

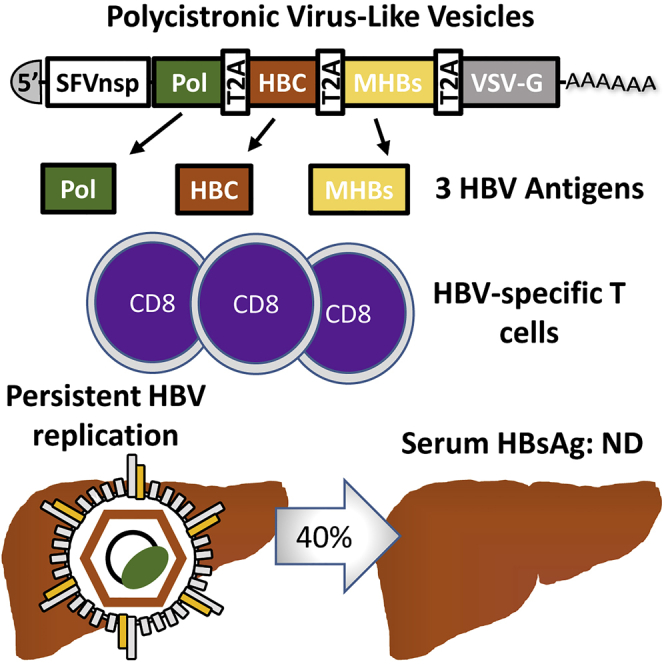
Summary
Infections with hepatitis B virus (HBV) can initiate chronic hepatitis and liver injury, causing more than 600,000 deaths each year worldwide. Current treatments for chronic hepatitis B are inadequate and leave an unmet need for immunotherapeutic approaches. We designed virus-like vesicles (VLV) as self-amplifying RNA replicons expressing three HBV antigens (polymerase, core, and middle surface) from a single vector (HBV-VLV) to break immune exhaustion despite persistent HBV replication. The HBV-VLV induces HBV-specific T cells in naive mice and renders them resistant to acute challenge with HBV. Using a chronic model of HBV infection, we demonstrate efficacy of HBV-VLV priming in combination with DNA booster immunization, as 40% of treated mice showed a decline of serum HBV surface antigen below the detection limit and marked reduction in liver HBV RNA accompanied by induction of HBsAg-specific CD8 T cells. These results warrant further evaluation of HBV-VLV for immunotherapy of chronic hepatitis B.
Subject Areas: Immunology, Virology, Medical Microbiology
Graphical Abstract
Highlights
-
•
Virus-like vesicles (VLV) designed to express three HBV antigens from a single vector
-
•
Expression of HBV polymerase, core, and middle S antigen-induced specific CD8+ T cells
-
•
A single immunization protects mice from acute HBV infection
-
•
VLV prime and DNA boost decreased HBV replication in a model of chronic hepatitis B
Immunology; Virology; Medical Microbiology
Introduction
Human hepatitis B virus (HBV), a partially double-stranded DNA virus of the genus Orthohepadnavirus, replicates in hepatocytes and causes liver inflammation and injury (Ganem and Prince, 2004, Seeger and Mason, 2015, Tang et al., 2018). Although most adults recover from the acute infection and become immune to subsequent exposures, 2%–6% of adults and 90% of infants develop chronic hepatitis B (CHB) and are at increased risk of developing liver cirrhosis or liver cancer (Ganem and Prince, 2004, Tang et al., 2018). Multiple factors, including vertical transmission to newborns before the immune system is fully formed and active suppression of the immune system by hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg), contribute to HBV persistence (Tsai et al., 2018). Worldwide, 240–350 million people are chronically infected with HBV and more than 600,000 die each year from HBV-associated liver diseases. In the United States, CHB prevalence is estimated at 850,000 and the rate of new infections remains about 1 per 100,000 population despite routine use of a prophylactic HBV vaccine (Schillie et al., 2018). Currently approved therapeutic approaches for CHB, including pegylated interferon-α (IFNα) and nucleoside/nucleotide analogues, may reduce and control HBV replication in a subset of patients (Tang et al., 2018). However, adverse effects of IFNα treatment, lack of complete HBV clearance, and development of drug resistance after prolonged treatment with nucleosides/nucleotides emphasize the unmet need for alternative therapeutic approaches that can lead to functional cure (Block et al., 2018, Schinazi et al., 2018, Tang et al., 2018).
Spontaneous clearance of HBV through immune-mediated mechanisms, particularly T cell-mediated immune responses, in acute and in some chronically infected patients provides a rationale for CHB immunotherapy (Kosinska et al., 2017). Ongoing efforts at preclinical and early clinical stages include activation of innate immunity through TLR ligands, treatment with immune checkpoint inhibitors, and/or immunization with HBV antigens delivered by plasmid DNA or viral vectors (Kosinska et al., 2017, Schinazi et al., 2018). DNA immunization is a safe and reliable method for induction of antibody responses, but this approach requires special delivery such as electroporation, and remains poorly immunogenic for T cells (Cova, 2017). On the other hand, viral vectors are highly immunogenic and efficient at mounting T cell responses, but their safety remains a concern even for attenuated and replication-deficient viral vectors (Kosinska et al., 2017).
Virus-like vesicles (VLVs), a form of self-amplifying RNA replicons, are a non-pathogenic and immunogenic alternative to viral vectors. VLVs have an excellent safety profile and induce T cell and antibody responses (Rose et al., 2014, Rose et al., 2008, Schell et al., 2011). The RNA-dependent RNA polymerase from Semliki Forest virus and glycoprotein from vesicular stomatitis virus (VSV-G) expressed from the same vector are essential components of the VLV platform that enables rapid and efficient production of VLVs in BHK-21 cells and expression of the desired antigens in the immunized host (Rose et al., 2014). A previous study showed that immunization with VLVs expressing HBV middle surface antigen (MHBs) induced HBV-specific CD8+ T cells and protected mice against experimental acute HBV infection (Reynolds et al., 2015). Furthermore, HBV-transgenic mice with low HBeAg produced HBV-specific CD8+ T cells after priming with plasmid DNA and boosting with the VLV expressing MHBs, suggesting that VLV immunization may break immune tolerance, a characteristic feature of CHB (Reynolds et al., 2015). Although that study demonstrated promise for VLV-based immunotherapy, it is evident from other studies that immunization with multiple HBV antigens to enhance and/or broaden T cell responses is necessary for therapeutic effect and HBV elimination (King et al., 2014, Martin et al., 2014, Obeng-Adjei et al., 2013).
In this report, we describe the design, production, immunogenicity, and immunotherapeutic potential of a polycistronic VLV vector that expresses three HBV antigens: polymerase (Pol), core (HBcAg), and MHBs. Our goal was to target multiple HBV antigens simultaneously and to elicit the broad and vigorous multi-specific immune responses needed for the resolution of CHB infection in humans. To that end, we co-expressed structural (MHBs and HBcAg) and non-structural (Pol) HBV proteins as a single polycistronic open reading frame. This approach enabled the induction of broad T cell responses to multiple antigens that were sufficient to protect mice from experimental acute HBV replication. In a murine model of experimental CHB driven by AAV-HBV transduction, prime immunization with the multiantigen VLV vector followed by two booster DNA immunizations induced HBsAg-specific CD8+ T cell responses and reduced serum HBsAg and liver HBV RNA.
Results
VLV Design and Production
Because HBV Pol and HBcAg are important targets for CD8+ T cells (Akbar et al., 2012, Mizukoshi et al., 2004, Zhuo et al., 2015), we aimed to incorporate these antigens into the new VLV vectors for immunization. The previously reported VLV vector (referred to as MT2A and depicted in Figure 1A) relied on the ribosome-skipping 2A peptide from Thosea asigna virus (T2A) to express both MHBs and VSV-G from the same subgenomic RNA (Reynolds et al., 2015). Using that vector as a backbone, we designed and constructed VLVs for expression of HBV polymerase alone (PolT2A) or as a polycistronic vector for expression of Pol, HBcAg, and MHBs by interspacing the HBV antigens with the ribosome skipping peptides (3xT2A) as depicted in Figure 1A. We replaced several key amino acids in the terminal protein domain of Pol (Y63A, W74A, Y147A, and Y173A) to abolish its DNA binding (primase) activity (Lanford et al., 1999). In addition, we deleted amino acids 538–544 encompassing the active site (residues YMDD) of the reverse transcriptase domain to remove its RNA-dependent DNA polymerase and DNA-dependent DNA polymerase activities. Furthermore, we introduced mutations into the RNAse H domain (amino acids 680–832) in order to abolish the ribonuclease H activity, as previously described (Kim et al., 1999). We altered the HBcAg sequence to improve processing and peptide presentation by substituting the disulfide bond-forming cysteine residues (C48S, C61S, C107S and C183S) and adding an N-terminal fusion with the 76-amino acid ubiquitin (Ub) peptide (Nassal et al., 1992, Zhuo et al., 2015). To produce VLVs for evaluation of antigen expression and immunogenicity, we transfected BHK-21 cells with the plasmid DNA expressing 3xT2A VLV RNA under the control of CMV promoter, collected the master VLV stocks, and used them for propagation and concentration of VLVs by ultrafiltration. The resulting VLV titers routinely exceeded 109 pfu/mL.
Figure 1.

Virus-like Vesicle Platform for Therapeutic HBV Vaccine Expressing Polymerase (Pol), Ubiquitinated Core (Ub-HBc), and Middle Surface (MHBs) Antigens
(A) Design of replicating VLV for expression of MHBs (MT2A), HBV Pol (PolT2A) single antigens, and for polycistronic expression of the three HBV antigens using different 2A self-cleaving peptides (3xT2A and Mix2A).
(B) Validation of antigen expression in BHK-21 cells after infection with PolT2A or 3xT2A VLV (MOI = 1) by immunofluorescence at 20 h post infection using the antibodies specific for HBV Pol and HBcAg.
(C) Validation of antigen expression in BHK-21 cells after infection with VLV expressing MHBs only (MT2A), Pol only (PolT2A), or the three antigens (3xT2A) by flow cytometry using antibodies specific for Pol, MHBs, HBcAg, and VSV-G. Geometric mean fluorescence intensity is shown for each antibody and VLV.
(D) Evaluation of antigen expression and VLV replication in BHK-21 cells transfected with plasmid DNA for 3xT2A or Mix2A VLV using 2A-peptide specific antibody in western blots. Anti-VSV-G and anti-actin were also used as controls. The data are representative of three independent experiments.
To mitigate potential risks of homologous recombination of the repeating T2A peptide sequences in the 3xT2A construct during VLV replication, we designed and evaluated an alternative construct, Mix2A, in which we replaced the two T2A peptides with the porcine teschovirus-1 (P2A) and equine rhinitis A virus (E2A) peptides (Liu et al., 2017) (Figure 1A). These peptides are structurally similar, but encoded by different nucleotide sequences. When produced in BHK-21 cells under identical conditions, the resulting titers were similar for Mix2A VLVs (2.2*109 pfu/mL) and 3xT2A VLVs (1.8*109 pfu/mL).
Validation of Antigen Expression
We evaluated expression of Pol and HBcAg in BHK-21 cells infected with PolT2A and 3xT2A VLVs by immunofluorescence (Figure 1B). Using HBV Pol specific mAb clone 2C8 (zu Putlitz et al., 1999), we observed a characteristic fine granular staining in the cytoplasm at 24 h after infection with either of the VLVs (Figure 1B, top row). Staining with the HBcAg-specific polyclonal antibody showed variable levels of HBcAg expression in most of the BHK-21 cells infected with 3xT2A, but not with the PolT2A VLV (Figure 1B, second row).
To compare expression levels of HBV antigens in BHK-21 cells infected with polycistronic VLVs 3xT2A versus VLVs expressing Pol or MHBs alone, all at MOI = 1 at 24 h post infection, we used intracellular staining with antibodies for Pol, HBcAg, and MHBs, followed by flow cytometry analyses (Figure 1C). Cells infected with 3xT2A VLVs showed slightly higher levels of Pol expression, compared with PolT2A VLVs. On the contrary, cells infected with 3xT2A VLVs showed slightly lower levels of MHBs expression compared with MT2A VLVs. As expected, no Pol staining was detected in cells infected with the MT2A VLVs and no MHBs staining was detected in cells infected with PolT2A VLVs. Staining for HBcAg was detectable only in cells infected with 3xT2A, albeit at relatively low levels. It is important to note that VSV-G, the only structural component of VLVs, was detectable in most BHK-21 cells after infection with all three VLV vectors above (Figure 1C).
We also compared antigen expression from 3xT2A or Mix2A VLVs by immunoblotting of BHK-21 cell lysates at 24 h post-infection at MOI = 1 (Figure 1D). Using a polyclonal anti-2A peptide antibody, which recognizes the GDVESNPG epitope shared by all 2A peptides, we detected bands corresponding to the predicted molecular weights of HBV polymerase (92 kDa), middle S antigen (32 kDa), and modified core (29 kDa), which is consistent with the expectation that each of the antigens is tagged with the 2A-peptide sequences at the C-terminus. We also observed additional bands at ∼16, 18, and 40 kDa in the lysates from Mix2A VLV-infected cells, which may be due to incomplete recognition of the 2A peptide sequences during translation or subsequent proteolytic degradation. In addition, we noted a high molecular weight (>250 kDa) and a 37-kDa non-specific bands present in lysates from non-infected and VLV-infected cells, which most likely are the result of the antibody cross-reactivity with the endogenous proteins in BHK-21 cells. VSV-G was detected by polyclonal anti-VSV-G antibody only in the VLV-infected cells with comparable intensity between 3xT2A and Mix2A.
Immunogenicity of 3xT2A and Mix2A VLV Vaccine Candidates in Naive Mice
Administration of 3xT2A or Mix2A VLVs at 108 pfu/mouse resulted in an apparent increase in frequency (Figure 2A) and absolute numbers (Figure 2B) of CD44hiCD62Llo effector CD4+ and CD8+ T cells in spleens at day 7 post-immunization. Furthermore, we quantified the frequencies (Figure 2C) and absolute numbers (Figure 2D) of CD8+ T cells that produce IFNγ after ex vivo restimulation with the peptides derived from the epitopes from within HBV Pol, HBcAg, and MHBs. The cumulative frequencies of the IFNγ+ (i.e., HBV-specific) CD8+ T cells exceeded by 2%; among those the CD8+ T cells that recognized the HBS-371 peptide were most prevalent. Furthermore, substantial numbers of CD8+ T cells responded to HBV peptide stimulation by producing IFNγ and TNF (Figures 2E and 2F), indicating that VLV may induce double-cytokine producing CD8+ T cells, which are efficient in non-cytolytic elimination of HBV from the infected hepatocytes (Guidotti et al., 1996). The 3xT2A vector showed a trend toward higher frequency and absolute numbers of MHBs-specific CD8+ T cells when compared with Mix2A, albeit with no statistically significant difference. Since the use of different 2A peptides in the Mix2A VLV construct did not provide any apparent advantage for HBV antigen immunogenicity in naive mice, we proceeded to use the simpler of the two constructs, 3xT2A, for efficacy studies.
Figure 2.

Comparison of Immunogenicity of the 3xT2A and Mix2A VLV in Naive C57BL/6J Mice
Accumulation of effector CD4+ and CD8+ T cells at day 7 after immunization with 3xT2A or Mix2A VLV.
(A) Representative plots for CD62L and CD44 staining after gating on CD4+ or CD8+ T cells.
(B) Absolute numbers of effector (CD44hiCD62Llo) T cells.
(C) Frequency of HBV-specific IFNγ-producing CD8+ T cells identified after ex vivo stimulation with the indicated peptides from Pol (HBP-44, HBP-396, HBP-419), HBcAg (HBC-93), and surface (HBS-353 and HBS-371) quantified by intracellular staining and flow cytometry.
(D) Numbers of IFNγ+ CD8+ T cells per spleen.
(E) Frequency of HBV-specific double cytokine (TNF+IFNγ+)-producing CD8+ T cells, respectively.
(F) Numbers of TNF+IFNγ+-double positive CD8+ T cells. Values in (C and E) are mean of 3–5 mice per group.
Immunization with 3xT2A VLV Protects Mice from Experimental Acute Challenge with AAV-HBV
To determine whether VLV-mediated expression of HBV Pol, HBcAg, and MHBs generates protective immunity against HBV, we immunized CB6F1/J mice with 3xT2A or empty vector VLV and administered an equivalent volume of PBS to a control group of mice. Six weeks later, we challenged these mice with HBV delivered to the liver via recombinant adeno-associated virus (AAV-HBV) encoding a 1.3-overlength HBV genome and monitored serum HBsAg and HBeAg as a measure of HBV replication for additional four weeks. HBsAg levels peaked in PBS and empty vector VLV control groups of mice 14 days after infection, but remained low in 3xT2A VLV-immunized mice throughout the experiment (Figure 3A). HBeAg levels steadily increased during infection in the PBS and empty vector VLV groups (Figure 3B) but barely rose above background at days 14 and 21 and declined by day 28 in 3xT2A VLV-immunized mice. Quantitative analyses of HBV RNA in the livers of mice euthanized on day 28 showed a statistically significant decrease in the 3xT2A VLV immunized group compared with empty vector VLV group (Figure 3C). 3xT2A VLV-immunized mice, but not mice immunized with the empty vector VLV, showed a statistically significant increase in HBsAg-specific cells in the spleens that produce IFNγ after stimulation with the dominant peptide epitopes (Figure 3D). Thus, immunization with the 3xT2A VLV expressing HBV Pol, HBcAg, and MHBs protects mice from experimental acute HBV replication.
Figure 3.

Efficacy of 3xT2A VLV as Prophylactic Vaccine in Acute HBV Infection
CB6F1 mice were immunized with 3xT2A VLV or empty vector VLV for 6 weeks prior to challenge with AAV-HBV.
(A) Levels of serum HBsAg (OD450) measured by specific ELISA from blood samples taken weekly.
(B) Weekly levels of HBeAg. The values are mean ± SEM (n = 5). Asterisks indicate significant difference (*, p < 0.05, **, p < 0.01) in 3xT2A-immunized mice compared with PBS or empty vector VLV.
(C) Abundance of HBV RNA in liver samples from mice euthanized on day 28 after challenge with AAV-HBV. TaqMan-based qRT-PCR was used to quantify HBV RNA and mouse GAPDH mRNA. The HBV RNA data were normalized to GAPDH and expressed as relative to the mean in the PBS group. Individual values and mean ± SD are shown.
(D) Frequency of HBV-specific IFNγ+ producing cells was measured by ELISPOT on day 28 after challenge with AAV-HBV after ex vivo stimulation of splenocytes with the indicated peptides. Individual values and mean + SD are shown.
Evaluation of 3xT2A VLV in an Experimental Model of Chronic HBV Infection
We explored the therapeutic potential of 3xT2A in the setting of the AAV-HBV mouse model of chronic HBV infection (Dion et al., 2013). To initiate this model, we transduced C57BL/6J male mice with AAV-HBV-1.2 (2*1011 genome copies of AAV-HBV particles per mouse) and monitored serum HBsAg levels every week. As expected, we observed a steady rise of serum HBsAg from 11.52 ± 2.40 ng/mL (mean ± SEM, n = 20) at day 7 to 70.04 ± 6.25 ng/mL (mean ± SEM, n = 20) at day 21 post-AAV-HBV transduction. Based on the measurements at day 21, we formed 2 groups of 10 mice each with equivalent levels of serum HBsAg prior to immunization with 3xT2A or empty vector VLV on day 28 post-AAV-HBV transduction. Figure 4A shows responses to the treatment through the full course of the experiment as the fold change of HBsAg levels (OD450) relative to day 28, whereas Figure 4B shows the concentrations of serum HBsAg in ng/ml after VLV prime. Both VLV vectors caused an apparent, but transient, decline in HBsAg levels at day 35, which may reflect host interferon response to VLVs. HBsAg levels rebounded in both groups by day 42 and remained essentially steady for the empty vector VLV group. By day 49, the average HBsAg levels of the 3xT2A VLV-immunized groups started to decline and become significantly lower at days 56 and 63. Importantly, by day 63, the concentration of HBsAg dropped below the limit of detection (3.12 ng/mL) in four out of ten 3xT2A VLV-immunized mice. These results indicate that 40% of the chronically infected animals responded to the treatment with 3xT2A VLV.
Figure 4.

Effects of 3xT2A VLV on HBsAg levels in Chronic AAV-HBV Model
Persistent HBV replication was established in C57BL/6 mice by delivery of HBV genome with AAV. The groups were balanced for HBsAg prior to immunization with empty vector VLV or 3xT2A at day 28 after AAV-HBV transduction. Booster DNA immunizations were carried out on days 65 and 84, as indicated by arrows, using a pool of 3 plasmids expressing MHBs, HBcAg, and HBV Pol for 3xT2A group or empty plasmid vectors for the empty vector VLV.
(A–C) (A) Fold change in HBsAg levels (OD450). Values are mean ± SEM (n = 10 for days 28–70 and n = 5 thereafter). Quantification of HBsAg (ng/mL) after VLV prime at day 28 (B) and DNA boost at day 65 (C). Individual values and median are shown by symbols and bars, respectively. Asterisks in A indicate significant difference between 3xT2A and empty vector VLV-immunized animals (*, p < 0.05, **, p < 0.01).
Because only 40% of the animals showed responses to 3xT2A VLV and considering that a single VLV immunization would be unlikely to overcome HBV tolerance in CHB, we added DNA immunization as a boost after blood collection on days 65 and 84 (Figure 4A). We administered a pool of three DNA plasmids expressing MHBs, HBcAg, and HBV Pol in pVAC1 vector (further referred as HBVpDNA) to the group of mice immunized with 3xT2A and empty vector plasmid to the group of mice immunized with empty vector VLV. HBsAg levels continued to decline in the group of mice treated with 3xT2A VLV and HBVpDNA and, with the exception of a single time point (day 98), were significantly lower (p < 0.05) than in the group of mice treated with empty vector VLV and DNA (Figure 4C).
To determine whether 3xT2A VLV-induced decreases in serum HBsAg were accompanied by reduction of HBV RNA in liver, we euthanized five animals in each group to collect liver samples on day 70 and the remaining five animals at the termination of the experiment on day 119. Quantitative analyses of HBV RNA showed no difference between the groups at day 70 (Figure 5A). However, at day 119 three out of five mice primed with 3xT2A VLV and boosted twice with HBVpDNA showed more than 100-fold reduction of liver HBV RNA. Remarkably, this group showed a very strong correlation between serum HBsAg and liver HBV RNA (R2 = 0.905, p = 0.013 for 3xT2A VLV plus HBVpDNA vs. R2 = 0.179, p = 0.478 for empty vector VLV plus control DNA), suggesting that the observed decrease of serum HBsAg is due to a reduction of HBV replication.
Figure 5.

Effects of 3xT2A VLV on Liver HBV RNA in Chronic AAV-HBV Model
Liver samples were collected at day 70 or day 119 from the animals immunized as shown in Figure 4 for qRT-PCR analyses.
(A) Abundance of HBV RNA was determined after normalization to mouse GAPDH mRNA and expressed relative to the mean of the empty vector VLV group (n = 5). Numbers of IFNγ+ CD8+ T cells per spleen at day 70 (B) and day 119 (C) were determined by intracellular flow cytometry after stimulation with HBV antigen peptides.
Finally, we analyzed HBV-specific CD8+ T cell responses in the immunized groups. At day 70, there was no significant increase over background in the numbers of IFNγ-producing CD8+ T cells (Figure 5B). There was also no apparent increase in the numbers of IFNγ-producing CD8+ T cells after restimulation with HBV Pol or HBcAg peptides at day 119. However, restimulation with HBsAg peptides at the same time point resulted in a statistically significant increase over background in the 3xT2A VLV-primed/HBVpDNA-boosted group of mice (Figure 5C).
Discussion
Our results demonstrate feasibility and potential of using the VLV platform for polycistronic expression of HBV antigens and immunotherapy for CHB. The HBV antigens expressed by 3xT2A and Mix2A VLV constitute three of the four major proteins expressed by HBV: the entire polymerase protein (HBV pol) with mutations to inactivate reverse transcriptase activity, ubiquitin-tagged core (UbHBc) with mutations to prevent the formation of stable disulfide-mediated dimers, and the middle surface antigen (MHBs). The combination of three different 2A peptides in Mix2A VLVs did not provide any obvious advantage over repeating T2A peptides in 3xT2A VLVs. The selected 3xT2A VLVs replicate to high titers (>109 pfu/mL), express the individual HBV proteins of interest (Figure 1), and induce slightly higher specific CD8+ T cell responses to each antigen than Mix2A VLV (Figure 2). Taking advantage of mouse strain-specific differences in persistence of AAV-mediated transduction of HBV (Huang et al., 2006), we demonstrated that a single immunization with 3xT2A VLVs provides protection of CB6F1 mice from an acute challenge (Figure 3). Using the AAV-HBV-transduced mice as a model of persistent HBV replication, we evaluated the utility of the 3xT2A VLV for CHB immunotherapy. Since persistent HBV replication in liver leads to tolerance to HBV antigens (Dion et al., 2013), we did not expect that a single immunization with 3xT2A VLV would be sufficient to clear the virus or induce high numbers of HBV-specific T cells. Thus, we used 3xT2A VLV for prime immunization followed by 2 booster immunizations with DNA plasmids expressing the same antigens. This immunization schedule resulted in sustained decline of serum HBsAg and liver HBV RNA in a subset of mice with established chronic HBV replication (Figures 4 and 5A). These data indicate that 3xT2A VLVs function not only as a prophylactic vaccine but could also be used as a component of comprehensive immunotherapy.
Immunotherapeutic approaches for CHB, including DNA vaccination and therapeutic vaccines based on viral vectors, promise to provide functional cure by reducing or eliminating the virus from infected hepatocytes (Kosinska et al., 2017, Schinazi et al., 2018). However, none of the experimental immunotherapies tested to date have shown sufficient efficacy in clinical trials (Kosinska et al., 2017). This suggests that these vaccines lacked sufficient potency, were not targeting the correct antigen(s), were targeting too few antigens or, perhaps, all the above. It appears that success depends on the induction of CD8+ T cells that are both cytolytic and non-cytolytic in action rather than induction of humoral responses by the present prophylactic vaccines (Kosinska et al., 2017, Webster et al., 2004). Although therapeutic vaccines using recombinant VSV Moshkani et al., 2019 or modified vaccinia virus have a potential to induce more robust T cell-mediated immune responses to HBV, concerns about their safety profile may represent an obstacle for clinical applications (Kosinska et al., 2017).
Immunotherapeutic vaccines expressing HBV Pol, HBcAg, and MHBs using VLV vectors address the safety concerns and provide sufficient efficacy by expressing multiple HBV antigens that are optimized for induction of T cell responses. Restriction of VLV replication by type I interferons enables 100% survival rate of mice even after intracranial administration of VLV (van den Pol et al., 2017). Inclusion of HBV Pol provides a number of highly immunogenic and conserved epitopes for CTL that can detect and eliminate HBV-infected cells and limit viral spread (Mizukoshi et al., 2004). Because expression of intact (wild-type) HBcAg from a single antigen-expressing VLV did not induce sufficient T cell responses to control HBV replication (Reynolds et al., 2015), the polycistronic constructs described in this report included ubiquitinated HBcAg with mutated cysteine residues. These changes may have improved peptide presentation to T cells through prevention of HBcAg dimerization and enhancement of proteolytic degradation (Zhuo et al., 2015). Even though MHBs are expressed in the third position in the 3xT2A and Mix2A vectors, it appeared as the dominant antigen, as demonstrated by the frequency of IFNγ+ and IFNγ+TNF+ CD8+ T cells after immunization (Figures 2C and 2D). Thus, the polycistronic design of the VLV vector induces broad T cell responses in naive mice with a potential to eliminate HBV through cytolytic and non-cytolytic mechanisms.
To evaluate prophylactic and therapeutic efficacy of 3xT2A VLV, we employed both acute and chronic models of HBV infection established by delivery of HBV genome DNA via liver-tropic AAV (Dion et al., 2013). This approach overcomes the species-specific restrictions in early steps in the HBV replication cycle and enables homogeneous transduction of mouse hepatocytes, HBV replication, and secretion of HBsAg (Dion et al., 2013). Delivery of HBV via AAV also provides more reproducible results than hydrodynamic delivery of HBV genome in plasmid DNA for acute challenge. Although the HBV-specific T cell responses are muted as the chronicity of infection causes T cell exhaustion and tolerance to HBV antigens, the mechanism and level of tolerance are different from those in the HBV transgenic mouse model (Dion et al., 2013, Huang et al., 2006, Sette et al., 2001). Thus, despite some limitations, including highly variable levels of viral load and HBsAg levels, the AAV-HBV model is more suitable for evaluation of immune-mediated clearance of HBV.
After determining that 3xT2A VLV can protect mice from acute AAV-HBV challenge (Figure 3), we evaluated the immunotherapeutic potency of 3xT2A in a mouse model of CHB. As a precaution that a single immunization with 3xT2A VLV might not be sufficient to break tolerance in the chronically infected mice, we added DNA booster immunization to the treatment protocol. We observed that priming of chronically infected mice with 3xT2A VLV, followed by booster DNA immunization, results in a statistically significant decline of serum HBsAg (Figure 4), with 40% of the mice dropping their HBsAg levels to the limit of detection and showing marked reduction of liver HBV RNA (Figure 5A). Such a partial response is not unheard of with immunotherapeutic approaches and may be due to variable levels of HBV replication or differences in the TCR repertoire of T cells responding to the viral antigens. Following this immunization schedule, a subset of mice also showed a statistically significant accumulation of HBsAg-specific IFNγ-producing CD8+ T cells in the spleens at later time points (Figures 5B and 5C). Although the design of the study described here did not allow us to be certain that the booster DNA immunizations would be necessary, our data clearly indicate that the multiantigen HBV-VLV shows promise as an immunotherapeutic tool for treatment of CHB.
Among other therapeutic vaccines for CHB evaluated in the HBV-AAV model, the adenovirus serotype 5 (Ad5)-based therapeutic vaccine (TG1050) is comparable to our VLV-based vector in approach and goals of inducing HBV-specific T cell responses (Martin et al., 2014). TG1050 expresses a fusion protein consisting of a deleted and mutated HBVpol, truncated HBcAg, and two regions of HBsAg (Martin et al., 2014). Similar to our observations with 3xT2A VLVs, the TG1050-treated group of mice showed a three-fold reduction in HBsAg levels compared to those in the control group. While the effects of TG1050 on individual animals were not reported, we observed that 40% of the animals in our study responded by decline of HBsAg to the limit of detection. TG1050 presently is in Phase I/II clinical trials (NCT02428400) in HBV-infected patients with the results of this trial expected in 2019.
In order to improve immunogenicity and efficacy of therapeutic vaccine candidates for chronic HBV infection, others have used a combination of approaches, such as adjuvants, addition of checkpoint inhibitors, and prime/boost regimens (Kosinska et al., 2017, Schinazi et al., 2018). Recent studies showed that an optimized, heterologous prime/boost strategy produced strong T and B responses to HBV in various preclinical models of CHB (Backes et al., 2016, Chuai et al., 2017, Liu et al., 2014). A combination of an HBsAg/HBcAg protein prime with TLR9 agonists followed by a boost with non-replicating modified vaccinia Ankara viruses, optimized for higher level expression of the same antigens, was able to overcome the high-level tolerance observed in HBV-transgenic mice (Backes et al., 2016).
Given the evidence of high efficacy in at least 40% of the chronically infected mice, we plan to develop the VLV-based immunotherapy further and initiate clinical trials to demonstrate safety and efficacy in CHB patients. Further improvement of VLV-based immunotherapy may include establishing a prime-boost strategy and/or combination with nucleoside/nucleotide treatment to lower HBV replication.
Limitations of the Study
Although the AAV-HBV model of persistent HBV replication is widely accepted as the state-of-the-art system to study immune-mediated clearance of HBV, its limitations include lack of reinfection of the host with circulating HBV and differences in mouse and human immune systems.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Lorraine Apuzzo, Mehdi Pouresmail, and Seyedehfatemeh Fazlhashemi at CaroGen Corporation for assistance with DNA cloning, production of VLV and characterization of the VLVs. We are also grateful to Drs. Raj Kalkeri, Kevin Walters, Krista Salley, Elizabeth Janci Wonderlich, and Jennifer Carl at Southern Research for their efforts on the chronic AAV-HBV model and to Shuguang Huang at Stat4ward for assistance with statistical analyses. This study was funded by Connecticut Innovations, Connecticut Bioscience Innovation Fund, Gerpang Healthcare, and NIH grants R43DK113858 (V.N.) and R01AI124006 (M.D.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author Contributions
Conceptualization, T.O.Y, S.W.M., B.A., J.K.R., M.D.R., and V.N.; Methodology, T.O.Y, S.W.M., M.D.R., and V.N.; Investigation, T.O.Y., M.M., M.M.K., M.H., B.R.M., X.M., S.M., C.C., and A.C.P.; Formal Analysis, T.O.Y., and S.W.M.; Writing – Original Draft, T.O.Y., and S.W.M; Writing – Review & Editing, T.O.Y., S.W.M., B.A., J.K.R., M.D.R., and V.N.; Funding Acquisition, B.A., M.D.R., and V.N.; Supervision, S.W.M., B.A., J.K.R., M.D.R., and V.N.
Declaration of Interests
T.O.Y, S.W.M., B.A., J.K.R., M.D.R., and V. N. have financial relationships with CaroGen Corporation.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.040.
Contributor Information
Timur O. Yarovinsky, Email: tyarovinsky@carogencorp.com.
Valerian Nakaar, Email: vnakaar@carogencorp.com.
Supplemental Information
References
- Akbar S.M.F., Chen S., Al-Mahtab M., Abe M., Hiasa Y., Onji M. Strong and multi-antigen specific immunity by hepatitis B core antigen (HBcAg)-based vaccines in a murine model of chronic hepatitis B: HBcAg is a candidate for a therapeutic vaccine against hepatitis B virus. Antiviral Res. 2012;96:59–64. doi: 10.1016/j.antiviral.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Backes S., Jäger C., Dembek C.J., Kosinska A.D., Bauer T., Stephan A.-S., Dišlers A., Mutwiri G., Busch D.H., Babiuk L.A. Protein-prime/modified vaccinia virus Ankara vector-boost vaccination overcomes tolerance in high-antigenemic HBV-transgenic mice. Vaccine. 2016;34:923–932. doi: 10.1016/j.vaccine.2015.12.060. [DOI] [PubMed] [Google Scholar]
- Block T.M., Alter H., Brown N., Brownstein A., Brosgart C., Chang K.-M., Chen P.-J., Cohen C., El-Serag H., Feld J. Research priorities for the discovery of a cure for chronic hepatitis B: report of a workshop. Antivir. Res. 2018;150:93–100. doi: 10.1016/j.antiviral.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuai X., Chen P., Chen H., Wang W., Deng Y., Ruan L., Li W., Tan W. Protective efficacy and hepatitis B virus clearance in mice enhanced by cell-mediated immunity with novel prime-boost regimens. J. Viral Hepat. 2017;24:337–345. doi: 10.1111/jvh.12649. [DOI] [PubMed] [Google Scholar]
- Cova L. Present and future DNA vaccines for chronic hepatitis B treatment. Expert Opin. Biol. Ther. 2017;17:185–195. doi: 10.1080/14712598.2017.1265940. [DOI] [PubMed] [Google Scholar]
- Dion S., Bourgine M., Godon O., Levillayer F., Michel M.-L. Adeno-associated virus-mediated gene transfer leads to persistent hepatitis B virus replication in mice expressing HLA-A2 and HLA-DR1 molecules. J. Virol. 2013;87:5554–5563. doi: 10.1128/JVI.03134-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem D., Prince A.M. Hepatitis B virus infection — natural history and clinical consequences. N. Engl. J. Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- Guidotti L.G., Ishikawa T., Hobbs M.V., Matzke B., Schreiber R., Chisari F.V. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- Huang L.-R., Wu H.-L., Chen P.-J., Chen D.-S. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. U S A. 2006;103:17862–17867. doi: 10.1073/pnas.0608578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Hong Y.B., Jung G. Hepatitis B virus: DNA polymerase activity of deletion mutants. Biochem. Mol. Biol. Int. 1999;47:301–308. doi: 10.1080/15216549900201323. [DOI] [PubMed] [Google Scholar]
- King T.H., Kemmler C.B., Guo Z., Mann D., Lu Y., Coeshott C., Gehring A.J., Bertoletti A., Ho Z.Z., Delaney W. A whole recombinant yeast-based therapeutic vaccine elicits HBV X, S and Core specific T cells in mice and activates human T cells recognizing epitopes linked to viral clearance. PLoS One. 2014;9:e101904. doi: 10.1371/journal.pone.0101904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinska A.D., Bauer T., Protzer U. Therapeutic vaccination for chronic hepatitis B. Curr. Opin. Virol. 2017;23:75–81. doi: 10.1016/j.coviro.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Lanford R.E., Kim Y.H., Lee H., Notvall L., Beames B. Mapping of the hepatitis B virus reverse transcriptase TP and RT domains by transcomplementation for nucleotide priming and by protein-protein interaction. J. Virol. 1999;73:1885–1893. doi: 10.1128/jvi.73.3.1885-1893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Kosinska A., Lu M., Roggendorf M. New therapeutic vaccination strategies for the treatment of chronic hepatitis B. Virol. Sin. 2014;29:10–16. doi: 10.1007/s12250-014-3410-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Chen O., Wall J.B.J., Zheng M., Zhou Y., Wang L., Ruth Vaseghi H., Qian L., Liu J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-02460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P., Dubois C., Jacquier E., Dion S., Mancini-Bourgine M., Godon O., Kratzer R., Lelu-Santolaria K., Evlachev A., Meritet J.-F. TG1050, an immunotherapeutic to treat chronic hepatitis B, induces robust T cells and exerts an antiviral effect in HBV-persistent mice. Gut. 2014;64:1961–1971. doi: 10.1136/gutjnl-2014-308041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukoshi E., Sidney J., Livingston B., Ghany M., Hoofnagle J.H., Sette A., Rehermann B. Cellular immune responses to the hepatitis B virus polymerase. J. Immunol. 2004;173:5863–5871. doi: 10.4049/jimmunol.173.9.5863. [DOI] [PubMed] [Google Scholar]
- Moshkani S., Chiale C., Lang S.M., Rose J.K., Robek M.D. A highly attenuated vesicular stomatitis virus-based vaccine platform controls hepatitis B virus replication in mouse models of hepatitis B. J. Virol. 2019;93 doi: 10.1128/JVI.01586-18. e01586–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassal M., Rieger A., Steinau O. Topological analysis of the hepatitis B virus core particle by cysteine-cysteine cross-linking. J. Mol. Biol. 1992;225:1013–1025. doi: 10.1016/0022-2836(92)90101-o. [DOI] [PubMed] [Google Scholar]
- Obeng-Adjei N., Hutnick N.A., Yan J., Chu J.S., Myles D.J.F., Morrow M.P., Sardesai N.Y., Weiner D.B. DNA vaccine cocktail expressing genotype A and C HBV surface and consensus core antigens generates robust cytotoxic and antibody responses in mice and Rhesus macaques. Cancer Gene Ther. 2013;20:652–662. doi: 10.1038/cgt.2013.65. [DOI] [PubMed] [Google Scholar]
- Reynolds T.D., Buonocore L., Rose N.F., Rose J.K., Robek M.D. Virus-like vesicle-based therapeutic vaccine vectors for chronic hepatitis B virus infection. J. Virol. 2015;89:10407–10415. doi: 10.1128/JVI.01184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose N.F., Buonocore L., Schell J.B., Chattopadhyay A., Bahl K., Liu X., Rose J.K. In vitro evolution of high-titer, virus-like vesicles containing a single structural protein. Proc. Natl. Acad. Sci. U S A. 2014;111:16866–16871. doi: 10.1073/pnas.1414991111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose N.F., Publicover J., Chattopadhyay A., Rose J.K. Hybrid alphavirus-rhabdovirus propagating replicon particles are versatile and potent vaccine vectors. Proc. Natl. Acad. Sci. U S A. 2008;105:5839–5843. doi: 10.1073/pnas.0800280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell J.B., Rose N.F., Bahl K., Diller K., Buonocore L., Hunter M., Marx P.A., Gambhira R., Tang H., Montefiori D.C. Significant protection against high-dose simian immunodeficiency virus challenge conferred by a new prime-boost vaccine regimen. J. Virol. 2011;85:5764–5772. doi: 10.1128/JVI.00342-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillie S., Vellozzi C., Reingold A., Harris A., Haber P., Ward J.W., Nelson N.P. Prevention of hepatitis B virus infection in the United States: recommendations of the advisory committee on immunization practices. MMWR Recomm. Rep. 2018;67:1–31. doi: 10.15585/mmwr.rr6701a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinazi R.F., Ehteshami M., Bassit L., Asselah T. Towards HBV curative therapies. Liver Int. 2018;38:102–114. doi: 10.1111/liv.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C., Mason W.S. Molecular biology of hepatitis B virus infection. Virology. 2015;479–480:672–686. doi: 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A.D., Oseroff C., Sidney J., Alexander J., Chesnut R.W., Kakimi K., Guidotti L.G., Chisari F.V. Overcoming T cell tolerance to the hepatitis B virus surface antigen in hepatitis B virus-transgenic mice. J. Immunol. 2001;166:1389–1397. doi: 10.4049/jimmunol.166.2.1389. [DOI] [PubMed] [Google Scholar]
- Tang L.S.Y., Covert E., Wilson E., Kottilil S. Chronic hepatitis B infection. JAMA. 2018;319:1802–1813. doi: 10.1001/jama.2018.3795. [DOI] [PubMed] [Google Scholar]
- Tsai K.-N., Kuo C.-F., Ou J.-H.J. Mechanisms of hepatitis B virus persistence. Trends Microbiol. 2018;26:33–42. doi: 10.1016/j.tim.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol A.N., Mao G., Chattopadhyay A., Rose J.K., Davis J.N. Chikungunya, influenza, nipah, and Semliki forest chimeric viruses with vesicular stomatitis virus: actions in the brain. J. Virol. 2017;91 doi: 10.1128/JVI.02154-16. e02154–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster G.J.M., Reignat S., Brown D., Ogg G.S., Jones L., Seneviratne S.L., Williams R., Dusheiko G., Bertoletti A. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J. Virol. 2004;78:5707–5719. doi: 10.1128/JVI.78.11.5707-5719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M., Song L., Tang Y., Dai S., Chen X., Yu Y., Zang G., Tang Z. Vaccination with ubiquitin-hepatitis B core antigen-cytoplasmic transduction peptide enhances the hepatitis B virus-specific cytotoxic T-lymphocyte immune response and inhibits hepatitis B virus replication in transgenic mice. Mol. Med. Rep. 2015;12:3591–3598. doi: 10.3892/mmr.2015.3834. [DOI] [PubMed] [Google Scholar]
- zu Putlitz J., Lanford R.E., Carlson R.I., Notvall L., de la Monte S.M., Wands J.R. Properties of monoclonal antibodies directed against hepatitis B virus polymerase protein. J. Virol. 1999;73:4188–4196. doi: 10.1128/jvi.73.5.4188-4196.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.