

Abstract
The progressive loss of cardiomyocytes caused by cell death leads to cardiac dysfunction and heart failure (HF). Rapamycin has been shown to be cardioprotective in pressure-overloaded and ischemic heart diseases by regulating the mechanistic target of rapamycin (mTOR) signaling network. However, the impact of rapamycin on cardiomyocyte death in chronic HF remains undetermined. Therefore, in the current study we addressed this issue using a rat myocardial infarction (MI)-induced chronic HF model induced by ligating the coronary artery. Following surgery, rats were randomly divided into six groups, including the sham-, vehicle- and rapamycin-operated groups, at 8 or 12 weeks post-MI. A period of 4 weeks after MI induction, the rats were treated with rapamycin (1.4 mg-kg-day) or vehicle for 4 weeks. Cardiac function was determined using echocardiography, the rats were subsequently euthanized and myocardial tissues were harvested for histological and biochemical analyses. In the cell culture experiments with H9c2 rat cardiomyocytes, apoptosis was induced using angiotensin II (100 nM; 24 h). Cardiomyocyte apoptosis and autophagy were assessed via measuring apoptosis- and autophagy-associated proteins. The activities of mTOR complex 1 (mTORC1) and mTORC2 were evaluated using the phosphorylation states of ribosomal S6 protein and Akt, respectively. The activity of the endoplasmic reticulum (ER) stress pathway was determined using the levels of GRP78, caspase-12, phospho-JNK and DDIT3. Echocardiographic and histological measurements indicated that rapamycin treatment improved cardiac function and inhibited cardiac remodeling at 8 weeks post-MI. Additionally, rapamycin prevented cardiomyocyte apoptosis and promoted autophagy at 8 weeks post-MI. Rapamycin treatment for 4 weeks inhibited the mTOR and ER stress pathways. Furthermore, rapamycin prevented angiotensin II-induced H9c2 cell apoptosis and promoted autophagy by inhibiting the mTORC1 and ER stress pathways. These results demonstrated that rapamycin reduced cardiomyocyte apoptosis and promoted cardiomyocyte autophagy, by regulating the crosstalk between the mTOR and ER stress pathways in chronic HF.
Keywords: mechanistic target of rapamycin signaling, rapamycin, apoptosis, autophagy, endoplasmic reticulum stress pathway, heart failure
Introduction
Heart failure (HF) is caused by any cardiac disorder that leads to an increased intracardiac pressure or a decreased cardiac output (1). Myocardial infarction (MI) is a common and increasingly recognized predisposition to HF (2,3). Following MI, neurohormonal systems are activated, via decreased cardiac output, to maintain hemodynamic stability (2). However, the sustained activation of neurohormonal systems results in maladaptive alterations, including cardiac hypertrophy and cardiomyocyte death, and can lead to loss of cardiomyocytes and contractile dysfunction (2,4). Slow and maintained cardiomyocyte death occurs in failing hearts and is a major pathophysiologic mechanism that underlies progressive cardiac dysfunction (2). Significant progress has been made in determining postinfarction therapeutic strategies. However, the incidence of HF continues to increase (5,6). Therefore, it is important to understand the underlying mechanisms associated with cardiomyocyte death to develop novel therapies for use in HF prevention and treatment.
Apoptosis is a highly choreographed form of cell death that is mediated by caspases (4). Gradual but sustained cardiomyocyte apoptosis occurs in failing hearts, leading to the progressive loss of cardiomyocytes and lethal HF (4,7-12). Angiotensin-converting enzyme inhibitors and β-receptor blockers are used in clinical practice, and the benefits of these treatments presumably result in part from reductions in cardiomyocyte apoptosis (10). Following MI, the activation of protein synthesis in response to neuroendocrine or growth factors serves an important role in cardiomyocyte hypertrophy and apoptosis (13,14). The endoplasmic reticulum (ER) is the main site of synthesized protein folding (13). Any cellular stressor leading to the accumulation of unfolded proteins in the ER will trigger ER stress (also known as the unfolded protein response) (13). Multiple stimuli, including hypoxia, oxidative injury and protein aggregates, have been identified in failing hearts and can trigger ER stress (15). The initial aim of the unfolded protein response is to reduce the accumulation of unfolded proteins and restore normal ER function (16). However, prolonged or severe ER stress can trigger processes leading to apoptosis (16). The initiation of ER stress is characterized by the upregulation of chaperones, including glucose-regulated protein 78 kDa (GRP78) (16), while prolonged or excessive ER stress promotes apoptosis through the transcriptional induction of DNA damage-inducible transcript 3 (DDIT3), or the activation of c-JUN NH2-terminal kinase (JNK)- and-or caspase-12-dependent pathways (17,18).
Autophagy is another type of cell death that is primarily a prosurvival mechanism by which intracellular components are digested and recycled by lysosomal degradation to maintain energy production and protein synthesis (4,19-22). Numerous forms of HF are implicated by the accumulation of aberrant proteins, which occurs due to the insufficient protein degradation (5,10,23). Autophagy is a major intracellular protein degradation mechanism that is often upregulated in cardio-myocytes within a failing heart (4,23). The protective effect of autophagy under conditions of stress, especially in ischemia, starvation and b-adrenergic stimulation, is exhibited by the remove of damaged or misfolded proteins, organelles and aggregates (4,19,24-27). Additionally, impaired autophagy has been indicated to serve a pathological role in the progression of HF (23,27,28). The mechanistic target of rapamycin (mTOR) kinase and Beclin-1 are two major regulators of autophagy (4). Under nutrient-rich conditions, mTOR inhibits autophagy by inactivating autophagy-associated genes. During starvation, the activity of mTOR is reduced by the class I PI3K-Akt pathway and autophagy is inhibited (4,23). Beclin-1 signaling positively regulates autophagy by facilitating autophagosome formation (4).
mTOR kinase is an important regulator that is associated with protein synthesis, autophagy and survival (29,30). The mTOR kinase is present in two distinct multiprotein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (29). mTORC1 detects upstream inputs and promotes protein synthesis, cell growth and ribosomal biogenesis through downstream effectors, including S6 kinase and eIF4E-binding protein-1, which phosphorylate ribosomal S6 protein (29,30). Stimuli of neurohormonal systems, including angiotensin II and b-adrenoceptor agonists, can cause the rapid and sustained activation of S6 kinase (31,32). Additionally, mTORC1 inhibits catabolic processes, including autophagy (30). The inhibition of mTORC1 signaling, by pharmacological or genetic approaches, increases cardiomyocyte autophagy (33,34). In comparison, the function of mTORC2 is not fully determined. Previous findings have demonstrated that mTORC2 triggers an anti-apoptotic response by phosphorylating the prosurvival kinase Akt-protein kinase B on Ser473 (30,35,36). Furthermore, the activities of mTORC1 and mTORC2 are closely linked, and they may be differently influenced by rapamycin (30). Rapamycin is an allosteric inhibitor of mTORC1 (37). However, mTORC2 is insensitive to rapamycin (29,35,36). Short-term rapamycin treatment has been indicated to acutely inhibit the activity of mTORC1 and stimulate mTORC2 signaling through a negative-feedback mechanism. However, prolonged rapamycin treatment reduces the level of mTORC2 by inhibiting its assembly (35,37). Due to its ability to suppress cell growth and proliferation, rapamycin has been used as an immunosuppressant, anticancer drug, and anti-restenosis agent in coronary arterial stents (37). In acute MI, rapamycin, or its analogue, limit cardiomyocyte death and infarct size and attenuate cardiomyocyte hypertrophy and cardiac remodeling (33,36,38). Although prolonged rapamycin pretreatment disrupts the pro-survival effect of mTORC2 on cardiomyocytes (36), rapamycin has been revealed to inhibit cardiomyocyte apoptosis (38). In clinical practice, HF develops from an old MI and follows a chronic course. Therefore, it is important to determine whether prolonged rapamycin treatment exhibits a potential therapeutic value in chronic postinfarction HF through the regulation of cardiomyocyte death.
Recent studies have demonstrated that inhibiting apoptosis and promoting autophagy in cardiomyocytes represent two potential therapeutic strategies for use in HF (19,39,40). Additionally, ER stress and the mTOR signaling network (mTORC1 and mTORC2 pathways) have been indicated to act in a coordinative manner to regulate a variety of cellular processes, including cell growth and survival (41-43). In the current study, using in vivo (MI-induced chronic HF rat model) and in vitro (angiotensin II-induced cardiomyocyte apoptosis model) experimental approaches, whether rapamycin impacts cardiomyocyte apoptosis and autophagy by affecting the crosstalk between mTOR signaling and ER stress pathways was assessed.
Materials and methods
Reagents
Rapamycin and chloroquine (diphosphate salt) were purchased from Sigma Aldrich (Merck KGaA). Angiotensin II was purchased from Phoenix Pharmaceuticals (Burlingame).
Animals
All animal procedures were conducted in accordance with the institutional guidelines for the care and use of laboratory animals by Jilin University, Jilin, China. All experimental procedures were approved by the Ethical Review Board of China-Japan Union Hospital of Jilin University. Male Wistar rats (age, 8 weeks; weight, 240-270 g) were obtained from the Center for Laboratory Animals, Medical College, Jilin University, China.
Postinfarction HF was generated following a method as previously described (39,44). Rats were subjected to sham surgery or surgery involving the ligation of the left anterior descending artery. Rats were then anesthetized using 100% oxygen containing 3% isoflurane, which was supplied using a rodent respirator. Following anesthetization, the thorax was opened in the left parasternal area, and MI was induced by ligating the left anterior descending coronary artery using a 3-0 suture between the pulmonary cone and the left atrium. Following surgery, rats were randomly divided into six groups, including the sham-, vehicle- and rapamycin-operated groups, at 8 weeks (n=6, n=8 and n=8, respectively) or 12 weeks post-MI (n=6, n=8 and n=8, respectively). After a period of 4 weeks, the successful induction of HF was confirmed using echocardiography, and the animals in the rapamycin- and vehicle-operated groups, at 8 weeks or 12 weeks post-MI, received an intraperitoneal injection of rapamycin (1.4 mg-kg-day) dissolved in dimethyl sulfoxide or vehicle control (equivalent volumes of dimethyl sulfoxide diluted in normal saline) for 4 weeks. The dose of rapamycin was selected based on the body surface area, as described previously, and this dose has been indicated to be effective and well tolerated in previous studies (45,46). At 8 and 12 weeks post-MI induction, body weight and echocardiography were recorded. Animals were then anesthetized using 100% oxygen containing 3% isoflurane and euthanized via a rapid exsanguination from the abdominal aorta and the removal of the hearts. Exsanguination was performed via an abdominal aortic catheter, which permitted the free flow of blood, and blood with a total volume of 7-9 ml per rat was rapidly removed until no longer bleeding. The hearts were then quickly harvested and washed with ice-cold normal saline, and subsequently blotted with medical gauze. The left ventricle was dissected and fixed in 4% paraformaldehyde for histological evaluation, or snap frozen for biochemical measurements.
Echocardiography
Rats were mildly anesthetized using 3% isoflurane, and transthoracic echocardiography was performed using a Vivid-i echocardiography machine (General Electric Company) equipped with an 11.5-MHz transducer. The investigators who conducted the echocardiography exam were blinded to the treatment groups.
Pathology
Histologic studies were conducted with 4% paraformaldehyde-fixed and paraffin-embedded left ventricular samples from rats of all six groups. Hematoxylin-eosin staining, Masson's trichrome staining and immunohistochemical staining for caspase-3 were conducted on cross sections of the left ventricle. Images were acquired with a microscope (Olympus Corporation) equipped with a digital camera and cellSens Dimension software (version 1.16; Olympus Corporation).
Cell culture
The rat cardiomyocyte line H9c2 cells were purchased from American Type Culture Collection (Manassas). H9c2 cells were cultured in Dulbecco's modified Eagle's medium with a 1:1 mixture of Ham's F-12 (DME-F-12 1:1; HyClone; GE Healthcare) supplemented with 10% fetal bovine serum (Biological Industries,), 100 U-ml penicillin (Gibco; Thermo Fisher Scientific, Inc.), and 100 mg-ml streptomycin (Gibco; Thermo Fisher Scientific, Inc.). The cells were cultured in a moist atmosphere with 5% CO2 and 95% air at 37°C.
Cell viability measurement
Cell viability was assessed with a Cell Counting Kit 8 assay (Dojindo Laboratories). In brief, H9c2 cells were plated in 96-well plates at a density of 10,000 cells-well. After 24 h, the cells were treated with graded concentrations of angiotensin II (0, 50, 100, 200, and 500 nM) for an additional 12, 24, or 36 h. Then, 10 µl of Cell Counting Kit 8 reagent was added to the culture medium. After incubating the cells for 30 min, the plates were read at OD 450 nm in a microplate spectrophotometer (BioTek Instruments, Inc.).
Caspase-3 activity assay
The caspase-3 activity assay kit (Beyotime Biotechnology) was used to confirm caspase-3 activity, which recognizes the substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA). The assay is mainly based on quantification of the yellow p-nitroaniline at a wavelength of 405 nm with a microplate spectrophotometer after cleavage from the substrate Ac-DEVD-pNA. The assay was carried out according to the kit instructions after preparing cell lysates from H9c2 cardiomyocytes.
Flow cytometry
Cell survival and apoptosis were assessed by flow cytometry with Annexin V-fluorescein isothiocyanate and propidium iodide (KeyGEN BioTECH) staining. Analysis of phosphatidylserine on the outer leaflet of apoptotic cell membranes was performed using Annexin V-fluorescein isothiocyanate and propidium iodide to identify apoptotic and necrotic cells, respectively. Briefly, the cells were collected with ethylenediaminetetraacetic acid-free trypsin and washed with PBS three times. Then, the cells were resuspended in 500 µl of HEPES buffer, mixed with 5 µl of Annexin V-fluorescein isothiocyanate and 5 µl of propidium iodide, and incubated at room temperature for 5 min in the dark. The cells were analyzed using a flow cytometer (BD Biosciences).
Immunofluorescence staining
Cells were seeded, treated, fixed, and stained directly in 24-well plates. The cells were then washed twice with PBS and fixed in ice-cold 100% methanol for 15 min at -20°C. After rinsing with PBS three times for 5 min each, the samples were incubated with 0.1% Triton X-100 at room temperature for 10 min, blocked with 1% bovine serum albumin at room temperature for 60 min, and incubated with rabbit anti-light chain 3B (1:200; Cell Signaling Technology, Inc.; cat. no. 3868) antibody at 4°C overnight. The samples were then washed three times with PBS, followed by incubation with Alexa Fluor 488-conjugated secondary antibodies (1:1,000; Cell Signaling Technology, Inc.; cat. no. 4412) at room temperature for 60 min after 4′,6-diamidino-2-phenyl-indole counterstaining for 5 min. Images were captured with a fluorescence microscope system (Olympus Corporation).
Western blot analysis
Heart samples (left ventricular tissue of an area far from the infarction site) and cardiomyocytes were homogenized in ice-cold lysis buffer consisting of Tris 50 mM (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate. The protein concentrations were determined by a bicinchoninic acid assay (KeyGEN BioTECH) using bovine serum albumin as a standard. Equal amounts of total protein were loaded for resolution by sodium dodecyl sulfate-polyacrylamide gels under reducing conditions. The primary antibodies against caspase-3 (1:1,000; cat. no. 9662), cleaved caspase-3 (1:500; cat. no. 4412), phospho-S6 ribosomal protein (Ser235-236) (1:1,000; cat. no. 4412), S6 ribosomal protein (1:1,000; cat. no. 4412), phospho-Akt (Ser473) (1:1,000; cat. no. 4412), light chain 3B (1:1,000; cat. no. 4412), SQSTM1-p62 (1:1,000; cat. no. 4412), Beclin-1 (1:1,000; cat. no. 3495), phospho-SAPK-JNK (Thr183-Tyr185) (1:1,000; cat. no. 4668), SAPK-JNK (1:1,000; cat. no. 9252) and GAPDH (1:1,000; cat. no. 4412) were purchased from Cell Signaling Technology, Inc. The primary antibody against caspase-12 (0.25 µg/ml; cat. no. AF1456-SP) was purchased from R&D Systems, Inc. The primary antibodies against DDIT3 (1:1,000; cat. no. ab179823) and GRP78 (1:2,000; cat. no. ab108613) were purchased from Abcam. The HRP-conjugated secondary antibody (1:5,000; cat. no. BS13278) was purchased from Bioworld Technology, Inc.
Statistical analysis
Data are expressed as the means ± S.E.M. Statistical analysis was performed with PASW Statistics 18 software (version 18.0.0; IBM, Inc.). Differences between two groups (the sham- and MI-operated groups at 4 weeks post-MI) were tested by two independent samples t-test. Differences among groups were tested by one-way ANOVA followed by post hoc comparisons with Student-Newman-Keuls (S) and Tukey's post hoc tests. Differences among groups for the long-term observations were tested by two-way ANOVA followed by post hoc comparisons with Student-Newman-Keuls (S) and Tukey's post hoc tests. Statistical graphing was performed using GraphPad Prism software (version 7.04; GraphPad Software, Inc., San Diego, CA, USA). For all analyses, P<0.05 was considered significantly different.
Results
Rapamycin improves cardiac function in rats with postinfarction HF
At 4 weeks following MI, a reduction in the echocardiographic ejection fraction and fractional shortening measurements was observed in MI animals compared with sham animals, which indicated the damage to cardiac function and initiation of chronic HF (Fig. 1A). At 8 weeks post-MI, impaired cardiac function was observed in vehicle-treated animals, and this impairment was prevented by rapamycin treatment (Fig. 1B). Ejection fraction measurements were significantly lower in the vehicle group compared with the sham group, whereas in the rapamycin group, ejection fraction measurements were higher compared with the vehicle group (Fig. 1B). Similar results were observed for fractional shortening measurements (Fig. 1B). The left ventricular internal end diastolic dimension and left ventricular internal end systolic dimension values were smaller in the rapamycin group compared with the vehicle group (Fig. 1B). To investigate the delayed effects of rapamycin on cardiac function, the same set of animal groups were observed at 12 weeks following MI (4 weeks after the end of rapamycin treatment). A significant improvement in cardiac function was still present in the rapamycin-treated group as assessed using the ejection fraction and fractional shortening measurements (Fig. 1C).
Figure 1.


Echocardiographic measurements of rats. (A) Echocardiographic EF and FS measurements of the sham- and MI-operated groups at 4 weeks following MI. (B) EF, FS, LVIDd and LVIDs values of the sham-, vehicle- and rapamycin-operated groups at 8 weeks following MI. *P<0.05 vs. the sham group; #P<0.05 vs. the vehicle group. Echocardiographic measurements of rats. (C) EF and FS measurements of rats at 8 and 12 weeks following MI. (D) Gross morphology of the hearts in the sham-, vehicle- and rapamycin-operated groups at 8 weeks following MI. (E) Hematoxylin and eosin staining of areas far from the infarction site. (F) Masson's trichrome staining of left ventricles. (G) Masson's trichrome staining of areas far from the infarction sites and cardiomyocyte size measurements. *P<0.05 vs. the sham group; #P<0.05 vs. the vehicle group. MI, myocardial infarction; Rap, rapamycin; EF, ejection fraction; FS, fractional shortening; LVIDd, left ventricular internal end diastolic dimension; LVIDs, left ventricular internal end systolic dimension.
At 8 weeks following MI, the size of the heart was slightly larger in the vehicle group compared with the sham group, and myocardial dilation was less significant in the rapamycin group compared with the vehicle group (Fig. 1D). Histopathology indicated that the left ventricle exhibited increased extracellular matrix synthesis in the vehicle group compared with the sham group, and this change was reduced in the rapamycin group (Fig. 1E). Rapamycin treatment also attenuated myocardial fibrosis of the left ventricle when compared with vehicle treatment at 8 weeks following MI (Fig. 1F). The results also demonstrated that cardiomyocyte size in an area distant from the site of infarction was largest in the vehicle group among all groups, and this hypertrophy was prevented by the rapamycin treatment (Fig. 1G).
Rapamycin inhibits the expression of cleaved caspase-3 and promotes cardiomyocyte autophagy in failing hearts
Cleaved caspase-3 serves a critical role during apoptosis. Cardiomyocyte apoptosis was evaluated in areas that were distant from the infarction site of left ventricles, using immunohistochemical staining for cleaved caspase-3. At 8 weeks following MI, the level of cleaved caspase-3 was higher in the vehicle group compared with the sham group, and this upregulation was partly prevented by rapamycin treatment (Fig. 2A). This result was also identified using western blot analysis (Fig. 2B and C). Additionally, the levels of cleaved caspase-3 in the vehicle group continued to increase up to 12 weeks following MI (Fig. 2B). During the 4 weeks after the end of rapamycin treatment, low but sustained inhibition of cleaved caspase-3 was observed in the rapamycin treatment group, as evaluated by immunoblot-ting for cleaved caspase-3 (Fig. 2B and C). Whether prolonged rapamycin treatment promoted sustained and protective autophagy was subsequently assessed by determining the conversion of light chain 3B I to light chain 3B II, an autophagosomal membrane protein used as a marker of autophagy, and the protein levels of p62 and Beclin-1. Beclin-1 is known to signal the onset of autophagy and p62 is a well-known substrate of autophagy, and is reduced by the lysosomal fusion of autophagosomes (23,47). The results indicated that the baseline levels of light chain 3B II and Beclin-1 were increased in the vehicle group, and enhanced by rapamycin treatment at 8 and 12 weeks post-MI (Fig. 2B and C). Additionally, the levels of p62 were increased in the vehicle group, and were decreased by rapamycin treatment at 8 weeks post-MI. No significant difference in the levels of p62 was identified between the vehicle- and rapamycin-operated groups at 12 weeks following MI.
Figure 2.

Rapamycin inhibits the expression of cleaved caspase-3 and promotes cardiomyocyte autophagy, in the hearts of rats with HF at 8 and 12 weeks following MI. (A) Immunohistochemical staining for cleaved caspase-3, which is dyed brown. The level of cleaved caspase-3 was higher in the vehicle group compared with the sham group, and this upregulation was partly prevented by rapamycin treatment. (B and C) Western blot analysis of caspase-3 and light chain 3B. *P<0.05 vs. the corresponding sham group; #P<0.05 vs. the corresponding vehicle group. HF, heart failure; Rap, rapamycin; MI, myocardial infarction; LC3B, light chain 3B.
Rapamycin inhibits the mTOR and ER stress pathways in rats with chronic HF
To further explore the mechanisms underlying the inhibitory effect of rapamycin on cardiomyo-cyte apoptosis, and the stimulatory effect on cardiomyocyte autophagy, the involvement of the mTOR signaling network and ER stress pathway was investigated. The activities of mTORC1 and mTORC2 were assessed as indicated by the phosphorylation states of their downstream targets, ribosomal S6 protein and Akt, respectively. At 8 weeks following MI, the phosphorylation of ribosomal S6 protein was activated in the vehicle group, indicating the activation of protein synthesis and cell growth following MI (Fig. 3A). Rapamycin administration inhibited the phosphorylation of ribosomal S6 protein compared with vehicle treatment at 8 weeks following MI (Fig. 3A). Furthermore, treatment with rapamycin for 4 weeks decreased the phosphorylation of Akt on Ser473 compared with the vehicle treatment, indicating that the prosurvival activity of mTORC2 was inhibited by rapamycin treatment (Fig. 3A). As shown in Fig. 2, although the prosurvival activity of mTORC2 was inhibited, the level of cleaved caspase-3 was still reduced by rapamycin treatment at 8 weeks following MI. After discontinuing rapamycin treatment for 4 weeks, the level of phosphoribosomal S6 protein was still decreased in the rapamycin-treated group compared with the vehicle group, whereas the inhibited phosphorylation of Akt on Ser473 was restored to baseline level at 12 weeks following MI (Fig. 3A). Additionally, the level of cleaved caspase-3 was still reduced by rapamycin treatment at 12 weeks following MI (Fig. 2). Therefore, the anti-apoptotic effects of rapamycin may be mediated through the inhibition of mTOR pathways, especially the mTORC1 pathway. Additionally, the inhibition of phosphoribosomal S6 protein by rapamycin was accompanied by the activation of cardiomyocyte autophagy by rapamycin at 8 and 12 weeks following MI (Fig. 2B and C, and Fig. 3A). The inhibitory effect of rapamycin on cardiomyocyte apoptosis and the promoting effect on cardiomyocyte autophagy may be mediated through the inhibition of mTOR pathways.
Figure 3.

Rapamycin inhibits the mTOR and endoplasmic reticulum stress pathways in hearts of HF rats at 8 and 12 weeks following MI. (A) Western blot of phospho-ribosomal S6 protein, total ribosomal S6, phospho-Akt and total Akt. (B) Western blot of GRP78, cleavage of caspase-12, phospho-JNK, total JNK and DDIT3. *P<0.05 vs. the corresponding sham group; #P<0.05 vs. the corresponding vehicle group. HF, heart failure; Rap, rapamycin; MI, myocardial infarction; RibS6, ribosomal S6 protein; GRP78, glucose-regulated protein 78 kDa; DDIT3, DNA damage-inducible transcript 3; JNK, c-JUN NH2-terminal kinase.
The mTOR and ER stress pathways are linked and they serve important roles in cell growth and cell survival (42). Prolonged or severe ER stress that is induced by the accumulation of unfolded proteins in the ER can lead to cell apoptosis (16). Whether the inhibition of mTOR by rapamycin prevented cardiomyocyte apoptosis through the regulation of the ER stress pathway was investigated in the current study. The effects of rapamycin on the ER stress pathway were determined using the levels of GRP78, caspase-12, phospho-JNK and DDIT3. At 8 weeks following MI, increased levels of GRP78, phospho-JNK and DDIT3 were demonstrated in the vehicle group, which coincided with the appearance of increased cleaved caspase-3 expression (Figs. 2 and 3B). Furthermore, rapamycin treatment reduced the levels of GRP78, phospho-JNK and DDIT3 compared with vehicle treatment (Fig. 3B). No significant differences in the levels of cleaved caspase-12 were revealed among the sham-, vehicle- and rapamycin-operated groups. The changes in the mTOR and ER stress pathways in hearts at 8 weeks following MI indicated that rapamycin may prevent cardiomyocyte apoptosis by inhibiting the ER stress pathway downstream of the mTORC1 pathway, which concomitantly promoted pro-survival cardiomyocyte autophagy.
Rapamycin prevents angiotensin II-induced H9c2 cell apoptosis
As a neuroendocrine factor, angiotensin II serves an important role in the pathogenesis of HF and induces protein synthesis and cardiomyocyte hypertrophy, which causes the activation of the ER stress and mTOR pathways (17,31, 48). Previous findings have demonstrated that angiotensin II induces modest levels of cardiomyocyte apoptosis (39,49), which is similar to the slight but persistent cardiomyocyte apoptosis observed in failing hearts. Therefore, in the current study, angiotensin II was used to create a pathological humoral environment for H9c2 cells. To determine the optimal concentration of angiotensin II for inducing apoptosis, an in vitro cell viability assay was performed, in which H9c2 cells were treated with graded concentrations of angiotensin II (50-500 nM) for 12, 24 and 36 h. Angiotensin II was indicated to reduce cell viability in a dose-independent and time-dependent manner, and the peak effect was identified at a dose of 100 nM and a time of 24-36 h (Fig. 4).
Figure 4.
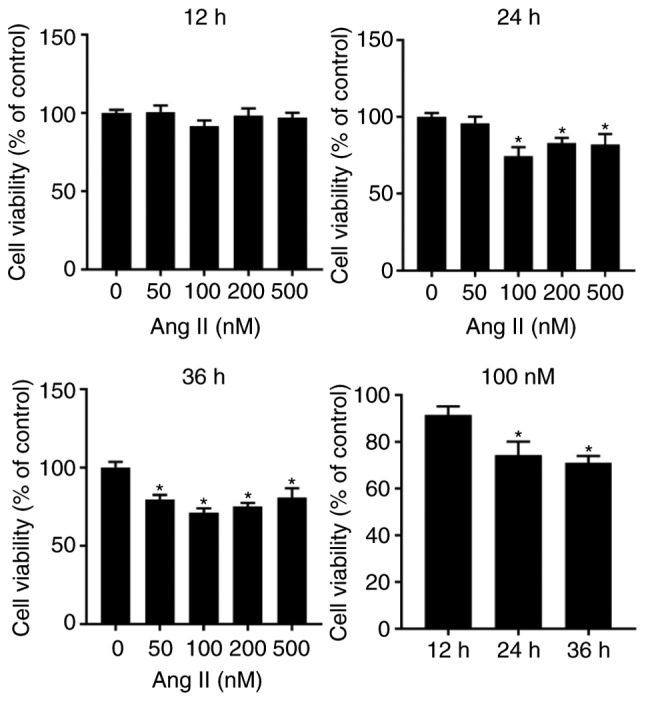
H9c2 cell viability as measured by the Cell Counting Kit 8 assay. *P<0.05 vs. the corresponding control group. Ang II, angiotensin II.
The effect of rapamycin on angiotensin II-induced H9c2 cell apoptosis was subsequently assessed. Long-term rapamycin treatment has been reported to inhibit mTORC2 function in a variety of cell types (35,36). Therefore, to investigate the roles of mTORC1 and mTORC2 in cell survival, H9c2 cells were pretreated with various concentrations of rapamycin (10,50 and 100 nM) or vehicle control (0.05% dimethyl sulfoxide) for 30 min (short term) or 24 h (long term) prior to treatment with angiotensin II (100 nM) for 24 h. Flow cytometry analysis demonstrated that rapamycin administration decreased basal and angiotensin II-induced H9c2 cell apoptosis in the groups treated for 30 min and 24 h, with a maximal inhibition observed at a dose of 10 nM (Fig. 5A). This antiapoptotic effect of rapamycin (10 nM) was reproduced in the caspase-3 activity assay (Fig. 5B) and western blot analysis of cleaved caspase-3 (Fig. 5C).
Figure 5.


Rapamycin prevented the basal and angiotensin II-induced H9c2 cell apoptosis in both the 30 min and 24 h rapamycin pretreatment groups. (A) Flow cytometry analysis of H9c2 cells in the 30 min (upper) and 24 h (lower) rapamycin pretreatment groups. (B) Caspase-3 activity assay of H9c2 cells. *P<0.05 vs. the corresponding control group; #P<0.05 vs. the corresponding angiotensin II-treated group. (C) Western blot of caspase-3 and cleaved caspase-3 in H9c2 cells. *P<0.05 vs. the corresponding control group; #P<0.05 vs. the corresponding angiotensin II-treated group. Ang II, angiotensin II; Rap, rapamycin.
Effects of rapamycin on H9c2 cell autophagic flux
To determine whether rapamycin-induced enhancement in cardiomyocyte autophagy is mediated by the upregulation of autophagosome formation or the inhibition of lysosomal degradation of autophagosomes, the effects of rapamycin on H9c2 cell autophagic flux were examined in an angio-tensin II-induced H9c2 cell apoptosis model in the presence or absence of chloroquine (50 µM, overnight), which is an inhibitor of lysosomal activity. As determined by the levels of light chain 3B II, rapamycin treatment increased basal and angiotensin II-induced autophagy, and these effects of rapamycin were further potentiated when chloroquine was added in the group treated for 30 min (Fig. 6A). These results indicated that the effect of rapamycin may not be due to the decreased degradation of autophagosomes. This is also supported by the results of the H9c2 cell autophagic flux assay using immuno-fluorescence staining for light chain 3B, which indicated that autophagy was significantly increased by rapamycin treatment when compared with vehicle treatment with or without the administration of angiotensin II in the 30 min group (Fig. 6B). Additionally, p62 expression was decreased and the levels of Beclin-1 were increased in rapamycin treatment group compared with the vehicle control group with or without angiotensin II pretreatment for 30 min (Fig. 6A).
Figure 6.


Rapamycin promoted the basal and angiotensin II-induced H9c2 cell autophagy in the 30 min rapamycin pretreatment groups. (A) Autophagy was assessed by western blot of light chain 3B II, p62 and Beclin-1 in H9c2 cells. (B) Immunofluorescence staining for autophagosomes in H9c2 cells, which was marked by Alexa Fluor 488-conjugated light chain 3B (green). The nuclei were stained by 4′,6-diamidino-2-phenylindole (blue). *P<0.05 vs. the corresponding control group; #P<0.05 vs. the corresponding angiotensin II-treated group. Ang II, angiotensin II; Rap, rapamycin; DAPI, 4′,6-diamidino-2-phenylindole.
Rapamycin inhibits the mTOR and ER stress pathways in H9c2 cells
As presented in Fig. 7A, the phosphorylation of ribosomal S6 protein was stimulated by angiotensin II alone (100 nM) for 24 h in 30 min and 24 h rapamycin pretreatment groups, indicating that angiotensin II increased protein synthesis in H9c2 cells via activation of the mTORC1 pathway (Fig. 7A). Pretreatment with rapamycin (10 nM) for 30 min or 24 h inhibited the basal and angiotensin II-induced phosphorylation of ribosomal S6 protein in H9c2 cells (Fig. 7A). Furthermore, treatment with rapamycin for 30 min stimulated the phosphorylation of Akt on Ser473 in H9c2 cells (Fig. 7A), and this stimulation was not present after 24 h of treatment with rapamycin (Fig. 7A). These results indicated that rapamycin treatment may prevent H9c2 cell apoptosis and promote autophagy through the inhibition of mTORC1 pathways. In the current study, it was revealed that administration of angiotensin II (100 nM) for 24 h induced GRP78 activation and an ER stress response in H9c2 cells, which coincided with H9c2 cell apoptosis (Fig. 7B). Furthermore, rapamycin treatment reduced basal and angiotensin II-induced GRP78 activation in the 30 min group (Fig. 7B). Rapamycin treatment also suppressed angiotensin II-induced DDIT3 activation and JNK phosphorylation in the 30 min group (Fig. 7B). The levels of phospho-JNK in H9c2 cells did not change following the administration of angiotensin II, and no caspase-12 cleavage was observed in H9c2 cells after the administration of angiotensin II or rapamycin (Fig. 7B).
Figure 7.

Rapamycin inhibits the mTOR and endoplasmic reticulum stress pathways in H9c2 cells. (A) Western blot analysis of phospho-ribosomal S6 protein, total ribosomal S6, phospho-Akt and total Akt in H9c2 cells. (B) Western blot of GRP78, cleavage of caspase-12, phospho-JNK, total JNK and DDIT3 in H9c2 cells. *P<0.05 vs. the corresponding control group; #P<0.05 vs. the corresponding angiotensin II-treated group. RibS6, ribosomal S6 protein; Ang II, angiotensin II; Rap, rapamycin; GRP78, glucose-regulated protein 78 kDa; DDIT3, DNA damage-inducible transcript 3; JNK, c-JUN NH2-terminal kinase.
Discussion
Following MI, thinning and distension are initially observed in infarcted and non-infarcted myocardium (44). Thinning of the infarcted wall is followed by resultant scar formation, while hypertrophy develops in non-infarcted myocardium in response to the increased stress and neuroendocrine or growth factors (44). Cardiomyocyte apoptosis occurs during the progression of myocardial hypertrophy and leads to cardiomyocyte loss and HF (4,7,10,11,17,50). Recently, the involvement of protein synthesis in cardiac remodeling and cardiomyocyte apoptosis has received considerable attention (5,10,23). It may be suggested that the sustained activation of protein synthesis with or without insufficient protein degradation can lead to ER stress and ER stress-induced cardiomyocyte apoptosis (15, 23). mTORC1 is a major regulator associated with protein synthesis and degradation (30). On the one hand, mTORC1 promotes protein synthesis by increasing ribosome production (30). On the other hand, mTORC1 is a key complex that inhibits autophagy, a major mechanism associated with cellular protein degradation, during stress and physiological and pathological conditions (23,51). The binding of rapamycin to its intracellular receptor, FKBP12, inhibits mTORC1 allosterically (37). Therefore, rapamycin is often used as an inhibitor of protein synthesis and cell growth to examine its effects on myocardial hypertrophy or cardiac remodeling in pressure-overloaded heart disease (52-55). However, the effects of rapamycin on cardiac function and cardiomyocyte apoptosis in chronic HF remain to be determined. The current study indicated that treatment with rapamycin can inhibit the phosphorylation of ribosomal S6 protein and promote cardiomyocyte autophagy, protecting cardiomyocytes from apoptosis by inhibiting the ER stress pathway and improving cardiac function in chronic HF. It can be suggested that the mTORC1 pathway may closely link to the ER stress pathway via regulating the balance between cellular protein synthesis and degradation. The inhibition of mTORC1 with rapamycin may inhibit ER stress by reducing the newly synthesized protein load and accelerating autophagy-induced unfolded protein degradation in the ER, and in turn protect cardiomyocytes from apoptosis, improving cardiac function in chronic HF.
ER is a highly dynamic organelle that serves important roles in protein synthesis and apoptosis (17). The increased protein synthesis in hypertrophic and failing hearts can lead to the accumulation of unfolded proteins in the ER, which in turn trigger ER stress and apoptosis (17). To prevent unfolded protein accumulation, cells upregulate the transcription of chaperones, including GRP78 and GRP94, reduce translation to attenuate the burden from newly synthesized proteins and degrade unfolded proteins that are accumulated in the ER (56). In the present study, ER stress was determined by assessing the upregulation of GRP78. In rats with HF and angiotensin II-treated H9c2 cells, GRP78 was upregulated, which coincided with cardiomyocyte apoptosis, and the inhibition of mTOR with rapamycin reduced the levels of GRP78 and cardiomyocyte apoptosis in the in vitro and in vivo setting. These results indicated that rapamycin may inhibit the ER stress pathway downstream of the mTOR pathway and prevent cardiomyocyte apoptosis caused by ER stress. Furthermore, prolonged or severe ER stress induces apoptosis through the DDIT3-, JNK- and-or caspase-12-dependent pathways (17,18). DDIT3, also known as CHOP, is a component downstream of ER stress pathways (16). DDIT3 protein overexpression has been indicated to induce apoptosis (16). JNK is a kinase that participates in signal transduction in response to stressors (18). ER stress activates JNK, and the sustained activation of the JNK pathway leads to apoptosis (18). Caspase-12 is localized in the ER membrane and is activated by ER stress but not death receptor- or mitochondrion-mediated apoptotic pathways (18). The results of the current study indicated that DDIT3 was induced in rats with failing hearts and in angiotensin II-treated H9c2 cells, and was suppressed by rapamycin treatment. Furthermore, the JNK pathway is closely associated with the mTORC1 pathway and has been indicated to be preferentially activated downstream of mTORC1 over other ER stress pathways (43). Although the activation of JNK was observed in failing hearts but not in angiotensin II-treated H9c2 cells, rapamycin treatment inhibited the phosphorylation of JNK in both cases. However, caspase-12 cleavage was not induced in failing hearts and angiotensin II-treated H9c2 cells. Although different ER stress inducers or courses may differentially impact the modulation of the three different ER stress pathways, failing hearts and angiotensin II-treated H9c2 cells increased ER stress, which contributed to cardiomyocyte apoptosis, while the inhibition of mTORC1 with rapamycin significantly prevented ER stress-induced cardiomyocyte apoptosis in vivo and in vitro.
The potential cardioprotective effects of rapamycin against cardiomyocyte hypertrophy during pressure overload have been previously demonstrated (52-55). In the context of chronic ischemic injury, Buss et al (33). demonstrated that everolimus prevented myocardial hypertrophy and cardiac remodeling through inhibition of the mTORC1 pathway. However, Völkers et al (36). revealed that prolonged rapamycin treatment (18 h) inhibited the prosurvival mTORC2 signaling and impaired the cell death-inhibiting effect of rapamycin in neonatal ventricular cardiomyocytes following an acute oxidative injury (50 µM H2O2 for 4 h). Although prolonged rapamycin treatment (1 week prior and 2 days following MI) inhibited mTORC2 signaling, a study indicated that the cardioprotective effects of rapamycin against myocardial hypertrophy and cardiomyocyte apoptosis were still observed in the mice suffering from chronic ischemic injury (38). In the present study, it was revealed that short-term rapamycin treatment inhibited the activity of mTORC1 and stimulated the activity of mTORC2 in H9c2 cells, while prolonged rapamycin treatment inhibited the mTORC1 and mTORC2 pathways in rats with chronic HF. Additionally, the activity of mTORC2 was restored to baseline level, and the activity of mTORC1 was still partly inhibited by rapamycin after discontinuing rapamycin treatment for 4 weeks in rats with chronic HF. This delayed inhibitory effect of rapamycin on mTORC1 may be associated with its allosteric inhibition of mTORC1 (mTORC1 needs to be newly synthesized). The cardioprotective effects of rapamycin on cardiac function and cardiomyocyte apoptosis were continuously observed in the rapamycin-treated groups at 8 and 12 weeks post-MI. These results indicated that mTORC1 inhibition and mTORC2 activation may contribute to the anti-apoptotic effect of short-term rapamycin treatment, but the inhibition of mTORC1 may be the primary mechanism for the cardioprotective effects of prolonged rapamycin treatment. The inhibition of protein synthesis and the activation of autophagy-induced protein renewal may be the major mechanism underlying the anti-apoptotic effect of rapamycin in chronic HF.
In conclusion, although the inhibitors of the neurohormonal systems, including angiotensin-converting enzyme inhibitors, β-receptor blockers and aldosterone antagonists have been widely used over the past 20 years, the incidence of chronic postinfarction HF remains high. The results of the current study demonstrated that rapamycin effectively prevented cardiomyocyte apoptosis, promoted cardiomyocyte autophagy and improved cardiac function via regulating the crosstalk between the mTORC1 and ER stress pathways in chronic postinfarction HF. However, the potential clinical application of rapamycin requires further investigation.
Acknowledgments
Not applicable.
Abbreviations
- HF
heart failure
- MI
myocardial infarction
- mTOR
mechanistic target of rapamycin
- mTORC1
mechanistic target of rapamycin complex 1
- mTORC2
mechanistic target of rapamycin complex 2
- ER
endoplasmic reticulum
- GRP78
glucose-regulated protein 78 kDa
- DDIT3
DNA damage-inducible transcript 3
- JNK
c-JUN NH2-terminal kinase
Funding
This study was supported by the National Natural Science Foundation of China (grant number 81570360); and the Fund of Key Laboratory of Myocardial Ischemia, Ministry of Education (grant number KF201815).
Availability of data and materials
The datasets used and-or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
PY and GG conceived and designed the study. GG, WC and MY performed the experiments. JL, HL and CW collected the data and prepared the Figures. GG analyzed the data and drafted the manuscript. PY edited the language of the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
All animal procedures were conducted in accordance with the institutional guidelines for the care and use of laboratory animals by Jilin University, Jilin, China. All experimental procedures were approved by the Ethical Review Board of China-Japan Union Hospital of Jilin University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Metra M, Teerlink JR. Heart failure. Lancet. 2017;390:1981–1995. doi: 10.1016/S0140-6736(17)31071-1. [DOI] [PubMed] [Google Scholar]
- 2.Cahill TJ, Choudhury RP, Riley PR. Heart regeneration and repair after myocardial infarction: Translational opportunities for novel therapeutics. Nat Rev Drug Discov. 2017;16:699–717. doi: 10.1038/nrd.2017.106. [DOI] [PubMed] [Google Scholar]
- 3.Teringova E, Tousek P. Apoptosis in ischemic heart disease. J Transl Med. 2017;15:87. doi: 10.1186/s12967-017-1191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 5.Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89:1401–1438. doi: 10.1007/s00204-015-1477-x. [DOI] [PubMed] [Google Scholar]
- 6.Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13:368–378. doi: 10.1038/nrcardio.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 8.Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, Bauer EP, Klövekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.CIR.0000051865.66123.B7. [DOI] [PubMed] [Google Scholar]
- 9.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abbate A, Biondi-Zoccai GG, Baldi A. Pathophysiologic role of myocardial apoptosis in post-infarction left ventricular remodeling. J Cell Physiol. 2002;193:145–153. doi: 10.1002/jcp.10174. [DOI] [PubMed] [Google Scholar]
- 11.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 12.Kostin S, Pool L, Elsässer A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn WP, Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 13.Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: Endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–642. doi: 10.1161/CIRCRESAHA.110.226803. [DOI] [PubMed] [Google Scholar]
- 14.Kolwicz SC, Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 16.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 17.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 18.Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/A:1016175429877. [DOI] [PubMed] [Google Scholar]
- 19.Takemura G, Kanamori H, Okada H, Miyazaki N, Watanabe T, Tsujimoto A, Goto K, Maruyama R, Fujiwara T, Fujiwara H. Anti-apoptosis in nonmyocytes and pro-autophagy in cardio-myocytes: Two strategies against postinfarction heart failure through regulation of cell death-degeneration. Heart Fail Rev. 2018;23:759–772. doi: 10.1007/s10741-018-9708-x. [DOI] [PubMed] [Google Scholar]
- 20.Kanamori H, Takemura G, Goto K, Maruyama R, Ono K, Nagao K, Tsujimoto A, Ogino A, Takeyama T, Kawaguchi T, et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol. 2011;300:H2261–H2271. doi: 10.1152/ajpheart.01056.2010. [DOI] [PubMed] [Google Scholar]
- 21.Kanamori H, Takemura G, Goto K, Maruyama R, Tsujimoto A, Ogino A, Takeyama T, Kawaguchi T, Watanabe T, Fujiwara T, et al. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc Res. 2011;91:330–339. doi: 10.1093/cvr/cvr073. [DOI] [PubMed] [Google Scholar]
- 22.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh R, Pattison JS. Macroautophagy and chaperone-mediated autophagy in heart failure: The known and the unknown. Oxid Med Cell Longev. 2018;2018:8602041. doi: 10.1155/2018/8602041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Jr, Gottlieb RA. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2010;3:365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Meyer GR, Martinet W. Autophagy in the cardiovascular system. Biochim Biophys Acta. 2009;1793:1485–1495. doi: 10.1016/j.bbamcr.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13:373–387. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 29.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. 2018;122:489–505. doi: 10.1161/CIRCRESAHA.117.311147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sadoshima J, Izumo S. Rapamycin selectively inhibits angiotensin II-induced increase in protein synthesis in cardiac myocytes in vitro. Potential role of 70-kD S6 kinase in angiotensin II-induced cardiac hypertrophy. Circ Res. 1995;77:1040–1052. doi: 10.1161/01.RES.77.6.1040. [DOI] [PubMed] [Google Scholar]
- 32.Simm A, Schlüter K, Diez C, Piper HM, Hoppe J. Activation of p70(S6) kinase by beta-adrenoceptor agonists on adult cardio-myocytes. J Mol Cell Cardiol. 1998;30:2059–2067. doi: 10.1006/jmcc.1998.0768. [DOI] [PubMed] [Google Scholar]
- 33.Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M, Izumo S, et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J Am Coll Cardiol. 2009;54:2435–2446. doi: 10.1016/j.jacc.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Xie J, Li R, Shi J, Sun J, Gu R, Ding L, Wang L, Xu B. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J Cell Mol Med. 2014;18:919–928. doi: 10.1111/jcmm.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt-PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 36.Völkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S, Joyo A, Ornelas L, Samse K, Thuerauf DJ, et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128:2132–2144. doi: 10.1161/CIRCULATIONAHA.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 38.Di R, Wu X, Chang Z, Zhao X, Feng Q, Lu S, Luan Q, Hemmings BA, Li X, Yang Z. S6K inhibition renders cardiac protection against myocardial infarction through PDK1 phos-phorylation of Akt. Biochem J. 2012;441:199–207. doi: 10.1042/BJ20110033. [DOI] [PubMed] [Google Scholar]
- 39.Liu M, Mao C, Li J, Han F, Yang P. Effects of the Activin A-follistatin system on myocardial cell apoptosis through the endoplasmic reticulum stress pathway in heart failure. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18020374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanamori H, Takemura G, Goto K, Tsujimoto A, Ogino A, Takeyama T, Kawaguchi T, Watanabe T, Morishita K, Kawasaki M, et al. Resveratrol reverses remodeling in hearts with large, old myocardial infarctions through enhanced autophagy-activating AMP kinase pathway. Am J Pathol. 2013;182:701–713. doi: 10.1016/j.ajpath.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Ji Y, Luo X, Yang Y, Dai Z, Wu G, Wu Z. Endoplasmic reticulum stress-induced apoptosis in intestinal epithelial cells: A feed-back regulation by mechanistic target of rapamycin complex 1 (mTORC1) J Anim Sci Biotechnol. 2018;9:38. doi: 10.1186/s40104-018-0253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274–282. doi: 10.1016/j.tcb.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012;19:310–320. doi: 10.1038/cdd.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldman S, Raya TE. Rat infarct model of myocardial infarction and heart failure. J Card Fail. 1995;1:169–177. doi: 10.1016/1071-9164(95)90019-5. [DOI] [PubMed] [Google Scholar]
- 45.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–1670. doi: 10.1161/01.CIR.0000057979.36322.88. [DOI] [PubMed] [Google Scholar]
- 46.Bishu K, Ogut O, Kushwaha S, Mohammed SF, Ohtani T, Xu X, Brozovich FV, Redfield MM. Anti-remodeling effects of rapamycin in experimental heart failure: Dose response and interaction with angiotensin receptor blockade. PLoS One. 2013;8:e81325. doi: 10.1371/journal.pone.0081325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schunkert H, Sadoshima J, Cornelius T, Kagaya Y, Weinberg EO, Izumo S, Riegger G, Lorell BH. Angiotensin II-induced growth responses in isolated adult rat hearts. Evidence for load-independent induction of cardiac protein synthesis by angiotensin II. Circ Res. 1995;76:489–497. doi: 10.1161/01.RES.76.3.489. [DOI] [PubMed] [Google Scholar]
- 49.Yan M, Yang S, Meng F, Zhao Z, Tian Z, Yang P. MicroRNA 199a-5p induces apoptosis by targeting JunB. Sci Rep. 2018;8:6699. doi: 10.1038/s41598-018-24932-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 53.Song X, Kusakari Y, Xiao CY, Kinsella SD, Rosenberg MA, Scherrer-Crosbie M, Hara K, Rosenzweig A, Matsui T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299:C1256–C1266. doi: 10.1152/ajpcell.00338.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikeda M, Ide T, Fujino T, Matsuo Y, Arai S, Saku K, Kakino T, Oga Y, Nishizaki A, Sunagawa K. The Akt-mTOR axis is a pivotal regulator of eccentric hypertrophy during volume overload. Sci Rep. 2015;5:15881. doi: 10.1038/srep15881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harston RK, McKillop JC, Moschella PC, Van Laer A, Quinones LS, Baicu CF, Balasubramanian S, Zile MR, Kuppuswamy D. Rapamycin treatment augments both protein ubiquitination and Akt activation in pressure-overloaded rat myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H1696–H1706. doi: 10.1152/ajpheart.00545.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/S0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and-or analyzed during the current study are available from the corresponding author on reasonable request.