

Abstract
Various types of cell death, including apoptosis, necrosis, necroptosis, and ferroptosis, are induced in renal tubular epithelial cells following exposure to environmental stresses and toxicants such as osmotic stress, ischemia/reperfusion injury, cisplatin, and cadmium. This is known to cause renal dysfunction, but the cellular events preceding stress-induced cell death in renal tubules are not fully elucidated. The activin receptor-like kinase (ALK) 4/5, also known as activin-transforming growth factor (TGF) β receptor, is involved in stress-induced renal injury. We, therefore, studied the role of ALK4/5 signaling in HK-2 human proximal tubular epithelial cell death induced by cisplatin, cadmium, hyperosmotic stress inducer, sorbitol, and the ferroptosis activator, erastin. We found that ALK4/5 signaling is involved in cadmium- and erastin-induced cell death, but not sorbitol- or cisplatin-induced apoptotic cell death. Cadmium exposure elevated the level of phosphorylated Smad3, and treatment with the ALK4/5 kinase inhibitors, SB431542 or SB505124, suppressed cadmium-induced HK-2 cell death. Cadmium-induced cell death was attenuated by siRNA-mediated ALK4 or Smad3 silencing, or by treatment with SIS3, a selective inhibitor of TGFβ1-dependent Smad3 phosphorylation. Furthermore, ALK4/5 signaling activated Akt signaling to promote cadmium-induced HK-2 cell death. In contrast, siRNA-mediated Inhibin-bA silencing or treatment with TGFβ1 or activin A had little effect on cadmium-induced HK-2 cell death. On the other hand, treatment with SB431542 or SB505124 attenuated erastin-induced ferroptosis by hyperactivating Nrf2 signaling in HK-2 cells. These results suggest that blockade of ALK4/5 signaling protects against cadmium- and erastin-induced HK-2 cell death via Akt and Nrf2 signaling pathways, respectively.
Subject terms: Kidney diseases, Metals
Introduction
Renal tubular cell death caused by environmental stresses or nephrotoxic agents has a critical role in nephropathy, and is therefore an important therapeutic target. Various types of cell death including apoptosis, necrosis, necroptosis, and ferroptosis, are reported to be involved in renal tubular cell death caused by nephrotoxic stresses or toxicants. For instance, sorbitol, which is a cause of diabetic nephropathy [1], induces apoptosis [2], whereas unusual exposure to cisplatin, an antitumor drug, or cadmium, an environmental pollutant metal, induces apoptosis, necrosis, or necroptosis in renal tubular cells [3–6]. In addition, ferroptosis and necroptosis are observed in renal tubular cells injured by ischemia/reperfusion [7, 8].
Apoptosis, the first identified mechanism of programmed cell death, is executed in a caspase-dependent or -independent manner involving chromatin condensation and karyorrhexis [9, 10]. In contrast, necrosis is originally recognized as unregulated cell death-exhibiting rupture of plasma membrane and leakage of cellular contents [9, 10], but recent reports have shown the existence of programmed necrotic cell death including necroptosis and ferroptosis [11, 12]. Necroptosis is a death receptor-triggered form of necrotic cell death, executed through insertion of oligomerized mixed lineage kinase domain-like protein into the plasma membrane [11]. Ferroptosis is another programmed necrotic cell death involving iron-dependent lipid peroxidation, and oxidative stress is believed to be a principal cause of ferroptosis [12]. However, the signaling pathways responsible for these distinct types of cell death in renal tubular cells have not yet been fully clarified.
Activin and transforming growth factor (TGF) β signaling are involved in cell proliferation, differentiation, migration, and cell death [13–15]. Activin and TGFβ mediate their signaling via a specific complex of type I/type II receptor serine/threonine kinases. Activin receptor-like kinase (ALK) 4 and ALK7 are type I receptors for activins, and ALK5 is a type I receptor for TGFβ. Once ligands bind to type II receptors, type I receptors are recruited to the ligand/type II receptor complex, leading to the phosphorylation and activation of type I receptors by type II kinases. In the canonical pathway, activated type I receptors phosphorylate the transcription factors Smad2/3, leading to the activation of Smad2/3 for the regulation of their target genes. The non-canonical pathway is Smad2/3-independent and may engage other signaling cascades such as Akt, extracellular signal-regulated kinase (ERK), p38, or c-Jun N-terminal kinase (JNK). In the context of renal pathology, activation or inactivation of ALK4/5 signaling has been reported to result in subsequent cellular damage [16–20].
To demonstrate the possible involvement of ALK4/5 signaling in renal tubular cell death associated with renal disease, we analyzed death of HK-2 human proximal tubular epithelial cells induced by the hyperosmotic stress inducer sorbitol, cisplatin, cadmium, and the ferroptosis activator erastin. We showed that ALK4/5 signaling has little effect on sorbitol- and cisplatin-induced apoptotic HK-2 cell death. In contrast, cadmium exposure activated ALK4/5 signaling in a ligand-independent manner and promoted apoptotic and necrotic cell death via Smad3 and Akt signaling in HK-2 cells. On the other hand, pharmacological inhibition of ALK4/5 signaling attenuated erastin-induced ferroptosis by hyperactivating Nrf2 signaling in HK-2 cells. These results indicated that ALK4/5 signaling is an important regulator of renal tubular cell death.
Materials and methods
Chemicals
Cadmium chloride (CdCl2) was obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Cisplatin and sorbitol were obtained from Wako (Osaka, Japan). U0126 was obtained from Calbiochem, Merck KGaA (Darmstadt, Germany). Cyclosporin A, deferoxamine, erastin, ferrostatin-1, necrostatin-1, SB431542, SB505124, SIS3, and Trolox were obtained from Cayman Chemical Company (Ann Arbor, MI, USA). Necrox-2 and Necrox-5 methanesulfonate were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). MK2206 was obtained from Active Biochem (Maplewood, NJ, USA). Necrostatin-1 inactive control and 1,5-dihydroxyisoquinoline were obtained from abcam plc (Cambridge, England). 7-Cl-O-Nec-1 was obtained from Focus Biomolecules (Plymouth Meeting, PA, USA). Z-VAD-FMK and RSL3 were obtained from AdooQ Bioscience (Irvine, CA, USA). TGF-β1 was obtained from Sigma-Aldrich (St. Louis, MO). Z-LEHD-FMK, Activin A, Human/Mouse/Rat Recombinant and Human/Mouse/Rat Activin A Quantikine ELISA Kit were obtained from R&D system (Minneapolis, MN, USA). Antibodies against phospho-Histone H2A.X, Caspase-3, Caspase-8 (1C12), Caspase-9, PARP, LC3B (D11) XP®, phospho-Smad2 (Ser465/467), Smad2 (D43B4) XP®, phospho-Smad3 (Ser423/425), Smad3 (C67H9), phospho-Smad1/5 (Ser463/465) (41D10), Smad1 (D59D7) XP®, Smad5, phospho-Akt (Thr308) (C31E5E), Akt (pan) (C67E7), NRF2 (D1Z9C) XP®, phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204), p44/42 MAPK (ERK1/2), phospho-p38 MAPK (Thr180/Tyr182), p38 MAPK, phospho-SAPK/JNK (Thr183/Tyr185), SAPK/JNK, phospho-FoxO1 (Tr24)/FoxO3a (Thr32), FOXO3a (75D8), BID, and Noxa (D8L7U) were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Inhibin-bA (E-1), BNIP3 (ANa40), Keap1 (G-2), actin (I-19), and lamin A/C (14/Lamin AC) antibodies were obtained from Santa Cruz Biotechnology. HO-1 antibody was obtained from Enzo life sciences, Inc. (Farmingdale, NY, USA). The siRNAs targeted against the human ALK4 (siRNA-1: Hs_ALK4_5 FlexiTube siRNA, SI00288127, siRNA-2: Hs_ALK4_6 FlexiTube siRNA, SI02622046), FOXO3a (Hs_FOXO3a_3 FlexiTube siRNA, SI04916366), INHBA (siRNA-1: Hs_INHBA_2 FlexiTube siRNA, SI00033950, siRNA-2: Hs_INHBA_4 FlexiTube siRNA, SI00033964), KEAP1 (Hs_KEAP1_5 FlexiTube siRNA, SI03246439), NFE2L2 (Hs_NFE2L2_7 FlexiTube siRNA, SI03246950), SMAD2 (Hs_SMAD2_6 FlexiTube siRNA, SI02757496), SMAD3 (siRNA-1: Hs_SMAD3_3 FlexiTube siRNA, SI00082495, siRNA-2: Hs_SMAD3_4 FlexiTube siRNA, SI00082502), and non-target siRNA (AllStars Negative Control siRNA) were purchased from Qiagen (Hilden, Germany).
Cell culture and treatments
HK-2 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and grown in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin (GIBCO, Invitrogen Corp., Carlsbad, CA, USA) in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Exponentially growing HK-2 cells were seeded at 1.9 to 4 × 105 cells/well in six-well culture plates and cultured for 1 day before each experiment. CdCl2 was dissolved in water and sterilized by filtration. Sorbitol was dissolved in serum-free medium and sterilized by filtration. Erastin and RSL3 were dissolved in sterilized dimethyl sulfoxide (DMSO). Cisplatin was dissolved in sterilized N,N-dimethylformamide (DMF). Cells were incubated in serum-free medium containing the appropriate concentration of CdCl2, sorbitol, cisplatin, erastin, or RSL3 for 1–36 h at 37 °C. SB431542, SB505124, SIS3, U0126, MK2206, cyclosporine A (CsA), desferrioxamine (DFO), Ferrostatin-1, Necrostatin-1, Necrostatin-1 inactive control, 7-Cl-O-Nec-1, Trolox, Necrox-2, Necrox-5, 1,5-dihydroxyisoquinoline (DiQ), Z-LEHD-FMK, and Z-VAD-FMK were dissolved in DMSO or DMF. After incubating cells in serum-free medium with DMSO, DMF (0.1 or 0.2%) or one of the inhibitors for 1 h, HK-2 cells were treated with 25 µm CdCl2, 500 mm sorbitol, 10 μg/ml cisplatin, 5 µm erastin, or 3 µm RSL3 for the indicated time.
Preparation of whole-cell lysates
After incubation, cells were washed with phosphate-buffered saline and lysed with sodium dodecyl sulfate-polyacrylamide gel Laemmli sample buffer. Cell lysates were collected, sonicated, and boiled for 5 min. Protein concentrations were determined using the RC DC Protein Assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Western blotting
Equal amounts of protein (20 μg) were subjected to sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (Hybond-ECL, Amersham Pharmacia Biotech, Buckinghamshire, England). The membrane was blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature. The membrane was then incubated overnight at 4 °C with the primary antibody, and protein was detected with a Phototope-HRP Western blot detection kit (Cell Signaling Technology, Inc.).
Gene knockdown of ALK4, FOXO3a, INHBA, KEAP1, NFE2L2, SMAD2, or SMAD3 by siRNA
Transfection of siRNA against human ALK4, FOXO3a, INHBA, KEAP1, NFE2L2, SMAD2, SMAD3, and non-target siRNA into HK-2 cells was carried out using Lipofectamine RNAiMAX (Invitrogen Corp.) according to the manufacturer’s instructions with some adjustments. The siRNAs were dissolved in nuclease-free water and diluted to 0.1 or 0.2 µm with 250 µl Opti-MEM (Invitrogen Corp.). Five microliters of Lipofectamine RNAiMAX was also diluted 50-fold with Opti-MEM. Equal volumes of these two solutions were mixed (500 µl total) and immediately added to 2 ml culture medium at the time of cell plating. After incubation for 24 h, cells were washed with medium and used for experiments.
Cadmium concentrations
The acid-digested samples were diluted with deionized water and measurements of cadmium concentrations were outsourced to IDEA Consultants, Inc. (Tokyo, Japan).
Measurement of activin A
Cell culture supernatants were collected after treatment with CdCl2. The concentration of activin A in the media was quantified by enzyme-linked immunosorbent assay (ELISA) (R&D systems, Minneapolis, MN) following the manufacturer’s instructions
Glutathione assay
The relative glutathione concentration in cell lysates was assessed using GSSG/GSH Quantification Kit (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. The glutathione concentration was normalized to the protein amount of cell lysates.
RNA isolation and reverse transcription-PCR
Total RNA was isolated with RNeasy (Qiagen), and first-strand cDNA was synthesized using the ReverTra Ace qPCR RT kit (TOYOBO, Osaka, Japan). Quantitative reverse transcription-PCR analysis was performed using the THUNDERBIRD® SYBR® qPCR Mix (TOYOBO) in StepOneTM Real-Time PCR system (Life Technologies Japan Ltd., Tokyo, Japan). The expression level was normalized to that of β-actin. PCR primers are listed in Supplemental Table S1.
AV and PI staining
Culture medium containing floating cells were aspirated and reserved. After trypsinization, cells were suspended in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 medium, and the culture medium was returned. The numbers of live, necrotic, and apoptotic cells were determined by the Tali® Image-Based Cytometer using a Tali® Apoptosis Kit-AV Alexa Fluor® 488 and Propidium Iodide (Life Technology Corp., Carlsbad, CA, USA) according to the manufacturer’s instructions.
Trypan blue exclusion assay
Culture medium was aspirated and reserved. After trypsinization, cells were suspended in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 medium, and the culture medium was returned. The mixture was centrifuged to pellet the cells. Cellular suspension and 0.4% trypan blue in Hank’s Balanced Salt Solution were mixed, and the number of viable cells was counted using a TC10TM Automated Cell Counter (Bio-Rad laboratories, Inc.).
Statistical analysis
Results are expressed as the mean ± SD. Statistical significance was determined by Student’s t test or Welch’s t test. A value of P < 0.05 was considered to be statistically significant.
Results
Characterization of HK-2 cell death induced by sorbitol, cisplatin, cadmium, and erastin
First, we characterized the types of HK-2 cell death induced by treatment with sorbitol, cisplatin, cadmium, and erastin. We used Annexin-V (AV)/propidium iodide (PI) staining and immunoblotting of cleaved-poly ADP-ribose polymerase-1 (PARP), cleaved-caspase-3, and the DNA damage marker histone H2A.X protein, phosphorylated at Ser139. In addition, the following substances known to modify cell death were used to evaluate the preferred types of HK-2 cell death: Z-VAD-FMK, a caspase inhibitor, CsA, a cyclophilin D-dependent mitochondrial permeability transition-triggered necrosis inhibitor [21, 22], Necrox-2 and Necrox-5, oxidative stress-induced necrosis inhibitors [23], necrostatin-1 (Nec-1), a necroptosis inhibitor [11], Trolox, an anti-oxidant; ferrostatin-1 (Fer-1) and DFO, the ferroptosis inhibitors.
Cell death induced by sorbitol and cisplatin
In HK-2 cells exposed to sorbitol and cisplatin, AV + /PI− cell number increased initially, and subsequently AV + /PI + cell number increased (Supplemental Figs. 1A, 2A). Consistently, accumulation of cleaved-PARP, cleaved-caspase-3, and phosphorylated H2A.X proteins (Supplemental Figs. 1B, 2B) and reduction of cell death by treatment with Z-VAD-FMK but not other substances, including CsA, Necrox-2, Necrox-5 (Supplemental Figs. 1C, 2C), Trolox, or Fer-1 (Supplemental Figs. 1D, 2D) were observed. These results suggest that treatment with sorbitol and cisplatin induces caspase-dependent apoptosis in HK-2 cells.
Cell death induced by cadmium
Following exposure to cadmium chloride (CdCl2), AV + /PI− cells increased, and then both AV + /PI + cells and AV−/PI + cells increased (Fig. 1a). The expression of cleaved-PARP, cleaved-caspase-3, and phosphorylated H2A.X proteins increased in a CdCl2 exposure time-dependent manner (Fig. 1b). Treatment with Z-VAD-FMK markedly suppressed CdCl2-induced cell death (Fig. 1c), but the caspase-independent apoptosis inhibitor DiQ failed to suppress cell death (Fig. 1d). In addition, treatment with CsA (Fig. 1c), Trolox, Fer-1, or DFO (Fig. 1e) showed a mild protective effect against CdCl2-induced cell death. Treatment with DFO, an iron chelator, did not affect the amount of cadmium absorbed into the cells (Fig. 1f). However, treatment with Necrox-2, Necrox-5 (Fig. 1c), Nec-1, or 7-Cl-O-Nec-1 (O-Nec-1), an active form of Nec-1 (Fig. 1g), did not reduce cell death induced by CdCl2 exposure. These results suggest that cadmium-induced HK-2 cell death appears to be a combination of caspase-dependent apoptosis and necrosis, rather than apoptosis only.
Fig. 1.

Characterization of HK-2 cell death induced by cadmium. a, b Cells were incubated with 25 μm CdCl2 (Cd) for the indicated time. Percentage of propidium iodide (PI) or Annexin-V (AV) positive cells were determined by Annexin-V and PI staining. Results are representative of at least three independent experiments a. Cell lysates were subjected to western blotting using the indicated antibodies. Immunoblots shown are representative of at least three independent experiments b. c–e, g Cells were incubated with 0.1% DMSO, 0.1% DMF, 50 μm Z-VAD-FMK, 2.5 μm CsA, 0.3 μm Necrox-2, 0.1 μm Necrox-5 c, 100 μm DiQ (d, 50 μm Trolox, 2 μm Fer-1, 100 μm DFO e, 20 μm necrostatin-1 (Nec-1), 20 μm 7-Cl-O-Nec-1 (O-Nec-1), or 20 μm necrostatin-1i (Nec-1i) g for 1 h and then incubated with or without 25 μm CdCl2 (Cd) for 30 h. The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean ± SD of at least three experiments with duplicate assays in each experiment. f Cells were incubated with or without 100 μm DFO for 1 h and then incubated with or without 10 μm CdCl2 (Cd) for 12 h. Concentration of cadmium per 1.0 × 105 cells was measured and reflects the mean ± SD of three experiments. *P < 0.05, **P < 0.01, significant difference between the samples
Cell death induced by erastin
Although erastin, a system Xc- (cysteine/glutamate antiporter) inhibitor, and RSL3, a glutathione peroxidase (Gpx) 4 inhibitor, are identified as ferroptosis activators [12, 24], erastin exposure and Gpx4 depletion also induce other types of death in different cell types [25–29]. In HK-2 cells exposed to erastin, AV + /PI + and AV−/PI + cell population increased, but AV + /PI− cell population did not clearly change (Fig. 2a). Whereas the expression of phosphorylated H2A.X increased, the levels of cleaved-PARP and cleaved-caspase-3 remained unchanged (Fig. 2b). Treatment with CsA, Necrox-2, or Necrox-5 attenuated erastin-induced cell death (Fig. 2c). In addition, treatment with Trolox, Fer-1, DFO (Fig. 2d), or the MEK inhibitor U0126, which disrupts ERK signaling (Fig. 2e) suppressed erastin-induced cell death, which was compatible with the characteristics of ferroptosis [10]. In contrast, the inhibitory effect of Z-VAD-FMK treatment on erastin-induced cell death was less marked (Fig. 2c). These results indicate that erastin exposure mainly induces ferroptosis in HK-2 cells. In contrast, characteristics of ferroptosis and apoptosis were observed in RSL3-induced HK-2 cell death (Supplemental Fig. 3). Finally, we also confirmed that erastin exposure induced more rapid and marked reduction of glutathione levels than CdCl2 exposure (Fig. 2f, g), which was consistent with a previous report [12]. Thus, we decided to use erastin as a ferroptosis inducer for subsequent experiments in HK-2 cells.
Fig. 2.

Characterization of HK-2 cell death induced by erastin. a, b Cells were incubated with 5 μm erastin for the indicated time. Percentage of propidium iodide (PI) or Annexin-V (AV)-positive cells were determined by Annexin-V and PI staining. Results are representative of at least three independent experiments a. Cell lysates were subjected to western blotting using the indicated antibodies. Immunoblots shown are representative of at least three independent experiments b. c–e Cells were incubated with 0.1% DMSO, 0.1% DMF, 50 μm Z-VAD-FMK, 2.5 μm CsA, 0.3 μm Necrox-2, 0.1 μm Necrox-5 c, 50 μm Trolox, 2 μm Fer-1, 100 μm DFO d, or 20 μm U0126 e for 1 h and then incubated with or without 5 μm erastin for 24 h. The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean ± SD of at least three experiments with duplicate assays in each experiment. f, g Cells were incubated with 5 μm erastin f or 25 μm CdCl2 (Cd) g for the indicated time, respectively. Glutathione concentration was measured, and each value reflects the mean ± SD of three experiments with duplicate assays in each experiment. *P < 0.05, **P < 0.01, significant difference between the samples
Role of ALK4/5 signaling in HK-2 cell death induced by sorbitol, cisplatin, cadmium, and erastin
Non-involvement of ALK4/5 signaling in cell death induced by sorbitol and cisplatin
Treatment with TGFβ1 and activin A induced marked phosphorylation of Smad2 at Ser465/467 and Smad3 at Ser423/425 in HK-2 cells, indicating activation of ALK4/5 signaling (Supplemental Fig. 4). However, treatment with sorbitol and cisplatin did not clearly induce phosphorylation of Smad2 and Smad3 (Supplemental Figs. 5A, 6A). Consistently, treatment with the ALK4/5 inhibitors, SB431542 or SB505124, failed to reduce the sorbitol- or cisplatin-induced expression of apoptosis markers and HK-2 cell death (Supplemental Figs. 5B–E, 6B–D).
Involvement of ALK4/5 signaling in cell death induced by cadmium and erastin
Exposure of HK-2 cells to CdCl2 increased the levels of phosphorylated Smad3 and, to a lesser extent, that of phosphorylated Smad2 (Fig. 3a), suggesting that ligand-induced phosphorylation of Smad2/3 is unlikely in this context. Furthermore, treatment with SB431542 or SB505124 attenuated the cadmium-induced elevation of phosphorylated Smad3, cleaved-PARP, cleaved-caspase-3, and phosphorylated H2A.X levels (Fig. 3b), and cell death without affecting the intracellular concentration of cadmium (Fig. 3c–e). Although treatment with erastin induced a transient phosphorylation of Smad3 (at 15 h exposure) (Fig. 4a), SB431542 or SB505124 also significantly suppressed erastin-induced HK-2 cells death (Fig. 4b, c). These results suggest that the ALK4/5 signaling is involved in HK-2 cell death induced by cadmium and erastin.
Fig. 3.

Role of ALK4/5 signaling in HK-2 cell death induced by cadmium. a Cells were incubated with 25 μm CdCl2 (Cd) for the indicated time. Cell lysates were subjected to western blotting using the indicated antibodies. Immunoblots shown are representative of at least three independent experiments. b–d Cells were incubated with 0.1% DMSO, 10 μm SB431542, or 10 μm SB505124 for 1 h and then incubated with or without 25 μm CdCl2 (Cd) for 15 h b or 30 h c, d. Cell lysates were subjected to western blotting using the indicated antibodies. Immunoblots shown are representative of at least three independent experiments b. The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean ± SD of at least three experiments with duplicate assays in each experiment c. Percentage of propidium iodide (PI) or Annexin-V (AV)-positive cells were determined by Annexin-V and PI staining. Results are representative of at least three independent experiments d. e Cells were incubated with 0.1% DMSO, 10 μm SB431542, or 10 μm SB505124 for 1 h and then incubated with or without 10 μm CdCl2 (Cd) for 12 h. Concentration of cadmium per 1.0 × 105 cells was measured and reflects the mean ± SD of three experiments. **P < 0.01, significant difference between the samples
Fig. 4.
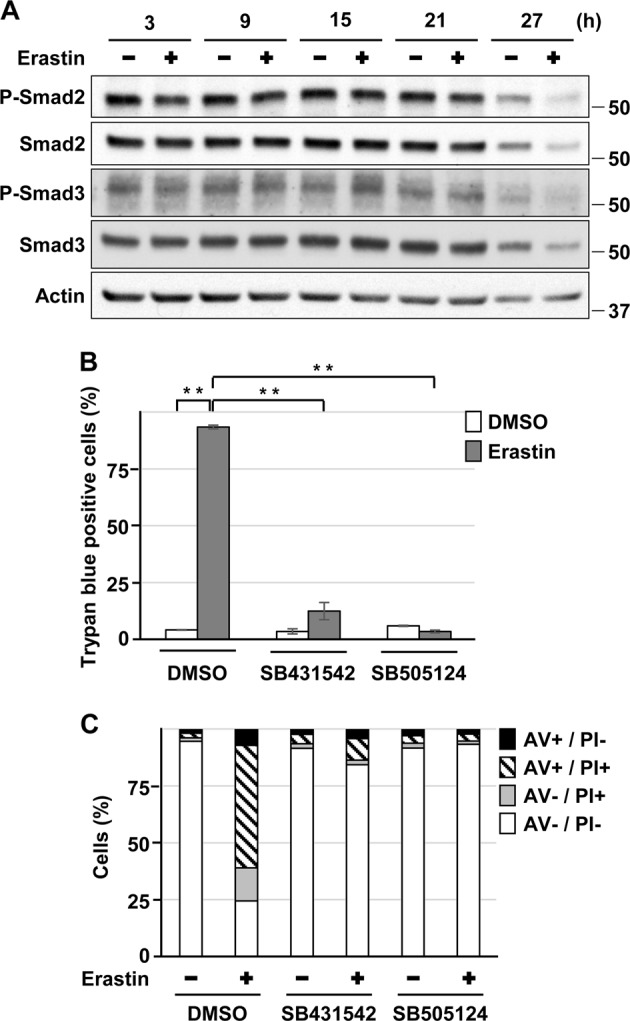
Role of ALK4/5 signaling in HK-2 cell death induced by erastin. a Cells were incubated with 5 μm erastin for the indicated time. Cell lysates were subjected to western blotting using the indicated antibodies. Immunoblots shown are representative of at least three independent experiments. b, c Cells were incubated with 0.1% DMSO, 10 μm SB431542, or 10 μm SB505124 for 1 h and then incubated with or without 5 μm erastin for 24 h b or 26 h c. The viability of cells was determined by trypan blue exclusion assay. Each value is the percentage of trypan blue-positive cells and reflects the mean ± SD of at least three experiments with duplicate assays in each experiment b. Percentage of propidium iodide (PI) or Annexin-V (AV) positive cells were determined by Annexin-V and PI staining. Results are representative of at least three independent experiments c. **P < 0.01, significant difference between the samples
Role of Smad3 in ALK4/5-mediated HK-2 cell death induced by cadmium
Knockdown of ALK4 or Smad3 with siRNAs attenuated the expression of phosphorylated Smad3 in HK-2 cells exposed to CdCl2 (Fig. 5a–c), and suppressed cadmium-induced cell death (Fig. 5d, e). Treatment with SIS3, a selective inhibitor of TGFβ1-dependent Smad3 phosphorylation, also suppressed cadmium-induced HK-2 cell death (Fig. 5f). In contrast, silencing of Smad2 with siRNA did not protect cells from cadmium toxicity (Supplemental Fig. 7A, B). TGFβ1 and TGFβ2 mRNA levels decreased following exposure of HK-2 cells to CdCl2 (Supplemental Fig. 7C, D). In contrast, mRNA and protein expression of Inhibin-bA, a monomer of activin A [30], increased in HK-2 cells exposed to CdCl2 (Fig. 5g, h), whereas the secretion of activin A into the culture medium was reduced (Fig. 5i). In addition, knockdown of Inhibin-bA with siRNAs failed to attenuate CdCl2-induced Smad3 phosphorylation (Fig. 5j) and HK-2 cell death (Fig. 5k). Furthermore, treatment with activin A or TGFβ1 did not increase CdCl2-induced cell death (Supplemental Fig. 7E, F). Taken together, these results suggest that ALK4/5/Smad3 signaling is involved in cadmium-induced HK-2 cell death independent of their ligands.
Fig. 5.

Role of Smad3 in ALK4/5-mediated HK-2 cell death induced by cadmium. a–e Cells transfected with control siRNA, ALK4 siRNA-1, ALK4 siRNA-2 a, b, d, Smad3 siRNA-1, or Smad3 siRNA-2 c, e were incubated with or without 25 μm CdCl2 (Cd) for 15 h a–c or 24 h d, e. Total RNAs were subjected to quantitative RT-PCR to determine ALK4 mRNA levels a. Cell lysates were subjected to western blotting using the indicated antibodies b, c. The viability of cells was determined by trypan blue exclusion assay d, e. f Cells were incubated with 0.1% DMSO, or 5 μm SIS3 for 1 h and then incubated with or without 25 μm CdCl2 (Cd) for 30 h. The viability of cells was determined by trypan blue exclusion assay. g, h Cells were incubated with 25 μm CdCl2 (Cd) for the indicated time. Total RNAs were subjected to quantitative RT-PCR to determine Inhibin-bA mRNA levels g. Cell lysates were subjected to western blotting using the indicated antibodies h. i Cells were incubated with or without 25 μm CdCl2 (Cd) for 12 h. Concentration of Activin A per 1.0 × 105 cells in the media was measured and reflects the mean ± SD of six experiments. j, k Cells transfected with control siRNA, Inhibin-bA siRNA-1, or Inhibin-bA siRNA-2 were incubated with or without 25 μm CdCl2 (Cd) for 15 h j or 24 h k. Cell lysates were subjected to western blotting using the indicated antibodies j. The viability of cells was determined by trypan blue exclusion assay k. Data of quantitative RT-PCR was normalized to β-actin expression and reflects the mean ± SD of at least three experiments. Immunoblots shown are representative of at least three independent experiments. Each value of trypan blue exclusion assay is the percentage of trypan blue-positive cells and reflects the mean ± SD of at least three experiments with duplicate assays in each experiment. *P < 0.05, **P < 0.01, significant difference between the samples
Role of Akt and FOXO3a in ALK4/5-mediated HK-2 cell death induced by cadmium
In addition to the Smad pathway, ALK4/5 can transmit their signals to non-canonical pathways such as Akt, p38, ERK1/2, and JNK [13–15]. Phosphorylation of Akt at Thr308, p38 at Thr180/Tyr182, ERK1/2 at Thr202/Tyr204, and JNK at Thr183/Tyr185, which lead to activation of their respective signaling cascades, were attenuated by SB431542 or SB505124 treatment in HK-2 cells exposed to CdCl2 (Fig. 6a). ALK4 depletion with siRNAs also partially suppressed CdCl2-induced phosphorylation of Akt, p38, and ERK but not of JNK in HK-2 cells (Supplemental Fig. 8A). These results revealed that ALK4/5 induces the activation of Akt, p38, and ERK signaling in HK-2 cells exposed to CdCl2.
Fig. 6.

Role of Akt and FOXO3a in ALK4/5-mediated HK-2 cell death induced by cadmium. a–c Cells were incubated with 0.1% DMSO, 10 μm SB431542, 10 μm SB505124 a, or 10 µm MK2206 a–c for 1 h and then incubated with or without 25 μm CdCl2 (Cd) for 15 h a or 30 h b, c. Cell lysates were subjected to western blotting using the indicated antibodies a. The viability of cells was determined by trypan blue exclusion assay b. Percentage of propidium iodide (PI) or Annexin-V (AV)-positive cells were determined by Annexin-V and PI staining c. d Cells transfected with control siRNA or FOXO3a siRNA were incubated with 0.1% DMSO or 10 µm SB431542 for 1 h and then incubated with or without 25 μm CdCl2 (Cd) for 36 h. The viability of cells was determined by trypan blue exclusion assay. Immunoblots shown are representative of at least three independent experiments. Each value of trypan blue exclusion assay is the percentage of trypan blue-positive cells and reflects the mean ± SD of at least three experiments with duplicate assays in each experiment. Results of Annexin-V and PI staining are representative of at least three independent experiments. *P < 0.05, **P < 0.01, significant difference between the samples
We have previously found that activation of Akt signaling promotes cadmium-induced cellular damage in HK-2 cells [31]. In addition, Akt phosphorylates a transcription factor forkhead box O (FOXO) 3a at Thr32, which results in the inactivation of FOXO3a by translocation from the nucleus [32], in HK-2 cells exposed to CdCl2 [33]. It has been reported that ALK5 activates Akt signaling and the resultant translocation of FOXO3a to cytoplasm promotes oxidative stress-induced cellular damage in rat mesangial cells treated with high glucose [34]. Consistent with our findings, treatment with MK2206, an Akt inhibitor, suppressed HK-2 cell death (Fig. 6b, c), and reduced levels of FOXO3a phosphorylated at Thr32 were observed in HK-2 cells treated with MK2206, SB431542, or SB505124 following exposure to CdCl2 (Fig. 6a). In contrast, we have reported that FOXO3a phosphorylated at Thr32 remains in nuclei of HK-2 cells treated with 10 µm CdCl2 [31], which was also observed in HK-2 cells treated with 25 µm CdCl2 (data not shown). In addition, we observed a synergistic effect of FOXO3a knockdown and SB431542 treatment on protection against CdCl2-induced HK-2 cell death without affecting the intracellular concentration of cadmium (Fig. 6d, Supplemental Fig. 8B, data not shown). These results indicate that although ALK4/5-mediated cell death by cadmium exposure depends on Akt signaling, the downstream target of ALK4/5/Akt does not appear to be FOXO3a phosphorylation in HK-2 cells.
Role of Nrf2 in ALK4/5-mediated HK-2 cell death induced by erastin
Treatment with SB431542, SB505124, or SIS3 suppressed erastin-induced HK-2 cell death (Fig. 7a). Consistently, co-incubation with TGFβ1 or activin A enhanced the cell death induced by erastin (Supplemental Fig. 9A, B). However, in contrast to cadmium-induced HK-2 cell death, treatment with MK2206 failed to suppress erastin-induced cell death (Fig. 7b), indicating an Akt signaling-independent mechanism for erastin-induced HK-2 cell death. It has been reported that the transcription factor NF-E2-related factor 2 (Nrf2) and its downstream target heme oxygenase (HO)-1 are involved in erastin-induced ferroptosis [35–39]. On the other hand, TGFβ signaling is known to downregulate Nrf2 signaling [40, 41]. Thus, we examined whether the disruption of ALK4/5 signaling induces hyperactivation of Nrf2 signaling and results in the suppression of erastin-induced ferroptosis in HK-2 cells. First, we confirmed the roles of Nrf2 and kelch-like ECH-associated protein 1 (Keap1), an adaptor protein that negatively regulates Nrf2 activity [42, 43], in erastin-induced ferroptosis of HK-2 cells. Treatment with erastin increased the expression of Nrf2 and HO-1 in a time-dependent manner (Fig. 7c) and Nrf2 knockdown suppressed erastin-induced HO-1 expression in HK-2 cells (Supplemental Fig. 9C), demonstrating the activation of Nrf2 signaling by erastin. Furthermore, Keap1 depletion increased the basal expression of Nrf2, and basal and erastin-induced expression of HO-1 (Supplemental Fig. 9D), and attenuated erastin-induced cell death, which was blocked by Nrf2 knockdown (Fig. 7d), indicating that Nrf2 signaling might protect HK-2 cells from erastin-induced ferroptosis. On the other hand, treatment with SB431542, SB505124, or SIS3 increased basal expression of Nrf2, and basal and erastin-induced expression of HO-1 without changing the level of phosphorylated ERK compared with U0126 treatment (Fig. 7e), indicating that pharmacological inhibition of ALK4/5 induces hyperactivation of Nrf2 signaling in HK-2 cells exposed to erastin. Consistently, Nrf2 depletion reversed the hyperactivation of Nrf2 signaling and the suppression of erastin-induced cell death induced by SB505124 (Fig. 7f, g) or SB431542 treatment (Supplemental Fig. 9E). Taken together, these results suggest that hyperactivated Nrf2 signaling is responsible for the attenuation of erastin-induced HK-2 cell death caused by blockage of ALK4/5 signaling.
Fig. 7.

Role of Nrf2 in ALK4/5-mediated HK-2 cell death induced by erastin. a, b Cells were incubated with 0.1% DMSO, 10 μm SB431542, 10 μm SB505124, 5 μm SIS3 a, or 10 µm MK2206 b for 1 h and then incubated with or without 5 μm erastin for 24 h. The viability of cells was determined by trypan blue exclusion assay. c Cells were incubated with 5 μm erastin for the indicated time. Cell lysates were subjected to western blotting using the indicated antibodies. d Cells transfected with control siRNA, Keap1 siRNA, or Nrf2 siRNA were incubated with or without 5 μm erastin for 24 h. The viability of cells was determined by trypan blue exclusion assay. e Cells were incubated with 0.1% DMSO, 10 µm SB431542, 10 µm SB505124, 5 µm SIS3, or 20 µm U0126 for 1 h and then incubated with or without 5 μm erastin for 20 h. Cell lysates were subjected to western blotting using the indicated antibodies. f, g Cells transfected with control siRNA or Nrf2 siRNA were incubated with 0.1% DMSO or 10 µm SB505124 for 1 h and then incubated with or without 5 μm erastin for 20 h f or for 24 h g. Cell lysates were subjected to western blotting using the indicated antibodies f. The viability of cells was determined by trypan blue exclusion assay g. Immunoblots shown are representative of at least three independent experiments. Each value of trypan blue exclusion assay is the percentage of trypan blue-positive cells and reflects the mean ± SD of at least three experiments with duplicate assays in each experiment. **P < 0.01, significant difference between the samples
It has been reported that cadmium-induced upregulation of TGFβ1 and downregulation of Nrf2 signaling have a role in the rat prostates damage [44]. However, Nrf2 knockdown showed little effect on the protective capacity of SB431542 against CdCl2-induced HK-2 cells death (Supplemental Fig. 9F), indicating an Nrf2-independent mechanism.
Discussion
In the present study, we showed the preferential types of death of HK-2 cells exposed to several chemicals. Our results showed induction of apoptosis by sorbitol and cisplatin, apoptosis and necrosis induced by cadmium, and ferroptosis induced by erastin. In addition, treatment with cadmium and erastin, but not with sorbitol or cisplatin, significantly increased the expression of phosphorylated Smad3 protein, and treatment with SB431542- or SB505124-attenuated cadmium- and erastin- but not sorbitol- or cisplatin-induced HK-2 cell death. We confirmed that CdCl2 exposure increased the levels of phosphorylated smad3, phosphorylated Akt, and phosphorylated FOXO3a proteins, and treatment with SB431542, SB505124, or SIS3 attenuated CdCl2-induced cytotoxicity in primary human renal proximal tubular epithelial cells (RPTEC) (Supplemental Fig. 10A, B). Furthermore, erastin exposure induced cytotoxicity in RPTEC, which was suppressed by SB431542, SB505124, or SIS3 treatment (Supplemental Fig. 10C). In addition, RSL3 exposure induced cytotoxicity in RPTEC, which was attenuated by SB431542, SB505124, SIS3, U0126, Trolox, or DFO treatment (Supplemental Fig. 10D). We also confirmed that CdCl2 exposure increased phosphorylated smad3 protein levels, and treatment with SB431542 or SIS3-attenuated CdCl2-induced cytotoxicity in mouse renal tubular cells (mProx24 cells), pig renal tubular cells (LLC-PK1 cells) or dog renal tubular cells (MDCK cells) (Supplemental Fig. 11A-F). Furthermore, erastin exposure induced cell death and morphological changes in rat renal tubular cells (NRK52E cells) or LLC-PK1 cells, which were suppressed by SB431542, SIS3, U0126, Fer-1, or DFO treatment (Supplemental Fig. 11G-I, data not shown). Taken together, our findings constitute the first demonstration, to our knowledge, that ALK4/5 signaling is responsible for cadmium- and erastin-induced cell death in the renal proximal tubular cells.
Given that cadmium induces cellular damage in the liver and nervous tissues [45–48], we also examined the role of ALK4/5 signaling in CdCl2-induced cell death in human hepatoma HepG2 cells (Supplemental Fig. 12) and human neuroblastoma SH-SY5Y cells (Supplemental Fig. 13). We found that CdCl2 exposure induced apoptotic and necrotic cell death in HepG2 cells, and necrotic but not apoptotic cell death in SH-SY5Y cells, indicating that the type of cadmium-induced cell death depends on the cellular context. Cadmium has been reported to induce lipid peroxidation [49, 50], and our present study demonstrated that cadmium-induced cytotoxicity was attenuated by ferroptosis inhibitors such as Fer-1, Trolox, or DFO in both HK-2 cells and SH-SY5Y cells, indicating that iron-dependent lipid peroxidation might contribute to cadmium-induced toxicity. In addition, cadmium increased the phosphorylation of Smad3 protein, and treatment with SB431542, SB505124, or SIS3 suppressed cadmium-induced damage in HepG2 and SH-SY5Y cells. Our data suggest that the pathological significance of cadmium-induced ALK4/5 signaling activation might extend beyond the regulation of cell death type. Furthermore, we found that cadmium exposure transiently increased the phosphorylation of bone morphogenetic protein (BMP)-specific Smad (Smad1/5), which is phosphorylated by BMP-specific type I receptors (ALK1/2/3/6) [14] in HK-2, HepG2, and SH-SY5Y cells (Supplemental Fig. 14A), indicating that cadmium also activates BMP signaling. Although treatment with LDN193189, a BMP type I receptor inhibitor, almost completely abolished CdCl2-induced phosphorylation of Smad1/5 in HK-2 cells (Supplemental Fig. 14B), CdCl2-induced death was not suppressed in HK-2, HepG2, or SH-SY5Y cells (Supplemental Fig. 14C), indicating that BMP signaling has little contribution to cell death induced by cadmium.
The activation of ALK4/5 signaling requires the interaction of ligands with their receptors [13–15, 30], and either elevation or reduction of activin A and TGFβ expression is observed in renal injury [16–20]. In the present study, exposure of HK-2 cells to CdCl2 elevated the levels of Inhibin-bA but not the levels of TGFβ1 or TGFβ2. However, although ALK5 or Smad3 knockdown attenuated cadmium-induced HK-2 cell death, Inhibin-bA knockdown or activin A or TGFβ1 treatment had little effect on it. Given that oxidative stressors such as cigarette smoke extract, hydrogen peroxide, and ozone induce a ligand-independent aberrant activation of a receptor-type tyrosine kinase [51–53], we hypothesize that cadmium may induce activation of ALK4/5 in a ligand-independent manner. Consistent with this possibility, Gentle et al. [54] demonstrated that ectopic expression of ligand-independent constitutively active ALK5 in the tubular epithelium alone is sufficient to induce cell death and acute tubular injury. In addition, Daehn et al. [55] also demonstrated that podocyte-specific expression of constitutively active ALK5 non-cell-autonomously induces endothelial cell death and focal segmental glomerular sclerosis. Further investigations are required to elucidate how cadmium activates and modifies ALK4/5 in HK-2 cells.
We examined how activated ALK4/5 mediates cadmium-induced HK-2 cell death and found that knockdown of Smad3, but not Smad2, attenuated cadmium-induced cell death. Consistent with our results, previous reports demonstrated that Smad3 and Smad2 have different roles in renal diseases, and Smad3 has therapeutic potential in diseases such as renal fibrosis [56–59]. Given that TGFβ increases the expression of Nox4, a member of NADPH oxidase, and induces oxidative stress via Smad3 [60, 61], we hypothesized that Nox4 might be involved in cadmium-induced stress response in HK-2 cells. However, little change was observed in Nox4 mRNA level in response to cadmium treatment, and Nox4 knockdown or treatment with DPI, a Nox inhibitor, failed to suppress CdCl2-induced HK-2 cell death (data not shown). Therefore, Nox4 does not have a major role in cell death induced by cadmium exposure.
We also found that ALK4/5/Akt and FOXO3a signaling facilitate cadmium-induced HK-2 cell death in parallel. Recently, we reported that Akt signaling elevates the expression of the transcription factor Snail and enhances cadmium-induced cellular damage in HK-2 cells [31], whereas Snail is a downstream target of Smad3 [62, 63], which raises the possibility that ALK4/5 signaling might upregulate Snail expression and facilitate cadmium-induced HK-2 cells death. On the other hand, although FOXO3a has been reported to promote cadmium-induced cytotoxicity by enhancing autophagic flux in mesenchymal stem cells (MSCs) [64], FOXO3a knockdown had little effect on the expression of LC3B, a marker of autophagy, in HK-2 cells exposed to CdCl2 (Supplemental Fig. 8B), indicating that the protective mechanism of FOXO3a knockdown against cadmium-induced toxicity in HK-2 cells might be different from that in MSCs. Further studies are needed to clarify the downstream targets of Smad3, Akt, and FOXO3a signaling.
We also examined the protective mechanism of ALK4/5 signaling inhibition against erastin-induced ferroptosis in HK-2 cells. Nrf2/HO-1 signaling has been reported to suppress ferroptosis in mouse renal tubular cells and hepatoma carcinoma cells [35, 36], but facilitate it in HT-1080 fibrosarcoma cells and breast cancer cell lines [37, 38]. In the present study, Keap1 depletion attenuated erastin-induced ferroptosis, and this effect was eliminated by Nrf2 knockdown, indicating that Nrf2 signaling might have protective potential against ferroptosis in HK-2 cells. In addition to these results, we found that disruption of ALK4/5 signaling hyperactivated Nrf2 signaling, and Nrf2 depletion blocked the protective effects of ALK4/5 signaling inhibition on erastin-induced ferroptosis, indicating that inhibition of ALK4/5 signaling might have a protective effect against ferroptosis via Nrf2 signaling in HK-2 cells. Interestingly, the Akt inhibitor MK2206 treatment attenuated cadmium- but not erastin-induced HK-2 cells death, whereas Nrf2 knockdown blocked the protective effect of SB431542 treatment on erastin- but not cadmium-induced HK-2 cell death, indicating that protective mechanisms of ALK4/5 signaling inhibition were different between cadmium-induced cell death (apoptosis and necrosis) and ferroptosis in HK-2 cells.
To elucidate how ALK4/5 signaling could modulate caspase-dependent apoptosis signaling in HK-2 cells exposed to CdCl2, we monitored the dynamics of some pro-apoptotic modulators (Supplemental Fig. 15). The expression of cleaved-caspase-8 and caspase-8-cleaved BH3 interacting domain death agonist (BID) increased in a CdCl2 exposure time-dependent manner, suggesting the activation of caspase-8 by CdCl2 exposure. On the other hand, cleaved-caspase-9 levels increased in HK-2 cells exposed to CdCl2, and inhibition of caspase-9 using Z-LEHD-FMK attenuated CdCl2-induced HK-2 cell death. In addition, CdCl2 exposure increased the expression of two members of pro-apoptotic BH3 only group, Noxa and BNIP3. Furthermore, treatment with SB431542 or MK2206 attenuated CdCl2-induced upregulation of pro-apoptotic modulators except for BNIP3. These findings indicate that ALK4/5 signaling might promote cadmium-induced cellular damages with a caspase-8 and caspase-9-dependent but BNIP3-independent manner in HK-2 cells.
In summary, the present study demonstrates that cadmium exposure activates ALK4/5 signaling in a ligand-independent manner to induce HK-2 cell death via Smad3 and Akt signaling. In addition, ALK4/5 signaling facilitates erastin-induced ferroptosis via Nrf2 signaling in HK-2 cells. Although in vivo or kidney proximal tubules cell death model studies as described by Skouta et al. [65] is necessary to improve the reliability of our new knowledge, these results raise the possibility that blockage of ALK4/5 signaling could be a potential therapeutic approach to attenuate cellular damage caused by cadmium exposure and ferroptosis in renal tubular epithelial cells.
Supplementary information
Acknowledgements
We thank Dr. Kenji Tanabe, Kouki Ishikawa, Yuta Komoike, Kaori Komoike, Takamitsu Miyayama, kennichi Matsumura, Tomoaki Mizuno, and Yutaka Eguchi for helpful discussion. Dr. Kenji Tanabe and Dr. Sekiko Taneda kindly provided cell lines for us. In this research work, we used instruments of Medical Research Institute (MRI), Tokyo Women’s Medical University. This study was supported by JSPS KAKENHI Grant Numbers 17K15495, and a Grant-in-Aid for medicine from the Takeda Science Foundation.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Edited by E. Baehrecke
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41418-019-0307-8) contains supplementary material, which is available to authorized users.
References
- 1.Tomlinson DR. Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia. 1999;42:1271–81. doi: 10.1007/s001250051439. [DOI] [PubMed] [Google Scholar]
- 2.Withanage K, Nakagawa K, Ikeda M, Kurihara H, Kudo T, Yang Z, et al. Expression of RASSF6 in kidney and the implication of RASSF6 and the Hippo pathway in the sorbitol-induced apoptosis in renal proximal tubular epithelial cells. J Biochem. 2012;152:111–9. doi: 10.1093/jb/mvs056. [DOI] [PubMed] [Google Scholar]
- 3.Lieberthal W, Triaca V, Levine J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs. necrosis. Am J Physiol. 1996;270:F700–8. doi: 10.1152/ajprenal.1996.270.4.F700. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol. 2015;26:2647–58. doi: 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu G, Zou H, Luo T, Long M, Bian J, Liu X, et al. Caspase-dependent and caspase-independent pathways are involved in cadmium-induced apoptosis in primary rat proximal tubular cell culture. PLoS ONE. 2016;11:e0166823. doi: 10.1371/journal.pone.0166823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shrestha S, Somji S, Sens DA, Slusser-Nore A, Patel DH, Savage E, et al. Human renal tubular cells contain CD24/CD133 progenitor cell populations: Implications for tubular regeneration after toxicant induced damage using cadmium as a model. Toxicol Appl Pharmacol. 2017;331:116–29. doi: 10.1016/j.taap.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111:16836–41. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W, Chen B, Wang Y, Meng C, Huang H, Huang XR, et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc Natl Acad Sci USA. 2018;115:E1475–E1484. doi: 10.1073/pnas.1716959115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hail N, Jr, Carter BZ, Konopleva M, Andreeff M. Apoptosis effector mechanisms: a requiem performed in different keys. Apoptosis. 2006;11:889–904. doi: 10.1007/s10495-006-6712-8. [DOI] [PubMed] [Google Scholar]
- 10.Majno G, Joris I. Apoptosis, oncosis, and necrosis. Overv Cell death Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 12.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 14.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–71. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 15.de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-beta signaling in cancer. J Natl Cancer Inst. 2000;92:1388–402. doi: 10.1093/jnci/92.17.1388. [DOI] [PubMed] [Google Scholar]
- 16.Huang M, Su L, Yang L, Zhu L, Liu Z, Duan R. Effect of exogenous TGF-β1 on the cadmium-induced nephrotoxicity by inhibiting apoptosis of proximal tubular cells through PI3K-AKT-mTOR signaling pathway. Chem Biol Interact. 2017;269:25–32. doi: 10.1016/j.cbi.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Maeshima A, Zhang YQ, Nojima Y, Naruse T, Kojima I. Involvement of the activin-follistatin system in tubular regeneration after renal ischemia in rats. J Am Soc Nephrol. 2001;12:1685–95. doi: 10.1681/ASN.V1281685. [DOI] [PubMed] [Google Scholar]
- 18.Maeshima A, Miya M, Mishima K, Yamashita S, Kojima I, Nojima Y. Activin A: autocrine regulator of kidney development and repair. Endocr J. 2008;55:1–9. doi: 10.1507/endocrj.KR-113. [DOI] [PubMed] [Google Scholar]
- 19.Miyajima A, Chen J, Lawrence C, Ledbetter S, Soslow RA, Stern J, et al. Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 2000;58:2301–13. doi: 10.1046/j.1523-1755.2000.00414.x. [DOI] [PubMed] [Google Scholar]
- 20.Böttinger EP. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27:309–20. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 22.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–30. [PubMed] [Google Scholar]
- 23.Kim HJ, Koo SY, Ahn BH, Park O, Park DH, Seo DO, et al. NecroX as a novel class of mitochondrial reactive oxygen species and ONOO− scavenger. Arch Pharm Res. 2010;33:1813–23. doi: 10.1007/s12272-010-1114-4. [DOI] [PubMed] [Google Scholar]
- 24.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dächert J, Schoeneberger H, Rohde K, Fulda S. RSL3 and Erastin differentially regulate redox signaling to promote Smac mimetic-induced cell death. Oncotarget. 2016;7:63779–92. doi: 10.18632/oncotarget.11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huo H, Zhou Z, Qin J, Liu W, Wang B, Gu Y. Erastin disrupts mitochondrial permeability transition pore (mPTP) and induces apoptotic death of colorectal cancer cells. PLoS ONE. 2016;11:e0154605. doi: 10.1371/journal.pone.0154605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoo SE, Chen L, Na R, Liu Y, Rios C, Van Remmen H, et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52:1820–7. doi: 10.1016/j.freeradbiomed.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakai O, Uchida T, Imai H, Ueta T, Amano S. Role of glutathione peroxidase 4 in conjunctival epithelial cells. Invest Ophthalmol Vis Sci. 2015;56:538–43. doi: 10.1167/iovs.14-15463. [DOI] [PubMed] [Google Scholar]
- 29.Canli Ouml, Alankuş YB, Grootjans S, Vegi N, Hültner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–48. doi: 10.1182/blood-2015-06-654194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedger MP, de Kretser DM. The activins and their binding protein, follistatin-Diagnostic and therapeutic targets in inflammatory disease and fibrosis. Bioessays. 2013;24:285–95. doi: 10.1016/j.cytogfr.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Fujiki K, Inamura H, Matsuoka M. Detrimental effects of Notch1 signaling activated by cadmium in renal proximal tubular epithelial cells. Cell Death Dis. 2014;5:e1378. doi: 10.1038/cddis.2014.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 33.Fujiki K, Inamura H, Matsuoka M. Phosphorylation of FOXO3a by PI3K/Akt pathway in HK-2 renal proximal tubular epithelial cells exposed to cadmium. Arch Toxicol. 2013;87:2119–27. doi: 10.1007/s00204-013-1077-6. [DOI] [PubMed] [Google Scholar]
- 34.Lu Q, Zhai Y, Cheng Q, Liu Y, Gao X, Zhang T, et al. The Akt-FoxO3a-manganese superoxide dismutase pathway is involved in the regulation of oxidative stress in diabetic nephropathy. Exp Physiol. 2013;98:934–45. doi: 10.1113/expphysiol.2012.068361. [DOI] [PubMed] [Google Scholar]
- 35.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF, et al. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Ren Physiol. 2018;314:F702–F714. doi: 10.1152/ajprenal.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6:24393–403. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124–37. doi: 10.1016/j.canlet.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 39.Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogensis. 2017;6:e371. doi: 10.1038/oncsis.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakin AV, Stourman NV, Sekhar KR, Rinehart C, Yan X, Meredith MJ, et al. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding Phase II detoxifying proteins. Free Radic Biol Med. 2005;38:375–87. doi: 10.1016/j.freeradbiomed.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 41.Ryoo IG, Ha H, Kwak MK. Inhibitory role of the KEAP1-NRF2 pathway in TGFβ1-stimulated renal epithelial transition to fibroblastic cells: a modulatory effect on SMAD signaling. PLoS ONE. 2014;9:e93265. doi: 10.1371/journal.pone.0093265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du L, Lei Y, Chen J, Song H, Wu X. Potential ameliorative effects of Qing Ye Dan against cadmium induced prostatic deficits via regulating Nrf-2/HO-1 and TGF-β1/Smad pathways. Cell Physiol Biochem. 2017;43:1359–68. doi: 10.1159/000481847. [DOI] [PubMed] [Google Scholar]
- 45.Xu S, Pi H, Chen Y, Zhang N, Guo P, Lu Y, et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013;4:e540. doi: 10.1038/cddis.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaikh ZA, Vu TT, Zaman K. Oxidative stress as a mechanism of chronic cadmium-induced hepatotoxicity and renal toxicity and protection by antioxidants. Toxicol Appl Pharmacol. 1999;154:256–63. doi: 10.1006/taap.1998.8586. [DOI] [PubMed] [Google Scholar]
- 47.Chen L, Liu L, Luo Y. Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J Neurochem. 2014;105:251–61. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- 48.Wang B, Du Y. Cadmium and its neurotoxic effects. Oxid Med Cell Longev. 2013;2013:898034. doi: 10.1155/2013/898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Lin SQ, He YL, Liu G, Wang ZY. Protective effects of quercetin on cadmium-induced cytotoxicity in primary cultures of rat proximal tubular cells. Biomed Environ Sci. 2013;26:258–67. doi: 10.3967/0895-3988.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 50.Xu L, Zhu Y, Xu J, Wu K, Li J, Xu W, et al. Effect of cadmium on lipid peroxidation and on some antioxidants in the liver, kidneys and testes of rats given diet containing cadmium-polluted radish bulbs. J Toxicol Pathol. 2013;26:359–64. doi: 10.1293/tox.2013-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan EM, Lanir R, Danielson AR, Goldkorn T. Epidermal growth factor receptor exposed to cigarette smoke is aberrantly activated and undergoes perinuclear trafficking. FASEB J. 2008;22:910–7. doi: 10.1096/fj.06-7729com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filosto S, Khan EM, Tognon E, Becker C, Ashfaq M, Ravid T, et al. EGF receptor exposed to oxidative stress acquires abnormal phosphorylation and aberrant activated conformation that impairs canonical dimerization. PLoS ONE. 2011;6:e23240. doi: 10.1371/journal.pone.0023240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu W, Wages PA, Devlin RB, Diaz-Sanchez D, Peden DB, Samet JM. Src-mediated EGF receptor activation regulates ozone-induced interleukin 8 expression in human bronchial epithelial cells. Environ Health Perspect. 2015;123:231–6. doi: 10.1289/ehp.1307379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gentle ME, Shi S, Daehn I, Zhang T, Qi H, Yu L, et al. Epithelial cell TGFβ signaling induces acute tubular injury and interstitial inflammation. J Am Soc Nephrol. 2013;24:787–99. doi: 10.1681/ASN.2012101024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, Barasch N, et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest. 2014;124:1608–21. doi: 10.1172/JCI71195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, et al. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945–53. doi: 10.1074/jbc.M102382200. [DOI] [PubMed] [Google Scholar]
- 57.Yuan W, Varga J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J Biol Chem. 2001;276:38502–10. doi: 10.1074/jbc.M107081200. [DOI] [PubMed] [Google Scholar]
- 58.Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J. 2006;393:601–7. doi: 10.1042/BJ20051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol. 2010;21:1477–87. doi: 10.1681/ASN.2009121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das R, Xu S, Quan X, Nguyen TT, Kong ID, Chung CH, et al. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am J Physiol Ren Physiol. 2014;306:F155–67. doi: 10.1152/ajprenal.00438.2013. [DOI] [PubMed] [Google Scholar]
- 61.Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, et al. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–94. doi: 10.1172/JCI200319270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo X, Jose PA, Chen SY. Response gene to complement 32 interacts with Smad3 to promote epithelial-mesenchymal transition of human renal tubular cells. Am J Physiol Cell Physiol. 2011;300:C1415–21. doi: 10.1152/ajpcell.00204.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang M, Pi H, Li M, Xu S, Zhang L, Xie J, et al. From the cover: autophagy induction contributes to cadmium toxicity in mesenchymal stem cells via AMPK/FOXO3a/BECN1 signaling. Toxicol Sci. 2016;154:101–14. doi: 10.1093/toxsci/kfw144. [DOI] [PubMed] [Google Scholar]
- 65.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–6. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.