

Abstract
Background & Aims
Activating mutation of the KRAS gene is common in some cancers, such as pancreatic cancer, but rare in other cancers. Chronic pancreatitis is a predisposing condition for pancreatic ductal adenocarcinoma (PDAC), but how it synergizes with KRAS mutation is not known.
Methods
We used a mouse model to express an activating mutation of Kras in conjunction with obstruction of the main pancreatic duct to recapitulate a common etiology of human chronic pancreatitis. Because the cell of origin of PDAC is not clear, Kras mutation was introduced into either duct cells or acinar cells.
Results
Although KrasG12D expression in both cell types was protective against damage-associated cell death, chronic pancreatitis induced p53, p21, and growth arrest only in acinar-derived cells. Mutant duct cells did not elevate p53 or p21 expression and exhibited increased proliferation driving the appearance of PDAC over time.
Conclusions
One mechanism by which tissues may be susceptible or resistant to KRASG12D-initiated tumorigenesis is whether they undergo a p53-mediated damage response. In summary, we have uncovered a mechanism by which inflammation and intrinsic cellular programming synergize for the development of PDAC.
Keywords: Cdkn1a, Cell of Origin, Acinar-to-Ductal Metaplasia, Pancreatic Duct Ligation
Abbreviations used in this paper: ADM, acinar-to-ductal metaplasia; CCK, cholecystokinin; CP, chronic pancreatitis; PanIN, pancreatic intraepithelial neoplasm; PDAC, pancreatic ductal adenocarcinoma; PDL, pancreatic duct ligation; SA-βgal, senescence-associated β-galactosidase
Graphical abstract

See editorial on page 645.
Summary.
In a mouse model of chronic obstructive pancreatitis, Kras mutation in pancreatic ducts led to rapid development of pancreatic ductal adenocarcinoma, whereas Kras mutation in acinar cells resulted in growth arrest and no neoplastic changes.
Inflammation plays a pivotal role in cancer initiation and progression in many organs.1 In the pancreas, chronic pancreatitis (CP) is a predisposing condition for pancreatic ductal adenocarcinoma (PDAC),2 the most common and deadly cancer of the pancreas, but the link between CP and PDAC is not known. PDAC is usually diagnosed in late-stage disease, leaving little information about how the cancer originated or progressed.
In older people, the presence of KRAS mutations in some cells of the healthy pancreas is not uncommon,3, 4 yet pancreatic cancer remains a relatively rare disease, suggesting that KRAS mutation alone is not sufficient for carcinogenesis. Mouse models support this hypothesis. In mice, widespread pancreatic expression of the KrasG12D mutation alone beginning during embryogenesis leads to PDAC only after long latency,5 suggesting that other, subsequent events that may be genetic, epigenetic, and/or microenvironmental are required. We found previously that introduction of KrasG12D expression in CK19+ epithelial cells resulted in neoplastic changes principally in the oral cavity, lungs, and stomach, 3 sites in which damage and inflammation are common.6 In this work, we directly test whether damage and inflammation in the mouse pancreas can promote KrasG12D-initiated pancreatic cancer, uncovering a mechanism by which inflammation and intrinsic cellular programming synergize for the development of PDAC.
PDAC is a disease of the exocrine pancreas that is composed of 2 predominant tissues, acini that produce zymogens and ducts that transport zymogens to the common bile duct and thence the duodenum. PDAC lesions express a number of ductal markers and have long been thought to arise from a ductal precursor. However, there is evidence from mouse models that acinar cells may be more susceptible than ductal cells to PDAC when tumor suppressor alleles are mutated concomitantly with an activating Kras mutation.7, 8, 9 This is likely due to the ability of acini to undergo a process of acinar-to-ductal metaplasia (ADM) in which they transdifferentiate into ductal cells in response to damage or growth factor signaling.10, 11, 12 How this etiology relates to the usual pathway of progression of human PDAC is not yet clear.
CP is one of the highest risk factors for human pancreatic cancer,13 but the underlying mechanism remains obscure. Although all etiologies of CP are not known, many are thought to occur via duct obstruction or defects in duct flow.14 Therefore, to determine the role of CP arising from duct impairment in pancreatic cancer initiation and progression, we induced duct obstruction in mice carrying an activating Kras mutation. Because it remains unclear whether PDAC arises from a ductal or an acinar progenitor cell, we investigated both sources in the setting of CP by using lineage tracing and cell type–specific KRASG12D induction. We found that chronic obstructive pancreatitis promotes KRASG12D-initiated pancreatic cancer in duct cells but not in acinar cells. Mechanistically, in the context of duct obstruction, KRASG12D protects both duct and acinar cells from the widespread cell loss that occurs immediately after duct obstruction. Acinar KrasG12D-expressing cells are able to undergo ADM distal to obstruction but up-regulate p53 and its target gene p21, undergoing growth arrest and failing to develop neoplastic changes. In contrast, ductal KrasG12D-expressing cells distal to obstruction lack a p53/p21 response and become proliferative after obstructive CP, leading to a dysplasia-carcinoma sequence of PDAC development.
Results
Obstructive Chronic Pancreatitis Promotes Pancreatic Ductal Adenocarcinoma Development From Ducts but not Acinar Cells Bearing a KrasG12DMutation
Ductal vs acinar origin of PDAC is unclear, but recent experiments in mice suggest that either cell type can give rise to PDAC if both copies of the Trp53 gene as well as 1 copy of the Kras gene are mutated simultaneously.7, 9 Whereas acinar cells developed PDAC when only 1 Trp53 allele was mutated in conjunction with Kras mutation, duct cells required both Trp53 alleles be mutated.7, 9 However, mutation of TP53 is thought to occur late in PDAC progression in humans, making it an unlikely initiating event. Because it is well-established in both humans and mice that Kras mutation is the initiating event in greater than 90% of PDAC, we compared neoplastic potential between acinar and ductal cells of the pancreas when Kras mutation alone was introduced in the setting of chronic obstructive pancreatitis. We introduced KrasG12D expression using the Cre-inducible LSL-KrasG12D allele15 combined with cell type–specific, tamoxifen-inducible CreERT alleles. When recombined, the LSL-KrasG12D allele expresses mutated Kras from the endogenous Kras locus. It is important to note that once the LSL-KrasG12D allele is recombined, all progeny of those cells will carry the activated KrasG12D allele even if they no longer express any Cre or CreERT protein. The Ptf1aCreERTM allele16 was used for inducible expression of KrasG12D in acinar cells, and the CK19CreERT2 allele17 was used for inducible expression of KrasG12D in ductal cells. It is critical to trace the lineage of cells expressing KrasG12D to understand how cancer arises. However, no antibody has thus far been developed that allows detection specifically of KrasG12D mutant protein in tissue sections. One antibody has been reported to distinguish KrasG12D on Western blots but is not recommended for immunohistochemistry by the manufacturer. We tested this antibody on tissue sections and found that it did label metaplastic and dysplastic ducts in mouse pancreas and did not label normal ducts. However, even metaplastic ducts arising in Kras+/+ mice were labeled by this antibody, indicating that it is not specific to mutant KRAS (data not shown). Because antibodies cannot distinguish wild-type from mutant KRAS mice, the R26REYFP reporter allele18 was bred into mice to label cells that had undergone recombination and therefore were likely to express KrasG12D. R26REYFP is an inefficient reporter undergoing recombination at lower rates than other reporters.19 We found that the R26REYFP allele under-reports phenotypes resulting from LSL-KrasG12D recombination in multiple organs6 including the pancreas (Figure 1), in which some pancreatic intraepithelial neoplasm (PanIN) lesions arising in Ptf1aCreERTM LSL-KrasG12D R26REYFP mice treated with tamoxifen are labeled with EYFP and some are not. Because of the apparent higher efficiency of LSL-KrasG12D recombination compared with R26REYFP recombination, we can be confident that most EYFP+ cells will be KRASG12D+, although a number of EYFP– cells will also be KRASG12D+. For this reason, we do not compare EYFP+ and EYFP– cells in the same mouse but rather compare EYFP+ cells in mice with and without the LSL-KrasG12D allele.
Figure 1.

Kras mutant phenotype occurs in EYFP+ and in EYFP– cells in Ptf1aCreERTMLSL-KrasG12DR26REYFPmice, reflecting broader recombination of the LSL-KrasG12Dallele than of the R26REYFPallele. Note the PanIN-like lesions with tall columnar morphology and basal nuclei, some of which are positive for EYFP (brown) and some of which are negative (arrows).
Mice were administered tamoxifen at 8–10 weeks of age for adult onset expression of KrasG12D. Approximately 4 weeks after tamoxifen administration, CP was induced via duct obstruction by using pancreatic duct ligation (PDL). Tamoxifen was administered at a dose shown to allow CreERT-mediated recombination only up to 1 month after administration because it was shown that higher doses resulted in continued CreERT-mediated recombination 2 months or more after injection.20 By waiting 4 weeks and using a low dosage of tamoxifen, there would be no tamoxifen-mediated recombination subsequent to PDL. This is critical because when acinar cells undergo ADM, they become ducts and could express duct-specific CreERT alleles if tamoxifen was still available to mediate recombination. This low dose of tamoxifen resulted in 26.4% ± 4.0% of acinar cells labeled with EYFP in Ptf1aCreERTM LSL-KrasG12D R26REYFP mice and 15.7% ± 2.1% of duct cells labeled in CK19CreERT2 LSL-KrasG12D R26REYFP mice. For PDL, a ligature was placed midway along the main pancreatic duct, allowing the head of the pancreas to remain unaffected while the tail region distal to ligation was subjected to effects of duct obstruction. We have previously shown that PDL results in apoptosis of acini distal to the site of ligation with concomitant inflammation and fibrosis.16, 21 Mice were followed for up to 4 months after PDL, until CK19CreERT2 LSL-KrasG12D mice had to be killed because of development of oral papillomas.
Although PDAC tumors have ductal characteristics, acinar cells have been proposed to be the cell of origin22 through a process of ADM in which acinar cells transdifferentiate into abnormal duct cells.10, 11, 12 To determine whether acinar cells and ductal cells had different propensities to develop pancreatic cancer in the context of obstructive CP, we compared Ptf1aCreERTM LSL-KrasG12D R26REYFP (acinar KrasG12D) and CK19CreERT2 LSL-KrasG12D R26REYFP (ductal KrasG12D) mice with control mice with no KrasG12D expression (Ptf1aCreERTM R26REYFP or CK19CreERT2 R26REYFP mice) (Figure 2). Ten acinar KrasG12D mice and 4 acinar control mice were all analyzed at 4 months after PDL (Table 1). Seven ductal KrasG12D mice and 4 ductal controls were analyzed between 2 and 4 months after PDL, the exact timing depending on oral papilloma development that necessitated euthanasia.
Figure 2.

Obstructive CP promotes tumorigenesis from duct cells but not from acinar cells. Mice were subjected to PDL 1 month after tamoxifen treatment and then followed for up to 4 months. (A–C) Pancreas distal to ligation (tail region) from mice with no Kras mutation (A) or KrasG12D mutation specifically in acinar cells (B) or ductal cells (C) 4 months after PDL. (A’, B’, and C’) show corresponding pancreata proximal to ligation (head region). (D) H&E staining of lesions in CK19CreERT2LSL-KrasG12DR26REYFP mouse 2 months after PDL. Note that no invasion was observed at this time point, but atypia occurred focally. (E and F) EYFP immunostaining CK19CreERT2LSL-KrasG12DR26REYFP mice showing early invasion. (G) Some CK19CreERT2LSL-KrasG12D lesions have high proliferative index as shown by Ki67 immunohistochemistry (G). (H and I) H&E staining of two Sox9-CreERT LSL-KrasG12DR26REYFP distal pancreata showing extensive invasion 8–10 months after PDL. (J–L) H&E staining of liver metastatic lesions from Sox9-CreERT LSL-KrasG12D mouse 10 months after PDL showing low magnification of a large lesion (J), a micrometastasis (K), and high magnification within a different large metastasis. Size bars, 100 μm. met, metastasis; WT, wild-type.
Table 1.
Mice Analyzed 2–10 Months After PDL
| Genotype | KrasG12D expression | Sex | Mo after PDL | Tail region pathology | Islets in tail |
|---|---|---|---|---|---|
| Ptf1aCreERT2R26REYFP | None | f | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2R26REYFP | None | f | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2R26REYFP | None | m | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2R26REYFP | None | m | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | f | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | f | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | f | 4 | No ducts | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | f | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | f | 4 | No ducts | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | f | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | m | 4 | No ducts | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | m | 4 | Normal duct epithelium | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | m | 4 | No ducts | Yes |
| Ptf1aCreERT2LSL-KrasG12DR26REYFP | Acinar | m | 4 | Normal duct epithelium | Yes |
| CK19CreERT2R26REYFP | None | m | 2 | Normal duct epithelium | Yes |
| CK19CreERT2R26REYFP | None | f | 2 | Normal duct epithelium | Yes |
| CK19CreERT2R26REYFP | None | f | 2.5 | Normal duct epithelium | Yes |
| CK19CreERT2R26REYFP | None | f | 4 | Normal duct epithelium | Yes |
| CK19CreERT2LSL-KrasG12DR26REYFP | Duct | f | 2 | Focal atypic ductal hyperplasia | Yes |
| CK19CreERT2LSL-KrasG12DR26REYFP | Duct | f | 2 | Focal atypic ductal hyperplasia | Yes |
| CK19CreERT2LSL-KrasG12DR26REYFP | Duct | m | 2.5 | CIS; focal invasion | Yes |
| CK19CreERT2LSL-KrasG12DR26REYFP | Duct | f | 3 | CIS; focal invasion | Yes |
| CK19CreERT2LSL-KrasG12DR26REYFP | Duct | m | 3 | CIS; focal invasion | Yes |
| CK19CreERT2LSL-KrasG12DR26REYFP | Duct | m | 4 | Early PDAC | Yes |
| aCK19CreERT2LSL-KrasG12DR26REYFP | Duct | m | 4 | Connective tissue only | Noa |
| Sox9-CreERT2 R26REYFP | None | m | 4 | Normal duct epithelium | Yes |
| Sox9-CreERT2 R26REYFP | None | f | 8 | No ducts | Yes |
| Sox9-CreERT2 R26REYFP | None | f | 8 | Normal duct epithelium | Yes |
| Sox9-CreERT2 R26REYFP | None | m | 8 | Normal duct epithelium | Yes |
| Sox9-CreERT2 R26REYFP | None | f | 10 | No ducts | Yes |
| Sox9-CreERT2 R26REYFP | None | f | 10 | Normal duct epithelium | Yes |
| Sox9-CreERT2 LSL-KrasG12DR26REYFP | Duct | m | 4 | Connective tissue only | Noa |
| Sox9-CreERT2 LSL-KrasG12DR26REYFP | Duct | m | 4 | Early invasion | Yes |
| Sox9-CreERT2 LSL-KrasG12DR26REYFP | Duct | f | 8 | Early PDAC | Yes |
| Sox9-CreERT2 LSL-KrasG12DR26REYFP | Duct | f | 8 | PDAC; liver metastasis | Yes |
| Sox9-CreERT2 LSL-KrasG12DR26REYFP | Duct | m | 8 | Early PDAC | Yes |
| Sox9-CreERT2 LSL-KrasG12DR26REYFP | Duct | f | 10 | PDAC | Yes |
| aSox9-CreERT2 LSL-KrasG12DR26REYFP | Duct | f | 8 | Connective tissue only | Noa |
CIS, carcinoma in situ.
Mice that were excluded from study because of loss of all pancreatic parenchyma distal to ligation.
By 4 months after PDL, all 8 mice lacking the LSL-KrasG12D allele completely lost acinar mass but maintained the main pancreatic duct and variable numbers of interlobular and intralobular ducts surrounded by fibrotic and steatotic regions (Figure 2A). In addition, islets and vasculature remained intact distal to ligation (data not shown). The head of the pancreas, proximal to the site of ligation, remained normal (Figure 2A’).
Introduction of KrasG12D expression into acinar cells of 10 mice followed by PDL resulted in a distal phenotype similar to mice bearing wild-type alleles of Kras, with no acinar cells remaining and no dysplastic lesions observed distal to ligation 4 months after PDL (Figure 2B). As in Kras wild-type pancreas, remaining distal ducts maintained a low cuboidal epithelium with no apparent cytologic abnormalities. In the head of the pancreas, ADMs and lesions similar to PanINs, thought to be precursors to PDAC, were abundant in acinar KrasG12D mice by 4 months after PDL, indicating that KrasG12D recombination occurred efficiently (Figure 2B’). Although it is possible that manipulation occurring during surgery contributed to the abundance of PanIN-like lesions proximal to ligation, these lesions also occur in mice that are not subjected to surgery23 (data not shown).
When KrasG12D mutation was introduced into ductal cells of 6 mice, cytologically and architecturally abnormal ductal lesions were found throughout the distal, post-ligation pancreas (Figure 2C) as early as 2 months after tamoxifen. Rare PanIN-like lesions and no ADMs were found in the head of the pancreas of ductal KrasG12D mice (Figure 2C’) in agreement with previous observations in mice without duct ligation or other insult.6 Two mice followed for 2 months after PDL developed focal atypic ductal hyperplasia (Table 1, Figure 2D). All ductal KrasG12D mice that survived to at least 2 months after PDL had cytologic and architectural atypia characteristic of carcinoma in situ in lesions distal to ligation (Figure 2D). Either focally or widespread throughout the distal pancreas, these lesions displayed nuclear hyperchromasia, loss of polarity, and increased nuclear/cytoplasmic ratio. At 2.5–4 months after ligation, varying numbers of lesions showed signs of early invasion with loss of basement membrane integrity and increased stromal cellularity (Figure 2C–F, Table 1). Invasion was evident by the morphology of the lesions and the presence of small groups of tumor cells, and occasional single cells, located near larger dysplastic lesions, consistent with having emerged from those lesions (Figure 2E and F). Many of the benign-appearing lesions were highly proliferative as indicated by Ki67 positivity, whereas regions of carcinoma in situ and early invasion contained sporadic Ki67+ cells (Figure 2G), consistent with invading cells having lower proliferation and highly proliferative cells being less invasive.24, 25 Thus, in the context of duct obstruction, ductal cells but not acinar cells expressing activated KRAS can undergo neoplasia and progression to dysplasia and local invasion.
Because only one CK19CreERT2 LSL-KrasG12D mouse could be followed for 4 months (because of development of oral papillomas), we used a second duct-specific CreERT driver, the Sox9-CreERT2 transgene.23, 26 Sox9-CreERT LSL-KrasG12D R26REYFP mice were followed for 4, 8, or 10 months after PDL. As seen with the CK19CreERT2 driver allele, six Sox9-CreERT2 LSL-KrasG12D mice exhibited complete loss of acinar mass distal to ligation, along with carcinoma in situ and early invasion similar to that seen at up to 4 months in CK19CreERT2 LSL-KrasG12D mice. However, by 8 and 10 months after ligation, lesions were more extensive, and there were larger numbers of invading cells (Figure 2H and I, Table 1). In ductal KrasG12D mice followed for 4–8 months after PDL, 20%–52% of lobules distal to ligation contained invasive PDAC lesions (Table 2). In 1 case followed for 10 months, a large tumor engulfed the entire distal region of the pancreas. In this mouse, multiple metastases and micrometastases were found in the liver. The lack of precursor lesions and multiple lesions within the liver and the morphology of tumors were consistent with metastatic PDAC and were likely not cholangiocarcinomas (Figure 2J–L). EYFP is an inefficient reporter not labeling all KrasG12D-expressing cells, and the large pancreatic tumor and the liver metastases were all negative for EYFP. Although this does not prove that the metastases derived from this large pancreatic tumor, the data are consistent with this link.
Table 2.
Frequency of Invasive PDAC Lesions
| Sample | Cre driver | No. of lobules |
% | Mo after PDL | |
|---|---|---|---|---|---|
| With invasive PDAC | Total quantified | ||||
| 782-2 | CK19 | 0 | 25 | 0 | 2 |
| 782-3 | CK19 | 0 | 15 | 0 | 2 |
| 764-2 | CK19 | 1 | 36 | 2.8 | 2.5 |
| 706-1 | CK19 | 3 | 18 | 12.5 | 3 |
| 791-1 | CK19 | 1 | 17 | 3.03 | 3 |
| 709-9 | CK19 | 6 | 21 | 28.9 | 4 |
| 865-2 | Sox9 | 2 | 12 | 20 | 4 |
| 875-3 | Sox9 | 8 | 35 | 22.9 | 8 |
| 883-2 | Sox9 | 6 | 14 | 52.6 | 8 |
| 886-3 | Sox9 | 3 | 7 | 35.7 | 8 |
| 886-2 | Sox9 | Tumor 1.5 × 1.2 × 0.7 cm | 10 | ||
Intriguingly, few of the CP-induced lesions in ductal KrasG12D mice exhibited the classic morphology of PanINs, the best characterized precursors of PDAC. PanINs are characterized, in part, by columnar mucinous cell morphology, often with papillary architecture.27 In ductal KrasG12D mice subjected to obstructive CP, lesions with increasing degrees of atypia comprised flat epithelial structures with little papillary or mucinous architecture (Figure 2D–I). In humans, well-differentiated PDAC lesions typically contain cuboidal or low columnar, mucinous cells, similar to PanIN precursors (Figure 3A). However, most human PDAC cases are moderately or poorly differentiated (Figure 3B and C), and these lesions with low cuboidal cells containing minimal mucin are morphologically similar to the lesions developing from duct cells in response to CP in our mouse model (Figure 3D–F).
Figure 3.

Comparison of human and mouse PDAC lesions. (A–C) Human PDAC exhibits a range of morphologies that can be broadly classified as well-differentiated (A), moderately differentiated (B), and poorly differentiated (C). (D–F) In the setting of obstructive CP, mouse lesions appear moderately (D and E) or poorly differentiated (F). Arrows, cancer cells.
The microenvironment around the dysplastic lesions in mice bore remarkable similarity to that of human PDAC. Collagen fibrils, visualized in blue by trichrome staining, were disorganized around regions of subepithelial invasion but showed the characteristic well-organized, dense parallel bundles around benign lesions (Figure 4A). Alcian blue was used to detect myxoid stroma around some of the dysplastic and invasive lesions (Figure 4B) as well as to show the low frequency of mucin-producing cells in epithelial lesions. As in human PDAC progression, early precursor lesions had little periostin or αSMA+ myofibroblasts surrounding them, with dense, localized accumulation of both occurring as carcinoma in situ developed (Figure 4C and D). On the other hand, macrophages were widely distributed throughout and showed characteristic morphologies of infiltrating macrophages in regions of edema and elongated morphologies of murine tissue-resident macrophages surrounding epithelial lesions (Figure 4E). As cancer progressed, myofibroblasts became more widely distributed around regions of extensive invasion (Figure 4G and H) as occurs in human PDAC.28 We also observed that fibronectin becomes reorganized into well-organized parallel fibrils at regions of invasion in human PDAC and in prostate cancer.29 In both ductal Kras models, regions of invading cells were aligned with parallel fibronectin fibrils (Figure 4F).
Figure 4.

Duct obstruction promotes PDAC with a microenvironment similar to that seen in human PDAC. All images are distal to ligation. (A) CK19CreERT2LSL-KrasG12D pancreas stained with Gomori’s trichrome, which stains fibrillar collagen blue. Arrowheads, dense, well-organized collagen bundles not in contact with dysplasia, whereas dysplastic and invasive regions are in contact with poorly organized collagen. (B) Alcian blue, pH 2.5, labels myxoid stroma (bracket) as well as a small number of ductal lesions (arrows). (C) Matrix remodeling protein periostin (brown) surrounding dysplastic and invasive lesions but scarce surrounding cytologically normal lesions (upper right corner). (D) Myofibroblasts labeled for αSMA (brown) are concentrated around dysplastic and invasive lesions but not cytologically normal lesions. (E) Macrophages labeled with F4/80 antibody (brown) are widely dispersed throughout distal pancreas. (F) Extracellular matrix protein fibronectin (brown) forming parallel bundles (bracket) around invading cells (arrowheads). (G and H) Macrophages (labeled for F4/80 in red) and myofibroblasts (labeled for αSMA in green) show the shift in distribution of myofibroblasts from cytologically normal lesions (G, lower right) with few surrounding myofibroblasts to atypic lesions with increased myofibroblast accumulation (G, upper left) to widespread distribution in areas of invasion (H), whereas macrophages are broadly distributed throughout. (G’ and H’) show αSMA labeling alone. (G” and H”) show F4/80 alone. Size bars, 100 μm.
KRASG12D Promotes Survival but not Proliferation in Acinar-derived Cells
To understand how CP promotes KRASG12D-initiated neoplasia in ductal cells but not in acinar cells, we examined an earlier time point at which all genotypes were morphologically similar. At 2 weeks after PDL, all pancreata distal to ligation exhibited comparable fibrosis and loss of acinar mass (Figure 5A–F; data not shown). Numerous ADM-like ductal lesions were present throughout this region regardless of genotype, whereas the head region proximal to ligation remained normal. Previous studies have shown that normal acinar cells undergo p53-dependent apoptosis within a week of duct ligation.21 To determine whether KrasG12D mutation protects against this early loss of acinar cells, we compared the relative number of remaining EYFP+ cells at 2 weeks after PDL in mice that were labeled with acinar-specific Ptf1aCreERTM or duct-specific CK19CreERT2, with and without KrasG12D mutation, with EYFP serving as a surrogate for KrasG12D expression as described above. To control for mouse-to-mouse differences in recombination efficiency, we normalized the percentage of EYFP+ cells in the distal affected portion of the pancreas to the percentage of EYFP+ cells in the unaffected head region (Figure 5G and H). The rapid, apoptotic loss of acinar cells without Kras mutation21 is reflected in loss of EYFP+ cells in Ptf1aCreERTM R26REYFP pancreas in which there is a 6-fold reduction (0.172% ± 0.053% tail-to-head labeling) in EYFP-labeled cells distal to ligation compared with the unaffected head region of the pancreas (Figure 5I). However, acinar KrasG12D mice exhibited a 1-to-1 ratio (1.02% ± 0.27% tail-to-head labeling) of labeled cells in the ligated tail compared with the unaffected head region of the pancreas, suggesting that Kras mutation protects acinar cells from undergoing early apoptotic loss (Figure 5I).
Figure 5.

Before morphologic differences, Kras mutation alters the abundance of acinar- and duct-derived cells 2 weeks after duct obstruction. (A and B) Gomori’s trichrome staining showing fibrillar collagen in blue in Ptf1aCreERTMKrasG12D (A) and in CK19CreERT2KrasG12D (B) mice. (C) Percent collagen+ area per genotype showing no statistically significant differences. (D–F) Pancreata from 3 different Ptf1aCreERTMKrasG12D mice showing loss of nearly all amylase staining distal to ligation 2 weeks after PDL. Note positive staining in region proximal to ligation in (D). (G and H) Ptf1aCreERTMLSL-KrasG12DR26REYFP pancreas (G) and CK19CreERT2LSL-KrasG12DR26REYFP pancreas (H) were labeled for the EYFP protein (brown) distal to ligation (tail). (G’ and H’) are corresponding proximal regions (heads) of the pancreata shown in (G) and (H). (I) Quantification of enrichment for EYFP+ (CreERT-activated) cells. Enrichment score is the ratio of the percent of non-endocrine epithelium that is EYFP+ in the tail region to the percent that is EYFP+ in the unaffected head region. (J) Similar to (I), but only ductal epithelium, not acinar epithelium, was quantified. Size bars, 100 μm. *P < .05; **P < .01.
Previous work21 demonstrated that duct cells do not undergo apoptosis after duct ligation. We found that epithelial cells labeled with EYFP expression by CK19CreERT2 but lacking an activating Kras mutation were enriched in the tail of the pancreas compared with the unaffected head region (5.61% ± 1.48% tail-to-head labeling; Figure 5I), but this was likely due to loss of acinar cells and therefore enrichment of duct-derived cells. When only duct cells, as opposed to all non-endocrine epithelium, were quantified, there was a reduction of approximately half of the EYFP+ cells without Kras mutation (0.461% ± 0.0018% tail-to-head labeling; Figure 5J). Introduction of a Kras mutation caused enrichment of duct-derived cells both when comparing total non-endocrine epithelium (17.81% ± 3.19% tail-to-head labeling; Figure 5I) and when comparing only ducts (1.70% ± 0.30% tail-to-head labeling; Figure 5J). Because Kras mutation was able to enrich both acinar- and duct-derived cells as well as inducing ADM during the first 2 weeks after duct ligation, we determined whether KRASG12D had cell type–specific effects subsequent to ADM. Using Ki67 labeling to mark proliferative cells, we found that KrasG12D mutation resulted in higher proliferation in duct-derived cells compared with acinar-derived cells (19.10% ± 0.75% of EYFP+ cells compared with 3.77% ± 1.13%) (Figure 6A–C). Two weeks after PDL, both acinar-derived and duct-derived Kras mutant cells had very low rates of apoptosis as measured by cleaved caspase 3+ cells, and the difference was not statistically significant (Figure 6D–F).
Figure 6.

Duct-derived cells are more highly proliferative than acinar-derived cells in response to Kras mutation distal to duct obstruction. (A–C) Ptf1aCreERTLSL-KrasG12DR26REYFP pancreata (A) and CK19CreERT2LSL-KrasG12DR26REYFP pancreata (B) were labeled for Ki67 (red) and EYFP (green) and quantified 2 weeks after PDL for the percent of EYFP+ cells that were also Ki67+ in the affected (distal) region of each pancreas (C). (A’ and B’) labeling for Ki67 alone. (A” and B”) labeling for EYFP alone. (D–F) Apoptosis in the same pancreata was quantified for the percent of EYFP+ cells that were also positive for cleaved caspase 3. *P < .05; ***P < .001.
As previously reported, wild-type acinar cells express p53 protein in response to duct obstruction, whereas wild-type ductal cells do not.21 Expression of KrasG12D did not change this differential response, with high p53 seen widely throughout acinar-derived epithelium but only low or no p53 detected immunohistologically in duct-derived epithelium (Figure 7A and B). One target of p53 that could explain the difference in proliferation between acinar-derived and duct-derived Kras mutant cells is the cell cycle inhibitor p21. In acinar KrasG12D mice, 13.86% ± 2.64% of EYFP+ cells were p21-positive, whereas only 1.04% ± 0.61% of EYFP+ cells were p21-positive in ductal KrasG12D mice (Figure 7C–E). No p53 or p21 protein was detected in normal acinar or ductal cells proximal to the site of ligation (data not shown). Sox9-CreERT2 LSL-KrasG12D R26REYFP mice had similar phenotypes to CK19CreERT2 LSL-KrasG12D R26REYFP mice with low levels of p21, p53, and cleaved caspase 3 and high levels of Ki67 (Figure 8).
Figure 7.

Mutant Kras induces p53 and p21 in acinar-derived but not duct-derived cells distal to duct obstruction. (A and B) Immunofluorescent labeling for p53 in Ptf1aCreERTLSL-KrasG12D cells (A) and in CK19CreERT2LSL-KrasG12D cells (B). Images were captured at identical gain and offset. (C–E) Ptf1aCreERTLSL-KrasG12DR26REYFP pancreata (C) and CK19CreERT2LSL-KrasG12DR26REYFP pancreata (D) were quantified (E) for the percent of EYFP+ cells that were also p21+ distal to ligation. *P < .05; **P < .01.
Figure 8.
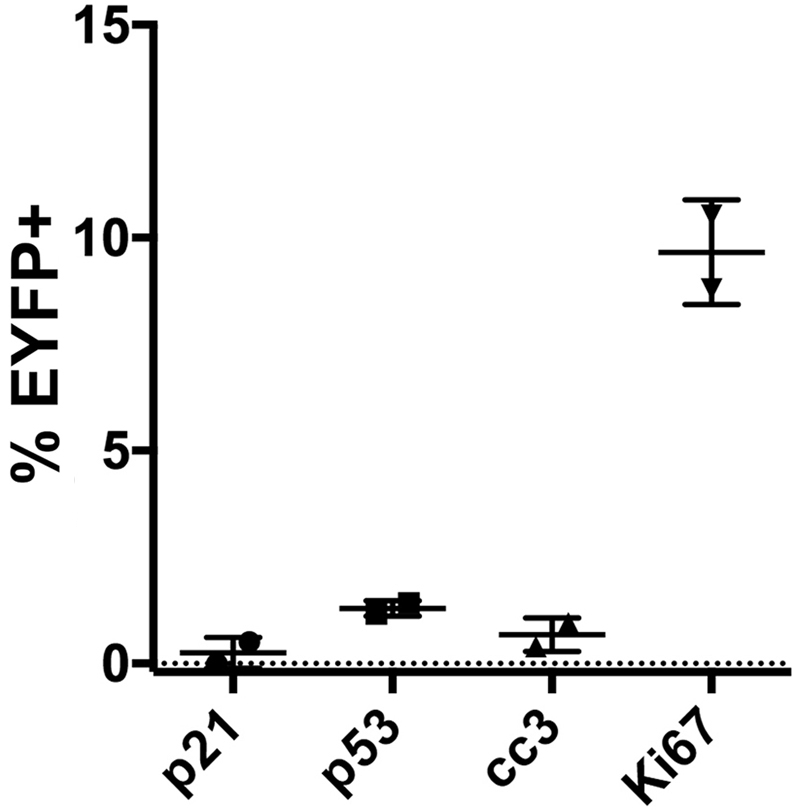
Sox9-CreERT2 LSL-KrasG12DR26REYFPmice phenocopy CK19CreERT2LSL-KrasG12DR26REYFPmice 2 weeks after PDL. Two Sox9-CreERT2 LSL-KrasG12D mice were injected with tamoxifen, and PDL was performed as described. Sections were analyzed distal to ligation 2 weeks after PDL for the indicated markers as described in Figures 6 and 7.
Because Kras mutation and Cdkn1a (encoding p21) expression have been linked to oncogene-induced senescence, we compared senescence-associated β-galactosidase (SA-βgal) activity in acinar-derived and duct-derived Kras mutant cells. No SA-βgal activity was seen in either acinar-derived or duct-derived cells with or without Kras mutation distal to ligation (Figure 9A–C), although islets showed weak positivity. As a positive control, PanIN-like lesions occurring in the head of pancreata in acinar KrasG12D mice exhibited SA-βgal activity (Figure 9D) as published.30, 31
Figure 9.

Kras mutation does not result in oncogene-induced senescence in the setting of obstructive CP. Cryosections of each genotype were stained for acidic βgal activity (blue) as an indicator of oncogene-induced senescence. Tissues shown are CK19CreERT2R26REYFP pancreas (A), CK19CreERT2LSL-KrasG12DR26REYFP pancreas (B), and Ptf1aCreERTLSL-KrasG12DR26REYFP pancreas (C) each at 2 weeks after PDL. (D) PanIN-like lesions in the head of the pancreas of Ptf1aCreERTLSL-KrasG12DR26REYFP mice 4 months after PDL. i, islet. Size bars, 100 μm.
The reduced proliferation of acinar-derived compared with duct-derived KRASG12D+ cells suggests that acinar cells may not give rise to tumors in the setting of CP because the cells are eliminated over time. To test this possibility, we determined whether EYFP+ cells remained 2–4 months after ligation. Indeed, in both Ptfa1CreERTM R26REYFP mice and Ptfa1CreERTM LSL-KrasG12D R26REYFP mice that had undergone tamoxifen injection and PDL, we found that some mice no longer contained EYFP+ acinar-derived cells distal to ligation, whereas all CK19CreERT2 R26REYFP and CK19CreERT2 LSL-KrasG12D R26REYFP mice retained EYFP+ duct-derived cells. However, with or without Kras mutation, acinar-derived cells were retained 4 months after PDL in some mice. In 2 of 4 of the Kras+/+ mice, acinar-derived EYFP+ cells comprised 6.7% and 9.8% of epithelial cells distal to ligation (Figure 10A). In 3 of 5 acinar KrasG12D mice retaining EYFP+ cells, 54.9% ± 8.4% of distal epithelial cells were EYFP+. In ductal control mice, 16.0% ± 2.3% of distal epithelium was EYFP+, and in ductal KrasG12D mice, 52.0% ± 6.4% was EYFP+, indicating an enrichment with mutant KRAS in cells of ductal origin (P < .01). Remaining acinar-derived as well as duct-derived EYFP+ cells were variably proliferative regardless of Kras status (Figure 10B). This may be in response to occurrence of areas of delamination in some distended ducts (Figure 10C). However, p21 was significantly more prevalent in acinar-derived EYFP+ cells (Figure 10D–G).
Figure 10.

Up to 4 months after PDL, many remaining lesions have recombined cells and varying degrees of proliferation. (A) Two to 4 months after PDL, recombined cells as measured by EYFP were still present in lesions from some acinar control and acinar KrasG12D mice and in all duct-derived control and KrasG12D pancreata. (B) Both acinar-derived and duct-derived lesions exhibited varying levels of proliferation. (C) Many dilated lesions of all genotypes exhibited loss of epithelial integrity associated with inflammatory infiltrates distal to ligation. Image shown is Ptf1aCreERTLSL-KrasG12D pancreas 4 months after obstruction. (D) Acinar-derived lesions continued to express p21 at a much higher level than did duct-derived lesions. Squares with white centers correspond to Sox9-CreERT2 LSL-KrasG12D mice, and solid black squares correspond to CK19CreERT2LSL-KrasG12D mice. (E–G) Images showing p21 (red) and EYFP (green) in acinar control (E), acinar KrasG12D (F), and duct KrasG12D (G). p21 alone is shown in corresponding images below (E’–G’). *P < .05; **P < .01; ***P < .001.
Discussion
The work presented here indicates that obstructive CP is a potent promoter of PDAC, suggesting that the local tissue microenvironment plays a major regulatory role in PDAC development. In studies of adult mouse pancreas, expression of KrasG12D from the endogenous Kras locus does not lead to carcinoma unless a tumor suppressor such as Trp53 and/or Fbw7 is also mutated.7, 8, 9 However, with KrasG12D expression in ducts, CP led to neoplastic changes including carcinoma in situ by 2 months. Focal areas of invasion were observed between 2.5 and 3 months, with widespread invasion by 4 months after obstruction. This rapid development of pancreatic cancer supports previous studies that invasion happens at a relatively early stage of PDAC development.32 Furthermore, PDAC lesions developed within a dynamic microenvironment that recapitulated that seen in the development of human PDAC.28 This microenvironment includes a gradual transition in accumulation of myofibroblasts and periostin protein as lesions progress from benign to malignant, whereas macrophages are prevalent throughout this transition.
KRAS is mutated in >90% of human PDAC tumors. Studies of human tissues and mouse models indicate that it acts as a cancer initiator that relies on other events to promote tumor development. We found in previous studies that effects of Kras mutation are very context-dependent in vivo, with tissues most exposed to environmental damage (oral cavity, lungs, and stomach) being much more susceptible to neoplasia than are the less exposed liver, kidney, and pancreas tissues.6 Here we have uncovered one mechanism by which damage and associated inflammation may promote transformation in some cells while preventing transformation in other cells. Obstructive CP induces apoptosis via p53 induction in acinar cells but not duct cells of the pancreas.21 We found that Kras mutation does not block p53 induction in acinar cells after obstructive CP but does block the early loss of acinar cells after duct obstruction, thus bypassing one of the functions of p53. Because loss of acinar cells was shown to result from p53-dependent apoptosis in Kras+/+ mice,21 the KrasG12D-dependent enrichment of acinar cells is likely due to decreased apoptosis. However, we cannot rule out that there is also a transient burst of proliferation during the first 2 weeks after duct obstruction. Despite protecting acinar-derived cells from early loss, induction of p53 and its target gene Cdkn1a, encoding the cell cycle inhibitor p21, correlated with decreased proliferation of acinar-derived cells even though they express KrasG12D. Ductal cells do not undergo p53 or p21 induction after duct obstruction and thus rapidly proliferate in response to KrasG12D expression. This proliferation then allows cell expansion and eventual neoplasia. Thus, one determinant of the ability of the KrasG12D mutation to initiate carcinogenesis may be whether the cells undergo a robust p53/p21 response.
In some mice, reduced proliferation in acinar-derived, Kras-mutated cells was associated with loss of these cells over time, and this may be one of the mechanisms by which acinar cells are less susceptible to carcinogenesis in the setting of obstructive CP. However, some acinar-derived mutant cells escaped this selection and were maintained up to 4 months after PDL. Although many of these cells continue to express p21, some cells become proliferative, perhaps in response to duct dilation and resultant loss of epithelial integrity. It is possible that these remaining mutant cells could, over time, acquire mutation of Trp53 or other tumor suppressor genes that would then lead to carcinogenesis. This would be consistent with data in the field that mutation of Trp53 in conjunction with Kras mutation rapidly leads to acinar-derived PDAC, whereas Kras mutation alone does not.7, 9 Our mouse model of acinar Kras mutation relies on loss of one Ptf1a allele, and this may also contribute to the inability of acinar-derived cells to undergo tumorigenesis. However, work from others indicates that loss of Ptf1a makes acinar cells more susceptible, not less susceptible, to tumorigenesis.33, 34 It is also unlikely that differences in Cre-mediated recombination in our acinar-derived and duct-derived cell models are responsible for the differences in carcinogenesis. Slightly higher percentages of acinar cells underwent Cre-mediated recombination, and there are many more acinar cells than duct cells in the pancreas, yet acinar cells were unable to undergo neoplastic changes. Another caveat to these experiments is that we are inducing CP and PDAC in the tail of the pancreas, whereas humans develop PDAC in the head of the pancreas in 60%–70% of cases.35
Our results and the work of others8, 21, 23 indicate that the 2 principal cells of the exocrine pancreas, acinar cells and duct cells, have variable responses to oncogenic and inflammatory stimuli. In terms of inflammatory responses, most prior work has studied the inflammatory response to cerulein, a cholecystokinin (CCK) analog that binds CCK receptors on mouse acinar cells and supraphysiologically stimulates zymogen secretion, provoking inflammation and ADM that gradually resolve when treatment is stopped.12 In the presence of mutant Kras, these ADM lesions gain a PanIN-like morphology and are maintained as such after cerulein administration has stopped.36 In Kras mutant mouse models, the high occurrence of acinar-derived PanIN-like lesions and low occurrence of duct-derived PanIN-like lesions in the presence of cerulein led to the conclusion that acinar cells are the most likely source of PDAC.23 However, cerulein does not directly target duct cells and does not appear to promote neoplastic events in duct cells.23 In addition, the cerulein-induced inflammation models are likely irrelevant to human disease because human acinar cells lack CCK receptors and are unresponsive to cerulein.37 Our obstructive CP model affects both acinar and ductal cells and models a common etiology of human CP. From this etiology, Kras mutation in duct cells but not in acinar cells was able to induce PDAC. Although introducing a Kras mutation embryonically allows PDAC to develop at >6 months of age in the FVB/N background5 and >10 months in C57BL/6J (data not shown), no other model has shown that adult-onset endogenously controlled expression of KrasG12D without further genetic modification is sufficient for PDAC development. In the setting of obstructive CP, we show that there is a clear difference in response of acinar cells and duct cells to Kras mutation. Whereas the susceptibility to PDAC was confined to cells of ductal origin distal to duct obstruction, PanIN-like lesions developed and appeared to be maintained in the head of the pancreata of acinar KrasG12D mice. Although these lesions occur even without surgical manipulation, we cannot rule out that the inflammation induced by duct ligation had an effect on the timing or abundance of these lesions.
The PDAC lesions developing in the context of obstructive CP did not appear to proceed through the classic PanIN series. Rather than columnar, mucinous precursor cells, benign lesions had low cuboidal cells (“flat” morphology) even as they exhibited increasing cellular atypia. Carcinoma in situ lesions showed extensive dysplasia, and the cancer itself was moderately to poorly differentiated. In humans, PDAC tumors show variable morphologies, some with well-differentiated ductal structures with columnar mucinous cells, whereas others consist of scattered clusters of moderately or poorly differentiated cells that may or may not be mucinous. These different morphologies may reflect different cells of origin. In a recent report, Lee et al9 found that acinar-derived tumors arising from KrasG12D mutation combined with targeted deletion of Trp53 (Ptf1aCreERT LSL-KrasG12D Trp53fl/fl mice) resulted in more frequent mucinous lesions than occurred in Sox9-CreERT LSL-KrasG12D Trp53fl/fl mice. Indeed, it has become appreciated that intraductal papillary mucinous neoplasm, once thought to be a separate and usually benign neoplasm, can be a precursor to PDAC.38 It has recently been suggested that atypical flat lesions, similar to those we see in our mouse model of CP-promoted PDAC, can also be precursors for human PDAC.39, 40 It is plausible that the differing morphologies of PDAC result from different etiologies with different types of precursor lesions.
Materials and Methods
Mice
Ptf1aCreERTM,16 CK19CreERT2,17 Sox9-CreERT2,23 LSL-KrasG12D,15 and R26REYFP 18 mice have been described previously. Cre-mediated recombination was performed at 8–12 weeks of age, with 3 intraperitoneal injections of 2.0 mg tamoxifen, once daily on alternating days (days 1, 3, and 5). One month later, PDL was performed as described,16 suturing around the main pancreatic duct in the body of the pancreas where the parenchyma narrows before again widening. Mice were allowed to recover and were followed for the times indicated. Two mice were excluded from the studies (but shown in Table 1) because of loss of all exocrine and endocrine tissue distal to ligation with only a small amount of connective tissue remaining, suggesting that necrosis may have occurred. All mouse procedures were approved by the Vanderbilt Institutional Animal Care and Use Committee.
Histology
Tissues were fixed in 4% paraformaldehyde, washed, then either cryopreserved or dehydrated and paraffin embedded. Sections were cut at a depth of 5 μm. For immunohistologic analyses, slides were deparaffinized and washed, and antigens were retrieved in 10 mmol/L sodium citrate, pH 6 for all except the GFP antibody, which was retrieved in 100 mmol/L Tris, pH 10. Retrieval was done in a pressure cooker (Cuisinart, Stamford, CT) set on high pressure for 15 minutes but left heating for 1 hour. Tissues were quenched in 3% hydrogen peroxide and then washed and blocked in phosphate-buffered saline + 5% normal goat or donkey serum + 1% bovine serum albumin. Slides were then incubated in the indicated antibody overnight at 4°C, detected with biotin-labeled secondary antibody and avidin-biotin amplification using Vector ABC Elite kits (Vector Laboratories, Burlingame, CA) and 3,3′-diaminobenzidine substrate (Vector Laboratories) according to manufacturer’s recommendation, and then counterstained with hematoxylin (Fisher Scientific, Pittsburgh, PA). Double immunofluorescence was performed by using TSA-plus kits (Perkin Elmer LAS, Boston, MA) according to manufacturer’s protocol except the tyramide reagent was diluted 1:1000 and counterstained with Toto3 (Molecular Probes, Eugene, OR). Antibodies used were rabbit anti-GFP (EYFP) (Novus Biologicals, Centennial, CO), mouse anti-αSMA (clone 1A4; Dako, Carpinteria, CA), rabbit anti-periostin (Abcam, Cambridge, England), rabbit anti-cleaved caspase 3 (Cell Signaling Technology, Boston, MA), rabbit anti-Ki67 (Abcam), rabbit anti-p53 (Novocastra Laboratories, Newcastle upon Tyne, UK), rabbit anti-p21(Abcam), goat anti-amylase (Santa Cruz Technology, Santa Cruz, CA), and rabbit anti-KrasG12D (Cell Signaling). SA-βgal activity on cryosections was done as published.41 Brightfield images were captured on an Axioskop40 microscope using an Axiocam MRC camera (Zeiss, Jena, Germany) except images in Figure 3, which were captured on a BX41 microscope using a U-CAM3 camera (Olympus, Center Valley, PA). Fluorescent images were captured on an LSM 510 META inverted confocal microscope (Zeiss).
Statistical Analyses
Trichrome analysis was performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center. Trichrome images were captured by using a high throughput SCN400 Slide Scanner (Leica Microsystems, Buffalo Grove, IL) and analyzed by the Tissue Image Analysis 2 software (Leica). Graphs were made and analyzed by using Prism (Graphpad, San Diego, CA). Student t tests were used to analyze labeled cell numbers between different genotypes. Numbers of mice analyzed are indicated in tables and by data points in graphs.
Acknowledgments
The authors thank Jamie Adcock for excellent surgical assistance, Joseph Roland for imaging assistance, and Susan Majka for helpful critiques of the manuscript.
Current affiliations: FCP: Department of Surgery, Weill Cornell Medical College, New York, New York; JNK: Department of Internal Medicine-Pediatrics, Loma Linda University, Loma Linda, California; JLK: Department of Cellular and Physiological Sciences, University of British Columbia, Vancouver, British Columbia, Canada.
Footnotes
Author contributions C. Shi and M. K. Washington acquired human tissues and analyzed pathology on all mouse and human tissues. F. C. Pan, J. N. Kim, C. Padmanabhan, C. T. Meyer, and A. L. Means performed experiments. M. Gannon, J. L. Kopp, and M. Sander provided antibody and mouse reagents and unpublished results. R. D. Beauchamp, C. V. Wright, and A. L. Means supervised the research. A.L. Means wrote the manuscript. All authors reviewed the manuscript and provided edits and comments.
Conflicts of interest The authors disclose no conflicts.
Funding Supported by NIH grants R21CA123061 (A.L.M.), P50CA095103 (A.L.M., C.S., M.K.W., C.V.W.), P30DK058404 (C.S., M.K.W.), R01DK105689 (M.G.), F32CA136124 (J.L.K.), P30CA068485 (R.D.B.); JDRF Advanced Postdoctoral Fellowship (J.L.K.); U01DK072473 (CVW), R01DK078803 (M.S.), R21CA194839 (M.S.); and VA Merit award 1I01BX003744-01 (M.G.). Core services used for cell imaging and tissue morphology were supported by Vanderbilt-Ingram Cancer CenterP30CA068485 and the Vanderbilt Digestive Diseases Research CenterP30DK08404.
References
- 1.Elinav E., Nowarski R., Thaiss C.A., Hu B., Jin C., Flavell R.A. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 2.Pinho A.V., Chantrill L., Rooman I. Chronic pancreatitis: a path to pancreatic cancer. Cancer Lett. 2014;345:203–209. doi: 10.1016/j.canlet.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 3.Luttges J., Reinecke-Luthge A., Mollmann B., Menke M.A., Clemens A., Klimpfinger M., Sipos B., Kloppel G. Duct changes and K-ras mutations in the disease-free pancreas: analysis of type, age relation and spatial distribution. Virchows Arch. 1999;435:461–468. doi: 10.1007/s004280050428. [DOI] [PubMed] [Google Scholar]
- 4.Luttges J., Schlehe B., Menke M.A., Vogel I., Henne-Bruns D., Kloppel G. The K-ras mutation pattern in pancreatic ductal adenocarcinoma usually is identical to that in associated normal, hyperplastic, and metaplastic ductal epithelium. Cancer. 1999;85:1703–1710. [PubMed] [Google Scholar]
- 5.Hingorani S.R., Petricoin E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A., Kawaguchi Y., Johann D., Liotta L.A., Crawford H.C., Putt M.E., Jacks T., Wright C.V., Hruban R.H., Lowy A.M., Tuveson D.A. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 6.Ray K.C., Bell K.M., Yan J., Gu G., Chung C.H., Washington M.K., Means A.L. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey J.M., Hendley A.M., Lafaro K.J., Pruski M.A., Jones N.C., Alsina J., Younes M., Maitra A., McAllister F., Iacobuzio-Donahue C.A., Leach S.D. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene. 2016;35:4282–4288. doi: 10.1038/onc.2015.441. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira R.M.M., Sancho R., Messal H.A., Nye E., Spencer-Dene B., Stone R.K., Stamp G., Rosewell I., Quaglia A., Behrens A. Duct- and acinar-derived pancreatic ductal adenocarcinomas show distinct tumor progression and marker expression. Cell Reports. 2017;21:966–978. doi: 10.1016/j.celrep.2017.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee A.Y.L., Dubois C.L., Sarai K., Zarei S., Schaeffer D.F., Sander M., Kopp J.L. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut. 2018;68:487–498. doi: 10.1136/gutjnl-2017-314426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaine S.A., Ray K.C., Anunobi R., Gannon M.A., Washington M.K., Means A.L. Adult pancreatic acinar cells give rise to ducts but not endocrine cells in response to growth factor signaling. Development. 2010;137:2289–2296. doi: 10.1242/dev.048421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Means A.L., Meszoely I.M., Suzuki K., Miyamoto Y., Rustgi A.K., Coffey R.J., Jr., Wright C.V., Stoffers D.A., Leach S.D. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 12.Strobel O., Dor Y., Alsina J., Stirman A., Lauwers G., Trainor A., Castillo C.F., Warshaw A.L., Thayer S.P. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133:1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raimondi S., Lowenfels A.B., Morselli-Labate A.M., Maisonneuve P., Pezzilli R. Pancreatic cancer in chronic pancreatitis: aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Kloppel G., Maillet B. Pathology of chronic pancreatitis. In: Beger H.G., Warshaw A.L., Buchler M.W., Carr-Locke D.L., Neoptolemos J.P., Russell C., Sarr M.G., editors. The Pancreas. Blackwell Science, Ltd; London: 1998. pp. 720–723. [Google Scholar]
- 15.Jackson E.L., Willis N., Mercer K., Bronson R.T., Crowley D., Montoya R., Jacks T., Tuveson D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan F.C., Bankaitis E.D., Boyer D., Xu X., Van de Casteele M., Magnuson M.A., Heimberg H., Wright C.V. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development. 2013;140:751–764. doi: 10.1242/dev.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Means A., Xu Y., Zhao A., Ray K., Gu G. A CK19-CreERT knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis. 2008;46:318–323. doi: 10.1002/dvg.20397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srinivas S., Watanabe T., Lin C.S., William C.M., Tanabe Y., Jessell T.M., Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J., Willet S.G., Bankaitis E.D., Xu Y., Wright C.V., Gu G. Non-parallel recombination limits Cre-LoxP-based reporters as precise indicators of conditional genetic manipulation. Genesis. 2013;51:436–442. doi: 10.1002/dvg.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reinert R.B., Kantz J., Misfeldt A.A., Poffenberger G., Gannon M., Brissova M., Powers A.C. Tamoxifen-induced Cre-loxP recombination is prolonged in pancreatic islets of adult mice. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scoggins C.R., Meszoely I.M., Wada M., Means A.L., Yang L., Leach S.D. p53-dependent acinar cell apoptosis triggers epithelial proliferation in duct-ligated murine pancreas. Am J Physiol Gastrointest Liver Physiol. 2000;279:G827–G836. doi: 10.1152/ajpgi.2000.279.4.G827. [DOI] [PubMed] [Google Scholar]
- 22.Rooman I., Real F.X. Pancreatic ductal adenocarcinoma and acinar cells: a matter of differentiation and development? Gut. 2012;61:449–458. doi: 10.1136/gut.2010.235804. [DOI] [PubMed] [Google Scholar]
- 23.Kopp J.L., von Figura G., Mayes E., Liu F.F., Dubois C.L., Morris JPt, Pan F.C., Akiyama H., Wright C.V., Jensen K., Hebrok M., Sander M. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao C.F., Xie Q., Su Y.L., Koeman J., Khoo S.K., Gustafson M., Knudsen B.S., Hay R., Shinomiya N., Vande Woude G.F. Proliferation and invasion: plasticity in tumor cells. Proc Natl Acad Sci U S A. 2005;102:10528–10533. doi: 10.1073/pnas.0504367102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hecht I., Natan S., Zaritsky A., Levine H., Tsarfaty I., Ben-Jacob E. The motility-proliferation-metabolism interplay during metastatic invasion. Scientific Reports. 2015;5:13538. doi: 10.1038/srep13538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopp J.L., Dubois C.L., Schaffer A.E., Hao E., Shih H.P., Seymour P.A., Ma J., Sander M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138:653–665. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hruban R.H., Takaori K., Klimstra D.S., Adsay N.V., Albores-Saavedra J., Biankin A.V., Biankin S.A., Compton C., Fukushima N., Furukawa T., Goggins M., Kato Y., Kloppel G., Longnecker D.S., Luttges J., Maitra A., Offerhaus G.J., Shimizu M., Yonezawa S. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–987. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 28.Shi C., Washington M.K., Chaturvedi R., Drosos Y., Revetta F.L., Weaver C.J., Buzhardt E., Yull F.E., Blackwell T.S., Sosa-Pineda B., Whitehead R.H., Beauchamp R.D., Wilson K.T., Means A.L. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab Invest. 2014;94:409–421. doi: 10.1038/labinvest.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erdogan B., Ao M., White L.M., Means A.L., Brewer B.M., Yang L., Washington M.K., Shi C., Franco O.E., Weaver A.M., Hayward S.W., Li D., Webb D.J. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216:3799–3816. doi: 10.1083/jcb.201704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carriere C., Gore A.J., Norris A.M., Gunn J.R., Young A.L., Longnecker D.S., Korc M. Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic Kras and impairs senescence in premalignant lesions. Gastroenterology. 2011;141:1091–1101. doi: 10.1053/j.gastro.2011.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guerra C., Schuhmacher A.J., Canamero M., Grippo P.J., Verdaguer L., Perez-Gallego L., Dubus P., Sandgren E.P., Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Rhim A.D., Mirek E.T., Aiello N.M., Maitra A., Bailey J.M., McAllister F., Reichert M., Beatty G.L., Rustgi A.K., Vonderheide R.H., Leach S.D., Stanger B.Z. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jakubison B.L., Schweickert P.G., Moser S.E., Yang Y., Gao H., Scully K., Itkin-Ansari P., Liu Y., Konieczny S.F. Induced PTF1a expression in pancreatic ductal adenocarcinoma cells activates acinar gene networks, reduces tumorigenic properties, and sensitizes cells to gemcitabine treatment. Molecular Oncology. 2018;12:1104–1124. doi: 10.1002/1878-0261.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krah N.M., De La O.J., Swift G.H., Hoang C.Q., Willet S.G., Chen Pan F., Cash G.M., Bronner M.P., Wright C.V., MacDonald R.J., Murtaugh L.C. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. Elife. 2015;4 doi: 10.7554/eLife.07125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kloppel G. Pathology of nonendocrine pancreatic tumors. In: Go V.L.W., editor. The Pancreas: Biology, Pathobiology, and Disease. Raven Press, Ltd; New York: 1993. pp. 871–897. [Google Scholar]
- 36.Morris JPt, Cano D.A., Sekine S., Wang S.C., Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji B., Bi Y., Simeone D., Mortensen R.M., Logsdon C.D. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology. 2001;121:1380–1390. doi: 10.1053/gast.2001.29557. [DOI] [PubMed] [Google Scholar]
- 38.Shi C., Hruban R.H. Intraductal papillary mucinous neoplasm. Hum Pathol. 2012;43:1–16. doi: 10.1016/j.humpath.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 39.Aichler M., Seiler C., Tost M., Siveke J., Mazur P.K., Da Silva-Buttkus P., Bartsch D.K., Langer P., Chiblak S., Durr A., Hofler H., Kloppel G., Muller-Decker K., Brielmeier M., Esposito I. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J Pathol. 2012;226:723–734. doi: 10.1002/path.3017. [DOI] [PubMed] [Google Scholar]
- 40.Esposito I., Konukiewitz B., Schlitter A.M., Kloppel G. Pathology of pancreatic ductal adenocarcinoma: facts, challenges and future developments. World J Gastroenterol. 2014;20:13833–13841. doi: 10.3748/wjg.v20.i38.13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Debacq-Chainiaux F., Erusalimsky J.D., Campisi J., Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nature Protocols. 2009;4:1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]