

Abstract
Clostridium difficile infection (CDI) is an antibiotic-induced microbiota shift disease of the large bowel. While there is a need for narrow-spectrum CDI antibiotics, it is unclear which cellular proteins are appropriate drug targets to specifically inhibit C. difficile. We evaluated the enoyl-acyl carrier protein reductase II (FabK) that catalyzes the final step of bacterial fatty acid biosynthesis. Bioinformatics showed C. difficile uses FabK as its sole enoyl-ACP reductase, unlike several major microbiota species. The essentiality of fabK for C. difficile growth was confirmed by failure to delete this gene using Clostron mutagenesis and by growth inhibition upon gene silencing with CRISPR-interference antisense to fabK transcription or by blocking protein translation. Inhibition of C. difficile’s FASII pathway could not be circumvented by supply of exogenous fatty acids, either during fabK’s gene silencing or upon inhibition of the enzyme with a phenylimidazole-derived inhibitor 1. The inability for fatty acids to bypass FASII inhibition is likely due to the function of transcriptional repressor FapR. Inhibition of FabK also inhibited spore formation, reflecting the enzyme’s role in de novo fatty acid biosynthesis for the formation of spore membrane lipids. Compound 1 did not inhibit growth of key microbiota species. These findings suggest that C. difficile FabK is a druggable target for discovering narrow-spectrum anti-C. difficile drugs to treat CDI but avoid collateral damage to the gut microbiota.
Keywords: Clostridium difficile, narrow-spectrum drug target, enoyl-ACP reductase, CRISPR-interference antisense, phenylimidazole-derived inhibitor
Graphical Abstract
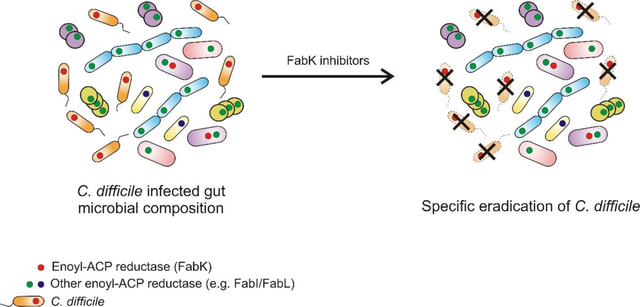
Clostridium difficile is a Gram-positive, spore forming anaerobe that is the leading cause of antibiotic associated diarrhea in developed countries. In the United States, in 2011, there were an estimated ~29,300 deaths from ~453,000 cases of C. difficile infections (CDIs),1 partly due to the emergence and spread of epidemic strains.2 Broad-spectrum antibiotics are the main risk factor for CDI, as they eradicate protective gastrointestinal microbiota species enabling C. difficile to colonize the large bowel.3 Following colonization, C. difficile produces toxins A and B that cause intestinal inflammation and diarrhea.3 Paradoxically, for >30 years the main treatments for CDI have been the antibiotics metronidazole and vancomycin, which are not narrow-spectrum and further disrupt the microbiota during therapy.4 This is thought to be a main contributing factor for why >20% of patients experience recurrent CDI after therapy.5 The recently approved antibiotic fidaxomicin is narrow-spectrum, but higher costs have limited its clinical use and resistance was reported during clinical trials.6–7 Besides antibiotics, other treatments in clinical use or under development include: fecal microbiota transplantation (FMT), inhibition of toxin activity by small molecules and antibodies (e.g. Bezlotoxumab) or inhibitors of germination and sporulation.8 The discontinued development of surotomycin after mixed Phase 3 study findings9–10 emphasize the need to maintain the CDI drug discovery pipeline with antibiotics, which are the standard-of-care for CDI. However, the discovery of narrow-spectrum antibiotics for CDI is largely serendipitous, because protein targets that are specific to C. difficile are either not experimentally confirmed or appear non-essential in genetic studies.11–12 Thus, there is a need to identify and validate narrow-spectrum targets to discover C. difficile selective antibacterials that could avoid killing critical gut microbiota species. For this purpose, we investigated the drug target enoyl-acyl carrier protein (enoyl-ACP) reductase FabK that is involved in de novo synthesis of fatty acids.
Bacterial type II fatty acid synthesis (FASII) is conducted by a series of individual enzymes, which is distinct from the mammalian FASI pathway that is carried out by a large multi-functional protein.13 This enzymatic difference has made the FASII pathway a valid target for discovering selective antibacterials. However, targeting FASII enzymes for antibiotic discovery in Gram-positive pathogens was previously questioned,14 as Streptococcus agalactiae was shown to use host lipids to bypass inhibition of its FASII pathway. More recent research clearly shows that not all bacteria readily use fatty acids to circumvent FASII inhibition, as this appears to be dictated by the type of genetic regulatory mechanism or whether unique lipids are required that cannot be obtained from the host.13, 15–17 For example, in Staphylococcus aureus and Bacillus subtilis, expression of FASII genes is controlled by the transcriptional repressor FapR, which is released from DNA binding by malonyl-CoA, the rate limiting substrate for fatty acid synthesis.13, 17 In the presence of exogenous lipids, the levels of malonyl-CoA remain high, preventing FapR from stopping expression of FASII genes. The continued activity of the pathway exhausts the cellular pool of ACP, thereby inhibiting downstream reactions that also require free ACP to manufacture phospholipids.17 This has led to one paradigm that FASII pathway is druggable in pathogens bearing FapR, as its inhibition is not readily circumvented by fatty acids.13
Various naturally occurring or synthetic molecules inhibit different enzymes in the FASII pathway, resulting in efficacy in murine infection models and in clinical trials in humans.13, 18–19 The enoyl-ACP reductase FabI, a NADPH dependent enzyme, has been the principal target for FASII inhibitors. It catalyzes reduction of trans-2-enoyl-acyl carrier protein (trans-2-enoyl-ACP) to acyl-ACP, which is the final step in fatty acid synthesis. However, FabI is not unique in all bacterial species as there are at least three other enoyl-ACP reductases i.e. FabK, FabL and FabV (Table 1 and Table S1). For example, S. pyogenes and S. pneumoniae carry the isoenzyme FabK as their sole enoyl reductase,20 while Enterococcus faecalis contains FabI along with FabK.21 Unlike many critical gut microbiota species, C. difficile expresses a single isozyme of FabK, which is controlled by FapR. Herein, we show that C. difficile FabK is an attractive target to discover narrow-spectrum antibiotics, whose cellular action may not be compromised by exogenous fatty acids.
Table-1:
Distribution of enoyl-ACP isoenzymes among gut microflora.
| Genus/Organisma | FabI | FabK | Repressor |
|---|---|---|---|
| Bacteriodes | + | ± | FabT |
| Bifidobacteriumb | Not Found | ||
| C. difficile | + | FapR | |
| Enterobacterium | + | ± | FapR |
| E. faecalis | + | + | FabT |
| E. faecium | + | FabT | |
| Faecalibacterium | + | FabT | |
| Lactobacillus | + | ± | FabT |
| Peptostreptococcus | + | FabT | |
| Propionibacterium | + | FabT | |
| Staphylococcus | + | FapR | |
| Streptococcusc | + | FabT |
Not shown: FabV is primarily found in Vibrio species, FabL in Bacillus species.
Yeast-like Fatty Acid synthase
Known to use endogenous fatty acids to bypass FASII
RESULTS
FabK is the sole enoyl-ACP isoenzyme in C. difficile.
Bioinformatic analysis was performed on genomes deposited in KEGG database/Genbank for different gut microorganisms. It revealed that C. difficile carried FabK as its sole enoyl-ACP reductase, in contrast to various important gut microbiota (Table 1 and Table S1). FabK was also the sole enoyl-ACP reductase in the related genus of Peptostreptococcus i.e. C. difficile is now classified in the Peptostreptococcaceae family.22 In contrast, the Gram-negative Bacteroides that makes up a large percent of gut flora23 contained the isoenzyme FabI, either with or without FabK. Similarly, Lactobacillus and Enterobacteriaceae species possessed FabI or both FabI and FabK isoenzymes. Enterococcus faecalis contained both FabI and FabK, while only FabI was found in E. faecium. Both the enoyl-ACP reductases in E. faecalis, as in B. subtilis, are able to compensate for each other.21, 24 Interestingly, Bifidobacterium species do not appear to carry any of the known enoyl-ACP reductase isoenzymes; rather, they possess a large holoenzyme complex whose protein sequence was similar to yeast FAS complex (See Supporting Information). Taken together, the heterologous carriage of enoyl-ACP enzymes among important gut flora, suggests that C. difficile FabK could be a narrow-spectrum target.
Expression of fabK is essential for C. difficile survival.
We next confirmed that fabK is essential for C. difficile growth, using gene disruption techniques. Strains and plasmids used are listed in Supporting Information (Table S2). Firstly, we could not isolate colonies with a disrupted fabK, after three separate attempts of inactivating fabK by insertional mutagenesis with Clostron retargeted to fabK.25 Therefore, growth inhibition was tested by two gene silencing strategies. Firstly, antisense nucleic acid fragments to fabK were cloned into the vector pMSPT under the control of the anhydrotetracycline (ATc) inducible Ptet promoter. The vector pMSPT was constructed from pRPF18526 by including a flanking paired termini nucleotides (see Supporting Information), which is predicted to enhance gene silencing by stabilizing the antisense RNA (asRNA), as reported for Escherichia coli.27 Induction of the asRNA to fabK’s mRNA inhibited growth of C. difficile CD630 (Figure 1A), in a dose-dependent manner at ATc concentrations ≥0.032 μg/mL. In contrast, cells containing the control empty vector survived. Analysis by RT-qPCR revealed that induction of asRNA, by ATc at 0.016 and 0.128 μg/mL, reduced the formation of cDNA from fabK’s mRNA by 4.0 ± 2.1 and 27.75 ± 11.7-fold, respectively (Figure 1B). However, transcript levels were unaffected for the up-stream (plsX, phosphate acyltransferase) and down-stream (fabD, malonyl CoA-ACP transacylase) genes in the operon; and for monocistronic fabZ (3-hydroxyacyl ACP dehydratase) (Figure S1). This suggests that asRNA designed to bind to fabK’s 5’ untranslated region inhibited translation initiation, by blocking the RBS, in a manner similar to natural antisensing.28
Figure-1: Impact of antisense fabK RNA on C. difficile CD630 growth and gene expression. (Panel A & C).

C. difficile CD630 strains carrying empty plasmid or plasmids encoding antisense fabK RNA were analyzed on BHI agar. From overnight cultures 3 μl was spotted on BHI agar plates containing ATc (A) and xylose (C). (Panel B & D) The mRNA levels for fabK was analyzed by RT-qPCR. Cultures were grown to OD600 ≈ 0.3 and antisense RNA expression induced with indicated concentrations of ATc (panel B) or xylose (panel D). Cells were harvested after 1 h of induction and the fold change calculated for differences of mRNA between strains containing the empty vector versus those with antisense cassettes.
To further confirm fabK’s essentiality we constructed a CRISPR interference vector, to silence gene transcription.29 The vector consisted of a constitutively expressed catalytically dead Cas9, from S. pyogenes, which lack endonuclease activity.29 The antisense cassette contained two antisense chimeras, each consisting of a target specific ~20-nucleotide base-pairing region to fabK, the Cas9-binding handle and the S. pyogenes terminator.29 Both guided RNAs (sgRNAs) targeted two different regions within fabK and were expressed from C. difficile’s xylose inducible pXyl promoter.30 Induction of sgRNAs to fabK inhibited growth of C. difficile CD630 (Figure 1C), in a dose dependent manner at xylose concentrations of 2–8% (w/v; g/L). Growth of CD630 containing the empty vector survived. From RT-qPCR analysis, exposure of cells to 1% (w/v; g/L) of xylose for 1 hour reduced transcription by 8.96 ± 4.4-fold (Figure 1D). While the mRNA levels for the upstream plsX were unchanged, the levels for down-stream fabD were reduced 6-fold (Figure S1). Transcription of fabZ was also unchanged (Figure S1). Overall, these results confirm the essentiality of C. difficile fabK for growth. It also demonstrates for the first time, the utility of dCas9 gene silencing for genetic manipulation of C. difficile.
Exogenous supply of fatty acids cannot bypass fabK’s essentiality in C. difficile.
A deciding factor for FabK as potential drug target is whether exogenous fatty acids can or cannot circumvent its inhibition. To test this, we first established the concentrations at which various fatty acids were not inhibitory to C. difficile growth. The test panel included fatty acids common in diets and the intestinal tract, including short chain fatty acids acetic and butyric acids.31 As indicated in Table 2, fatty acids of varying saturation and chain lengths (from C4 to C20) had varying effects on growth at 32 μM to >512 μM. Growth was inhibited by dodecanoic acid, linolenic and arachidonic acid at 32 μM; linoleic acid and myristic acid at 64 μM; and oleic acid at 128 μM. C. difficile grew in all other fatty acids at concentrations up to 512 μM. Fatty acids were combined at sub-inhibitory concentrations to create a fatty acid mix. Growth inhibition by antisenses to fabK was then tested in the presence of sub-inhibitory concentrations of individual or mixed fatty acids. However, exogenous fatty acids could not rescue cell viability, during induction of asRNA to fabK (Figure 2).
Table-2:
Inhibition concentrations of C. difficile CD630 by different fatty acids
| Fatty acid | Abbreviation | MIC (μM) |
|---|---|---|
| Acetic acid | C2:0 | >512 |
| Butyric acid | C4:0 | >512 |
| Octanoic acid | C8:0 | >512 |
| Decanoic acid | C10:0 | >512 |
| Dodecanoic acid | C12:0 | 32 |
| Myristic acid | C14:0 | 64 |
| Palmitic acid | C16:0 | >512 |
| Stearic acid | C18:0 | >512 |
| Oleic acid | C18:1Δ9 | 128 |
| Linoleic acid | C18:2Δ9,12 | 64 |
| Linolenic acid | C18:3Δ9,12,15 | 32 |
| Arachidonic acid | C20:4Δ5,10,11,14 | 32 |
Figure-2: Effect on cell growth in presence of antisense fabK RNA and exogenous fatty acids.

Overnight cultures of C. difficile CD630 harbouring respective plasmids were analyzed on BHI agar plates with respective concentrations of fatty acids. Antisense RNA expression was induced by 0.256 μg/mL ATc (top panel) or 1% (w/v) xylose (bottom panel). Cell viability was tested in presence of individual fatty acids i.e. decanoic acid (256 μM), dodecanoic acid (8 μM), myristic acid (16 μM), palmitic acid (256 μM), stearic acid (256 μM), oleic acid (32 μM), linoleic acid (16 μM), linolenic acid (8 μM) and arachidonic acid (8 μM). The fatty acid mixture was used at sub-inhibitory concentrations of individual fatty acids; in some cases partial growth recovery was observed with the xylose inducible Crispr-interference; with addition of 1% (w/v) xylose, cells were more sensitive to oleic acid and linoleic acid.
C. difficile FabK is druggable target.
We investigated the druggability of C. difficile FabK, in the presence and absence of fatty acids, using previously reported FabK inhibitors (i.e. phenylimidazole derivatives). The phenylimidazole related compounds are the best validated inhibitors of FabK and were being studied by Meiji Seika Kaisha Ltd for streptococcal infections.32–34 Compounds from the phenylimidazole series were reported to: inhibit S. pneumoniae FabK, and not E. coli FabI; and to bind FabK’s active site as shown by co-crystal structures with SpFabK (PDB code 2Z6J) and mapping of resistance mutants.32–34 As the protein sequences of SpFabK (UniProt: Q9FBC5) and CdFabK (UniProt: C9YKB7) are homologous (54% identity and 70% similarity), we first showed that a lead phenylimidazole analog, herein designated as compound 1 (Figure 3A), retained in silico binding interactions in a homology model of CdFabK.33 We therefore synthesized 1 and further evaluated its cellular action. Against S. pyogenes ATCC 19615, 1 had activities of <0.0625 μM in broth, but it was less active against C. difficile CD630 (MICs >64 μM). Since C. difficile expresses efflux pumps,35 1 was retested in the presence of the pump inhibitor verapamil (100 μg/mL), which increased the activity of 1 to 16 μM (Table 3). Addition of palmitic acid (100 μM) or the fatty acid mixture did not alter the activity of 1 against CD630 (MIC= 16 μM). Compound 1 showed activity against C. difficile R20291, a representative of the clinically dominant ribotype 027, even in the absence of verapamil; this suggests heterogeneity in efflux pump activity among C. difficile. The MICs of 1 against R20291 were 8 and 2 μM in the absence and presence of palmitic acid or fatty acid mix, respectively. In contrast, 100 μM of palmitic acid or 50 μM of oleic acid ablated the activity of 1 against S. pyogenes (MIC= 1–4 μM and 4–8 μM or 16–64 and 64–128 fold decreases in activity with palmitic acid or oleic acid, respectively). To further examine the inability of fatty acids to bypass FASII inhibition in C. difficile, we tested cerulenin, an inhibitor of FabF (3-oxoacyl-ACP synthase). Cerulenin’s MIC (16 μM) was not altered by palmitic acid, oleic acid or the fatty acid mix, unlike its activity against S. pyogenes (MIC= 16 μM, which increased to 64 μM upon addition of palmitic acid or oleic acid). Interestingly, 1 did not show antimicrobial activity against various species, including Bacteroides and Lactobacillus species, that carry different homologs of enoyl-ACP reductase isoenzymes (Table-3).
Figure-3: Characterization of compound 1 activity against CdFabK.

(A) Chemical structure of phenylimidazole derivative 1. (B) Homology modeling of 1 in active site of FabK. The comparative model of the C. difficile FabK (green) is shown aligned with the experimental model of S. pneumoniae FabK. Compound 1 is shown docked into the C. difficile FabK active site, closely recapitulating the experimental structure of the bound compound, which is an analog 1. Active site residues are visible within 3 angstroms of the inhibitor, showing 100% identity between the binding sites of the two enzymes. (C) Minimum inhibitory concentrations (MICs) for 1 against C. difficile CD630 overexpressing FabK. MIC’s for C. difficile CD630 harboring respective plasmids were analyzed in BHI broth with respective concentration of compounds. The MIC’s against empty vector control pRPF185 (red bars) and FabK overexpressing pPRPF185-fabK (green bars) are shown for 4 biological replicates. The data included MIC’s that were >128 μM, as reflected by error bars beyond 128. (D) Sigmoidal plot showing inhibition of CdFabK (purple line) and SpFabK (black line) to NADH by 1. The assay was performed with fixed concentration of cofactor and substrate but three-fold dilution of the 1 ranging from 100 to 0.005 and 33 to 0.0017 μM respectively for CdFabK and SpFabK. NADH fluorescence was measured at 340/460 nm and the IC50 values were determined through Hill curve analysis in Graph pad prism 7.
Table-3:
Minimum inhibitory concentrations (MICs)a for compound 1 against various bacteria
| Organism | Enoyl-ACP reductase | MIC (μM) - Palmitic acid + Palmitic acid |
|
|---|---|---|---|
| C. difficile CD630 | FabK | 16 | 16 |
| C. difficile R20291 | FabK | 8 | 2 |
| S. pyogenes ATCC 19615 | FabK | <0.0625 | 4 |
| C. perfringens HM310 | FabV | >64 | >64 |
| B. ovatus HM222 | FabI & FabK | >64 | >64 |
| L. reuteri LTH 5448 | FabI | >64 | >64 |
| S. aureus Newman | FabI | >64 | >64 |
Vancomycin’s MIC against test strains ranged from 0.25–1 μM. With exception of S. pyogenes MICs were done with addition of verapamil (100 μg/mL).
Cellular inhibition of C. difficile FabK by compound 1 was investigated by target overexpression. When C. difficile fabK was placed under the control of the Ptet promoter, its induction in C. difficile CD630 increased the MIC’s of 1 in a dose dependent manner (Figure 3C). In the absence of ATc inducer, the MIC of 1 was 8 – 16 μM, but increased to 64 - >128 μM following addition of ≥0.064 μg/mL ATc. In contrast, against the control strain carrying the empty vector, the MIC of 1 was not significantly altered (i.e. MIC of 8 – 16 μM and 4 – 8 μM, with and without ATc, respectively). As further controls, overexpression of fabK did not affect the activities of metronidazole (MIC = 0.4 μM), vancomycin (MIC = 0.4 μM) and fidaxomicin (MIC <0.2 μM). This further confirms that target overexpression is an applicable strategy for testing the cellular activity of FASII inhibitors; which is also consistent with target overexpression being a mechanism for clinical resistance to the FabI inhibitor, triclosan.36
We confirmed that 1 inhibited the enoyl reductase activity of CdFabK, and did so in a dose dependent manner, with an IC50 of 3.31 ± 0.046 μM (Figure 3D). CdFabK was not inhibited by the FabI inhibitor triclosan (IC50 >100 μM). Under the same assay conditions, 1 was a significantly more potent against FabK from S. pneumoniae (IC50 = 67 ± 0.042 nM; ~49.4 fold better IC50 compared to CdFabK), but was ~ 28-fold higher than previously reported observation.34 Although, enzyme concentrations used in our assay (~50 nM for the monomer enzyme) was similar to that previously reported (2 μg/mL or ~60 nM for the enzyme monomer),34 the reason for this discrepancy is not immediately clear. The resulting Hill coefficients from the IC50 logistic curve fits (see Methods) for 1 against CdFabK and SpFabK were 0.901 (0.761 – 1.064, 95% CI) and 1.137 (0.923 – 1.409, 95% CI), respectively. These values are not significantly greater or lesser than unity (Hill slope of 1.000), suggesting that 1 displayed normal inhibitory behavior without binding cooperativity, aggregation, micellar formation or insolubility. It also suggests that while FabK is a functional dimer, its active sites function independently.33, 37
Inhibition of FASII reduces spore production.
Sporulation by C. difficile is important for endogenous CDI recurrence and dissemination of this pathogen. Studies in B. subtilis show that de novo synthesis of fatty acids and membrane lipids is highly active during sporulation, to form the inner and outer membranes of spores.38–39 To test if depletion of FASII mRNA affects spore production, we analyzed the rate of sporulation in the presence or absence of asRNA to fabK’s mRNA or with empty vector (pMSPT). Induction of asRNA with 0.016 and 0.128 μg/mL of ATc significantly reduced spore formation by ~2-log10, whereas spore formation was unaffected in cells containing the empty vector control (Figure 4A). The spore index (number of spores/total number of viable cells) for asRNA to fabK was comparable to cells expressing an asRNA to spoIIID (Figure 4A), encoding an essential regulator of mother-cell specific gene expression that ensures correct spore formation. We next tested cerulenin at (1× and 4× MIC), showing it reduced spore numbers by ~2 logs (Figure 4B), comparable to the positive control acridine orange (113 μM). In contrast, vancomycin (0.2 and 1 μM) was inactive, as it does not primarily inhibit sporulation.40–41
Figure-4: Effect of antisense fabK on sporulation.

(Panel A, B & C) Sporulation was analyzed over 5 days as the total number of spores/total population (spore forming and vegetative cells). (A) Colonies obtained from BHI agar plate with 0.1% (w/v) taurocholate were inoculated into BHI medium and grown until OD600 ≈ 0.3, at which antisense RNA expression was induced by addition of 0, 0.016 or 0.128 μg/mL of ATc or 113 μM acridine orange (AO). (B) Comparison of inhibition of sporulation by cerulenin (CRN; MIC=16 μM) and vancomycin (VAN; MIC=0.25 μM). DMSO and AO were negative and positive controls.
DISCUSSION
While there is an important need for narrow-spectrum antibacterials to treat CDI, it remains uncertain how to design inhibitors specifically targeting C. difficile, to reduce collateral damage to important gut microbes. We approached identifying a narrow-spectrum drug target for C. difficile by assessing known macromolecular synthetic pathways that contain proven antibacterial drug targets. In this regard, FabK was identified as the sole enoyl-ACP reductase in C. difficile strains. Enoyl-ACP reductase displays extensive sequence and structural diversity among bacteria, with there being four different enoyl-ACP isoenzymes, FabI, FabL, FabV and FabK.42 Out of the four isoenzymes FabI, FabL and FabV are members of the short chain dehydrogenase reductase (SDR) superfamily, and are related by structure and mechanism; whereas FabK is a TIM barrel flavin containing protein with no structural similarity to the members of SDR superfamily.42 While this precludes the discovery of broad-spectrum inhibitors of enoyl-ACP reductases, it is suitable for identifying narrow-spectrum antibacterials. Indeed, phenylimidazole derivatives, such as compound 1, were reported to specifically inhibit FabK with no inhibition of FabI.34 This suggests that structural differences of FabK to SDR enzymes could be exploited to derive selective inhibitors for C. difficile. Challenging this concept is that gut flora such as Bacteroides species carry an SDR enzyme, alone or in combination with FabK. Previously, it was reported that in species carrying two enoyl-ACP reductases, inactivation of one isoenzyme is not sufficient to shunt the FASII pathway.24, 43–44 Although, compound 1 showed anti-C. difficile activity and did not inhibit representative gut species, studies will be required to determine if this is due to: an inability to inhibit both isoenzymes in gut species; or whether poor penetration across the outer membrane prevented cellular activity against Gram-negative Bacteroides. Indeed, poor penetration into Gram-negatives by lipophilic compounds, such as compound 1, may also contribute to species selectivity for future FabK inhibitors. Nonetheless, the lower activity of 1 against CdFabK, compared to SpFabK, suggests that co-crystallization and biophysical analysis of its molecular interactions will be needed to guide optimization of phenylimidazole analogs for C. difficile.
Our attempts to delete CdfabK using Clostron were unsuccessful, while the same technique successfully deleted the non-essential gene for D-proline racemase,11 highlighting fabK’s essentiality. Consequently, growth was inhibited by gene silencing of fabK and by compound 1. These results support findings from Himar1 mutagenesis of C. difficile, where eleven fatty acid biosynthesis genes, including fabK, could not be deleted to produce viable colonies.45 Importantly, sporulation was reduced upon genetic inhibition of fabK and chemical inhibition of the FASII pathway by cerulenin. This might suggest that FASII inhibition could reduce episodes of recurrence that arise from endogenous spores that survive in the colon; it is estimated that half of the recurrent CDI cases occur from endogenous spores, while others are due to reinfection with spores from the environment.46
Another factor favoring FabK as a drug target is its FASII pathway appears to be controlled by the FapR regulator, explaining why exogenous fatty acids could not prevent inhibition by either compound 1 or antisenses to fabK. Conversely, S. pyogenes that bears the FabT repressor, bypassed inhibition with compound 1, when supplied with exogenous fatty acids. Nonetheless, it is unlikely that C. difficile is immune to developing genetic resistance to FabK inhibitors, either by mutations that decrease binding to the target site or allow cells to use of exogenous fatty acids without depleting ACP. This was shown in S. aureus, where mutations to the initiation enzyme FabD decreased its catalytic activity, thereby increasing available free ACP for the synthesis of phospholipids from host fatty acids.47 For FabK inhibitors with future clinical potential, it will be required to determine whether high concentrations of inhibitors in the intestines may prevent the emergence of resistance.
CONCLUSIONS
We provided the first experimental evidence that CdFabK is a potential drug target, showing that its cellular inhibition may not be readily bypassed by fatty acids, including those from dietary and host origins. This may be due to the function of the FapR regulator in C. difficile, which we speculate that like S. aureus, plays a role in preventing bypass of FASII inhibition by exogenous fatty acids. The low homology of CdFabK to other enoyl-ACP reductases may further permit the rational design of species specific inhibitors, afforded by compound 1 as a chemical starting point. However, as FabK moves forward in a structure-based drug discovery process, it will be critical to optimize for molecules that are more potent, soluble, non-absorbed from the intestinal tract and not protein bound to fully leverage inhibition of C. difficile vegetative cells and sporulation.41, 48 We anticipate that such C. difficile specific FabK inhibitors will not only inactivate this pathogen, but also allow the resident microbiota to repopulate the large bowel to prevent recurrent CDI.
MATERIALS AND METHODS
Strains, growth conditions and chemicals.
C. difficile was grown in Brain Heart Infusion (BHI) agar or broth, supplemented with 15 μg/mL thiamphenicol when appropriate. Cultures were grown at 37°C in an anaerobic chamber (Don Whitley A35 anaerobic chamber). Strains and plasmids used in this study are in Tables S2. E. coli was grown in Luria-Bertani (LB) medium, supplemented with required antibiotics when appropriate (ampicillin 100 μg/mL, kanamycin 50 μg/mL or chloramphenicol 35 μg/mL). S. pyogenes was grown in BHI medium supplemented with 10 g/L tryptone and 5 g/L yeast extract (BHITY) at 37 °C in presence of 5% CO2. S. aureus was grown in Muller Hinton (MH) broth. Fatty acids were purchased from: Sigma (decanoic acid, dodecanoic acid, butyrate, octanoic acid, oleic acid and stearic acid), Merck Millipore (arachidonic acid), Acros organics (acetic acid, linoleic acid, myristic acid and palmitic acid) and Chem Impex Inc (linolenic acid).
Construction of anti-sense and target overexpression vectors.
The vector pMSPT was used to express ATc inducible asRNAs to the mRNA of fabK or spo0A. Further details on the design of pMSPT is in the Supporting Information. Vector pMSPT was derived by cloning a paired termini region, synthesized by GenScript in pUC57, into BamHI/SacI sites of pRPF185. Antisense fragments were designed and synthesized to span 50-bases pairs upstream and downstream of the start codon, to mimic naturally occurring bacterial asRNAs that block translation by binding to the ribosome binding site. The antisense fragments were cloned into the SphI/XhoI sites of pMSPT. We constructed the first Crispr-interference vector for C. difficile and provide further details in the Supporting Information. Essentially, we first modified vector PMTL8415149 by introducing C. difficile pXyl promoter30 for xylose inducible gene expression, followed by cloning dcas9 from S. pyogenes under a xylose inducible promoter. The sgRNA to fabK was synthesized by GenScript and cloned into SalI restriction site of the constructed vector pXWxyl-dcas9.
Overexpression of fabK was achieved by cloning the gene into pRPF185. The plasmids and primers used in this study are in Table S2 and Table S3. The plasmid pRPF185 was digested with BamHI/SacI and a similarly digested PCR amplicon of fabK was ligated into the vector. The derived construct, termed pRPF185-fabK, contained the gene with its own ribosomal binding site, expressed from the Ptet promoter.
Insertional inactivation of fabK.
To genetically delete fabK in the CD630 derivative strain JIR8094, we applied Group IIA ClosTron mutagenesis. Essentially, the intron retargeting cassette was designed by Perutka algorithm at www.clostron.com and a 344 bp target region synthesized and cloned into pMTL007C-E2 by DNA2.0 Inc. The resulting plasmid pMTL007C-E2ΔfabK was conjugated into C. difficile JIR8094 and mutagenesis attempted as described.25
Susceptibility tests and effects on growth.
Growth inhibition following induction of asRNA to fabK mRNA was performed by agar dilution method as described previously.50 Essentially, serial dilutions of anhydrotetracycline (ATc) were added to molten BHI agar and spotted with C. difficile to a final inoculum of ~105 CFU/spot, using a Bench top pipettor (Sorenson BioScience Inc). Plates were incubated for 24 h, before recording the lowest concentration of ATc that inhibited growth. Susceptibility to test compounds was determined by microbroth dilution in 96-well microtiter plates under anaerobic growth condition as described.37 For S. pyogenes and S. aureus aerobic growth conditions were maintained. The MIC was determined as the lowest compound concentration inhibiting visible growth at 24 h.
Quantitative Reverse-Transcription PCR Analysis.
RT-qPCR experiments were done as described previously40 with slight modifications. Briefly, C. difficile CD630 was grown in BHI broth to OD600 ~0.2 (T0). Next, ATc was added to final concentrations of 0.016 or 128 μg/mL and cultures incubated for 1 h before adding two volumes of RNA protect (Qiagen) solution and harvesting by centrifugation. Total RNA was extracted from cell pellets using: 700 μl of Qiazol lysis reagent (Qiagen) followed by lysing cells in a Fastprep® cell disrupter for 5 min and then isolating RNA with RNeasy mini kit (Qiagen). From one μg of RNA, complementary DNA (cDNA) was synthesized using qScript cDNA supermix (Quanta biosciences). qRT-PCR was done using qScript SYBR Green RT-qPCR master mix (Quanta biosciences) in Applied Biosystems ViiA7 RT-PCR system. Results were calculated using the 2−ΔΔCt method51, with transcript levels normalized to 16S rRNA. The primers used are listed in Table S3.
Synthesis of FabK inhibitor, phenylimidazole-derivative.
Compound 1 was synthesized by following reported procedures32, 34 with modifications (Scheme S1, see Supporting Information).52–53 The structure was characterized by1H and13C NMR, and HRMS. Purity was determined to be >99% by reverse phase C18-HPLC. General chemistry procedures, synthetic scheme including reagents and conditions, full characterization data of compound 1 and its associated1H and13C NMR spectra, HRMS, and HPLC chromatogram, are provided in Supporting Information.
Computational modelling of CdFabK.z
A composite homology model of the CdFabK enzyme was created using the published structures of the S. pneumoniae (pdb ID 2z6j) and Porphyromonas gingivalis (pdb ID 4IQL) FabK enzymes as templates.33, 37 P. gingivalis loop positions were used for missing loop regions in the S. pneumoniae template. The Prime software package available in the Schrödinger molecular modeling suite (Schrödinger Release 2018–1: Prime, Schrödinger, LLC, New York, NY,USA) was used to build and refine the model.54 The CdFabK sequence from strain 630 was used for the model sequence. Secondary structures were predicted, and sequences were aligned automatically using ClustalW (high sequence identity) with manual adjustment as necessary. The energy-based option was used for model building, wherein residues not matching the templates, including structural discontinuities, are constructed and refined using iterative energy calculations. The resulting CdFabK model is believed to be high-quality due to the high degree of homology to the template structures. Following generation of the CdFabK homology model, compound 1 was docked into the active site using the Glide docking program with XP (extra precision) settings.55–57 The unbiased docking recapitulated the experimental binding pose of the phenylimidazole analog from the SpFabK structure.
Protein expression and purification.
The fabK genes from C. difficile CD630 or S. pneumoniae R6 were cloned within the NdeI/BamHI sites of pET15b. The CdfabK construct was codon optimized, synthesized, and cloned by GenScript (Piscataway, NJ), whereas SpfabK was obtained by PCR using primers in Table-S3. Proteins were overexpressed in E. coli BL21-Gold (DE3) cultivated at 37°C in Terrific Broth with 100 μg/mL ampicillin by transferring 1% (v/v) overnight culture into fresh medium. Cells were grown until OD600 of ~ 0.6 and protein expression induced with 0.1 mM IPTG. To enhance the expression, 0.5 mM flavin mononucleotide (FMN) was added along with IPTG. After 18 h at 18°C, cells were harvested by centrifugation at 18000 × g for 15 min and the pellet resuspended in buffer A (20 mM Tris buffer [pH 7.4] with 300 mM NH4Cl). Cell suspensions were supplemented with 1 mM DTT, 5% glycerol and 10 mM imidazole, 0.5 mg/mL lysozyme, 10 μg/mL DNaseI, 5 mM MgCl2, 0.5% Triton-X 100, 25 mM sucrose (for CdFabK stabilization), and EDTA-free Protease inhibitor. In case of CdFabK, the Tris in buffer A was replaced with 50 mM HEPES buffer (pH 8.0) with 10 μg/mL benzamidine. Cells were lysed by pulsed sonication with 50% amplitude for 8 min, lysates centrifuged at 39000 × g for 15 min and the supernatant was passed through 0.22 μM filters. Proteins were purified by affinity chromatography on HisTrap HP column (GE Life Sciences) and eluted with 500 mM imidazole. Eluted proteins were further purified by gel filtration on Superdex 200 PG (GE Life Sciences) using respective buffer A with 18% glycerol and 3 mM DTT. After purification, concentrated proteins were stored at −80°C in 35% glycerol.
IC50 determination.
Enzyme reactions were carried out in an assay buffer consisting of 100 mM HEPES pH 8.0, 500 mM NH4Cl, 10% glycerol, and 0.125 mg/mL γ-Globulins; 10% DMSO with 150 μM crotonyl-CoA (substrate) and 150 μM NADH (cofactor). Compounds were incubated with 50 nM of enzyme in three-fold dilutions ranging from 100 to 0.005 μM and 100 to 0.0017 μM for CdFabK and SpFabK, respectively. After 10 min, the reaction was started by adding substrate and cofactor. Reaction kinetics were monitored for 10 min by measuring NADH fluorescence at 340/460 nm. Experiments were carried out in duplicates for each enzyme. IC50 values were calculated using the four-parameter logistic (Hill) curve analysis in GraphPad Prism 7; using the equation y=bottom + (top - bottom)/(1+10^((LogIC50 - x)*HillSlope)), where x is the logarithm of concentration and y is the response.
Sporulation assay.
This assay was performed as described previously41 to assess the effect of inhibiting FASII on sporulation. Briefly, C. difficile CD630 harboring respective plasmids were grown overnight on BHI agar plate with 0.1% (w/v) sodium taurocholate and 15 μg/mL of thiamphenicol. Colonies were inoculated in fresh BHI broth and grown to OD600 ≈ 0.3, before adding ATc at concentrations (0.016 and 0.128 μg/mL) that did not inhibit growth in broth. Total viable count and heat-resistant spores were analyzed over 5 days.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institute of Allergy and Infectious Diseases of the National Institutes of Health for funding R21AI126755. We thank Uyen T. Dang for help in preparing pRPF185-fabK. We also thank Drs. Shan Goh (University of Hertfordshire) and Liam Good (University of London) for advice in constructing the paired-termini in vector PMSPT.
Footnotes
SUPPORTING INFORMATION
Fatty acid biosynthesis in Bifidobacterium species; Sequences and general strategy of constructing anti-sense expression vectors; General chemistry procedure and synthesis of compound 1;1H and13C NMR, HRMS and HPLC chromatogram of compound 1; Table S1: Enoyl-ACP isoenzymes among gut microflora; Table S2: Strains and plasmids used for molecular studies; Table S3: Primers used in this study; Figure S1: Impact of antisense fabK RNA on C. difficile CD630 gene expression; References.
REFERENCES
- 1.Lessa FC; Mu Y; Bamberg WM; Beldavs ZG; Dumyati GK; Dunn JR; Farley MM; Holzbauer SM; Meek JI; Phipps EC; Wilson LE; Winston LG; Cohen JA; Limbago BM; Fridkin SK; Gerding DN; McDonald LC, Burden of Clostridium difficile infection in the United States. N Engl J Med 2015, 372 (9), 825–34. DOI: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freeman J; Bauer MP; Baines SD; Corver J; Fawley WN; Goorhuis B; Kuijper EJ; Wilcox MH, The changing epidemiology of Clostridium difficile infections. Clin Microbiol Rev 2010, 23 (3), 529–49. DOI: 10.1128/CMR.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rupnik M; Wilcox MH; Gerding DN, Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 2009, 7 (7), 526–36. DOI: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 4.Lewis BB; Buffie CG; Carter RA; Leiner I; Toussaint NC; Miller LC; Gobourne A; Ling L; Pamer EG, Loss of Microbiota-Mediated Colonization Resistance to Clostridium difficile Infection With Oral Vancomycin Compared With Metronidazole. J Infect Dis 2015, 212 (10), 1656–65. DOI: 10.1093/infdis/jiv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vardakas KZ; Polyzos KA; Patouni K; Rafailidis PI; Samonis G; Falagas ME, Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents 2012, 40 (1), 1–8. DOI: 10.1016/j.ijantimicag.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Bartsch SM; Umscheid CA; Fishman N; Lee BY, Is fidaxomicin worth the cost? An economic analysis. Clin Infect Dis 2013, 57 (4), 555–61. DOI: 10.1093/cid/cit346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstein EJ; Citron DM; Sears P; Babakhani F; Sambol SP; Gerding DN, Comparative susceptibilities to fidaxomicin (OPT-80) of isolates collected at baseline, recurrence, and failure from patients in two phase III trials of fidaxomicin against Clostridium difficile infection. Antimicrob Agents Chemother 2011, 55 (11), 5194–9. DOI: 10.1128/AAC.00625-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jarrad AM; Karoli T; Blaskovich MA; Lyras D; Cooper MA, Clostridium difficile drug pipeline: challenges in discovery and development of new agents. J Med Chem 2015, 58 (13), 5164–85. DOI: 10.1021/jm5016846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daley P; Louie T; Lutz JE; Khanna S; Stoutenburgh U; Jin M; Adedoyin A; Chesnel L; Guris D; Larson KB; Murata Y, Surotomycin versus vancomycin in adults with Clostridium difficile infection: primary clinical outcomes from the second pivotal, randomized, double-blind, Phase 3 trial. J Antimicrob Chemother 2017, 72 (12), 3462–3470. DOI: 10.1093/jac/dkx299. [DOI] [PubMed] [Google Scholar]
- 10.Boix V; Fedorak RN; Mullane KM; Pesant Y; Stoutenburgh U; Jin M; Adedoyin A; Chesnel L; Guris D; Larson KB; Murata Y, Primary Outcomes From a Phase 3, Randomized, Double-Blind, Active-Controlled Trial of Surotomycin in Subjects With Clostridium difficile Infection. Open Forum Infect Dis 2017, 4 (1), ofw275 DOI: 10.1093/ofid/ofw275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu X; Hurdle JG, The Clostridium difficile proline racemase is not essential for early logarithmic growth and infection. Can J Microbiol 2014, 60 (4), 251–4. DOI: 10.1139/cjm-2013-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jadhav A; Ezhilarasan V; Sharma Prakash O.; Pan A, Clostridium-DT(DB): a comprehensive database for potential drug targets of Clostridium difficile. Comput Biol Med 2013, 43 (4), 362–7. DOI: 10.1016/j.compbiomed.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Parsons JB; Rock CO, Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery? Curr Opin Microbiol 2011, 14 (5), 544–9. DOI: 10.1016/j.mib.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinster S; Lamberet G; Staels B; Trieu-Cuot P; Gruss A; Poyart C, Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 2009, 458 (7234), 83–6. DOI: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 15.Balemans W; Lounis N; Gilissen R; Guillemont J; Simmen K; Andries K; Koul A, Essentiality of FASII pathway for Staphylococcus aureus. Nature 2010, 463 (7279), E3; discussion E4. DOI: 10.1038/nature08667. [DOI] [PubMed] [Google Scholar]
- 16.Kingry LC; Cummings JE; Brookman KW; Bommineni GR; Tonge PJ; Slayden RA, The Francisella tularensis FabI enoyl-acyl carrier protein reductase gene is essential to bacterial viability and is expressed during infection. J Bacteriol 2013, 195 (2), 351–8. DOI: 10.1128/JB.01957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsons JB; Frank MW; Subramanian C; Saenkham P; Rock CO, Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc Natl Acad Sci U S A 2011, 108 (37), 15378–83. DOI: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hafkin B; Kaplan N; Murphy B, Efficacy and Safety of AFN-1252, the First Staphylococcus-Specific Antibacterial Agent, in the Treatment of Acute Bacterial Skin and Skin Structure Infections, Including Those in Patients with Significant Comorbidities. Antimicrob Agents Chemother 2015, 60 (3), 1695–701. DOI: 10.1128/AAC.01741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan N; Albert M; Awrey D; Bardouniotis E; Berman J; Clarke T; Dorsey M; Hafkin B; Ramnauth J; Romanov V; Schmid MB; Thalakada R; Yethon J; Pauls HW, Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob Agents Chemother 2012, 56 (11), 5865–74. DOI: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heath RJ; Rock CO, A triclosan-resistant bacterial enzyme. Nature 2000, 406 (6792), 145–6. DOI: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 21.Zhu L; Bi H; Ma J; Hu Z; Zhang W; Cronan JE; Wang H, The two functional enoyl-acyl carrier protein reductases of Enterococcus faecalis do not mediate triclosan resistance. MBio 2013, 4 (5), e00613–13. DOI: 10.1128/mBio.00613-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yutin N; Galperin MY, A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environmental microbiology 2013, 15 (10), 2631–41. DOI: 10.1111/1462-2920.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckburg PB; Bik EM; Bernstein CN; Purdom E; Dethlefsen L; Sargent M; Gill SR; Nelson KE; Relman DA, Diversity of the human intestinal microbial flora. Science 2005, 308 (5728), 1635–8. DOI: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bi H; Zhu L; Wang H; Cronan JE, Inefficient translation renders the Enterococcus faecalis fabK enoyl-acyl carrier protein reductase phenotypically cryptic. J. Bacteriol 2014, 196 (1), 170–9. DOI: 10.1128/JB.01148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuehne SA; Minton NP, ClosTron-mediated engineering of Clostridium. Bioengineered 2012, 3 (4), 247–54. DOI: 10.4161/bioe.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fagan RP; Fairweather NF, Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 2011, 286 (31), 27483–93. DOI: 10.1074/jbc.M111.263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakashima N; Tamura T; Good L, Paired termini stabilize antisense RNAs and enhance conditional gene silencing in Escherichia coli. Nucleic Acids Res 2006, 34 (20), e138 DOI: 10.1093/nar/gkl697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dryselius R; Aswasti SK; Rajarao GK; Nielsen PE; Good L, The translation start codon region is sensitive to antisense PNA inhibition in Escherichia coli. Oligonucleotides 2003, 13 (6), 427–33. DOI: 10.1089/154545703322860753. [DOI] [PubMed] [Google Scholar]
- 29.Larson MH; Gilbert LA; Wang X; Lim WA; Weissman JS; Qi LS, CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc 2013, 8 (11), 2180–96. DOI: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nariya H; Miyata S; Kuwahara T; Okabe A, Development and characterization of a xylose-inducible gene expression system for Clostridium perfringens. Appl Environ Microbiol 2011, 77 (23), 8439–41. DOI: 10.1128/AEM.05668-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National-Research-Council, In Diet and Health: Implications for Reducing Chronic Disease Risk, National Academy Press: Washington (DC), 1989. DOI: 10.17226/1222. [DOI] [PubMed] [Google Scholar]
- 32.Kitagawa H; Ozawa T; Takahata S; Iida M; Saito J; Yamada M, Phenylimidazole derivatives of 4-pyridone as dual inhibitors of bacterial enoyl-acyl carrier protein reductases FabI and FabK. J Med Chem 2007, 50 (19), 4710–20. DOI: 10.1021/jm0705354. [DOI] [PubMed] [Google Scholar]
- 33.Saito J; Yamada M; Watanabe T; Iida M; Kitagawa H; Takahata S; Ozawa T; Takeuchi Y; Ohsawa F, Crystal structure of enoyl-acyl carrier protein reductase (FabK) from Streptococcus pneumoniae reveals the binding mode of an inhibitor. Protein science: a publication of the Protein Society 2008, 17 (4), 691–9. DOI: 10.1110/ps.073288808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozawa T; Kitagawa H; Yamamoto Y; Takahata S; Iida M; Osaki Y; Yamada K, Phenylimidazole derivatives as specific inhibitors of bacterial enoyl-acyl carrier protein reductase FabK. Bioorg Med Chem 2007, 15 (23), 7325–36. DOI: 10.1016/j.bmc.2007.08.050. [DOI] [PubMed] [Google Scholar]
- 35.Ngernsombat C; Sreesai S; Harnvoravongchai P; Chankhamhaengdecha S; Janvilisri T, CD2068 potentially mediates multidrug efflux in Clostridium difficile. Sci Rep 2017, 7 (1), 9982 DOI: 10.1038/s41598-017-10155-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer AC; Kishony R, Opposing effects of target overexpression reveal drug mechanisms. Nat Commun 2014, 5, 4296 DOI: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hevener KE; Santarsiero BD; Lee H; Jones JA; Boci T; Johnson ME; Mehboob S, Structural characterization of Porphyromonas gingivalis enoyl-ACP reductase II (FabK). Acta Crystallogr F Struct Biol Commun 2018, 74 (Pt 2), 105–112. DOI: 10.1107/S2053230X18000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pedrido ME; de Ona P; Ramirez W; Lenini C; Goni A; Grau R, Spo0A links de novo fatty acid synthesis to sporulation and biofilm development in Bacillus subtilis. Mol Microbiol 2013, 87 (2), 348–67. DOI: 10.1111/mmi.12102. [DOI] [PubMed] [Google Scholar]
- 39.Schujman GE; Grau R; Gramajo HC; Ornella L; de Mendoza D, De novo fatty acid synthesis is required for establishment of cell type-specific gene transcription during sporulation in Bacillus subtilis. Mol Microbiol 1998, 29 (5), 1215–24. [DOI] [PubMed] [Google Scholar]
- 40.Babakhani F; Bouillaut L; Gomez A; Sears P; Nguyen L; Sonenshein AL, Fidaxomicin inhibits spore production in Clostridium difficile. Clin Infect Dis 2012, 55 Suppl 2, S162–9. DOI: 10.1093/cid/cis453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu X; Cherian PT; Lee RE; Hurdle JG, The membrane as a target for controlling hypervirulent Clostridium difficile infections. J Antimicrob Chemother 2013, 68 (4), 806–15. DOI: 10.1093/jac/dks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massengo-Tiasse RP; Cronan JE, Diversity in enoyl-acyl carrier protein reductases. Cell Mol Life Sci 2009, 66 (9), 1507–17. DOI: 10.1007/s00018-009-8704-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahata S; Iida M; Osaki Y; Saito J; Kitagawa H; Ozawa T; Yoshida T; Hoshiko S, AG205, a novel agent directed against FabK of Streptococcus pneumoniae. Antimicrob Agents Chemother 2006, 50 (8), 2869–71. DOI: 10.1128/AAC.00270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seefeld MA; Miller WH; Newlander KA; Burgess WJ; DeWolf WE Jr.; Elkins PA; Head MS; Jakas DR; Janson CA; Keller PM; Manley PJ; Moore TD; Payne DJ; Pearson S; Polizzi BJ; Qiu X; Rittenhouse SF; Uzinskas IN; Wallis NG; Huffman WF, Indole naphthyridinones as inhibitors of bacterial enoyl-ACP reductases FabI and FabK. J Med Chem 2003, 46 (9), 1627–35. DOI: 10.1021/jm0204035. [DOI] [PubMed] [Google Scholar]
- 45.Dembek M; Barquist L; Boinett CJ; Cain AK; Mayho M; Lawley TD; Fairweather NF; Fagan RP, High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio 2015, 6 (2), e02383 DOI: 10.1128/mBio.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chilton CH; Pickering DS; Freeman J, Microbiologic factors affecting Clostridium difficile recurrence. Clin Microbiol Infect 2018, 24 (5), 476–482. DOI: 10.1016/j.cmi.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 47.Morvan C; Halpern D; Kenanian G; Hays C; Anba-Mondoloni J; Brinster S; Kennedy S; Trieu-Cuot P; Poyart C; Lamberet G; Gloux K; Gruss A, Environmental fatty acids enable emergence of infectious Staphylococcus aureus resistant to FASII-targeted antimicrobials. Nature communications 2016, 7, 12944 DOI: 10.1038/ncomms12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cherian PT; Wu X; Yang L; Scarborough JS; Singh AP; Alam ZA; Lee RE; Hurdle JG, Gastrointestinal localization of metronidazole by a lactobacilli-inspired tetramic acid motif improves treatment outcomes in the hamster model of Clostridium difficile infection. J Antimicrob Chemother 2015. DOI: 10.1093/jac/dkv231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heap JT; Pennington OJ; Cartman ST; Minton NP, A modular system for Clostridium shuttle plasmids. J Microbiol Methods 2009, 78 (1), 79–85. DOI: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 50.Cherian PT; Wu X; Yang L; Scarborough JS; Singh AP; Alam ZA; Lee RE; Hurdle JG, Gastrointestinal localization of metronidazole by a lactobacilli-inspired tetramic acid motif improves treatment outcomes in the hamster model of Clostridium difficile infection. J Antimicrob Chemother 2015, 70 (11), 3061–9. DOI: 10.1093/jac/dkv231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak KJ; Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25 (4), 402–8. DOI: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Chelucci G; Falorni M; Giacomelli G, Synthesis of 1-Substituted 2-[(2S)-2-Pyrrolidinyl]pyridine from L-Proline. Synthesis 1990, 1990, 1121–1122. DOI: 10.1055/s-1990-27109. [DOI] [Google Scholar]
- 53.Rawling T; McDonagh AM; Tattam B; Murray M, Synthesis of unsymmetrical biaryl ureas from N-carbamoylimidazoles: kinetics and application. Tetrahedron Letters 2012, 68, 6065–6070. DOI: 10.1016/j.tet.2012.05.002 [DOI] [Google Scholar]
- 54.Jacobson MP; Pincus DL; Rapp CS; Day TJ; Honig B; Shaw DE; Friesner RA, A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55 (2), 351–67. DOI: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 55.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of medicinal chemistry 2006, 49 (21), 6177–96. DOI: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 56.Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL, Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. Journal of medicinal chemistry 2004, 47 (7), 1750–9. DOI: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 57.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of medicinal chemistry 2004, 47 (7), 1739–49. DOI: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.