

Abstract
Allosteric inhibitors of glutaminase (GAC), such as BPTES, CB-839 and UPGL00019, have great promise as inhibitors of cancer cell growth, but potent inhibitors with drug-like qualities have been difficult to achieve. Here, a small library of GAC inhibitors based on the UPGL00019 core is described. This set of derivatives was designed to assess if one or both of the phenylacetyl groups flanking the UPGL00019 core can be replaced by smaller simple aliphatic acyl groups without loss in potency. We found that one of the phenylacetyl moieties can be replaced by a set of small aliphatic moieties without loss in potency. We also found that enzymatic potency co-varies with the VDW volume or the maximum projection area of the groups used as replacements of the phenylacetyl moiety and used literature X-ray data to provide an explanation for this finding.
Keywords: GAC, UPGL00019, CB-839, UPGL00019 derivatives, Novel glutaminase inhibitors, BPTES
Graphical Abstract
Most cancer cells engage in an altered metabolic program that includes an addiction to glutamine. This glutamine addiction is satisfied in part by the enzymatic action of kidney type glutaminase (GLS), which has two splice variants: KGA, and the shorter variant GAC.1 Cumulative evidence suggests that GAC in particular is an important target for anticancer therapy. Upregulation of GAC, is seen in a number of human tumor cell lines and correlates with increased proliferative rates and in certain instances with tumor progression.2–9
Evidence suggest that KGA/GAC exist as a dimer in the inactive state, and as tetramer in the active state.10 BPTES was the first allosteric small molecule inhibitor of KGA/GAC reported.11 It binds to KGA/GAC at the interface between two symmetrical dimers apparently stabilizing an inactive tetrameric form of the protein.10,12–13 Since its disclosure, a number of BPTES-based analogs have been reported with CB-839 being the most advanced.14
In an earlier report we described a series of KGA/GAC inhibitors with heteroatom substituted cyclic spacers as surrogates for the straight chain spacers seen in BPTES and CB-839.15 These analogs were prepared in an effort to improve potency by minimizing the entropic penalty for binding flexible linkers impose and also improve the physicochemical properties of the BPTES-class of compounds by reducing rotational bonds and improving logP.
Among the compounds we disclosed was UPGL00019 (Figure 1), a compound with a 4-hydroxypiperidine linker and high potency in enzymatic and cell assays. In our earlier report we also showed that removal of phenyl moieties from this compound, reduces potency (Figure 1, UPGL00020).15
Figure 1.
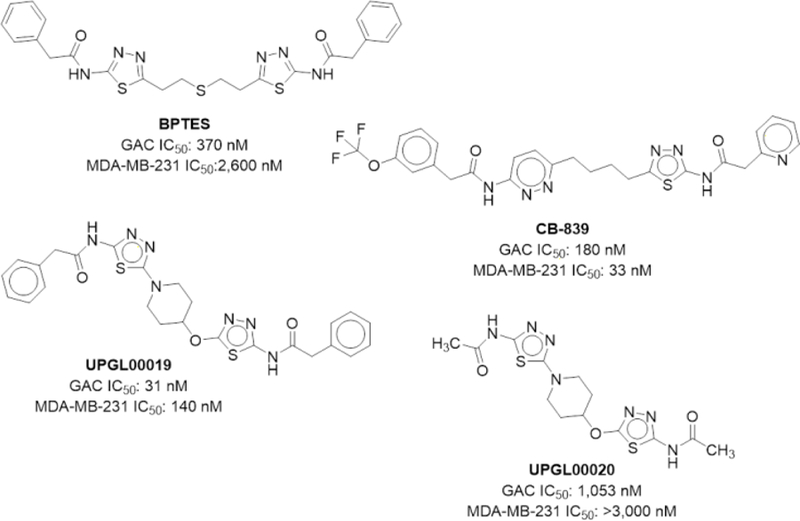
BPTES, CB-839 and 4-hydroxypiperidine-containing analogs UPGL00019 and UPGL00020
We were interested in identifying drug-like Lipinski/Veber compliant derivatives of UPGL00019 that have equal or better potency than the parent, and in which one or both of the phenyl moieties are replaced by simple low MW non-aromatic surrogates.16–17 We were also interested in understanding terminal group requirements and their relation to potency. Available x-ray structures of KGA and GAC in complex with BPTES (PDB: 3UO9, 3V0Z), or GAC in complex with CB-839 (PDB: 5HL1), UPGL00019 (PDB: 5I94) and other analogs show that the terminal phenylacetyl groups present in the compounds are mobile and always have higher b-factors than the core of the compound they are attached to. Furthermore, these groups have variable orientations in the x-ray structures which cannot be explained by simple differences in overall compound structure. For example, overlaying the 3VOZ and 3UO9 x-ray structures (Fig 2A) the terminal phenyls of BPTES are seen to have high b-factors and occupy different areas in the allosteric pocket but the BPTES cores (thiadiazoles and flexible chain) in both structures align perfectly and occupy identical space. An overlay of the 3UO9 and 5I94 similarly shows that the phenyl groups are highly mobile but the compounds’ cores occupy identical space (Fig 2B). The apparent mobility of the aryl substituents would suggest that they should be dispensable with respect to potency yet the SAR we and others have disclosed suggests otherwise.
Figure 2.
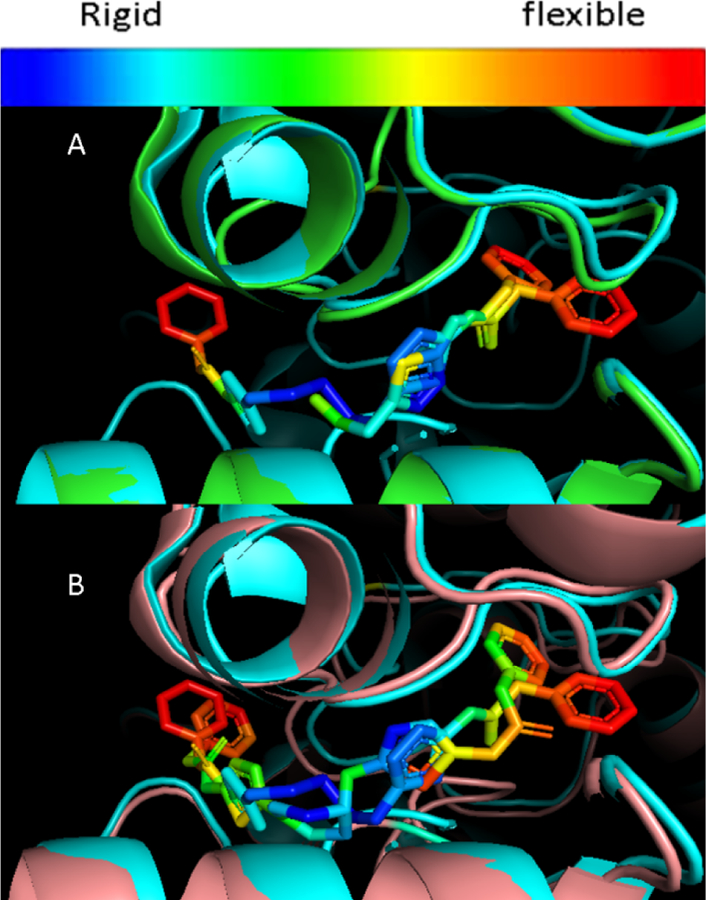
Overlay of 3UO9 (green) and 3VOZ (cyan) (A), 3UO9 and 5I94 (clay) (B) X-rays. Heat map of b-factors for BPTES and UPGL00019
To achieve our goals we decided on a two stage strategy. In the first stage we would pursue the synthesis of UPGL00019 derivatives that contained one phenylacetic acid moiety and one simple small non-aromatic acid moiety. In this stage, our objective was to identify small simple non-aromatic moieties that could serve as possible replacements for one of the UPGL00019 phenylacetic acid moieties and which could yield derivatives with equal or better potency than the parent molecule in enzyme and cell assays. In the second stage of our strategy, the best stage-1 replacement moieties identified would be combined with the UPGL00019 core to form new Lipinski compliant derivatives that hopefully would be equipotent or more potent than UPGL00019.
The allosteric pocket where BPTES and UPGL00019 bind is symmetric. It is formed at the interface of KGA/GAC dimers via the antiparallel arrangement of identical residues from each of the monomers participating in the dimer. As such, although UPGL00019 derivatives with one phenylacetic acid moiety and one smaller group are inherently asymmetric, the symmetry of the binding pocket makes the inherent asymmetry of such derivatives irrelevant with respect to their binding orientation in the pocket and precludes potency differences between them because of differential binding orientation alone.
In the first stage of our strategy we prepared derivatives 6a–f with one phenyl acetic acid group and one simple aliphatic acyl group from key intermediate 3, which we have previously described, as shown in scheme 1.15 Each derivative was then tested for potency against our GAC biochemical assay and against the MDA-MB-231 cancer cell line.18
Scheme 1.
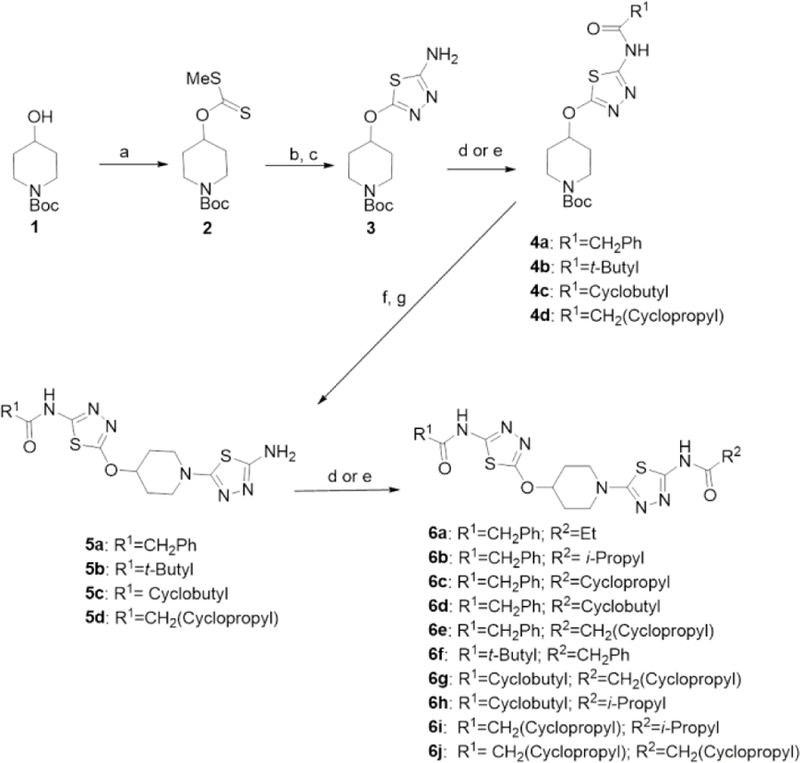
Synthesis of UPGL00019 derivatives. Reagents and conditions: a) NaH, CS2, MeI, THF, rt; b) NH2NH2 MeOH, rt; c) BrCN, Et3N, MeOH, rt; d) R1/R2-acyl chloride or R1/R2-acyl anhydride, Et3N, DMF, rt; e) R1/R2-acid HATU, Et3N, DMF, rt; f) 4N HCl in Dioxane, rt; g) 2-bromo-4-aminothiadiazole, K2CO3, DMSO, 50–55 °C or 2-bromo-4-aminothiadiazole, Et3N, EtOH, 80 °C.
With the exception of 6a, all of the stage-1 derivatives had similar potency to that of UPGL00019 in enzymatic assays, suggesting that –isopropyl, -cyclopropyl, -cyclobutyl, -CH2(cyclopropyl) and -tert-butyl groups are good surrogates for the -CH2Phe moieties present in that compound (Table 1).
Table 1.
Group characteristics and potencies for UPGL00019, UPGL00020 and derivatives 6a–j
| Cmpd | R1 | Acyl-R1 VDW volumea (Å3) | Acyl-R1 MPAa (Å2) | R2 | Acyl-R2 VDW volumea (Å3) | Acyl-R2 MPAa (Å2) | R1+R2 VDW volume (Å3) | R1+R2 MPA (Å2) | GACb IC50 (nM) | GAC pIC50 | MDA-MB-231b IC50 (nM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UPGL00019 | 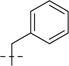 |
107.2 | 29.8 | 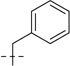 |
107.2 | 29.8 | 214.4 | 59.6 | 31 | 7.51 | 140 |
| UPGL00020 | 36.4 | 12.4 | 36.4 | 12.4 | 72.8 | 24.8 | 1,053 | 5.98 | >3,000 | ||
| 6a | 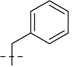 |
107.2 | 29.8 | 53.4 | 16.7 | 160.6 | 46.5 | 103 | 6.99 | 227 | |
| 6b | 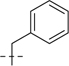 |
107.2 | 29.8 | 70.6 | 19.4 | 177.8 | 49.2 | 27 | 7.57 | 253 | |
| 6c | 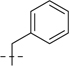 |
107.2 | 29.8 | 58.7 | 19.4 | 165.9 | 49.2 | 10 | 8.00 | 627 | |
| 6d | 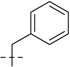 |
107.2 | 29.8 | 75.9 | 23.4 | 183.1 | 53.2 | 17 | 7.77 | 159 | |
| 6e | 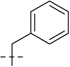 |
107.2 | 29.8 | 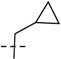 |
75.7 | 23.0 | 182.9 | 52.8 | 29 | 7.54 | 179 |
| 6f | 88.1 | 23.8 |  |
107.2 | 29.8 | 195.3 | 53.6 | 38 | 7.42 | 589 | |
| 6g | 75.9 | 23.4 |  |
75.7 | 23.0 | 151.6 | 46.4 | 162 | 6.79 | 384 | |
| 6h | 75.9 | 23.4 | 70.6 | 19.4 | 146.5 | 42.8 | 240 | 6.62 | 419 | ||
| 6i | 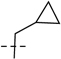 |
75.7 | 23.0 | 70.6 | 19.4 | 146.3 | 42.4 | 200 | 6.70 | 396 | |
| 6j | 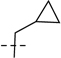 |
75.7 | 23.0 | 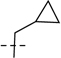 |
75.7 | 23.0 | 151.4 | 46.0 | 157 | 6.80 | 630 |
Acyl-R1 and acyl-R2 Van der Walls (VDW ) volumes and maximum projected areas (MPA) were calculated using Chemaxon’s Chemicalize
IC50 values are the result of a single experiment run in triplicate
With the exception of 6f, which had MW that exceeded the Lipinski cut-off (MW≤500), derivatives also had physicochemical properties well within the Lipinski/Veber drug-like space and all compounds had excellent ligand efficiency (LE) (Table 2).
Table 2.
Physicochemical properties of UPGL00019, UPGL00020 and derivatives 6a–j
| Cmpd | MW | cLogPa | NRB | LEb |
|---|---|---|---|---|
| UPGL00019 | 535.6 | 4.2 | 9 | 0.28 |
| UPGL00020 | 383.5 | 0.5 | 5 | 0.33 |
| 6a | 473.6 | 3.0 | 8 | 0.31 |
| 6b | 487.6 | 3.6 | 8 | 0.32 |
| 6c | 485.6 | 3.1 | 8 | 0.34 |
| 6d | 499.6 | 3.6 | 8 | 0.32 |
| 6e | 499.6 | 3.3 | 9 | 0.31 |
| 6f | 501.6 | 4.1 | 8 | 0.31 |
| 6g | 463.6 | 2.7 | 8 | 0.31 |
| 6h | 451.6 | 3.0 | 7 | 0.31 |
| 6i | 451.6 | 2.7 | 8 | 0.31 |
| 6j | 463.6 | 2.4 | 9 | 0.31 |
Calculated using Chemaxon’s Chemicalize web portal
LE:=∆G/N=−1.4log(IC50)/N
From the simple aliphatic moieties investigated, -cyclobutyl, -isopropyl and the -CH2(cyclopropyl) groups provided derivatives with best potency in enzyme and cell assays (Table 1). As such, these groups were chosen for synthesis of derivatives where both phenylacetyl moieties were replaced by them.
Compound asymmetry is not a factor with respect to binding orientation. As such, in order to efficiently explore the viability of these groups as replacements for both of the phenylacetic acid groups we pursued the synthesis of ‘mixed’ derivatives 6g-i in which the UPGL00019 core is flanked by combinations of the best groups identified. Testing of these derivatives revealed that they were 5–8 fold less potent than UPGL00019 in the biochemical assay, 2–3 fold less potent in the MDA-MB-231 cell assay and all had potency similar to compound 6j, a compound we had prepared during our initial SAR investigation in the broader class.15
The mobility of the phenyl groups seen in the available crystal structures of BPTES-class derivatives led us to hypothesize that potency in the UPGL00019 series, and possibly the BPTES-class in general, may be driven simply by the volume (physical and/or rotational) displaced by the acyl groups flanking the UPGL00019 core.
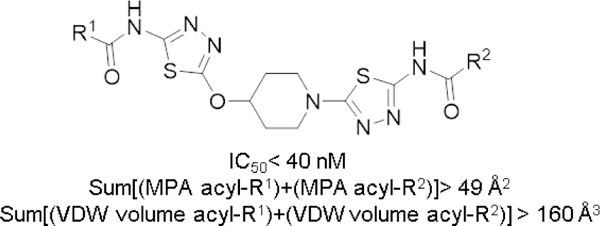
We calculated the Van der Walls (VDW) volumes and maximum projection area (MPA) for each of the acyl-R1 and acyl-R2 groups present in UPGL00019, UPGL00020 and derivatives 6a–j and found that these parameters, correlate very well with potency. As the cumulative VDW volume or the cumulative MPA values of the acyl-R1 and acyl-R2 groups flanking the UPMP00019 core increase the potency of derivatives increases and vice versa (Table 1, Figure 3).19 The data suggests that acyl-R1/R2 groups with cumulative VDW volume equal or greater than 160 Å3 and/or cumulative MPA values greater than 49 Å2 provide UPGL00019 derivatives that are equipotent or more potent than the parent.
Figure 3.

Correlation of pIC50 with the cumulative values of VDW volume or MPA of the acyl-R1 and acyl-R2 groups of UPGL000019 and its derivatives.
The symmetric BPTES binding pocket is formed in part by two antiparallel identical loops (Ser314-Asp327), each from the monomers that form a dimer. To understand the correlation between the size/mobility of the groups flanking the UPGL00019 core and potency we overlayed the UPGL00019-GAC X-ray structure (PDB 5I94) with publicly available X-ray structures of KGA in complex with non-potent BPTES derivatives that lack phenylacetic acid moieties (PDBs: 3VP2, 3VP3, 3VP4).12 This overlay (Figure 4) showed that in the 3VP2, 3VP3 and 3VP4 structures the Gly315-Lys320 segment of the Ser314-Asp327 loop is particularly disordered and, based on the positioning of the resolved residues, is at a different location from the location of the same segment in the 5I94 structure. In the latter structure, the Gly315-Lys320 segments from either side of the symmetric pocket are “pushed back” by the phenylacetyl groups of UPGL00019 leading to a drastically different orientation of the amino acids involved in this segment. A similar “pushed back” orientation for the Gly315-Lys320 segment can be seen in the BPTES-GAC (PDB: 3UO9), CB-839-GAC (5HL1), BPTES-KGA (3V0Z), and other publicly available x-ray structures with potent inhibitors.20
Figure 4.
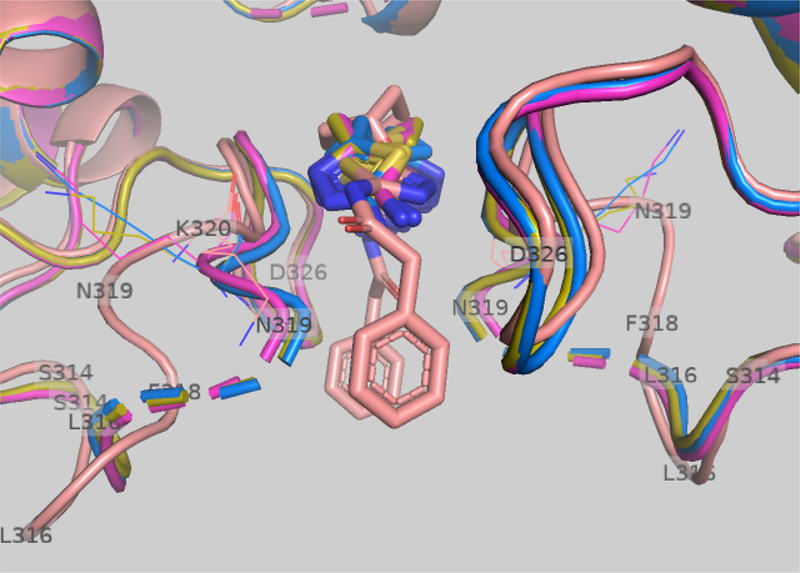
Overlay of 5I94 (brown), 3VP2 (magenta), 3VP3 (blue), and 3VP4 (green) x-ray structures.
The crystallographic data appears to suggest that symmetric or asymmetric substitution of the UPGL00019 core with acyl groups can provide derivatives equipotent to the parent as long as these groups have adequate cumulative size (physical and/or rotational) to effectively “push back” the Gly315-Lys320 segment at both sides of the symmetric binding pocket. This aligns well with our observation that UPGL00019 derivative potency co-varies with the cumulative VDW volume and/or the MPA values of the acyl moieties flanking its core.
In summary, we prepared a small library of UPGL00019 derivatives in an effort to identify Lipinski/Veber compliant derivatives of this compound and to understand how the observed mobility and size of its terminal phenylacetic acid groups imparts potency. We found that activity of derivatives correlates with cumulative terminal group size (VDW volume and/or MPA). Examination of available x-ray data appears to suggest that larger groups through their size and mobility are better able to deflect a 5 amino acid segment composed by residues Gly315-Lys320.
Supplementary Material
Aknowledgements
Funding support was provided by the University of Pittsburgh Innovation Institute and NIH grants R01GM108340, R01CA201402 and R01GM1222575.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data to this article can be found on line
References
- 1.Elgadi KM; Meguid RA; Qian M; Souba WW; Abcouwer SF, Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genomics 1999, 1 (2), 51–62. [DOI] [PubMed] [Google Scholar]
- 2.Szeliga M; Obara-Michlewska M, Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem Int 2009, 55 (1–3), 71–5. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Gomez C; Campos-Sandoval JA; Alonso FJ; Segura JA; Manzanares E; Ruiz-Sanchez P; Gonzalez ME; Marquez J; Mates JM, Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem J 2005, 386 (Pt 3), 535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang F; Zhang Q; Ma H; Lv Q; Zhang T, Expression of glutaminase is upregulated in colorectal cancer and of clinical significance. Int J Clin Exp Pathol 2014, 7 (3), 1093–100. [PMC free article] [PubMed] [Google Scholar]
- 5.Pan T; Gao L; Wu G; Shen G; Xie S; Wen H; Yang J; Zhou Y; Tu Z; Qian W, Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem Biophys Res Commun 2015, 456 (1), 452–8. [DOI] [PubMed] [Google Scholar]
- 6.Altman BJ; Stine ZE; Dang CV, From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 2016, 16 (10), 619–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matre P; Velez J; Jacamo R; Qi Y; Su X; Cai T; Chan SM; Lodi A; Sweeney SR; Ma H; Davis RE; Baran N; Haferlach T; Su X; Flores ER; Gonzalez D; Konoplev S; Samudio I; DiNardo C; Majeti R; Schimmer AD; Li W; Wang T; Tiziani S; Konopleva M, Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget 2016, 7 (48), 79722–79735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang Y; Stine ZE; Xia J; Lu Y; O’Connor RS; Altman BJ; Hsieh AL; Gouw AM; Thomas AG; Gao P; Sun L; Song L; Yan B; Slusher BS; Zhuo J; Ooi LL; Lee CG; Mancuso A; McCallion AS; Le A; Milone MC; Rayport S; Felsher DW; Dang CV, Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest 2015, 125 (6), 2293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassago A; Ferreira AP; Ferreira IM; Fornezari C; Gomes ER; Greene KS; Pereira HM; Garratt RC; Dias SM; Ambrosio AL, Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A 2012, 109 (4), 1092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y; Erickson JW; Stalnecker CA; Katt WP; Huang Q; Cerione RA; Ramachandran S, Mechanistic Basis of Glutaminase Activation: A key enzyme that promotes glutamine metabolism in cancer cells. J Biol Chem 2016, 291 (40), 20900–20910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newcomb RW, Selective Inhibition of Glutaminase by Bis-thiadiazoles. US2002/0115698 A1 2002.
- 12.Thangavelu K; Pan CQ; Karlberg T; Balaji G; Uttamchandani M; Suresh V; Schuler H; Low BC; Sivaraman J, Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A 2012, 109 (20), 7705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeLaBarre B; Gross S; Fang C; Gao Y; Jha A; Jiang F; Song JJ; Wei W; Hurov JB, Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 2011, 50 (50), 10764–70. [DOI] [PubMed] [Google Scholar]
- 14.Zimmermann SC; Duvall B; Tsukamoto T, Recent Progress in the Discovery of Allosteric Inhibitors of Kidney-Type Glutaminase. J Med Chem 2019, 62 (1), 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDermott LA; Iyer P; Vernetti L; Rimer S; Sun J; Boby M; Yang T; Fioravanti M; O’Neill J; Wang L; Drakes D; Katt W; Huang Q; Cerione R, Design and evaluation of novel glutaminase inhibitors. Bioorg Med Chem 2016, 24 (8), 1819–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipinski CA; Lombardo F; Dominy BW; Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001, 46 (1–3), 3–26. [DOI] [PubMed] [Google Scholar]
- 17.Veber DF; Johnson SR; Cheng HY; Smith BR; Ward KW; Kopple KD, Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002, 45 (12), 2615–23. [DOI] [PubMed] [Google Scholar]
- 18. Assay details have been disclosed previously and can be found in reference 15.
- 19. Acyl-R VDW volumes and MPAs were calculated by substracting the calculated VDW volume or MPA value of aminothiadiazole from the calculated VDW volume or the MPA value of the corresponding acyl-R-aminothiazole moiety present in each derivative. VDW volumes and MPA values were calculated using Chemaxon’s Chemicalize.
- 20. A “pushed back” orientation of this segment can be seen also in x-ray structures: 5FI7,5JYP,5JYO, 5WJ6, 5FI7, 5FI6 and 5FI2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.