

Abstract
Autism spectrum disorder (ASD) is a prevalent neurodevelopmental disorder characterized by social deficits and associated restrictive and/or repetitive behaviors. The breadth of ASD symptoms is paralleled by the multiplicity of genes that have been implicated in its etiology. Initial findings revealed numerous ASD risk genes that contribute to synaptic function. More recently, genomic and gene expression studies point to altered chromatin function and impaired transcriptional control as additional risk factors for ASD. The consequences of impaired transcriptional alterations in ASD involve consistent changes in synaptic gene expression and cortical neuron specification during brain development. The multiplicity of genetic and environmental factors associated with ASD risk and their convergence onto common molecular pathways in neurons point to ASD as a disease of gene regulatory networks.
Autism spectrum disorder (ASD) is a heterogeneous neurodevelopmental disorder affecting more than 1% of individuals [1]. The main manifestations of ASD are impaired social communication and interaction, repetitive behaviors, and/or restricted interests. The scale of social impairment in individuals with ASD is highly variable and ranges from subtle to most severe conditions that can leave patients unable to lead an independent life. This wide range of symptom severity implies a different degree of impairment of the neuronal networks that regulate social interaction and behavior.
The initial hints about possible neurobiological causes for the etiology of ASD came from human genetic studies that revealed an association between ASD and mutations in genes encoding different components of the synaptic machinery [2]. Clinical data further showed that mutations in the same gene can result in phenotypic variability with a wide range of clinical presentations, as observed in individuals with SHANK3 haploinsufficiency or Phelan-McDermid syndrome [3]. These early findings led to the widely accepted view of ASD as a “synaptopathy” characterized by abnormal neuronal circuit formation during brain development followed by impaired behavior [4] (Figure 1).
Figure 1. Converging pathways in ASD.
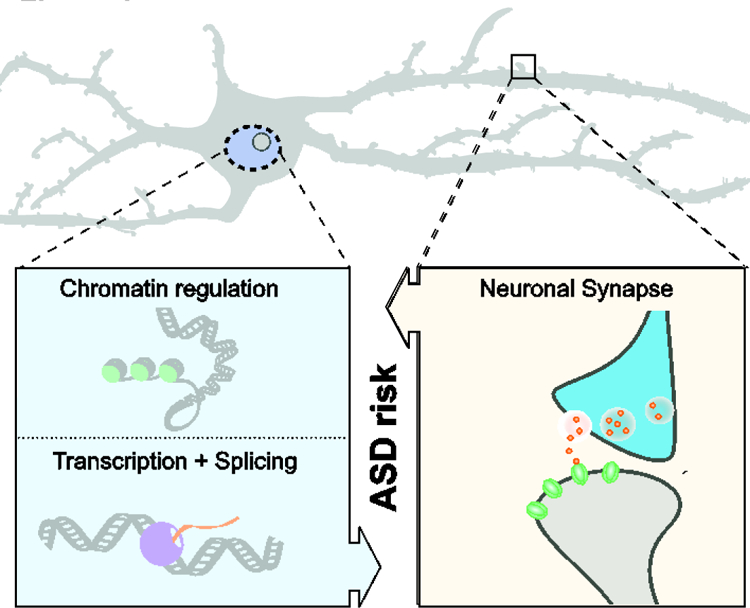
Scheme shows the two major pathways implicated in ASD risk based on genomic studies and gene expression analysis of affected individuals. (Left) Dysregulation of gene expression at the level of chromatin modifications, chromatin remodeling, regulation of transcription, and RNA splicing, as well as (right) alterations in synapse development and function are strongly associated with ASD risk. Recent data suggest a convergence of the two pathways in ASD pathology, where changes in neuronal gene expression regulation during fetal brain development preferentially affect genes important for synapse function and neuronal differentiation, and conversely, changes in genes important for synaptic function and neuronal specification indirectly affect neuronal gene expression regulation.
Several recent large-scale sequencing studies, however, led to a shift in our understanding of the genetic mechanisms contributing to the synaptic impairments in ASD. Studies of thousands of families with children with ASD led to the identification of a large number of novel high-confidence genes conferring risk for the disease [5–8]. Many of the affected genes encoded functionally distinct regulators of gene expression that range from chromatin modifiers to bona fide RNA transcription factors [5–8] (Table 1, Figure 1). The role of impaired transcriptional regulation in ASD was furthermore underscored by studies that link ASD with genetic variation in non-coding gene regulatory sequences such as promoters [9]. Machine learning approaches applied to whole-genome sequencing data from 1,902 families identified the strongest association between ASD risk and de novo mutations in evolutionary conserved loci, including transcription factor binding sites, located within distal promoter regions [9].
Table 1. High-confidence ASD risk genes encoding chromatin and transcription regulators.
For each gene, the references for the whole-exome sequencing (WES) studies that implicated the gene in risk with a false discovery rate (FDR) < 0.1, the associated condition reported in the Online Mendelian Inheritance in Man® database (OMIM), the inheritance indicated in OMIM, the OMIM ID, and the references (PMIDs) to mouse models and gene expression studies in mouse models are indicated. For inheritance, AD indicates autosomal dominant, AR autosomal recessive, XLD X-linked dominant, and XLR X-linked recessive. Please, note that all WES studies indicated focused on autosomal genes supposedly acting as haploinsufficient.
| Gene | WES study for ASD association | Condition (OMIM) | Inherita nce | OMI M ID | Mouse models | Gene Expression data | |
|---|---|---|---|---|---|---|---|
| Chromatin regulator | ADNP | De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Helsmoortel-van der Aa syndrome | AD | 615873 | Hacohen-Kleiman et al., 2018; Malishkevich et al., 2015; Vulih-Shultzman et al., 2007 | Hacohen-Kleiman et al., 2018 |
| ARID1B | De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Coffin-Siris syndrome 1 | AD | 135900 | Celen et al., 2017; Jung et al., 2017; Shibutani et al., 2017 | Celen et al., 2017; Shibutani et al., 2017 | |
| ASH1L | De Rubeis et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Mental retardation, autosomal dominant 52 | AD | 617796 | Zhu et al., 2016 | ||
| ASXL3 | De Rubeis et al., 2014; Satterstrom et al., 2018 | Bainbridge-Ropers syndrome | AD | 615485 | |||
| CHD2 | Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Epileptic encephalopathy, childhood-onset | AD | 615369 | Kim et al., 2018 | Kim et al., 2018 | |
| CHD8 | De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Gompers et al., 2017; Jung et al., 2018; Katayama et al., 2016; Platt et al., 2017; Suetterlin et al., 2018 | Gompers et al., 2017; Jung et al., 2018; Katayama et al., 2016; Platt et al., 2017; Suetterlin et al., 2018 | ||||
| CREBBP | Satterstrom et al., 2018 | Rubinstein-Taybi syndrome 1 | AD | 180849 | Merk et al., 2018; Zheng et al., 2016 | ||
| KDM6B | Sanders et al., 2015; Satterstrom et al., 2018 | Park et al., 2014 | Park et al., 2014 | ||||
| KMT2C | De Rubeis et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Kleefstra syndrome 2 | AD | 617768 | |||
| KMT2E | Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | ||||||
| MBD5 | Sanders et al., 2015; Satterstrom et al., 2018 | Mental retardation, autosomal dominant 1 | AD | 156200 | Camarena et al., 2014 | ||
| NSD1 | Satterstrom et al., 2018 | Sotos syndrome 1 | AD | 117550 | |||
| PHF12 | Satterstrom et al., 2018 | ||||||
| PHF2 | Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | ||||||
| PHF21A | Satterstrom et al., 2018 | ||||||
| SETD5 | De Rubeis et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Mental retardation, autosomal dominant 23 | AD | 615761 | Deliu et al., 2018 Moore et al., 2019 | Deliu et al., 2018 Moore et al., 2019 | |
| SKI | Satterstrom et al., 2018 | Shprintzen-Goldberg syndrome | AD | 182212 | |||
| SMARCC2 | Satterstrom et al., 2018 | Tuoc et al., 2017; Tuoc et al., 2013 | |||||
| SUV420H1/K MT5B | De Rubeis et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Mental retardation, autosomal dominant 51 | AD | 617788 | |||
| ZMYND8 | Satterstrom et al., 2018 | ||||||
| Transcription regulator | BCL11A | De Rubeis et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Dias-Logan syndrome | AD | 617101 | Dias et al., 2016 | Dias et al., 2016 |
| DEAF1 | Satterstrom et al., 2018 | Dyskinesia, seizures, and intellectual developmental disorder/Mental retardation, autosomal dominant 24 | AR/AD | 602635/615828 | Luckhart et al., 2016; Vulto-van Silfhout et al., 2014 | ||
| DYRK1A | De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Mental retardation, autosomal dominant 7 | AD | 614104 | Arque et al., 2009; Arque et al., 2008; Benavides-Piccione et al., 2005; Fotaki et al., 2002; Fotaki et al., 2004; Raveau et al., 2018 | ||
| FOXP1 | Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Mental retardation with language impairment and with or without autistic features (FOXP1 syndrome) | AD | 613670 | Araujo et al., 2015; Araujo et al., 2017; Bacon et al., 2015; Frohlich et al., 2017; Usui et al., 2017 | Araujo et al., 2015; Araujo et al., 2017; Usui et al., 2017 | |
| FOXP2 | Satterstrom et al., 2018 | Speech-language disorder-1 | AD | 602081 | Chen et al., 2016; French et al., 2018; Medvedeva et al., 2018; Shu et al., 2005; Xu et al., 2018 | Medvedeva et al., 2018; Vernes et al., 2011 | |
| MECP2 | Rett syndrome/Mental retardation, X-linked syndromic/Lubs type (MECP2 duplication syndrome) | XLD/XLR | 312750/300260 | Guy et al., 2007; Hao et al., 2015; Lu et al., 2016; Moretti et al., 2006; Shahbazian et al., 2002; Sztainberg et al., 2015 | |||
| MYT1L | De Rubeis et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Mental retardation, autosomal dominant 39 | AD | 616521 | |||
| NCOA1 | Satterstrom et al., 2018 | ||||||
| POGZ | De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | White-Sutton syndrome | AD | 616364 | |||
| SATB1 | Satterstrom et al., 2018 | Balamotis et al., 2012 | |||||
| SIN3A | Satterstrom et al., 2018 | Witteveen-Kolk syndrome | AD | 613406 | |||
| TBL1XR1 | Satterstrom et al., 2018 | Mental retardation, autosomal dominant 41/Pierpont syndrome | AD | 616944/602342 | |||
| TBR1 | De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 | Intellectual developmental disorder with autism and speech delay | AD | 606053 | Fazel Darbandi et al., 2018; Huang et al., 2014 | Fazel Darbandi et al., 2018; Huang et al., 2014 | |
| TCF20 | Satterstrom et al., 2018 | ||||||
| TCF4 | Satterstrom et al., 2018 | Pitt-Hopkins syndrome | AD | 610954 | Crux et al., 2018; Kennedy et al., 2016 | Kennedy et al., 2016 | |
| TCF7L2 | Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2018 |
A number of large-scale gene expression studies comparing postmortem brains from individuals with ASD and matched unaffected controls underscore the role of impaired epigenetic/transcriptional regulation along with synapse dysfunction in ASD [10–14]. The importance of transcriptional dysregulation in ASD receives additional support from abnormal patterns of RNA splicing and isoform usage in the brains of individuals with ASD. This may have a particular impact on brain development and physiology, since alternative splicing occurs more frequently in the brain than in any other tissue [15]. One specific group of genes carrying microexons (3 to 27 base pairs) is found to be not only preferentially expressed in the brain but also frequently dysregulated in ASD [12,15,16]. Overall, genes showing isoform dysregulation in ASD are likewise enriched in regulators of gene expression and synaptic function [13]. Besides alterations in gene expression and isoform usage, widespread changes in RNA editing in brains from individuals with ASD [17] further point to a broad spectrum of impairments that can contribute to transcriptional dysregulation in ASD.
The possible causal role of perturbed epigenetic and transcriptional processes in ASD has been gaining strong support from animal studies that employ genetically engineered mice lacking/bearing specific ASD risk genes. The haploinsufficiency or neuron-specific targeting of ASD risk genes encoding the chromatin remodelers Arid1b [18–20] and Chd8 [21–23], the histone methyltransferases Ehmt1 [24,25], and Setd5 [26,27], and the transcriptional regulators Foxp1 [28–31] and Foxp2 [32–35] result in ASD-like behavioral phenotypes in mice (Table 1). Transcriptomic analyses of the brains of mice with haploinsufficiency or cell-type specific ablation of chromatin modifiers or transcription regulators revealed consistent changes in the expression of synaptic genes [18,24,26,27,29,36–38] (Table 1), suggesting impaired transcriptional regulation as one of the key mechanisms for altered synaptic gene expression and function in ASD (Figure 1).
One of the peculiar aspects of the ASD-related transcriptional changes deals with the selective impairment of the expression of genes of extended length (>100kb). Transcriptome analysis in the human and mouse brain revealed a significant enrichment in the expression of long genes as compared to any other organ [39,40]. Additionally, genes of extended length are enriched in genes encoding synaptic proteins and ion channels, including those that have been linked to ASD [39,41,42]. The efficiency of gene transcription, as defined by the abundance of the full-length mRNA transcript as well as the pattern of RNA splicing, depends greatly on the fidelity of RNA elongation [43]. The latter is governed by numerous factors that collectively support the movement of the RNA polymerase at a defined speed of RNA elongation [44]. Therefore, it is very likely that the transcription of genes of extended length is more sensitive to changes in the transcriptional machinery as compared to shorter genes. Recent data revealed the particular dependency of ASD risk genes of extended length on the Top2b-dependent transcriptional elongation process [41]. Moreover, long genes linked to ASD appear to be distinct from other genes by harboring expanded enhancer domains [45], and by being particularly sensitive to transcriptional repression in response to pharmacological inhibitors of the bromodomain - containing proteins of the BET family [42].
The impaired transcription of genes of extended length as well as other structural and functional alterations associated with transcriptional regulation in ASD may also lead to erroneous timing of gene expression during the tightly regulated developmental trajectories of neurons in the developing brain. The differentiation of specific neuronal subtypes during brain development is governed by complex gene regulatory networks [46], with each cell type acquiring a unique expression profile dictating their morphological and phenotypic specialization [47–49]. Time course analysis of developing human brain tissue revealed that late cortical development in the fetus is characterized by widespread changes in gene expression patterns, including increased neuron subtype-specific signatures and the expression of genes associated with synapse development and neuronal functions [50]. These data suggest that the convergence of the varied ASD risk genes onto the same behavioral outcome may reflect the defective timing of neuronal subtype specification and associated circuit formation during pre- and post-natal brain development. In other words, genetic lesions associated with ASD act as a form of “chaotropic” agents that, by acting on multiple pathways during the extremely precise differentiation processes in the fetal brain, affect the stability of neuronal networks. As a consequence, the social challenges that occur imminently after birth confront a neuronal network with impaired robustness and hence increased susceptibility to activity-driven changes that may lead to the establishment of the disease phenotype. This notion is in line with evidence of post-natal or adult activity-driven transcriptional changes in the brain of different mouse models of the disease [24,42,51,52]. The described scenario views attenuated brain robustness, rather than defects in a specific gene or cell type, as an underlying mechanism for ASD pathophysiology.
The robustness of cell differentiation reflects the so-called “canalization” process towards a specific outcome from uncertain starting conditions [53] (Figure 2). In Waddington’s “epigenetic landscape”, environmental signals lead to the establishment of “valleys” that guide the direction of the differentiation processes to the finite cell type [53] (Figure 2A). Projected into this landscape (Figure 2B), the process of neuronal differentiation and sub-specification is reflected in the trajectories of a multipotent neuronal progenitor cell. Exposed to a combination of external and internal signals (growth factors, signaling molecules, transcriptional regulators), the progenitor cell is guided through the differentiation process acquiring new features that ultimately will define its distinct neuronal identity and function. This process of neuron sub-type specification is governed by high “ridges” that prevent divergence during the differentiation process and stabilize the newly acquired phenotypes.
Figure 2. Robustness of cell/neuron differentiation during development.
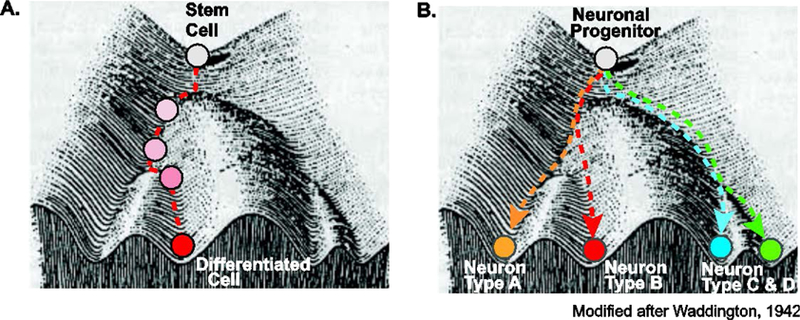
Modified scheme of Waddington’s “epigenetic landscape” [53] illustrates cell differentiation during development. (A) Multipotent stem cells and (B) neuronal progenitor cells (white circle) follow a robust developmental trajectory or “canalization” towards a specific outcome. In this “epigenetic landscape”, distinct extrinsic and intrinsic signals lead to the establishment of “valleys” and “ridges” that ensure robustness and guide the differentiation processes towards distinct differentiated cell types (A) or neuronal subtypes (B).
The Waddington landscape reflects the state of gene regulatory networks that operate within cells [54]. In turn, the notion of a stable dynamical state invites the comparison to the so-called “attractor state” which, in material physics, is defined as a place where the dynamical system is exerting a minimal amount of energy [54]. The attractor state, which has been widely discussed in the context of gene network regulation during development [54], represents a defined outcome of numerous interactions within any given network, from transcriptional networks to intercellular interactions. Each of the interactions within such a network represents a single dimension and, accordingly, multiple interactions yield highly complex multidimensional manifold. The topography of the manifold is molded into a conformation of “ridges” and “valleys”, where unstable, high energy states occupy the top of the “ridges” and low energy states form the energetically favorable attractor states or “valleys” [55] (Figure 3). Thus, the low energy level of the attractor state contributes to its stability and protection against environmental perturbation [56]. The stability of a given attractor state can be attenuated by introducing systemic alterations (genetic mutations and/or environmental insults) that “lower” the protective “ridges” of the attractor state or increase the instability of the network by altering numerous different network components or by targeting a key regulator of the network [57,58]. Both of these scenarios may yield abnormal cell or tissue function.
Figure 3. Epigenetic landscape of normal brain development and ASD.
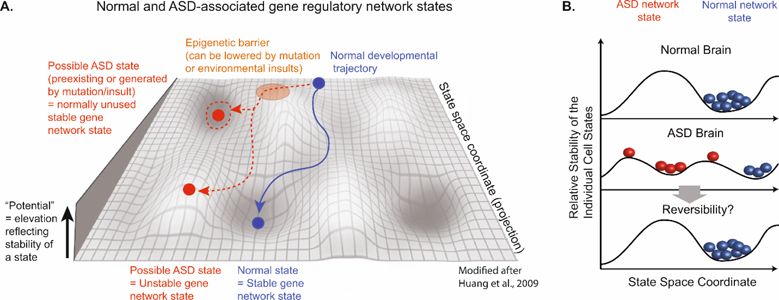
(A) Modified scheme from Huang et al, 2009 [53] of an epigenetic landscape during brain development. The landscape is a schematic projection of a complex gene network into a two-dimensional state space. The y axis represents the relative stability of individual cell states where higher positions indicate less stability. The valleys represent stable attractor states that occupy the low-energy stable basin and are resistant to perturbations. Normal developmental trajectory (blue) progresses from back to front towards a stable attractor state, which represents a distinct neuronal state, and is prevented from entering unused “abnormal attractors” (red dashed circle) along the path due to epigenetic barriers (orange area). Mutations or environmental insults can lower this barrier, thus opening access to unused attractors that encode an abnormal phenotype = ASD attractor state (red dashed arrow). Alternatively, the ASD gene network state may reflect an unstable neuronal state that hinders the formation of stable neuronal networks. (B) Model of ASD network states and their potential reversibility. Scheme shows highly simplified version of proposed model for normal (blue) and ASD (red) gene network states. The y axis represents the relative stability of individual cell states where higher positions indicate less stability. The x axis represents the specific space coordinate of a given neuronal network state. Future potential therapies could be aimed at trying to reverse symptoms of ASD by targeting the ASD network states (red, novel ASD attractor state or unstable state) followed by their conversion into a normal stable neuronal network state (blue).
Notably, it has been shown that undifferentiated progenitor cells can reach the same finite differentiation state in response to a set of different stimuli [54]. Despite the differences in individual stimulus-induced pathways, the distinct gene expression processes that follow the signal eventually converge onto the same attractor state/gene expression profile [54]. The same principle could be applicable to neurodevelopmental disorders like ASD, where distinct alterations within the neuronal gene network may lead to a similar outcome such as i.e. the dysregulation of synaptic gene expression and associated functions in ASD. Since diseases are not a hardwired part of our evolution, the disease-associated attractor states could be highly individual and reflect subtle differences in the energy landscape of the interacting components. The impaired attractor state model has been discussed in the context of malignant transformation, where genetic mutations and environmentally induced expression changes of distinct genes trigger the formation of immortal and invasive states characteristic of cancer cells [56,58,59]. Following this model, it is possible that the different genetic and environmental factors contributing to ASD risk, while targeting different sets of genes during the critical phase of fetal and early post-natal brain development, may converge on a common neuronal “attractor state” and induce a switch to a new phenotypic state. This newly acquired state may either become stabilized in form of a “new attractor state” (Figure 3) or remain permanently unstable, hence generating multiple different, slightly heterogeneous phenotypes (Figure 3). In summary, it could be feasible to view ASD as a pathological and perhaps unstable attractor state, where slight variations in the input factors drive the severity of the clinical manifestations. The important aspect of this model is that it directs much of the attention from individual genes towards genetically and/or environmentally impaired gene regulatory networks and associated alterations in neuronal function during development. This very concept of the attractor state bears a certain futility as the multiplicity of interactions may preclude the identification of key driver genes or cell types.
The idea of ASD reflecting an unstable neuronal regulatory network state may be particularly interesting from a therapeutic point of view since it suggests a possible reversibility of the phenotype (Figure 3B). This scenario is supported by recent data showing that some of the transcriptional and/or behavioral changes in mice are reversed by restoring expression or function of ASD risk genes in the adult brain [42,60–65]. Collectively, these data suggest the exciting possibility that ASD-associated neuronal network states could be reverted to their predestine state (Figure 3B) and allow the amelioration of social deficits. This concept is supported by intriguing examples of reported temporary alleviation of ASD symptoms in humans in response to strong perturbations, such as increased body temperature during fever [66], and suggest potential novel therapeutic approaches could be aimed at rewiring neuronal gene networks/attractor states in ASD.
Acknowledgements.
This work was supported by the NIH Director New Innovator Award DP2 MH100012 (A.S) and Ruth L. Kirschstein NRSA fellowship F31MH111147 (J.M.S), and the Beatrice and Samuel A. Seaver Foundation (to S.D.R and A.S). Josefa M. Sullivan is a Seaver Graduate Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement.
Nothing to declare.
References
- 1.Christensen DL, Baio J, Van Naarden Braun K, Bilder D, Charles J, Constantino JN, Daniels J, Durkin MS, Fitzgerald RT, Kurzius-Spencer M, et al. : Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years--Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summ 2016, 65:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourgeron T: From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 2015, 16:551–563. [DOI] [PubMed] [Google Scholar]
- 3.De Rubeis S, Siper PM, Durkin A, Weissman J, Muratet F, Halpern D, Trelles MDP, Frank Y, Lozano R, Wang AT, et al. : Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol Autism 2018, 9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zoghbi HY: Postnatal neurodevelopmental disorders: meeting at the synapse? Science 2003, 302:826–830. [DOI] [PubMed] [Google Scholar]
- 5.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. : Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. : The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins RL, Grove J, Klei L, et al. : Novel genes for autism implicate both excitatory and inhibitory cell lineages in risk. bioRxiv 2018. [Google Scholar]; * This is the largest whole-exome sequencing study on ASD to date. By analyzing sequence data from >35,000 individuals, the authors identified novel risk genes for ASD. The genes include regulators of gene expression and genes involved in synaptic and neuronal communication, most of which are expressed in both excitatory and inhibitory neurons in the human cortex.
- 8.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. : Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87:1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An JY, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, et al. : Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 2018, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study has analyzed whole-genome sequencing data of 1,902 quartet families - each including a child affected with ASD, one unaffected sibling control, and their parents - and identified de novo mutations in noncoding regions that contribute to ASD risk, with the strongest contributors being mutations in the distal promoter regions and ~2000 nucleotides upstream of the transcriptional start sites of coding genes.
- 10.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH: Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474:380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J, et al. : Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallieres M, Tapial J, Raj B, O’Hanlon D, et al. : A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 2014, 159:1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. : Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study has integrated RNAseq and genotyping data from postmortem brains from over 1,600 individuals diagnosed with ASD, schizophrenia, or bipolar disorder, or unaffected individuals. The authors found transcriptome-wide changes at the gene and isoform level emphasizing the importance of splicing and isoform-level gene regulation in psychiatric disorders.
- 14.Lombardo MV, Courchesne E, Lewis NE, Pramparo T: Hierarchical cortical transcriptome disorganization in autism. Mol Autism 2017, 8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. : Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quesnel-Vallieres M, Dargaei Z, Irimia M, Gonatopoulos-Pournatzis T, Ip JY, Wu M, Sterne-Weiler T, Nakagawa S, Woodin MA, Blencowe BJ, et al. : Misregulation of an Activity-Dependent Splicing Network as a Common Mechanism Underlying Autism Spectrum Disorders. Mol Cell 2016, 64:1023–1034. [DOI] [PubMed] [Google Scholar]; * This study shows that mice haploinsufficient for the neuronal splicing regulator nSR100/SRRM4 have reduced splicing of microexons that significantly overlap those found dysregulated in postmortem brains from individuals with ASD by Irimia et al., 2014. These molecular alterations are accompanied by social deficits and altered synaptic transmission and neuronal excitability.
- 17.Tran SS, Jun HI, Bahn JH, Azghadi A, Ramaswami G, Van Nostrand EL, Nguyen TB, Hsiao YE, Lee C, Pratt GA, et al. : Widespread RNA editing dysregulation in brains from autistic individuals. Nat Neurosci 2019, 22:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study has used RNAseq data from 69 postmortem brains of individuals with ASD or unaffected controls to analyze for the first time the levels of A-to-I RNA editing on a genome-wide scale and across three brain regions. The analyses revealed a global bias for hypoediting in ASD brains, which was shared across brain regions and involved many synaptic genes. Furthermore, the authors described a role for the Fragile X proteins in RNA editing and, consistently, convergent patterns of RNA editing dysregulation between samples from cases with idiopathic ASD and Fragile X syndrome.
- 18.Celen C, Chuang JC, Luo X, Nijem N, Walker AK, Chen F, Zhang S, Chung AS, Nguyen LH, Nassour I, et al. : Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung EM, Moffat JJ, Liu J, Dravid SM, Gurumurthy CB, Kim WY: Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat Neurosci 2017, 20:1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study has shown that Arid1b heterozygous mice have reduced number of cortical GABAergic interneurons, as a result of impaired proliferation of progenitors in the ganglionic eminence during development. The mice also show cognitive and social deficits that could be rescued by adult-initiated treatment with a positive allosteric modulator of the GABAA receptor.
- 20.Shibutani M, Horii T, Shoji H, Morita S, Kimura M, Terawaki N, Miyakawa T, Hatada I: Arid1b Haploinsufficiency Causes Abnormal Brain Gene Expression and Autism-Related Behaviors in Mice. Int J Mol Sci 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung H, Park H, Choi Y, Kang H, Lee E, Kweon H, Roh JD, Ellegood J, Choi W, Kang J, et al. : Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat Neurosci 2018, 21:1218–1228. [DOI] [PubMed] [Google Scholar]
- 22.Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, Suyama M, Takumi T, Miyakawa T, Nakayama KI: CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016, 537:675–679. [DOI] [PubMed] [Google Scholar]; * This is the first characterization of a mouse model of CHD8 haploinsufficiency. The study has shown that Chd8 heterozygous mice have repetitive behaviors and social deficits, accompanied by small changes in the expression of a large number of genes.
- 23.Suetterlin P, Hurley S, Mohan C, Riegman KLH, Pagani M, Caruso A, Ellegood J, Galbusera A, Crespo-Enriquez I, Michetti C, et al. : Altered Neocortical Gene Expression, Brain Overgrowth and Functional Over-Connectivity in Chd8 Haploinsufficient Mice. Cereb Cortex 2018, 28:2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, Tarakhovsky A, Greengard P: Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 2009, 64:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balemans MC, Huibers MM, Eikelenboom NW, Kuipers AJ, van Summeren RC, Pijpers MM, Tachibana M, Shinkai Y, van Bokhoven H, Van der Zee CE: Reduced exploration, increased anxiety, and altered social behavior: Autistic-like features of euchromatin histone methyltransferase 1 heterozygous knockout mice. Behav Brain Res 2010, 208:47–55. [DOI] [PubMed] [Google Scholar]
- 26.Deliu E, Arecco N, Morandell J, Dotter CP, Contreras X, Girardot C, Kasper EL, Kozlova A, Kishi K, Chiaradia I, et al. : Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat Neurosci 2018, 21:1717–1727. [DOI] [PubMed] [Google Scholar]; * This is the first characterization of a mouse model of SETD5 haploinsufficiency. Setd5 heterozygous mice show cognitive deficits, delayed ultrasonic vocalizations, and behavioral inflexibility, as well as alteration in the expression of postsynaptic density proteins.
- 27.Moore SM, Seidman JS, Ellegood J, Gao R, Savchenko A, Troutman TD, Abe Y, Stender J, Lee D, Wang S, et al. : Setd5 haploinsufficiency alters neuronal network connectivity and leads to autistic-like behaviors in mice. Transl Psychiatry 2019, 9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Araujo DJ, Anderson AG, Berto S, Runnels W, Harper M, Ammanuel S, Rieger MA, Huang HC, Rajkovich K, Loerwald KW, et al. : FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev 2015, 29:2081–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Araujo DJ, Toriumi K, Escamilla CO, Kulkarni A, Anderson AG, Harper M, Usui N, Ellegood J, Lerch JP, Birnbaum SG, et al. : Foxp1 in Forebrain Pyramidal Neurons Controls Gene Expression Required for Spatial Learning and Synaptic Plasticity. J Neurosci 2017, 37:10917–10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usui N, Araujo DJ, Kulkarni A, Co M, Ellegood J, Harper M, Toriumi K, Lerch JP, Konopka G: Foxp1 regulation of neonatal vocalizations via cortical development. Genes Dev 2017, 31:2039–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bacon C, Schneider M, Le Magueresse C, Froehlich H, Sticht C, Gluch C, Monyer H, Rappold GA: Brain-specific Foxp1 deletion impairs neuronal development and causes autistic-like behaviour. Mol Psychiatry 2015, 20:632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medvedeva VP, Rieger MA, Vieth B, Mombereau C, Ziegenhain C, Ghosh T, Cressant A, Enard W, Granon S, Dougherty JD, et al. : Altered social behavior in mice carrying a cortical Foxp2 deletion. Hum Mol Genet 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.French CA, Vinueza Veloz MF, Zhou K, Peter S, Fisher SE, Costa RM, De Zeeuw CI: Differential effects of Foxp2 disruption in distinct motor circuits. Mol Psychiatry 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YC, Kuo HY, Bornschein U, Takahashi H, Chen SY, Lu KM, Yang HY, Chen GM, Lin JR, Lee YH, et al. : Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat Neurosci 2016, 19:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shu W, Cho JY, Jiang Y, Zhang M, Weisz D, Elder GA, Schmeidler J, De Gasperi R, Sosa MA, Rabidou D, et al. : Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc Natl Acad Sci U S A 2005, 102:9643–9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hacohen-Kleiman G, Sragovich S, Karmon G, Gao AYL, Grigg I, Pasmanik-Chor M, Le A, Korenkova V, McKinney RA, Gozes I: Activity-dependent neuroprotective protein deficiency models synaptic and developmental phenotypes of autism-like syndrome. J Clin Invest 2018, 128:4956–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gompers AL, Su-Feher L, Ellegood J, Copping NA, Riyadh MA, Stradleigh TW, Pride MC, Schaffler MD, Wade AA, Catta-Preta R, et al. : Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci 2017, 20:1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martens MB, Frega M, Classen J, Epping L, Bijvank E, Benevento M, van Bokhoven H, Tiesinga P, Schubert D, Nadif Kasri N: Euchromatin histone methyltransferase 1 regulates cortical neuronal network development. Sci Rep 2016, 6:35756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME: Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zylka MJ, Simon JM, Philpot BD: Gene length matters in neurons. Neuron 2015, 86:353–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. : Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501:58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan JM, Badimon A, Schaefer U, Ayata P, Gray J, Chung CW, von Schimmelmann M, Zhang F, Garton N, Smithers N, et al. : Autism-like syndrome is induced by pharmacological suppression of BET proteins in young mice. J Exp Med 2015, 212:1771–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study shows that pharmacological dysregulation of transcriptional initiation/elongation via inhibition of the BET proteins triggers autistic-like behaviors in developing but not adult mice. Social behavioral changes, which are associated with a specific suppression of synapse development and neuronal functional genes of extended length, are reversible following the cessation of the treatment.
- 43.Sims RJ 3rd, Belotserkovskaya R, Reinberg D: Elongation by RNA polymerase II: the short and long of it. Genes Dev 2004, 18:2437–2468. [DOI] [PubMed] [Google Scholar]
- 44.Jonkers I, Lis JT: Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 2015, 16:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao YT, Kwon DY, Johnson BS, Fasolino M, Lamonica JM, Kim YJ, Zhao BS, He C, Vahedi G, Kim TH, et al. : Long genes linked to autism spectrum disorders harbor broad enhancer-like chromatin domains. Genome Res 2018, 28:933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study identified a set of long genes containing broad enhancer-like chromatin domains (BELDs) spanning across their gene bodies. These domains are associated with high transcriptional activities and frequent chromatin interactions. BELD genes are enriched in synaptic genes and risk genes for neurodevelopmental disorders, including ASD.
- 46.Hobert O: Regulation of terminal differentiation programs in the nervous system. Annu Rev Cell Dev Biol 2011, 27:681–696. [DOI] [PubMed] [Google Scholar]
- 47.Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. : Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 2008, 135:749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. : Molecular Architecture of the Mouse Nervous System. Cell 2018, 174:999–1014 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study reports a census of brain cell types based on single-cell RNAseq data across 19 regions of the mouse brain, for a total of half a million of cells.
- 49.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. : Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174:1015–1030 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study catalogs the transcriptional profiles of 690,000 single cells from 9 different regions of the adult mouse brain. Data is compiled in and easily accessed through the useful web-based tool, DropViz.
- 50.Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. : Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Flagship paper from the PsychENCODE Consortium that details transcriptomic and epigenomic changes in human postmortem brain tissue across development, from early embryonic ages to 64 years. Analysis of these samples illuminate how cell subtype composition and gene expression networks transform over time in the human brain.
- 51.Osenberg S, Karten A, Sun J, Li J, Charkowick S, Felice CA, Kritzer M, Nguyen MVC, Yu P, Ballas N: Activity-dependent aberrations in gene expression and alternative splicing in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A 2018, 115:E5363–E5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogers TD, Anacker AMJ, Kerr TM, Forsberg CG, Wang J, Zhang B, Veenstra-VanderWeele J: Effects of a social stimulus on gene expression in a mouse model of fragile X syndrome. Mol Autism 2017, 8:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waddington CH: Canalization of Development and the Inheritance of Acquired Characters. Nature 1942, 150:563–565. [Google Scholar]
- 54.Huang S, Eichler G, Bar-Yam Y, Ingber DE: Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys Rev Lett 2005, 94:128701. [DOI] [PubMed] [Google Scholar]
- 55.Macarthur BD, Ma’ayan A, Lemischka IR: Systems biology of stem cell fate and cellular reprogramming. Nat Rev Mol Cell Biol 2009, 10:672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang S, Ernberg I, Kauffman S: Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin Cell Dev Biol 2009, 20:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munoz-Descalzo S, de Navascues J, Arias AM: Wnt-Notch signalling: an integrated mechanism regulating transitions between cell states. Bioessays 2012, 34:110–118. [DOI] [PubMed] [Google Scholar]
- 58.Li Q, Wennborg A, Aurell E, Dekel E, Zou JZ, Xu Y, Huang S, Ernberg I: Dynamics inside the cancer cell attractor reveal cell heterogeneity, limits of stability, and escape. Proc Natl Acad Sci U S A 2016, 113:2672–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang S, Kauffman S: How to escape the cancer attractor: rationale and limitations of multi-target drugs. Semin Cancer Biol 2013, 23:270–278. [DOI] [PubMed] [Google Scholar]
- 60.Guy J, Gan J, Selfridge J, Cobb S, Bird A: Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315:1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Landmark paper suggesting for the first time a potential reversibility of the neurological phenotype of a mouse model for Rett syndrome, a severe neurodevelopmental disorder associated with ASD caused by mutations in the transcriptional regulator MECP2.
- 61.Sztainberg Y, Chen HM, Swann JW, Hao S, Tang B, Wu Z, Tang J, Wan YW, Liu Z, Rigo F, et al. : Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 2015, 528:123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, Feng G: Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016, 530:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This exciting study shows that normalizing expression of Shank3 in adult Shank3-deficient mice rescues both synaptic and behavioral phenotypes, suggesting the neuronal network state may be more plastic than originally thought.
- 63.Creson T, Rojas C, Hwaun E, Vaissiere T, Kilinc M, Holder JL, Tang J, Colgin LL, Miller CA, Rumbaugh G: Re-expression of SynGAP Protein in Adulthood Improves Translatable Measures of Brain Function and Behavior in a Model of Neurodevelopmental Disorders. bioRxiv 2018:474965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, Foust KD, Kaspar BK, Bird A, Mandel G: Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci 2013, 33:13612–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson L, Guy J, McKay L, Brockett E, Spike RC, Selfridge J, De Sousa D, Merusi C, Riedel G, Bird A, et al. : Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain 2012, 135:2699–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grzadzinski R, Lord C, Sanders SJ, Werling D, Bal VH: Children with autism spectrum disorder who improve with fever: Insights from the Simons Simplex Collection. Autism Res 2018, 11:175–184. [DOI] [PubMed] [Google Scholar]; * Analysis of parent-reported symptom improvement in children with ASD during fever indicates various behaviors temporarily improve in 17% of children. Interestingly, the children with symptom improvement had more severe ASD clinical presentations.