

Summary
Sustained treatment of estrogen receptor (ER)-positive breast cancer with ER-targeting drugs results in ER mutations and refractory unresponsive cancers. Androgen receptor (AR), which is expressed in 80%–95% of ER-positive breast cancers, could serve as an alternate therapeutic target. Although AR agonists were used in the past to treat breast cancer, their use is currently infrequent due to virilizing side effects. Discovery of tissue-selective AR modulators (SARMs) has renewed interest in using AR agonists to treat breast cancer. Using translational models, we show that AR agonist and SARM, but not antagonist, inhibit the proliferation and growth of ER-positive breast cancer cells, patient-derived tissues, and patient-derived xenografts (PDX). Ligand-activated AR inhibits wild-type and mutant ER activity by reprogramming the ER and FOXA1 cistrome and rendering tumor growth inhibition. These findings suggest that ligand-activated AR may function as a non-canonical inhibitor of ER and that AR agonists may offer a safe and effective treatment for ER-positive breast cancer.
Subject Areas: Molecular Biology, Molecular Mechanism of Gene Regulation, Cancer
Graphical Abstract
Highlights
-
•
Androgen receptor (AR) agonists inhibit estrogen receptor (ER)-positive breast cancer
-
•
Activating AR reprograms ER and FOXA1 cistrome, resulting in ER inhibition
-
•
AR agonist alters the phosphoproteome signature consistent with growth inhibition
Molecular Biology; Molecular Mechanism of Gene Regulation; Cancer
Introduction
Worldwide, over 2 million women were diagnosed with breast cancer in 2018, and over 600,000 died of breast cancer (Bray et al., 2018). In the United States, an estimated 266,000 women were diagnosed and approximately 40,000 women died from breast cancer in 2018 (Siegel et al., 2018). Estrogen receptor-α (ER)-positive breast cancer subtype constitutes the majority of breast cancers. ER plays an important role in classifying breast cancer, in defining proliferative characteristics, and in developing a treatment regimen (Andersen and Poulsen, 1989, Musgrove and Sutherland, 2009, Perou et al., 2000, Rossi et al., 2015).
ER is a ligand-dependent transcription factor that is predominantly activated by estrogens. ER-positive breast cancers that are dependent on ER for growth are treated with ER antagonists or inhibitors of estradiol-synthesizing enzyme, aromatase (AI) (Eastell et al., 2008, Robertson et al., 2013, Vogel et al., 2006). Compared with ER-negative or triple-negative breast cancer (TNBC), ER-positive breast cancers typically have a lower proliferation index and a well-differentiated phenotype (Heise and Gorlich, 1982, Neifeld et al., 1982, Perou et al., 2000, Stewart et al., 1982). Women suffering from ER-positive breast cancers suffer from osteoporosis, muscle wasting, and poor quality of life, resulting from extended ER blockade.
Prolonged treatment of cancers with inhibitors or antagonists results in mutations in the target protein and activation of resistance pathways (Balbas et al., 2013, Hara et al., 2003, Sasaki et al., 2011). For example, continued treatment of ER-positive breast cancers with ER antagonists or AI results in resistance, often due to mutations in the ER ligand-binding domain (LBD) (Fuqua et al., 1993, Karnik et al., 1994, Musgrove and Sutherland, 2009, Robinson et al., 2013). Clinical studies have estimated that over 30% of breast cancers treated with tamoxifen become refractory and recur as a resistant cancer and over 40% of recurrent breast cancers express mutated ER (Chandarlapaty et al., 2016, Early Breast Cancer Trialists' Collaborative et al., 2015, Magnani et al., 2017). Mutant ERs that have escaped the hormonal axis and have become hormone independent fail to respond to endocrine therapy, and, consequently, these patients will need to be treated with cell cycle inhibitors such as CDK4/6 inhibitors or cytotoxic agents. Such cancers require new non- or less-toxic effective endocrine therapies.
Androgen receptor (AR) is expressed in over 80%–90% of ER-positive breast cancer (Collins et al., 2011, Garay and Park, 2012, Hu et al., 2011, Narita et al., 2006, Niemeier et al., 2010). Until ER-targeted treatment options were made available, breast cancer was treated with steroidal androgens such as dihydrotestosterone (DHT) (Adair and Herrmann, 1946, Kennedy, 1958) or even with estrogens such as diethylstilbestrol (DES) (Gordan et al., 1963). AR expression in ER-positive breast cancer is associated with improved overall survival (OS) and disease-free survival (DFS) (Vera-Badillo et al., 2014). Studies have shown that co-expression of AR and steroidogenic enzymes such as 5-α-reductase that synthesize active DHT correlated with better progression-free survival (PFS) and OS (Sultana et al., 2014). Other evidences suggest that the AR may increase breast cancer growth or even have a role in the development of tamoxifen resistance (Barton et al., 2017, Bronte et al., 2018, Ciupek et al., 2015, Danforth et al., 2010, De Amicis et al., 2010, Kaaks et al., 2005, Liao and Dickson, 2002).
Recent preclinical and clinical studies indicate that AR could be a growth promoter in tamoxifen-resistant breast cancer, but a growth inhibitor in tamoxifen-sensitive breast cancer. Although AR antagonist enzalutamide increased proliferation of parental breast cancer cell line MCF-7, it inhibited proliferation of tamoxifen-resistant MCF-7 cells (Creevey et al., 2019). Similarly, tamoxifen-resistant clinical specimens that had higher AR:ER ratio had aggressive disease and poor prognosis (Cao et al., 2019). On the contrary, AR-positive ER-positive breast cancer had smaller tumors, better prognosis, lower tumor grade, and better disease-free survival after chemotherapy (Aleskandarany et al., 2016, Witzel et al., 2013). Preclinical studies in parental MCF-7 and ZR-75-1 cells demonstrated antiproliferative effects with AR agonists (Kandouz et al., 1999, Poulin et al., 1988). These conflicting evidences from literature can be comprehensively resolved using translational patient-derived tissues and controlled clinical trials.
In this study, we found that proliferation and growth of patient-derived xenografts (PDX) and tissues that express wild-type and mutant ER were inhibited by AR agonist and tissue-selective AR modulator (SARM) (Dalton et al., 1998) but not by an AR antagonist. Ligand-activated AR inhibited growth of these tumors by reprogramming the ER and FOXA1 cistrome and by altering the phosphokinome signature, resulting in inhibition of ER function. Overall, the results provide an evidence for a tumor suppressive role for ligand-activated AR and create an opportunity to treat ER-positive breast cancer with a less-toxic hormonal approach.
Results
The SARM enobosarm (GTx-024 or ostarine) is an AR agonist that binds to and activates the AR with EC50 at less than 10 nM (Narayanan et al., 2014, Ponnusamy et al., 2017b). Enobosarm was evaluated in clinical trials and was shown to increase lean mass and physical function without having significant virilizing side effects (Crawford et al., 2016, Dalton et al., 2011, Dobs et al., 2013). Preclinical studies described in this manuscript were conducted with a non-metabolizable SARM to eliminate any confounding results obtained due to potential metabolism of steroidal androgens into weaker androgen or estrogen metabolites (Jin and Penning, 2001, Oliveira et al., 2007). Moreover, clinical trials have shown enobosarm to be an effective treatment for breast cancer (https, 2018, Overmoyer, 2015).
SARM Inhibits Breast Cancer Cell Proliferation
To determine the role of AR in breast cancer, we analyzed the TCGA dataset for survival of breast cancer patients expressing higher vs lower AR (Figure 1A). Kaplan-Meier plot of the TCGA dataset demonstrated that breast cancer patients expressing higher AR correlated with longer survival than patients with breast cancer expressing lower AR (hazard ratio of 0.52 and log rank P of 1.1 e−10). This suggests that AR expression might have a beneficial role in breast cancer. Further analysis of the dataset suggested that patients with luminal A and B breast cancers expressing higher AR had a significantly improved survival, whereas ER-negative breast cancer patients had no significant survival benefit.
Figure 1.

AR Agonist Inhibits Proliferation and Growth of Wild-Type and Mutant ER-positive Xenograts
(A) Higher AR expression correlates with patient survival. Patients who have high or low expression of AR in the TCGA dataset were compared for survival. Hazard ratio = 0.52 (0.44–0.61). Log rank P = 1.1 × 10−16.
(B) Enobosarm inhibits the proliferation of ZR-75-1 cells. ZR-75-1 breast cancer cells plated in growth medium (n = 4/treatment) were treated with indicated doses of enobosarm for 12 days, with medium changed and re-treated every third day. After 12 days of treatment, cells were harvested, and the number of cells was counted.
(C) Enobosarm inhibits HCI-7 tumor growth. HCI-7 PDX was surgically implanted as 1 mm3 fragments under the mammary fat pad in NSG female mice (n = 6-10/group) that were ovariectomized and supplemented with estradiol. Once the tumors reached 100–200 mm3, the mice were randomized and treated with vehicle (DMSO:PEG-300 (15%:85%)), enobosarm (10 mpk p.o.), or enzalutamide (30 mpk p.o.). Tumor volume was measured weekly. At sacrifice, tumors were removed, weighed (right panel), and stored for further analysis.
(D) Enobosarm inhibits growth of HCI-13 PDX. HCI-13 PDX was surgically implanted as 1 mm3 fragments under the mammary fat pad in intact female NSG mice (n = 6-10/group). Once the tumors reached 100–200 mm3, the mice were randomized and treated with vehicle (DMSO:PEG-300 (15%:85%)) or enobosarm (10 mpk p.o.). Tumor volume was measured weekly. At sacrifice, tumors were removed, weighed (middle panel), and stored for further analysis. Tumors that were stored in formalin were further processed and immunohistochemistry was performed for the proliferation marker Ki-67 (right panel).
(E) AR agonist DHT inhibits growth of HCI-13 PDX. HCI-13 PDX was performed as indicated above. The mice were randomized and treated with vehicle (DMSO:PEG-300 (15%:85%)), DHT (10 mpk s.c.), or enzalutamide (30 mpk p.o.), or fulvestrant (200 mpk s.c. twice weekly). Tumor volume was measured twice weekly. At sacrifice, tumors were removed, weighed (middle panel), and stored for further analysis. Ki-67 was performed in formalin-fixed paraffin-embedded tumors (right panel).
* = p < 0.05; ** = p < 0.01; *** = p < 0.001. HCI, Huntsman Cancer Institute; AR, androgen receptor; ER, estrogen receptor; NSG, NOD SCID Gamma; PDX, patient-derived xenograft; OVX, ovariectomy; mpk, milligram per kilogram body weight; DHT, dihydrotestosterone.
We conducted studies using various preclinical and translational models to understand the role of AR and its mechanism of action in ER-positive breast cancer. Proliferation of ZR-75-1 cells that express AR and ER was reduced dose-dependently by the AR agonist enobosarm after twelve days of treatment, providing an evidence for anti-proliferative effects of an AR agonist in ER-positive breast cancer (Figure 1B).
Enobosarm Inhibits Wild-Type ER-positive Breast Cancer PDX Growth
Enobosarm was tested in a PDX expressing wild-type ER. From the numerous PDXs available in Dr. Alana Welm's laboratory (DeRose et al., 2011), we identified two ER-positive AR-positive PDXs, HCI-7 and HCI-13 (Table S1; Figure S1 top blot; HCI-9 is an ER-negative AR-positive PDX). HCI-7 tumor fragments were implanted under the mammary fat pad of female NSG mice that were ovariectomized and supplemented with an estradiol pellet. Once the tumors reached 100–200 mm3, the mice were randomized and treated orally with vehicle, 10 mg/kg enobosarm, or 30 mg/kg enzalutamide. The enzalutamide dose was selected based on previously published data (Cochrane et al., 2014, D'Amato et al., 2016, Park et al., 2016, Pollock et al., 2016). The growth and tumor weight (measured at the conclusion of the study) of HCI-7 were inhibited significantly by enobosarm but not by enzalutamide (Figure 1C). Immunohistochemistry staining of the HCI-7 tumors confirmed ER and AR expression and also significant inhibition of proliferation marker Ki-67 in enobosarm-treated tumors (Figure S1).
One characteristic of AR agonists, but not antagonists, is their ability to stabilize AR protein expression (Kemppainen et al., 1992, Zhou et al., 1995). We used AR expression to ensure that enobosarm behaved as an agonist in our PDXs. Results show that AR was stabilized by enobosarm in HCI-7 tumors, indicating that the tumors were exposed to enobosarm and that enobosarm behaved as an agonist (Figure S1).
As the presence of integral murine-stromal cellular infiltration is an issue in PDXs that have undergone passages in immunodeficient mouse strains, we chose Ku80 as a marker for human epithelial cells and H&E evaluation as an approach to quantifying murine stromal content and human epithelial cells (Allard et al., 2014, Maykel et al., 2014, Schneeberger et al., 2016, Tentler et al., 2012). These stains clearly enable us to differentiate between the epithelial and stromal cells and will provide us with the percentage of different cell types present in the PDXs. Ku80 and H&E staining showed that the tumors have predominantly epithelial cells with minimal mouse stromal infiltration in a ratio of around 91:9% of epithelial:stromal cells (Figure S1).
AR Agonist Inhibits Growth of Mutant ER-positive PDX
We discovered by internal sequencing as well as from literature that HCI-13 (an invasive lobular breast cancer) PDX expresses an ER that is mutated in the LBD at Y537 (Sikora et al., 2014). This mutation frequently occurs in ER-positive breast cancers that have been treated with ER antagonists or aromatase inhibitors (Jeselsohn et al., 2018, Toy et al., 2017). HCI-13 was obtained from a patient who was treated with and relapsed from drugs ranging from ER-targeted therapeutics to chemotherapy (Table S1).
To determine if an AR agonist will have the ability to inhibit growth of a mutant ER-positive breast cancer, HCI-13 tumor fragments were implanted under the mammary fat pad in NSG mice. Once the tumors attained 100–200 mm3, the animals were randomized and treated with vehicle or enobosarm. Enobosarm significantly inhibited growth (Figure 1D left), tumor weights (Figure 1D middle), and Ki-67 (Figure 1D right).
We conducted additional xenograft studies with HCI-13 tumors where the animals were treated with steroidal androgen, DHT (10 mg/kg/day subcutaneously), competitive AR antagonist, enzalutamide (30 mg/kg/day orally), and the ER degrader, fulvestrant (200 mg/kg/twice weekly subcutaneously) (Guo et al., 2018). Consistent with the effect of a SARM, DHT significantly inhibited tumor growth, whereas the AR antagonist enzalutamide failed to inhibit tumors (Figure 1E left). Fulvestrant also inhibited tumor growth significantly. Tumor weights recorded at the end of the study, and Ki67 confirmed the results observed in tumor growth (Figure 1E middle and right).
We measured drug concentration in the serum of animals treated with various drugs using standardized LC-MS/MS methods. Steady-state drug concentrations were above their target engagement concentration, suggesting that drug exposure was not a limiting factor in HCI-13 tumor-bearing animals (Figure S2A).
H&E and Ku80 staining indicated that the HCI-13 tumors have tumor epithelial cells with limited mouse stromal cell infiltration (84:16) (Figure S2B). For all tumors (HCI-7 and 13), morphology observed via H&E was within the range typical of breast carcinoma.
Ex Vivo Culture with Tumor Specimens Indicates Heterogeneity of Response to ER and AR Ligands
Breast cancer is heterogeneous in its genomic profile as well as in its response to treatments. To determine the effect of enobosarm and fulvestrant on growth inhibition, we cultured breast cancer specimens obtained from patients, on dental sponges. These patient specimens were collected as indicated in the methods under an Institutional Review Board (IRB) approval. The characteristics of these patient specimens are provided in Table S1. Specimens were treated with vehicle, 1 μM enobosarm, or 100 nM fulvestrant. Three days after treatment, RNA was isolated from the tissues and expression of ER- and AR-target genes was measured (Figure S2C). Expression of ER and AR was confirmed using immunohistochemistry. All the specimens, except specimen 1,075, express the two targets with greater than 80% expression observed (Figure S3). Fulvestrant inhibited the ER function as measured by the expression of pS2 in five of eight specimens, whereas enobosarm inhibited the ER function in three of eight specimens (Figure S2C). FKBP5, an AR-target gene, was used to ensure that the AR in these tumors was functional.
Enobosarm Inhibits HCI-13 Breast Cancer Growth by Inhibiting ER Function
To determine global gene-expression pattern in response to enobosarm, RNA from HCI-13 tumors was subjected to Affymetrix microarray. In total, 3,029 genes were differentially regulated by enobosarm in HCI-13 tumors compared with vehicle-treated tumors. Enobosarm upregulated 1,792 genes and downregulated 1,237 genes. Heatmap of the differentially regulated genes clearly indicates a shift in the expression pattern of genes due to enobosarm treatment (Figure 2A).
Figure 2.

Gene Expression Study in HCI-13 PDX Indicates Inhibition of ER Pathway by an AR Agonist
(A–C) RNA was isolated from HCI-13 PDX xenografts treated with vehicle or enobosarm (Figure 1D) and microarray was performed (n = 3-4/group). Genes that were different in enobosarm-treated group (q < 0.05) are represented in the heatmap (A). Representative ER- and AR-target genes and the most up- and down-regulated genes are shown in panel (B). Canonical pathway, upstream regulators, and diseases represented by the enriched genes obtained from Ingenuity Pathway Analysis (IPA) are shown in panel (C).
(D) Genes enriched in the AR pathway were fed into TCGA database and Kaplan-Meier survival plots were created. * = q < 0.05. ER, estrogen receptor; AR, androgen receptor; PDX, patient-derived xenograft.
Ingenuity pathway analysis (IPA) showed that the ER-target genes were highly enriched followed by the AR-target genes in enobosarm-treated specimens (p values of 6.66−11 vs 2.83−7; Figure 2C). A subset of the ER-target genes was down-regulated by enobosarm, whereas all the AR-target genes were up-regulated (Figure 2B). Although ER-target genes such as TFF1, PGR, GREB1, and NRIP1 were down-regulated by enobosarm, other ER-target genes such as CTSD and CCND1 were not inhibited by enobosarm. These results provide evidence that enobosarm functions in breast cancer by at least partially inhibiting the ER-signaling pathway to reduce cancer growth.
The genes enriched for the AR pathway were fed into TCGA database to determine the consequence of altering the AR pathway by an AR agonist. AR pathway genes correlated with a significant increase in survival of breast cancer patients (hazard ratio of 0.64 and log rank P of 1.1 × 10−8) (Figure 2D).
To ensure that enobosarm is not an ER antagonist and the effects are mediated through AR, an ER competitive ligand binding assay (Figure S4A) and an ER transactivation assay (Figure S4B) were performed. Both results indicate that enobosarm has no direct interaction with ER, which is in concordance with earlier published results (Yin et al., 2003).
Chromatin Immunoprecipitation-Sequencing (ChIP-Seq) Analysis Demonstrates that Enobosarm Reprograms ER and AR Cistromes
To determine if the effect of enobosarm on ER function is due to any direct effect on ER binding to DNA, ChIP-sequencing for ER was performed in the tumor samples obtained from animals shown in Figure 1D. ER binding to 1,148 regions (q < 0.05) on the DNA was reprogrammed by enobosarm, with 572 regions statistically enriched with ER and 576 regions depleted of ER (Figure 3A), whereas Principal component analysis (PCA) (Figure 3B) and unsupervised hierarchical clustering (Figure 3D) show the distinct distribution of individual samples, an indication that enobosarm modified the DNA-binding pattern of ER in HCI-13. The motifs that were enriched by the ER represent androgen response element (ARE) and FOXA1 response elements (FOXA1RE), whereas the regions that were depleted of ER represent estrogen response element (ERE) and FOXA1RE (Figure 3A right). Although the DNA regions depleted of ER by enobosarm favor the inhibition of the ER-target gene expression pattern, the enrichment of ER at AREs is surprising and has not been previously reported. Figure S5 shows representative regions enriched by and depleted of ER. Figure S6A shows the heatmap of individual tumor specimens. Variability between individual samples can be attributed to the inherent variability between xenograft specimens. Repeating the studies in a cell line model under controlled conditions might provide a robust redistribution outcome.
Figure 3.

ChIP-sequencing Shows Reprogramming of ER Binding after Enobosarm Treatment
(A) Chromatin immunoprecipitation (ChIP) assay was performed for ER in tumors treated with vehicle (n = 4) or 10 mg/kg/day enobosarm (n = 3) (tumors from animals shown in Figure 1D). Next-generation sequencing was performed to determine the genome-wide binding of ER to the DNA. Heatmap of significantly different peaks (q < 0.05) is shown as average of the individual tumor samples. The top enriched motifs are shown to the right of the heatmap.
(B) Principal Component Analysis (PCA) plot of vehicle- and enobosarm-treated samples that corresponds to ER-ChIP peaks is shown.
(C) Pie charts showing the distribution of ER enrichment in enobosarm-treated HCI-13 samples.
(D) Unsupervised hierarchical clustering.
(E) ChIP assay was performed with ER antibody in HCI-13 specimens treated with vehicle or enobosarm and, real-time PCR was performed with the primers and TaqMan probe to the specified regions. AR, androgen receptor; ER, estrogen receptor; ChIP, chromatin immunoprecipitation; ARE, androgen response elements; ERE, estrogen response element; FOXA1RE, Forkhead box A1 response element.
Between 50% and 60% of the ER-enriched and depleted sites were mapped to distal regulatory regions, whereas only around 2%–3% of the sites were mapped to promoter regions (Figure 3C). Interestingly, although the intron and exon binding percentage match with previous reports, the proportion of ER bound to promoters and distal regulatory elements are distinct from that observed in response to estrogens or with a constitutively active ER (Jeselsohn et al., 2018). Other studies have indicated that the ER cistrome comprises about 30%–40% distal regulatory regions and 7%–22% proximal promoter regions, whereas AR-regulated ER cistrome in this study comprises of 50%–60% and 2%–3% of these regions, respectively. ER binding to pS2 ERE, PSA (KLK3) promoter ARE, and PSA enhancer ARE was validated by ChIP real-time PCR (Figure 3E).
It was interesting to observe that an AR agonist such as enobosarm reprogrammed ER cistrome by depleting EREs of ER and enriching the AREs with ER. It is important to recognize that as enobosarm neither binds to ER nor alters ER activity (Kearbey et al., 2007, Narayanan et al., 2008); its effect on ER cistrome is mediated by activating AR. It is likely that ER is following AR or FOXA1 toward the respective response elements. We performed ChIP-Seq for AR in HCI-13 PDX treated with vehicle (n = 4) or enobosarm (n = 3). Enobosarm significantly enriched 5,156 sites, and no binding was observed in the absence of enobosarm (Figure 4A). As expected, the individual samples were distinctly positioned in the PCA plot (Figure 4B), and the motifs enriched by AR represent AREs and FOXA1RE (Figure 4A right). Figure S6B shows the heatmap of individual tumor specimens. The unsupervised hierarchical clustering shown in Figure 4C distinctly clustered the vehicle- and the enobosarm-treated samples.
Figure 4.

ChIP-sequencing Shows Rearrangement Binding of AR after Enobosarm Treatment
(A) Chromatin immunoprecipitation (ChIP) assay was performed for AR in tumors treated with vehicle (n = 4) or 10 mg/kg/day enobosarm (n = 3) (tumors from animals shown in Figure 1D). Next-generation sequencing was performed to determine the genome-wide binding of AR to the DNA. Heatmap of significantly different peaks (q < 0.05) is shown as average of the individual tumor samples. The top enriched and depleted motifs are shown to the right of the heatmap.
(B) Principal Component Analysis (PCA) plot of vehicle- and enobosarm-treated samples that corresponds to AR-ChIP peaks is shown.
(C) Unsupervised hierarchical clustering.
(D) Pie charts showing the distribution of AR enrichment in enobosarm-treated HCI-13 samples.
(E) AR and ER co-occupied sites represented as Venn diagram. AR, androgen receptor; ChIP, chromatin immunoprecipitation; ARE, androgen response elements; FOXA1RE, Forkhead box A1 response element.
Mapping the genomic regions bound by AR in HCI-13 indicates that genomic distribution is consistent with that of previously mapped region in LNCaP prostate cancer cells (Figure 4D) (Toropainen et al., 2016). We determined the overlap between ER and AR at statistically enriched sites to be 385 sites, which represent a remarkable 67% of the gained ER sites. AR alone occupies 4,777 out of 5,156 enriched sites, whereas ER alone occupies only 188 out of 572 ER-enriched sites (Figure 4E). This shows that new ER binding in the presence of an AR agonist is highly dependent on the AR occupancy. This warrants further exploration of a potential association between the two proteins on the ARE.
To determine if the AR cistrome in HCI-13 (where an agonist was growth inhibitory) in response to an agonist overlaps with AR cistrome in different models, a database search was performed. Most, if not all, of the AR cistromes found in the database search were identified in prostate cancer models. Table S3 shows a significant overlap between AR cistrome in HCI-13 and the AR cistromes in the database (TArbs = Tumor AR-binding sites (Pomerantz et al., 2015)).
Enobosarm Reprograms the FOXA1 Cistrome
It is interesting to observe that FOXA1RE motifs are represented in both enriched and depleted ER cistrome in response to enobosarm. Enrichment of FOXA1RE in the AR cistrome is not surprising, as previous studies have shown that the ligand-activated ER and AR bind to cis elements with FOXA1 binding adjacent to the ER and AR (Carroll et al., 2005, Zhao et al., 2016). Previous studies have also shown that FOXA1 binding is required for ER and AR to interact with the DNA at many genomic loci and that knockdown of FOXA1 reduces the association of ER and AR with chromatin (Carroll et al., 2005, Zhao et al., 2016). These results suggest that FOXA1 binding to DNA is an important event required for the AR and ER to function as transcription factors. Considering that FOXA1 is a critical pioneering transcription factor, we evaluated its interaction with DNA in the presence of vehicle and enobosarm treatment in HCI-13 PDX. Although FOXA1 is enriched in 4,946 DNA regions, it is depleted in 840 regions with a total of 5,786 DNA regions statistically altered in enobosarm-treated samples compared with the vehicle-treated samples (Figure 5A). Figure S6C shows the heatmap of individual tumor specimens. The PCA plot and hierarchical clustering distinctly segregates the samples belonging to the individual groups, suggesting that enobosarm clearly modifies FOXA1-chromatin interaction (Figures 5B and 5C). Enriched FOXA1 cistrome motifs represent ARE and FOXA1RE, whereas the depleted cistrome motifs represent ERE and FOXA1RE (Figure 5A right). This is consistent with the ER and AR ChIP-seq results. FOXA1 genomic binding regions are similar to that of ER and AR DNA binding regions and are consistent with earlier publications (Figure 5D). Out of the 4,946 gained FOXA1 sites, 2007 (40.6%) overlap with the gained AR-binding sites (Figure 5E). These results suggest a significant overlap between the gained AR and FOXA1 binding. Representative peaks for AR and FOXA1 are presented in Figure S7. Strikingly, out of the 840 sites with reduced FOXA1 binding, 474 overlap with lost ER-binding sites (83% of total ER sites and 56% of total FOX sites) (Figure 5E).
Figure 5.

ChIP-sequencing Shows Reprogramming of FOXA1 Binding to the DNA after Enobosarm Treatment
(A) ChIP assay was performed for FOXA1 in tumors treated with vehicle (n = 4) or 10 mg/kg/day enobosarm (n = 4) (tumors from animals shown in Figure 1D). Next-generation sequencing was performed to determine the genome-wide binding of FOXA1 to the DNA. Heatmap of significantly different peaks (q < 0.05) is shown as average of the individual tumor samples. The top enriched and depleted motifs are shown to the right of the heatmap.
(B) Principal Component Analysis (PCA) plot of vehicle- and enobosarm-treated samples that corresponds to FOXA1-ChIP peaks is shown.
(C) Unsupervised hierarchical clustering.
(D) Pie charts showing the distribution of FOXA1 enrichment in enobosarm-treated HCI-13 samples.
(E) AR and FOXA1 and ER and FOXA1 co-occupied sites represented as Venn diagram. FOXA1, Forkhear box A1; ChIP, chromatin immunoprecipitation; ARE, androgen response elements; GRE, glucocorticoid response elements; FOXA1RE, Forkhead box A1 response element.
The above results were replotted to address whether the AR and FOXA1 were occupied or depleted from the ER-enriched or depleted sites in the presence of enobosarm (Figure S8A). Interestingly, ER enrichment is matched by the AR and FOXA1 enrichment, suggesting that all three transcription factors occupy the same sites. Similar to the enriched sites, ER-depleted sites were also depleted of FOXA1 binding (Figure S8A).
AR Agonist Inhibits ER-target Gene and ER-DNA Binding in HCI-7 Xenografts
Considering the impact on ER function observed in the presence of an AR agonist in ER-positive breast cancer PDX HCI-13, we performed experiments to confirm the gene expression and DNA occupancy results in a different ER-positive breast cancer model. RNA from HCI-7 tumors shown in Figure 1C was isolated and expression of ER and AR-target genes was measured by real-time PCR. Consistent with the HCI-13 PDX results, enobosarm increased the expression of AR-target gene FKBP5 and significantly reduced the expression of ER-target genes TFF1 (pS2) and PGR (PR) (Figure 6A).
Figure 6.

Enobosarm Effects on Gene Expression and ER Recruitment Are Reproducible in Different Models
(A) Enobosarm activates AR-target genes and inhibits ER-target genes in HCI-7 xenografts. RNA was extracted from vehicle- or enobosarm-treated HCI-7 tumor tissues (n = 4/group), and expression of the indicated genes was measured by real-time PCR.
(B) Enobosarm inhibits ER recruitment to pS2 promoter in HCI-7 tumors. HCI-7 tumors that were treated with vehicle or enobosarm (n = 3/group) were snap-frozen at the time of collection. Tumors were formalin fixed, homogenized, and ER immunoprecipitated with an ER antibody. Real-time PCR was performed for the pS2 promoter EREs. *p < 0.05.
We further evaluated the DNA binding efficiency of ER in the presence of enobosarm in HCI-7 PDX tumor samples by ChIP-real time PCR (Figure 6B). ER occupancy on the pS2 promoter in HCI-7 PDX was inhibited by enobosarm (Figure 6B). These results are consistent with the results observed in HCI-13 and suggest that these results are not model dependent.
Enobosarm Has No Effect on an AR-positive ER-negative Breast Cancer PDX
The results in ER-positive models indicate that the tumor suppressive function of AR is through indirect ER-inhibitory properties. If this is true, AR agonists should have no effect on ER-negative breast cancers. AR-positive ER-negative breast cancer HCI-9 tumor fragments were implanted under the mammary fat pad of NSG mice. Once the tumors grew to 100–200 mm3, the animals were randomized and treated orally with vehicle or enobosarm. Enobosarm did not alter the tumor growth, indicating that the AR agonist was not effective in HCI-9 PDX that does not express ER (Figure 7A). This result was confirmed with another AR-positive TNBC PDX (data not shown). Enobosarm treatment increased the AR protein expression, indicating that it functioned as an agonist in this tumor model and the drug was delivered to the tumors (Figure 7B). The results of immunohistochemistry indicate that the tumors have minimal mouse stromal cells and the tumors predominantly comprised of epithelial cells with a ratio of 86:14% epithelial: stromal cells (Figures 7C and 7D). Also, enobosarm had no effect on Ki-67 levels in the tumors. To ensure that the AR is functional, we extracted RNA from the tumors and performed real-time PCR for AR-target genes. Enobosarm significantly increased the expression of all AR-target genes that were measured, indicating that the AR is functional and responds to an AR agonist (Figure 7E). Finally, we performed preliminary ChIP-Seq with vehicle- and enobosarm-treated HCI-9 tumors to determine if the sites occupied by AR in HCI-13 are also occupied by AR in HCI-9 (Figure S8B). Although the HCI-9 ChIP-Seq was not performed in replicates, the preliminary results show the overlapping AR cistrome in ER-negative and -positive breast cancers.
Figure 7.

Enobosarm Does Not Inhibit Growth of an ER-negative AR-positive HCI-9 PDX
(A) AR-positive, but ER-negative, HCI-9 PDX was surgically implanted as 1 mm3 fragments under the mammary fat pad in intact NSG mice (n = 8–10/group). Once the tumors reached 100–200 mm3, the mice were randomized and treated with vehicle (DMSO:PEG-300 (15%:85%)) or enobosarm (10 mpk p.o.). Tumor volume was measured thrice weekly.
(B) AR Western blot was performed from the tumor specimens shown on the left. Densitometric quantification of the bands is provided at the bottom of the blot.
(C) HCI-9 tumors were formalin-fixed and immunostained with the indicated antibodies. Representative images of n = 4–5/group/stain are shown. Ki-67 staining was quantified and represented as bar graph (n = 5/group). Scale is provided in one of the images (50 μm).
(D) Table showing the percent of stromal and epithelial cells in the xenografts.
(E) Enobosarm increases AR-target genes. HCI-9 tumor fragments from vehicle- or enobosarm-treated samples (n = 4/group) were homogenized and RNA isolated. Expression of AR-target genes was measured by real-time PCR and normalized to GAPDH expression. Data are represented as fold change from vehicle-treated samples.
Collectively, these results suggest that the AR mediates its anti-proliferative effects by indirectly inhibiting the growth promoting function of ER.
Co-localization of AR and ER in Luminal B Breast Cancer Cells
In order to determine the nuclear reactivity of AR and ER and potential co-localization in breast cancer specimens, immunohistochemistry was performed in several luminal B breast cancer specimens. Because this subtype is the faster growing of the two luminal types, luminal B subtype was chosen over luminal A. Relatively abundant nuclear immunoreactivity of both AR and ER was detected in all the breast cancer cases examined in this study (Figure S9 representative images). In addition, several cases examined had moderate levels of cytoplasmic immunoreactivity for both markers. As levels of immunoreactivity of both markers exceeded 60% in all the cases examined, a relatively high percentage of carcinoma cells were immunohistochemically positive for both ER and AR. The patterns of immunolocalization were also similar between these two markers above. Overall, the number of carcinoma cells immunohistochemically positive for AR in any one case exceeded those which were positive for ER. However, the semi-quantitative analysis of immunohistochemistry did preclude us from being able to state conclusively that AR was expressed at greater levels than ERα. It was also possible to detect the cases in which AR immunoreactivity was weaker or absent while ER immunoreactivity present, although less frequent.
Protein-Pathway Activation Mapping Shows Inhibition of Oncogenic and Induction of Tumor-Suppressor Protein Phosphorylation by Enobosarm
To determine the effect of enobosarm on protein signaling, we performed RPPA-based analysis in HCI-13 tumors treated with vehicle or enobosarm to measure the phosphorylation and total levels of protein in key signaling pathways known to be involved in tumorigenesis and metastatic progression. Enobosarm inhibited the phosphorylation of various oncogenic proteins such as pERK, PKC ʐ, RSK3, Ezrin, BCL2, ELF4G, and ER (Figure S10). Enobosarm also inhibited the expression of proliferation marker Ki67. Alternatively, enobosarm increased the phosphorylation of tumor suppressor proteins such as p53, p27, and ACC. AR phosphorylation was also increased in enobosarm-treated samples. Enobosarm also increased the phosphorylation of STAT5, which could be a tumor suppressor or an oncogene depending on the context (Figure S10). These results demonstrate that activating the AR with an agonist promotes the alteration of appropriate pathways that facilitate tumor growth inhibition. These results were confirmed using Western blots for phospho Ser81 AR and phospho Ser118 ER (Figure S10 lower blots).
Discussion
Although breast cancer was often successfully treated with steroidal androgens until the development of ER-targeted therapies, unwanted virilizing effects have limited their clinical use. SARMs that tissue-selectively activate the AR are currently being developed for multiple indications, including muscle wasting, osteoporosis, muscular dystrophy, breast cancer, and urinary incontinence. Enobosarm, which has been tested in the clinic (Crawford et al., 2016, Dobs et al., 2013), has demonstrated pharmacologic effects consistent with selective AR targeting. In clinical trials, enobosarm increased lean body mass by more than a kilogram and performance of various muscles (Dobs et al., 2013) and has not shown virilizing effects at the 3 mg dose. In addition, enobosarm treatment has demonstrated clinical benefit in women with ER-positive breast cancer who previously responded to hormonal therapies (San Antonio breast cancer symposium, 2015) (Overmoyer, 2015). Women who were extensively treated with ER-targeted therapies could be benefited by these secondary bone and muscle building effects of AR agonists such as the SARMs.
The results obtained in this study are encouraging. A tumor (HCI-13) that relapsed and continued to grow in the presence of a range of therapeutics was inhibited by AR agonists. This result and the result from ex vivo studies support the use of AR agonists even after the tumors relapse from other treatment options. The mutation represented in HCI-13, Y537S, is one of the common mutants found in the clinic (Katzenellenbogen et al., 2018).
The unique property of inhibiting the ER function by activating the AR demonstrates the complex interaction between various nuclear receptors and their associated proteins. The microarray results indicate that the inhibition of ER pathway by an AR agonist could provide greater benefit to patients in whom the oncogenic pathway is constitutively active. This beneficial effect is further enhanced by the increase in the phosphorylation of various tumor suppressors and inhibition of the phosphorylation of oncogenes.
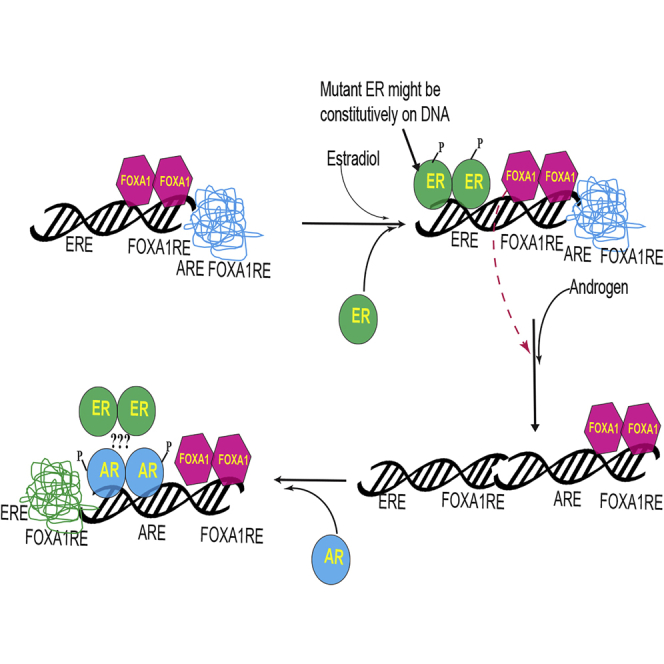
The ChIP-Seq results suggest that the AR, ER, and FOXA1 colocalized in the presence of AR agonist and shift from ER cistrome to AR cistrome. As FOXA1 pioneering transcription factor is important for the function of both AR and ER and has overlapping binding sites with ARE and ERE (Carroll et al., 2005, Wang et al., 2007), it is possible that the activated AR might sequester FOXA1 from the FOXA1REs adjacent to the EREs to open up the nucleosome and facilitate its binding to ARE. Based on these results, we propose a model (Figure 8) wherein the absence of androgen or estrogen, the DNA is in a condensed conformation where the EREs, AREs, and FOXA1REs are unbound. FOXA1 occupies FOXA1REs in the absence of estrogens or androgens, and upon estradiol exposure, FOXA1 opens the chromatin and binds to its response elements adjacent to the EREs. This facilitates the ER to bind to its response elements and promote the transcription of the target genes. While this is ongoing, AR is not bound to AREs. However, when an AR agonist binds to the AR, it changes conformation and proceeds to bind to AREs that may or may not be near pre-existing FOXA1 sites. Strikingly, ER binding is significantly reduced at its original sites and is significantly gained at new sites highly overlapping with the AR gained sites. The mechanism for this reprogramming is not clear at the molecular level, but likely involves FOXA1, which is lost at many of the original ER-binding sites and gained at many of the new AR-binding sites and so might be due to squelching. Future studies will need to determine whether the ER's interaction with AREs is due to a complex formed with AR or through FOXA1. The stoichiometry and interaction sites of ER and AR in these AR agonist-induced complexes also remains uncertain as is the role that squelching of FOXA1 or other co-factors may play in this nuclear hormone receptor reprogramming.
Figure 8.

Model Depicting the Regulation of ER Function by AR Ligands
The model shows the redistribution of transcription factors AR, ER, and FOXA1 when AR is activated by an agonist. AR, androgen receptor; ER, estrogen receptor; ARE, androgen response element; ERE, estrogen response element; FOXA1RE, FOXA1 response element.
Clinical trial results with enobosarm (presented in San Antonio Breast Cancer Symposium in 2015 and 2016) already demonstrated a favorable outcome in patients who have relapsed from hormonal therapies. Twenty-two patients with breast cancer were treated with 9 mg of enobosarm. Of the 17 AR-positive patients, 35.3% achieved clinical benefit after six months of treatment with 9 mg enobosarm administered once daily. These results will be published in the future.
Overall, these mechanism-based preclinical and translational studies support the use of an AR agonist to treat refractory hormone receptor-positive breast cancer. Various advantages that an AR agonist will confer include tumor growth inhibition, muscle mass increase, increase in bone mass, better cognition, improvement in sexual function, and reduction in the incidence of urinary incontinence (Ho et al., 2004, Kearbey et al., 2009, Ponnusamy et al., 2017a, Ponnusamy et al., 2017b). Tissue-selective AR agonism might offer an alternative hormonal approach for hormone receptor-positive breast cancers.
Limitations of the Study
Although the results support the mechanism of action and the tumor suppressive role of AR in ER-positive breast cancer, we recognize several limitations in this work. First, the ChIP-Seq and mechanistic studies were conducted in one PDX and with one SARM. Future studies need to be conducted in multiple ER-positive PDXs using different AR agonists to validate the findings. Secondly, because the concept was tested in one wild-type ER-positive and one mutant ER-positive PDX, the results need to be validated in additional PDXs of each type. Other ER-LBD mutants need to be included in such studies to deduce a comprehensive conclusion. Finally, the role of AR antagonists needs to be evaluated in detail using PDX models to understand why under certain conditions, especially in cellular models, they inhibit ER-positive breast cancer growth.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank UTHSC molecular resource center for their help with microarray experiments. The authors thank Dr. Dejian Ma, department of pharmaceutical sciences, UTHSC Mass Spectrometry core for his help with the LC-MS/MS measurement of drug concentration. The studies were partially funded by a research grant from GTx, Inc.
Author Contributions
SP, SA, and TT performed all animal experiments, ex vivo sponge cultures, RNA isolation and gene expression studies, and ChIP-PCR assay. RN conceived and designed the experiments and managed the overall project. LSS managed the clinical aspects of the project with the help of BG, MDF, FEP, MPB, RO, and REF. HS and KMM designed the IHC part, which were executed by FG. HL and MB planned and managed the ChIP-Seq experiments, whereas AFT and PKR performed the experiments. XQ, YX, and DLJ provided bioinformatics and statistical support. EFP planned the RPPA studies, whereas MP performed the experiments.
Declaration of Interests
RN is a consultant to GTx, Inc., Memphis. This role has not influenced the conduct of experiments and the interpretation of results.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.038.
Data and Code Availability
Accession number for the sequencing data from public deposition is GSE128018. Microarray data was deposited in GEO and the accession number is GSE126318.
Supplemental Information
References
- Adair F.E., Herrmann J.B. The use of testosterone propionate in the treatment of advanced carcinoma of the breast. Ann. Surg. 1946;123:1023–1035. [PubMed] [Google Scholar]
- Aleskandarany M.A., Abduljabbar R., Ashankyty I., Elmouna A., Jerjees D., Ali S., Buluwela L., Diez-Rodriguez M., Caldas C., Green A.R. Prognostic significance of androgen receptor expression in invasive breast cancer: transcriptomic and protein expression analysis. Breast Cancer Res. Treat. 2016;159:215–227. doi: 10.1007/s10549-016-3934-5. [DOI] [PubMed] [Google Scholar]
- Allard J., Li K., Lopez X.M., Blanchard S., Barbot P., Rorive S., Decaestecker C., Pochet R., Bohl D., Lepore A.C. Immunohistochemical toolkit for tracking and quantifying xenotransplanted human stem cells. Regen. Med. 2014;9:437–452. doi: 10.2217/rme.14.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J., Poulsen H.S. Immunohistochemical estrogen receptor determination in paraffin-embedded tissue. Prediction of response to hormonal treatment in advanced breast cancer. Cancer. 1989;64:1901–1908. doi: 10.1002/1097-0142(19891101)64:9<1901::aid-cncr2820640924>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Balbas M.D., Evans M.J., Hosfield D.J., Wongvipat J., Arora V.K., Watson P.A., Chen Y., Greene G.L., Shen Y., Sawyers C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton V.N., Christenson J.L., Gordon M.A., Greene L.I., Rogers T.J., Butterfield K., Babbs B., Spoelstra N.S., D'Amato N.C., Elias A. Androgen receptor supports an anchorage-independent, cancer stem cell-like population in triple-negative breast cancer. Cancer Res. 2017;77:3455–3466. doi: 10.1158/0008-5472.CAN-16-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Bronte G., Rocca A., Ravaioli S., Puccetti M., Tumedei M.M., Scarpi E., Andreis D., Maltoni R., Sarti S., Cecconetto L. Androgen receptor in advanced breast cancer: is it useful to predict the efficacy of anti-estrogen therapy? BMC Cancer. 2018;18:348. doi: 10.1186/s12885-018-4239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Xiang G., Liu F., Xu C., Liu J., Meng Q., Lyu S., Wang S., Niu Y. A high AR: ERalpha or PDEF: ERalpha ratio predicts a sub-optimal response to tamoxifen therapy in ERalpha-positive breast cancer. Cancer Chemother. Pharmacol. 2019;84:609–620. doi: 10.1007/s00280-019-03891-6. [DOI] [PubMed] [Google Scholar]
- Carroll J.S., Liu X.S., Brodsky A.S., Li W., Meyer C.A., Szary A.J., Eeckhoute J., Shao W., Hestermann E.V., Geistlinger T.R. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Chandarlapaty S., Chen D., He W., Sung P., Samoila A., You D., Bhatt T., Patel P., Voi M., Gnant M. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016;2:1310–1315. doi: 10.1001/jamaoncol.2016.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciupek A., Rechoum Y., Gu G., Gelsomino L., Beyer A.R., Brusco L., Covington K.R., Tsimelzon A., Fuqua S.A. Androgen receptor promotes tamoxifen agonist activity by activation of EGFR in ERalpha-positive breast cancer. Breast Cancer Res. Treat. 2015;154:225–237. doi: 10.1007/s10549-015-3609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane D.R., Bernales S., Jacobsen B.M., Cittelly D.M., Howe E.N., D'Amato N.C., Spoelstra N.S., Edgerton S.M., Jean A. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014;16:R7. doi: 10.1186/bcr3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins L.C., Cole K.S., Marotti J.D., Hu R., Schnitt S.J., Tamimi R.M. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod. Pathol. 2011;24:924–931. doi: 10.1038/modpathol.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford J., Prado C.M., Johnston M.A., Gralla R.J., Taylor R.P., Hancock M.L., Dalton J.T. Study design and rationale for the phase 3 clinical development program of enobosarm, a selective androgen receptor modulator, for the prevention and treatment of muscle wasting in cancer patients (POWER trials) Curr. Oncol. Rep. 2016;18:37. doi: 10.1007/s11912-016-0522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creevey L., Bleach R., Madden S.F., Toomey S., Bane F.T., Vareslija D., Hill A.D., Young L.S., McIlroy M. Altered steroid milieu in AI resistant breast cancer facilitates AR mediated gene expression associated with poor response to therapy. Mol. Cancer Ther. 2019;18:1731–1743. doi: 10.1158/1535-7163.MCT-18-0791. [DOI] [PubMed] [Google Scholar]
- D'Amato N.C., Gordon M.A., Babbs B., Spoelstra N.S., Carson Butterfield K.T., Torkko K.C., Phan V.T., Barton V.N., Rogers T.J., Sartorius C.A. Cooperative dynamics of AR and ER activity in breast cancer. Mol. Cancer Res. 2016;14:1054–1067. doi: 10.1158/1541-7786.MCR-16-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton J.T., Barnette K.G., Bohl C.E., Hancock M.L., Rodriguez D., Dodson S.T., Morton R.A., Steiner M.S. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial. J. Cachexia Sarcopenia Muscle. 2011;2:153–161. doi: 10.1007/s13539-011-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton J.T., Mukherjee A., Zhu Z., Kirkovsky L., Miller D.D. Discovery of nonsteroidal androgens. Biochem. Biophys. Res. Commun. 1998;244:1–4. doi: 10.1006/bbrc.1998.8209. [DOI] [PubMed] [Google Scholar]
- Danforth K.N., Eliassen A.H., Tworoger S.S., Missmer S.A., Barbieri R.L., Rosner B.A., Colditz G.A., Hankinson S.E. The association of plasma androgen levels with breast, ovarian and endometrial cancer risk factors among postmenopausal women. Int. J. Cancer. 2010;126:199–207. doi: 10.1002/ijc.24709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Amicis F., Thirugnansampanthan J., Cui Y., Selever J., Beyer A., Parra I., Weigel N.L., Herynk M.H., Tsimelzon A., Lewis M.T. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010;121:1–11. doi: 10.1007/s10549-009-0436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose Y.S., Wang G., Lin Y.C., Bernard P.S., Buys S.S., Ebbert M.T., Factor R., Matsen C., Milash B.A., Nelson E. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011;17:1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobs A.S., Boccia R.V., Croot C.C., Gabrail N.Y., Dalton J.T., Hancock M.L., Johnston M.A., Steiner M.S. Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol. 2013;14:335–345. doi: 10.1016/S1470-2045(13)70055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists' Collaborative Group. Dowsett M., Forbes J.F., Bradley R., Ingle J., Aihara T., Bliss J., Boccardo F., Coates A., Coombes R.C. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet. 2015;386:1341–1352. doi: 10.1016/S0140-6736(15)61074-1. [DOI] [PubMed] [Google Scholar]
- Eastell R., Adams J.E., Coleman R.E., Howell A., Hannon R.A., Cuzick J., Mackey J.R., Beckmann M.W., Clack G. Effect of anastrozole on bone mineral density: 5-year results from the anastrozole, tamoxifen, alone or in combination trial 18233230. J. Clin. Oncol. 2008;26:1051–1057. doi: 10.1200/JCO.2007.11.0726. [DOI] [PubMed] [Google Scholar]
- Fuqua S.A., Chamness G.C., McGuire W.L. Estrogen receptor mutations in breast cancer. J. Cell Biochem. 1993;51:135–139. doi: 10.1002/jcb.240510204. [DOI] [PubMed] [Google Scholar]
- Garay J.P., Park B.H. Androgen receptor as a targeted therapy for breast cancer. Am. J. Cancer Res. 2012;2:434–445. [PMC free article] [PubMed] [Google Scholar]
- Gordan G.S., Graham W.P., 3rd, Goldman L., Papac R., Sheline G.E., Vaeth J., Witt J. Hormonal treatment of disseminated cancer of the female breast. Calif. Med. 1963;98:189–194. [PMC free article] [PubMed] [Google Scholar]
- Guo S., Zhang C., Bratton M., Mottamal M., Liu J., Ma P., Zheng S., Zhong Q., Yang L., Wiese T.E. ZB716, a steroidal selective estrogen receptor degrader (SERD), is orally efficacious in blocking tumor growth in mouse xenograft models. Oncotarget. 2018;9:6924–6937. doi: 10.18632/oncotarget.24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T., Miyazaki J., Araki H., Yamaoka M., Kanzaki N., Kusaka M., Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–153. [PubMed] [Google Scholar]
- Heise E., Gorlich M. Estradiol receptor activity in human breast cancer, disease free interval and survival time. Anticancer Res. 1982;2:33–36. [PubMed] [Google Scholar]
- Ho M.H., Bhatia N.N., Bhasin S. Anabolic effects of androgens on muscles of female pelvic floor and lower urinary tract. Curr. Opin. Obstet. Gynecol. 2004;16:405–409. doi: 10.1097/00001703-200410000-00009. [DOI] [PubMed] [Google Scholar]
- https://finance.yahoo.com/news/gtx-announces-top-line-results-120000738.html. 2018. https://finance.yahoo.com/news/gtx-announces-top-line-results-120000738.html
- Hu R., Dawood S., Holmes M.D., Collins L.C., Schnitt S.J., Cole K., Marotti J.D., Hankinson S.E., Colditz G.A., Tamimi R.M. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin. Cancer Res. 2011;17:1867–1874. doi: 10.1158/1078-0432.CCR-10-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R., Bergholz J.S., Pun M., Cornwell M., Liu W., Nardone A., Xiao T., Li W., Qiu X., Buchwalter G. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. 2018;33:173–186.e5. doi: 10.1016/j.ccell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Penning T.M. Steroid 5alpha-reductases and 3alpha-hydroxysteroid dehydrogenases: key enzymes in androgen metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2001;15:79–94. doi: 10.1053/beem.2001.0120. [DOI] [PubMed] [Google Scholar]
- Kaaks R., Berrino F., Key T., Rinaldi S., Dossus L., Biessy C., Secreto G., Amiano P., Bingham S., Boeing H. Serum sex steroids in premenopausal women and breast cancer risk within the European Prospective Investigation into Cancer and Nutrition (EPIC) J. Natl. Cancer Inst. 2005;97:755–765. doi: 10.1093/jnci/dji132. [DOI] [PubMed] [Google Scholar]
- Kandouz M., Lombet A., Perrot J.Y., Jacob D., Carvajal S., Kazem A., Rostene W., Therwath A., Gompel A. Proapoptotic effects of antiestrogens, progestins and androgen in breast cancer cells. J. Steroid Biochem. Mol. Biol. 1999;69:463–471. doi: 10.1016/s0960-0760(99)00069-2. [DOI] [PubMed] [Google Scholar]
- Karnik P.S., Kulkarni S., Liu X.P., Budd G.T., Bukowski R.M. Estrogen receptor mutations in tamoxifen-resistant breast cancer. Cancer Res. 1994;54:349–353. [PubMed] [Google Scholar]
- Katzenellenbogen J.A., Mayne C.G., Katzenellenbogen B.S., Greene G.L., Chandarlapaty S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer. 2018;18:377–388. doi: 10.1038/s41568-018-0001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearbey J.D., Gao W., Fisher S.J., Wu D., Miller D.D., Dalton J.T. Effects of selective androgen receptor modulator (SARM) treatment in osteopenic female rats. Pharm. Res. 2009;26:2471–2477. doi: 10.1007/s11095-009-9962-7. [DOI] [PubMed] [Google Scholar]
- Kearbey J.D., Gao W., Narayanan R., Fisher S.J., Wu D., Miller D.D., Dalton J.T. Selective Androgen Receptor Modulator (SARM) treatment prevents bone loss and reduces body fat in ovariectomized rats. Pharm. Res. 2007;24:328–335. doi: 10.1007/s11095-006-9152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen J.A., Lane M.V., Sar M., Wilson E.M. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. J. Biol. Chem. 1992;267:968–974. [PubMed] [Google Scholar]
- Kennedy B.J. Fluoxymesterone therapy in advanced breast cancer. N. Engl. J. Med. 1958;259:673–675. doi: 10.1056/NEJM195810022591404. [DOI] [PubMed] [Google Scholar]
- Liao D.J., Dickson R.B. Roles of androgens in the development, growth, and carcinogenesis of the mammary gland. J. Steroid Biochem. Mol. Biol. 2002;80:175–189. doi: 10.1016/s0960-0760(01)00185-6. [DOI] [PubMed] [Google Scholar]
- Magnani L., Frige G., Gadaleta R.M., Corleone G., Fabris S., Kempe H., Verschure P.J., Barozzi I., Vircillo V., Hong S.P. Acquired CYP19A1 amplification is an early specific mechanism of aromatase inhibitor resistance in ERalpha metastatic breast cancer. Nat. Genet. 2017;49:444–450. doi: 10.1038/ng.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maykel J., Liu J.H., Li H., Shultz L.D., Greiner D.L., Houghton J. NOD-scidIl2rg (tm1Wjl) and NOD-Rag1 (null) Il2rg (tm1Wjl) : a model for stromal cell-tumor cell interaction for human colon cancer. Dig. Dis. Sci. 2014;59:1169–1179. doi: 10.1007/s10620-014-3168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove E.A., Sutherland R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- Narayanan R., Ahn S., Cheney M.D., Yepuru M., Miller D.D., Steiner M.S., Dalton J.T. Selective androgen receptor modulators (SARMs) negatively regulate triple-negative breast cancer growth and epithelial:mesenchymal stem cell signaling. PLoS One. 2014;9:e103202. doi: 10.1371/journal.pone.0103202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R., Coss C.C., Yepuru M., Kearbey J.D., Miller D.D., Dalton J.T. Steroidal androgens and nonsteroidal, tissue-selective androgen receptor modulator, S-22, regulate androgen receptor function through distinct genomic and nongenomic signaling pathways. Mol. Endocrinol. 2008;22:2448–2465. doi: 10.1210/me.2008-0160. [DOI] [PubMed] [Google Scholar]
- Narita D., Raica M., Suciu C., Cimpean A., Anghel A. Immunohistochemical expression of androgen receptor and prostate-specific antigen in breast cancer. Folia Histochem. Cytobiol. 2006;44:165–172. [PubMed] [Google Scholar]
- Neifeld J.P., Lawrence W., Jr., Brown P.W., Banks W.L., Terz J.J. Estrogen receptors in primary breast cancer. Arch. Surg. 1982;117:753–757. doi: 10.1001/archsurg.1982.01380300001001. [DOI] [PubMed] [Google Scholar]
- Niemeier L.A., Dabbs D.J., Beriwal S., Striebel J.M., Bhargava R. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod. Pathol. 2010;23:205–212. doi: 10.1038/modpathol.2009.159. [DOI] [PubMed] [Google Scholar]
- Oliveira A.G., Coelho P.H., Guedes F.D., Mahecha G.A., Hess R.A., Oliveira C.A. 5alpha-Androstane-3beta,17beta-diol (3beta-diol), an estrogenic metabolite of 5alpha-dihydrotestosterone, is a potent modulator of estrogen receptor ERbeta expression in the ventral prostrate of adult rats. Steroids. 2007;72:914–922. doi: 10.1016/j.steroids.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Overmoyer, B. (2015). Phase 2 open label, multinational, randomized, parallel design study investigating the efficacy and safety of GTx-024 on metastatic (MET) or locally advanced (LA) ER+/AR+ breast cancer (BC) in postmenopausal (PM) women. Paper presented at: San Antonio Breast Cancer Symposium OT2-01-06 (San Antonio).
- Park B.Y., Grisham R.N., den Hollander B., Thapi D., Berman T., de Stanchina E., Zhou Q., Iyer G., Aghajanian C., Spriggs D.R. Tumor inhibition by enzalutamide in a xenograft model of ovarian cancer. Cancer Invest. 2016;34:517–520. doi: 10.1080/07357907.2016.1242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou C.M., Sorlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Pollock J.A., Wardell S.E., Parent A.A., Stagg D.B., Ellison S.J., Alley H.M., Chao C.A., Lawrence S.A., Stice J.P., Spasojevic I. Inhibiting androgen receptor nuclear entry in castration-resistant prostate cancer. Nat. Chem. Biol. 2016;12:795–801. doi: 10.1038/nchembio.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz M.M., Li F., Takeda D.Y., Lenci R., Chonkar A., Chabot M., Cejas P., Vazquez F., Cook J., Shivdasani R.A. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015;47:1346–1351. doi: 10.1038/ng.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy S., Sullivan R.D., Thiyagarajan T., Tillmann H., Getzenberg R.H., Narayanan R. Tissue selective androgen receptor modulators (SARMs) increase pelvic floor muscle mass in ovariectomized mice. J. Cell Biochem. 2017;118:640–646. doi: 10.1002/jcb.25751. [DOI] [PubMed] [Google Scholar]
- Ponnusamy S., Sullivan R.D., You D., Zafar N., He Yang C., Thiyagarajan T., Johnson D.L., Barrett M.L., Koehler N.J., Star M. Androgen receptor agonists increase lean mass, improve cardiopulmonary functions, and extend survival in preclinical models of duchenne muscular dystrophy. Hum. Mol. Genet. 2017;26:2526–2540. doi: 10.1093/hmg/ddx150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin R., Baker D., Labrie F. Androgens inhibit basal and estrogen-induced cell proliferation in the ZR-75-1 human breast cancer cell line. Breast Cancer Res. Treat. 1988;12:213–225. doi: 10.1007/BF01805942. [DOI] [PubMed] [Google Scholar]
- Robertson J.F., Ferrero J.M., Bourgeois H., Kennecke H., de Boer R.H., Jacot W., McGreivy J., Suzuki S., Zhu M., McCaffery I. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 2013;14:228–235. doi: 10.1016/S1470-2045(13)70026-3. [DOI] [PubMed] [Google Scholar]
- Robinson D.R., Wu Y.M., Vats P., Su F., Lonigro R.J., Cao X., Kalyana-Sundaram S., Wang R., Ning Y., Hodges L. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S., Basso M., Strippoli A., Dadduzio V., Cerchiaro E., Barile R., D'Argento E., Cassano A., Schinzari G., Barone C. Hormone receptor status and HER2 expression in primary breast cancer compared with synchronous axillary metastases or recurrent metastatic disease. Clin. Breast Cancer. 2015;15:307–312. doi: 10.1016/j.clbc.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Sasaki T., Koivunen J., Ogino A., Yanagita M., Nikiforow S., Zheng W., Lathan C., Marcoux J.P., Du J., Okuda K. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–6060. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger V.E., Allaj V., Gardner E.E., Poirier J.T., Rudin C.M. Quantitation of murine stroma and selective purification of the human tumor component of patient-derived xenografts for genomic analysis. PLoS One. 2016;11:e0160587. doi: 10.1371/journal.pone.0160587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Sikora M.J., Cooper K.L., Bahreini A., Luthra S., Wang G., Chandran U.R., Davidson N.E., Dabbs D.J., Welm A.L., Oesterreich S. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 2014;74:1463–1474. doi: 10.1158/0008-5472.CAN-13-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J., King R., Hayward J., Rubens R. Estrogen and progesterone receptors: correlation of response rates, site and timing of receptor analysis. Breast Cancer Res. Treat. 1982;2:243–250. doi: 10.1007/BF01806937. [DOI] [PubMed] [Google Scholar]
- Sultana A., Idress R., Naqvi Z.A., Azam I., Khan S., Siddiqui A.A., Lalani E.N. Expression of the androgen receptor, pAkt, and pPTEN in breast cancer and their potential in prognostication. Transl Oncol. 2014 doi: 10.1016/j.tranon.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentler J.J., Tan A.C., Weekes C.D., Jimeno A., Leong S., Pitts T.M., Arcaroli J.J., Messersmith W.A., Eckhardt S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toropainen S., Niskanen E.A., Malinen M., Sutinen P., Kaikkonen M.U., Palvimo J.J. Global analysis of transcription in castration-resistant prostate cancer cells uncovers active enhancers and direct androgen receptor targets. Sci. Rep. 2016;6:33510. doi: 10.1038/srep33510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W., Weir H., Razavi P., Lawson M., Goeppert A.U., Mazzola A.M., Smith A., Wilson J., Morrow C., Wong W.L. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017;7:277–287. doi: 10.1158/2159-8290.CD-15-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Badillo F.E., Templeton A.J., de Gouveia P., Diaz-Padilla I., Bedard P.L., Al-Mubarak M., Seruga B., Tannock I.F., Ocana A., Amir E. Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J. Natl. Cancer Inst. 2014;106:djt319. doi: 10.1093/jnci/djt319. [DOI] [PubMed] [Google Scholar]
- Vogel V.G., Costantino J.P., Wickerham D.L., Cronin W.M., Cecchini R.S., Atkins J.N., Bevers T.B., Fehrenbacher L., Pajon E.R., Jr., Wade J.L., 3rd Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295:2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- Wang Q., Li W., Liu X.S., Carroll J.S., Janne O.A., Keeton E.K., Chinnaiyan A.M., Pienta K.J., Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzel I., Graeser M., Karn T., Schmidt M., Wirtz R., Schutze D., Rausch A., Janicke F., Milde-Langosch K., Muller V. Androgen receptor expression is a predictive marker in chemotherapy-treated patients with endocrine receptor-positive primary breast cancers. J. Cancer Res. Clin. Oncol. 2013;139:809–816. doi: 10.1007/s00432-013-1382-8. [DOI] [PubMed] [Google Scholar]
- Yin D., He Y., Perera M.A., Hong S.S., Marhefka C., Stourman N., Kirkovsky L., Miller D.D., Dalton J.T. Key structural features of nonsteroidal ligands for binding and activation of the androgen receptor. Mol. Pharmacol. 2003;63:211–223. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]
- Zhao J.C., Fong K.W., Jin H.J., Yang Y.A., Kim J., Yu J. FOXA1 acts upstream of GATA2 and AR in hormonal regulation of gene expression. Oncogene. 2016;35:4335–4344. doi: 10.1038/onc.2015.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.X., Lane M.V., Kemppainen J.A., French F.S., Wilson E.M. Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol. Endocrinol. 1995;9:208–218. doi: 10.1210/mend.9.2.7776971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession number for the sequencing data from public deposition is GSE128018. Microarray data was deposited in GEO and the accession number is GSE126318.