

Cancer stem cells (CSCs) achieved prominence as a concept to explain the inexorable growth of cancers, but the paucity of robust CSC markers for solid tumors has seen the field wane in recent years. Wang et al. now report that a cell surface protein PROCR (Protein C Receptor) enriches for CSCs in about half of triple negative breast cancers and show that inhibition of receptor function by a nanobody profoundly decreases tumor growth, thus highlighting PROCR as a potential therapeutic target.
Triple negative breast cancers (TNBCs) constitute about 15–20% of breast cancers and are defined by the absence of expression for HER2 and the hormone receptors, estrogen receptor (ER) and progesterone receptor (PR). This breast cancer subgroup is highly heterogeneous in terms of biology, molecular features and response to treatment, and thus requires more effective biomarkers for better stratification. Furthermore, there are few therapeutic options besides chemotherapy available to patients with aggressive TNBC due to a lack of targetable molecules.
The CSC model provides one mechanism for understanding intratumoral heterogeneity, resistance to radio- and chemo-therapy, and tumor recurrence.1 CSCs refer to the subset of cancer cells that establish the cellular heterogeneity inherent within the tumor and sustain tumorigenesis. These cells are capable of self-renewal and differentiation, and can reconstitute the entire tumor upon engraftment. Interestingly, the first evidence supporting the CSC theory for solid tumors came from studies on breast cancer by Clarke and colleagues.2 However, there appear to be no universal markers for CSCs in breast and other cancers, with the CSC phenotype varying between individual patient tumors of a given subtype.1 Moreover, little overlap was seen between ALDH+ and CD44hi/CD24lo CSC-enriched populations within the same breast tumor.3
PROCR, also known as Endothelial Protein C Receptor (EPCR), encodes a cell surface protein that is expressed on stem cells in multiple tissues. In the context of human breast tissue, this gene was identified by expression profiling to be highly enriched in the CD44+ population of fractionated normal tissue as well as breast tumors,4 suggesting that it might be a useful marker for the prospective isolation of CSCs.4,5 This concept, however, did not gain momentum at the time, perhaps due to the large variation in CD44 expression observed across different breast cancer subtypes. The study by Wang et al. has significantly clarified this area through identification of PROCR as a specific marker of CSCs in TNBCs.6 They first compared the tumor-initiating capacity of Procr+ tumor cells in three different mouse models of breast cancer. Although Procr was expressed on a small subset of tumor cells in all models, it specifically enriched for CSCs in the Wnt1-driven transgenic model, which harbors an expanded population of stem-like cells.7 Interestingly, Procr was previously shown by Zeng and colleagues to mark a rare subset of cycling mammary stem cells in the mouse mammary gland8 (Fig. 1), suggesting that stem cells could be the cell of origin in Wnt1-driven tumors. Whether PROCR plays an analogous role in normal human breast tissue remains unknown but its expression is confined to the basal layer.6
Fig. 1.
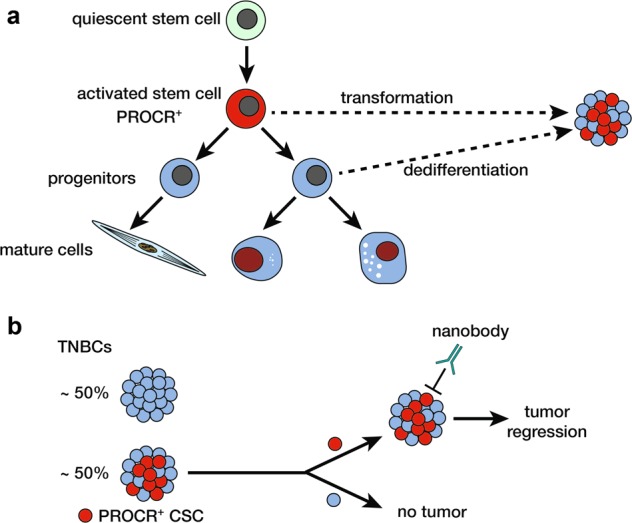
a Proposed mammary epithelial differentiation hierarchy based on fractionation and transplantation studies, comprising quiescent10 and activated stem cells,8 the latter of which are marked by PROCR. b PROCR+ CSCs, present in ~50% of TNBCs, can regenerate an entire tumor that recapitulates the parental tumor while PROCR− cells lack tumor-forming potential upon transplantation. Treatment of mice harboring PROCR+ tumors with a specific anti-PROCR nanobody leads to profound tumor regression
Extending their study to specimens from breast cancer patients, Wang et al. next showed that PROCR was highly expressed in approximately half of TNBCs in their patient cohorts. Notably, there was an inverse correlation between PROCRhi and BRCA1 mutant TNBCs. Using transplantation assays, CSCs were demonstrated to be highly enriched in the PROCR+ population of PROCR-expressing triple-negative tumors. Indeed, a single PROCR+ cancer cell was capable of reconstituting a tumor that recapitulated the original PDX tumor (Fig. 1). In contrast to the classical CSC hypothesis positing that only a small fraction of cancer cells behave as CSCs,1 up to 45% of PROCR+ cells were present in primary TNBCs and PDXs in this study. It is plausible that highly aggressive tumors harbor a more substantial population of CSCs.
Functional studies via shRNA knockdown or CRISPR/CAS9 knockout revealed that PROCR is essential for the growth of PDX tumors.6 To explore the effect of targeting PROCR function using a more clinically feasible strategy, Wang et al. developed an inhibitory nanobody against an extracellular region of PROCR. This antibody proved to be very effective as a single agent in delaying tumor growth in recipient mice transplanted with PROCR+ cells isolated from breast tumors, when treatment was commenced shortly after engraftment. This was accompanied by a decrease in the PROCR+ CSC subset as well as in mesenchymal characteristics. Importantly, the antibody was also effective in the setting of established tumors, in which the nanobody significantly inhibited the growth of TNBC PDXs, either alone or in combination with two standard-of-care treatments for TNBC, paclitaxel and doxorubicin. In combination therapy, the antibody led to a sustained response and almost completely blocked tumor relapse. These findings for established tumors have significant implications for breast cancer patients.
Questions for future studies in this area relate to tumor heterogeneity and the PROCR-mediated signal transduction pathways relevant to CSCs. For example, while a single PROCR+ cell was able to regenerate tumors in the transplantation assay, CSCs among PROCR+ cells in PROCR+ human breast tumors were estimated at approximately 1:100. This may in part reflect the xenotransplantation environment but also suggests that the PROCR+ population is not homogeneous. Single-cell profiling of PROCR+ cells in PROCR+ tumors may help to unravel the heterogeneity within this cell population and further define PROCR+ CSCs. Furthermore, there was no apparent overlap between PROCR+ CSCs and other markers of breast CSCs,1,3 consistent with potential heterogeneity amongst CSC populations. While promising as a functional marker, further work will be needed to establish how robustly PROCR identifies tumor-propagating cells in primary TNBCs. Interestingly, PROCR+ cancer cells show similarity to PROCR+ bipotent stem cells in the normal mouse mammary gland.8 Genetic lineage tracing using mouse models mimicking PROCR+ TNBC will help determine whether PROCR+ stem cells can serve as cells of origin of cancer. PROCR has previously been shown by this group to regulate a complex set of signaling pathways including MEK-ERK, PI3K-AKT-mTOR and RhoA-ROCK,9 but the key pathways activated by this single-pass transmembrane receptor and those necessary for conferring CSC features remain to be elucidated. Interestingly, PROCR is a Wnt target gene8 and gain-of-function mutations in β-catenin or inactivating mutations in APC result in enhanced Wnt signaling in diverse cancer types, although these mutations do not play a prominent role in breast cancer. Nevertheless, Wnt pathway activation may still occur in TNBCs. In addition, it is plausible that specific ligand(s) bind PROCR on the membrane to activate downstream signaling pathways. Irrespective of the precise mechanisms, the study by Wang et al. provides compelling evidence for an important role of PROCR in a substantial subset of TNBCs and the potential of anti-PROCR therapy.6
References
- 1.Visvader JE, Lindeman GJ. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Proc. Natl Acad. Sci. USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ginestier C, et al. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shipitsin M, et al. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Schaffner F, et al. PLoS ONE. 2013;8:e61071. doi: 10.1371/journal.pone.0061071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang D, et al. Cell Res. 2019;29:832–845. doi: 10.1038/s41422-019-0225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shackleton M, et al. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, et al. Nature. 2015;517:81–84. doi: 10.1038/nature13851. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, et al. J. Biol. Chem. 2018;293:1413–1424. doi: 10.1074/jbc.M117.814046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu NY, et al. Nat. Cell Biol. 2017;19:164–176. doi: 10.1038/ncb3471. [DOI] [PubMed] [Google Scholar]