the absence of association. Thus, it may be that the relative absence of confounders allowed us to unmask the association between IαI and endothelial injury during early sepsis. Alternatively, IαI effects may be superseded by other factors that influence endothelial activation in later sepsis stages. Admission levels of IαI and soluble adhesion factors have been independently found to predict morbidity and survival in sepsis [18, 20] and our work suggests that there may be a causative connection between the two.
In conclusion, we show that IαI protects endothelia from injury caused by sepsis and the activated complement factor C5a. This find provides additional mechanistic insights into the effects of IαI in sepsis and supports the use of IαI as a treatment in sepsis.
Acknowledgements
The authors thank Drs. Michael B. Fessler and John House for constructive criticism, and Ms. Ligon Perrow for excellent animal husbandry. This work was supported, in part, by the Division of Intramural Research, National Institute of Environmental Health Sciences, NIH. Yow-Pin Lim gratefully acknowledges support from NIAID grant 2R42AI062095.
Footnotes
Conflict of interest Y-PL is the President and Chief Scientific Officer at ProThera Biologics, LLC, a company which explores the use of IαI in several systemic inflammatory diseases, including sepsis.
References
- 1.Angus DC et al. (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29(7):1303–1310 [DOI] [PubMed] [Google Scholar]
- 2.Kaukonen KM et al. (2014) Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. Jama 311(13):1308–1316 [DOI] [PubMed] [Google Scholar]
- 3.Ferreira FL et al. (2001) Serial evaluation of the SOFA score to predict outcome in critically ill patients. Jama 286(14):1754–1758 [DOI] [PubMed] [Google Scholar]
- 4.Schmidt EP et al. (2012) The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 18(8):1217–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ince C et al. (2016) The endothelium in sepsis. Shock 45(3):259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosmann M, Ward PA (2012) Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol 946:147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Czermak BJ et al. (1999) Protective effects of C5a blockade in sepsis. Nat Med 5(7):788–792 [DOI] [PubMed] [Google Scholar]
- 8.Huber-Lang M et al. (2001) Role of C5a in multiorgan failure during sepsis. J Immunol 166(2):1193–1199 [DOI] [PubMed] [Google Scholar]
- 9.Goldenberg NM et al. (2011) Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med 3(88):88ps25. [DOI] [PubMed] [Google Scholar]
- 10.Albrecht EA et al. (2004) C5a-induced gene expression in human umbilical vein endothelial cells. Am J Pathol 164(3):849–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyan-Aung U, Haskard DO, Lee TH (1991) Vascular cell adhesion molecule-1 and eosinophil adhesion to cultured human umbilical vein endothelial cells in vitro. Am J Respir Cell Mol Biol 5(5):445–450 [DOI] [PubMed] [Google Scholar]
- 12.Kyan-Aung U et al. (1991) Endothelial leukocyte adhesion molecule-1 and intercellular adhesion molecule-1 mediate the adhesion of eosinophils to endothelial cells in vitro and are expressed by endothelium in allergic cutaneous inflammation in vivo. J Immunol 146(2):521–528 [PubMed] [Google Scholar]
- 13.Bochner BS et al. (1991) Adhesion of human basophils, eosinophils, and neutrophils to interleukin 1-activated human vascular endothelial cells: contributions of endothelial cell adhesion molecules. J Exp Med 173(6):1553–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh K et al. (2010) Inter-alpha inhibitor protein administration improves survival from neonatal sepsis in mice. Pediatr Res 68(3):242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang S et al. (2002) Administration of human inter-alpha-inhibitors maintains hemodynamic stability and improves survival during sepsis. Crit Care Med 30(3):617–622 [DOI] [PubMed] [Google Scholar]
- 16.Jourdain M et al. (1997) Effects of inter-alpha-inhibitor in experimental endotoxic shock and disseminated intravascular coagulation. Am J Respir Crit Care Med 156(6):1825–1833 [DOI] [PubMed] [Google Scholar]
- 17.Garantziotis S et al. (2007) Inter-alpha-trypsin inhibitor attenuates complement activation and complement-induced lung injury. J Immunol 179(6):4187–4192 [DOI] [PubMed] [Google Scholar]
- 18.Opal SM et al. (2007) Longitudinal studies of inter-alpha inhibitor proteins in severely septic patients: a potential clinical marker and mediator of severe sepsis. Crit Care Med 35(2):387–392 [DOI] [PubMed] [Google Scholar]
- 19.Skibsted S et al. (2013) Biomarkers of endothelial cell activation in early sepsis. Shock 39(5):427–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zonneveld R et al. (2014) Soluble adhesion molecules as markers for sepsis and the potential pathophysiological discrepancy in neonates, children and adults. Crit Care 18(1):204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jesmin S et al. (2012) Time-dependent alterations of VEGF and its signaling molecules in acute lung injury in a rat model of sepsis. Inflammation 35(2):484–500 [DOI] [PubMed] [Google Scholar]
- 22.Amatschek S et al. (2011) CXCL9 induces chemotaxis, chemorepulsion and endothelial barrier disruption through CXCR22-mediated activation of melanoma cells. Br J Cancer 104(3):469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zsebo KM et al. (1988) Vascular endothelial cells and granulopoiesis: interleukin-1 stimulates release of G-CSF and GM-CSF. Blood 71(1):99–103 [PubMed] [Google Scholar]
- 24.Fuste B et al. (2004) Granulocyte colony-stimulating factor increases expression of adhesion receptors on endothelial cells through activation of p38 MAPK. Haematologica 89(5):578–585 [PubMed] [Google Scholar]
- 25.Burg J et al. (2002) GM-CSF expression by human lung microvascular endothelial cells: in vitro and in vivo findings. Am J Physiol Lung Cell Mol Physiol 283(2):L460–L467 [DOI] [PubMed] [Google Scholar]
- 26.Chandrasekar B et al. (2004) Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem 279(19):20221–20233 [DOI] [PubMed] [Google Scholar]
- 27.Brueckmann M et al. (2007) Prognostic value of platelet-derived growth factor in patients with severe sepsis. Growth Factors 25(1):15–24 [DOI] [PubMed] [Google Scholar]
- 28.Monsinjon T et al. (2003) Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. Faseb J 17(9):1003–1014 [DOI] [PubMed] [Google Scholar]
- 29.Levy MM et al. (2014) Surviving sepsis campaign: association between performance metrics and outcomes in a 7.5-year study. Crit Care Med 40(11):1623–1633. [DOI] [PubMed] [Google Scholar]
- 30.Vallet B (2003) Bench-to-bedside review: endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? Crit Care 7(2):130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubli S et al. (2003) Endothelium-dependent vasodilation in the skin microcirculation of patients with septic shock. Shock 19(3):274–280 [DOI] [PubMed] [Google Scholar]
- 32.Davis JS et al. (2009) Sepsis-associated microvascular dysfunction measured by peripheral arterial tonometry: an observational study. Crit Care 13(5):R155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coletta C et al. (2014) Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: preclinical studies in a murine model of cecal ligation and puncture. Crit Care 18(5):511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duffy MJ et al. (2011) Impaired endothelium-dependent vasodilatation is a novel predictor of mortality in intensive care. Crit Care Med 39(4):629–635 [DOI] [PubMed] [Google Scholar]
- 35.Smith WB, Gamble JR, Vadas MA (1994) The role of granulocyte-macrophage and granulocyte colony-stimulating factors in neutrophil transendothelial migration: comparison with interleukin-8. Exp Hematol 22(3):329–334 [PubMed] [Google Scholar]
- 36.Lindahl P et al. (1997) Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277(5323):242–245 [DOI] [PubMed] [Google Scholar]
- 37.Motro B et al. (1990) Pattern of interleukin 6 gene expression in vivo suggests a role for this cytokine in angiogenesis. Proc Natl Acad Sci USA 87(8):3092–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pintucci G et al. (2002) Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. Faseb J 16(6):598–600 [DOI] [PubMed] [Google Scholar]
- 39.Colotta F et al. (1994) Expression of monocyte chemotactic protein-1 by monocytes and endothelial cells exposed to thrombin. Am J Pathol 144(5):975–985 [PMC free article] [PubMed] [Google Scholar]
- 40.Amalakuhan B et al. (2016) Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine 88:267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laudes IJ et al. (2004) Disturbed homeostasis of lung intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 during sepsis. Am J Pathol 164(4):1435–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oertli B et al. (1998) Mechanisms of hyaluronan-induced upregulation of ICAM-1 and VCAM-1 expression by murine kidney tubular epithelial cells: hyaluronan triggers cell adhesion molecule expression through a mechanism involving activation of nuclear factor-kappa B and activating protein-1. J Immunol 161(7):3431–3437 [PubMed] [Google Scholar]
- 43.Wuthrich RP (1992) Intercellular adhesion molecules and vascular cell adhesion molecule-1 and the kidney. J Am Soc Nephrol 3(6):1201–1211 [DOI] [PubMed] [Google Scholar]
- 44.Kanayama N et al. (1997) The role of chondroitin sulfate chains of urinary trypsin inhibitor in inhibition of LPS-induced increase of cytosolic free Ca2+ in HL60 cells and HUVEC cells. Biochem Biophys Res Commun 238(2):560–564 [DOI] [PubMed] [Google Scholar]
- 45.El Maradny E et al. (1994) Urinary trypsin inhibitor has a protective effect on the amnion. Gynecol Obstet Invest 38(3):169–172 [DOI] [PubMed] [Google Scholar]
- 46.Kanayama N et al. (1995) Urinary trypsin inhibitor prevents uterine muscle contraction by inhibition of Ca++ influx. Am J Obstet Gynecol 173(1):192–199 [DOI] [PubMed] [Google Scholar]
- 47.Kanayama N et al. (1998) Urinary trypsin inhibitor suppresses vascular smooth muscle contraction by inhibition of Ca2+ influx. Biochim Biophys Acta 1381(2):139–146 [DOI] [PubMed] [Google Scholar]
- 48.Kanayama S et al. (2007) Bikunin suppresses expression of pro-inflammatory cytokines induced by lipopolysaccharide in neutrophils. J Endotoxin Res 13(6):369–376 [DOI] [PubMed] [Google Scholar]
- 49.Chaaban H et al. (2015) Inter-alpha inhibitor protein and its associated glycosaminoglycans protect against histone-induced injury. Blood 125(14):2286–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ni K et al. (2019) Intravascular heavy chain-modification of hyaluronan during endotoxic shock. Biochem Biophys Rep 17:114–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ni K et al. (2018) Rapid clearance of heavy chain-modified hyaluronan during resolving acute lung injury. Respir Res 19(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Unnewehr H et al. (2013) Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. J Immunol 190(8):4215–4225 [DOI] [PubMed] [Google Scholar]
- 53.Flierl MA, Schreiber H, Huber-Lang MS (2006) The role of complement, C5a and its receptors in sepsis and multiorgan dysfunction syndrome. J Invest Surg 19(4):255–265 [DOI] [PubMed] [Google Scholar]
- 54.Laudes IJ et al. (2002) Expression and function of C5a receptor in mouse microvascular endothelial cells. J Immunol 169(10):5962–5970 [DOI] [PubMed] [Google Scholar]
- 55.Chen BC, Lin WW (2001) PKC- and ERK-dependent activation of I kappa B kinase by lipopolysaccharide in macrophages: enhancement by P2Y receptor-mediated CaMK activation. Br J Pharmacol 134(5):1055–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molor-Erdene P et al. (2005) Urinary trypsin inhibitor reduces LPS-induced hypotension by suppressing tumor necrosis factor-alpha production through inhibition of Egr-1 expression. Am J Physiol Heart Circ Physiol 288(3):H1265–1271 [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi H et al. (2003) A Kunitz-type protease inhibitor, bikunin, inhibits ovarian cancer cell invasion by blocking the calcium-dependent transforming growth factor-beta 1 signaling cascade. J Biol Chem 278(10):7790–7799 [DOI] [PubMed] [Google Scholar]
- 58.Monk PN, Partridge LJ (1993) Characterization of a complement-fragment-C5a-stimulated calcium-influx mechanism in U937 monocytic cells. Biochem J 295(Pt 3):679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hawlisch H et al. (2005) C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity 22(4):415–426 [DOI] [PubMed] [Google Scholar]
- 60.Perumal NB, Kaplan MH (2011) Regulating Il9 transcription in T helper cells. Trends Immunol 32(4):146–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bader BL et al. (1998) Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 95(4):507–519 [DOI] [PubMed] [Google Scholar]
- 62.Adair JE et al. (2009) Inter-alpha-trypsin inhibitor promotes bronchial epithelial repair after injury through vitronectin binding. J Biol Chem 284(25):16922–16930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lennon FE, Singleton PA (2011) Hyaluronan regulation of vascular integrity. Am J Cardiovasc Dis 1(3):200–213 [PMC free article] [PubMed] [Google Scholar]