

ABSTRACT
Pulmonary hypertension is defined as a resting mean pulmonary artery pressure of 25 mm Hg or above. This review deals with pulmonary arterial hypertension (PAH), a type of pulmonary hypertension that primarily affects the pulmonary vasculature. In PAH, the pulmonary vasculature is dynamically obstructed by vasoconstriction, structurally obstructed by adverse vascular remodeling, and pathologically non-compliant as a result of vascular fibrosis and stiffening. Many cell types are abnormal in PAH, including vascular cells (endothelial cells, smooth muscle cells, and fibroblasts) and inflammatory cells. Progress has been made in identifying the causes of PAH and approving new drug therapies. A cancer-like increase in cell proliferation and resistance to apoptosis reflects acquired abnormalities of mitochondrial metabolism and dynamics. Mutations in the type II bone morphogenetic protein receptor (BMPR2) gene dramatically increase the risk of developing heritable PAH. Epigenetic dysregulation of DNA methylation, histone acetylation, and microRNAs also contributes to disease pathogenesis. Aberrant bone morphogenetic protein signaling and epigenetic dysregulation in PAH promote cell proliferation in part through induction of a Warburg mitochondrial-metabolic state of uncoupled glycolysis. Complex changes in cytokines (interleukins and tumor necrosis factor), cellular immunity (T lymphocytes, natural killer cells, macrophages), and autoantibodies suggest that PAH is, in part, an autoimmune, inflammatory disease. Obstructive pulmonary vascular remodeling in PAH increases right ventricular afterload causing right ventricular hypertrophy. In some patients, maladaptive changes in the right ventricle, including ischemia and fibrosis, reduce right ventricular function and cause right ventricular failure. Patients with PAH have dyspnea, reduced exercise capacity, exertional syncope, and premature death from right ventricular failure. PAH targeted therapies (prostaglandins, phosphodiesterase-5 inhibitors, endothelin receptor antagonists, and soluble guanylate cyclase stimulators), used alone or in combination, improve functional capacity and hemodynamics and reduce hospital admissions. However, these vasodilators do not target key features of PAH pathogenesis and have not been shown to reduce mortality, which remains about 50% at five years. This review summarizes the epidemiology, pathogenesis, diagnosis, and treatment of PAH.
Introduction
Pulmonary hypertension is defined as a resting mean pulmonary artery pressure (mPAP) of 25 mm Hg or above. The classification system proposed by the Fifth World Symposium on Pulmonary Hypertension attempts to guide the clinical approach to pulmonary hypertension by dividing patients into five groups: group 1—pulmonary hypertension due to pulmonary vascular disease; group 2—pulmonary hypertension due to left heart disease; group 3—pulmonary hypertension due to lung disease or hypoxia; group 4—pulmonary hypertension due to chronic thromboembolic disease; and group 5—a miscellaneous collection of pulmonary hypertension syndromes caused by a variety of disorders, including hemolytic anemias and sarcoidosis (fig 1).1 In principle, patients in each of these groups share pathophysiology, prognosis, and therapeutic response; in reality, tremendous heterogeneity exists within each group.
Fig 1.
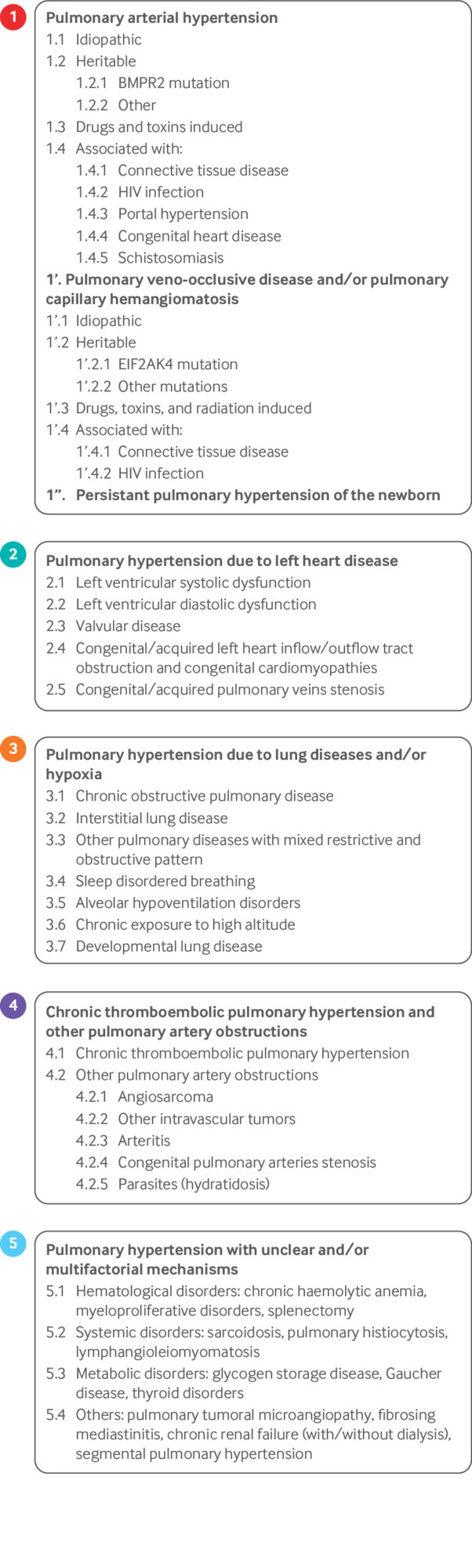
Updated World Symposium on Pulmonary Hypertension classification of pulmonary hypertension (2013). Adapted with permission from Simonneau G, et al. J Am Coll Cardiol 2013;62(25 Suppl):D34-411
This review focuses on group 1 pulmonary hypertension, also known as pulmonary arterial hypertension (PAH). PAH is a disease of the cardiopulmonary unit, affecting the pulmonary arterial and venous circulation and the right ventricle. Obstructive, hyperproliferative, vascular lesions, vasoconstriction of pre-capillary arterioles, and venous obstruction (in some forms of group 1 disease) increase pulmonary vascular resistance (PVR), increase right ventricular afterload, and promote right ventricular failure (RVF), which is the leading cause of death in PAH.2 Current treatments for PAH are primarily pulmonary vasodilators. They ameliorate symptoms and reduce hospital admissions, but they are expensive and not curative.
This review summarizes the epidemiology, diagnostic evaluation, and treatment of PAH. It also examines recent advances in basic science, noting potential therapeutic targets and future research questions.
Sources and selection criteria
We identified references for this review by doing a PubMed search for years 2007-17. We only included peer reviewed articles written in English. We used the following search terms in combination with the term “pulmonary hypertension”: “mechanisms”, “experimental models”, “diagnosis”, “epidemiology”, “survival”, “pulmonary arterial hypertension”, “guidelines”, “classification”, “imaging”, “hemodynamics”, and “therapy”. We included articles on the basis of the quality of study design and size, favoring randomized controlled trials, reports from large registries, and guidelines. For the pathogenesis section, we selected papers in reputable journals and highlighted evidence in which concordant data were available from more than one research group. We also included highly cited papers written before 2007. We excluded case reports and papers in non-peer reviewed journals. We screened approximately 1000 articles of evidence classes I-IV and included about 740 for detailed review.
Epidemiology and natural history of PAH
The incidence of PAH ranges from 2.0 to 7.6 cases per million adults per year, and its prevalence varies from 11 to 26 cases per million adults (table 1). The incidence of PAH is fourfold higher in women than in men, but survival is paradoxically worse in men with PAH.13 14 Nearly half of the patients have idiopathic PAH (IPAH), heritable PAH, or anorexigen induced PAH, with connective tissue disease associated PAH (APAH) being the second most common subgroup. The National Institutes of Health (NIH) registry from the 1980s was the first major epidemiological study of PAH.3 Subsequently, 10 major registries have described the epidemiology of PAH (table 1).4 5 6 7 8 9 10 11 12 These modern registries have provided two novel insights.
Table 1.
Characteristics of pulmonary arterial hypertension (PAH) registries
| Registry | Date of enrollment | Sample size, No | Patient population, % | Mean (SD) age, years | Incidence and prevalence | One year survival, % | Five year survival, % |
|---|---|---|---|---|---|---|---|
| NIH3 | 1981-88; prospective | 187 | IPAH; heritable; drug induced | 36 (15) | NA | 68 | 34 |
| US-PHC4 | 1982-2006; retrospective | 578 | IPAH: 44; CTD: 30; CHD: 11; POPH: 7; heritable: 4; anorexigen: 3; HIV: 1 | 48 (14) | NA | 86 | 61 |
| Scottish5 | 1986-2001; retrospective | 374 | IPAH: 47; CTD: 30; CHD: 23 | Men:50 (13); women:52 (12) | PAH: incidence 7.6 cases/MAI/year and prevalence 26 cases; IPAH: incidence 2.6 cases/MAI/year and prevalence 9 cases/MAI | NA | NA |
| Mayo Clinic6 | 1995-2004; prospective | 484 | IPAH/heritable: 56; CTD: 24; POPH: 10.4; CHD: 9.2; HIV: 0.4 | 52 (15) | NA | 81.1 | 47.9 |
| Chinese7 | 1999-2004; prospective | 72 | IPAH: 94.4; heritable: 5.6 | 35.9 (12.2) | NA | 68 | 20.8 |
| French8 | 2002-03; prospective | 674 | IPAH: 39.2; CTD: 15.3; CHD: 11.3; anorexigen: 9.5; HIV: 6.2; heritable: 3.9; POPH: 0.4 | 50 (15) | PAH: incidence 2.4 cases/MAI/year and prevalence 15 cases/MAI; IPAH: incidence 1.0 cases/MAI/year and prevalence 5.9 cases/MAI | 87 | NA |
| REVEAL9 | 2006-07; prospective | 2525 | IPAH: 46.2; CTD: 25.3; CHD: 9.9; POPH: 5.3; anorexigen: 5.2; heritable: 2.9; HIV: 1.9 | 53 (14) | PAH: incidence 2.0 cases/MAI/year and prevalence 10.6 cases/MAI; IPAH: Incidence – 0.9 cases/MAI/year | Incident: 86.3; prevalent: 90.4 | Incident: 61.2; prevalent: 65.4 |
| UK and Ireland10 | 2001-09; prospective | 482 | IPAH: 93; heritable: 5; anorexigen: 2 | 50 (17) | Incidence 1.1 cases/MAI/year and prevalence 6.6 cases/MAI | 93 | 60 |
| New Chinese registry11 | 2008-11; prospective | 956 | CHD: 43; IPAH: 35; CTD: 19 | 36 (13) | NA | IPAH: 92.1; CTD: 85.4 | NA |
| COMPERA12 | 2007-11; prospective | 587 | IPAH: 97; anorexigen: 2; heritable: 1 | 71 (16) | NA | 92 | NA |
CTD=connective tissue disease; CHD=congenital heart disease; IPAH=idiopathic pulmonary arterial hypertension; MAI=million adult inhabitants; NA=not available; NIH=National Institutes of Health; POPH=portopulmonary hypertension; US-PHC=United States Pulmonary Hypertension Connections.
Firstly, in the current era, patients with IPAH, heritable PAH, or anorexigen associated PAH are older than those in the NIH registry (mean age at diagnosis 45-65 versus 36 years). Potential explanations include increased awareness of PAH due to the availability of PAH specific therapies and widespread use of Doppler echocardiography, leading to wider recognition of PAH in older patients. However, as the mean age of the group 1 cohort increases, the risk of misdiagnosis increases, as group 2 pulmonary hypertension in patients with heart failure and preserved ejection fraction (HFpEF) is much more prevalent and shares several echocardiographic abnormalities.
Secondly, long term survival in patients with PAH has significantly improved in the past two decades. The median survival is now six years, compared with 2.8 years in the 1980s.15 Similarly, one year survival rates for PAH patients range from 86% to 90%, improved from 65% in the 1990s.16 17 18 Improved awareness of PAH, availability of PAH specific therapies, long term anticoagulation therapy, and better management of RVF are possible reasons for improved survival.
Nonetheless, PAH still causes a substantial clinical and economic burden. Although PAH related hospital admissions decreased between 2001 and 2012, the mean charge and length of stay per PAH related admission have increased with no significant decline in inpatient mortality.19
Diagnosis of PAH
PAH is defined as a resting mPAP of 25 mm Hg or above, pulmonary capillary wedge pressure (PCWP) below 15 mm Hg, and PVR above 3 Wood units20), in the absence of more prevalent causes of pulmonary hypertension such as left heart disease, chronic lung disease,21 or venous thromboembolism.
Most patients are referred for evaluation for pulmonary hypertension after detection of increased right ventricular systolic pressure (RVSP) by Doppler ultrasound. RVSP is calculated from Bernoulli’s principle, on the basis of the velocity of the tricuspid regurgitant jet (RVSP=4V2, where V is the maximum tricuspid regurgitant jet velocity) plus the estimated right atrial pressure (RAP). RAP is determined by observing the jugular venous pressure or, more commonly, by assessing the dimensions of the inferior vena cava, at baseline and in response to sniffing. On the basis of comparison with right heart catheterization (RHC), an inferior vena cava maximum diameter above 19 mm or a collapse of 30% or less on sniff testing implies an RAP above 10 mm Hg.22 The tricuspid regurgitant estimate of RVSP can underestimate or overestimate pulmonary artery systolic pressure and does not measure mPAP or indicate whether the pulmonary hypertension relates to intrinsic pulmonary vascular disease.23
Hence, echocardiographic findings suggestive of pulmonary hypertension should prompt referral to specialized centers for further evaluation to confirm the hemodynamic abnormality and classify the patient into the correct pulmonary hypertension group.24 This is important, as treatment and prognosis vary considerably by the cause of pulmonary hypertension. Figure 2 summarizes the diagnostic algorithm for PAH. Pulmonary function tests and overnight oximetry are needed to evaluate for chronic hypoxic lung disease. The ventilation/perfusion scan is the diagnostic test of choice for excluding chronic pulmonary thromboembolic pulmonary hypertension (CTEPH), as it is more sensitive than a computed tomography pulmonary angiogram in identifying distal CTEPH.25 An invasive pulmonary angiogram or computed tomography angiogram of the chest may be done for patients with a high probability ventilation/perfusion scan for confirmatory diagnosis. Exclusion of CTEPH is important, as it can be cured by surgical pulmonary thromboendarterectomy in suitable candidates. Serological testing helps to identify associated causes of PAH, such as connective tissue disease or cirrhosis. RHC is mandatory for confirming the diagnosis of PAH and is required before PAH targeted treatment is started. RHC also assesses the severity of pulmonary hypertension, evaluates potential left heart disease, and identifies the subset of patients who respond favorably to acute vasodilators. Figure 3 illustrates the test results in a typical patient with PAH.
Fig 2.

Diagnostic testing algorithm for pulmonary arterial hypertension. 6MWT=six minute walk test; ABGs=arterial blood gases; ANA=antinuclear antibody serology; BNP=brain natriuretic peptide; CBC=complete blood count; COPD=chronic obstructive pulmonary disease; CPET=cardiopulmonary exercise test; CT=computed tomography; CTD=connective tissue disease; CXR=chest x ray; DLCO=diffusion capacity of the lungs for carbon monoxide; ECG=electrocardiogram; LHD= left heart disease; HRCT=high resolution computed tomography of the chest; LFT=liver function tests; MRI=magnetic resonance imaging; PA=pulmonary artery; PAP/CO=pulmonary artery pressure and cardiac output; PEA=pulmonary endarterectomy; PFT=pulmonary function tests; PH=pulmonary hypertension; RHC=right heart catheterization; RV=right ventricle; RVE=right ventricular enlargement; TEE=transesophageal echocardiography; TR=tricuspid regurgitation; Tx=treatment; TTE=transthoracic echocardiogram; V/Q scan=ventilation/perfusion scintigram. This figure is modified to reflect the authors’ practice but was based on a figure in McLaughlin VV, et al. J Am Coll Cardiol 2009;53:1573-161924
Fig 3.

Images obtained from patient with pulmonary arterial hypertension (PAH). (A) R wave predominance, ST segment depression, and T wave inversion in V1 and V2 suggestive of right ventricular hypertrophy. (B) Chest radiograph showing enlarged pulmonary artery and pruning of distal pulmonary vasculature. (C) cardiac magnetic resonance imaging showing severe right ventricular (RV) dilatation, RV hypertrophy, and flattening of interventricular septum in short axis view. (D) Apical four chamber view echocardiography showing severe RV dilatation and short axis view showing flattened D shaped interventricular septum. (E) Tricuspid regurgitation velocity* is proportional to right ventricular systolic pressure and estimated by Bernoulli's equation. (F) Ventilation/perfusion scan showing patchy perfusion defects (“moth eaten” appearance). (G) Pulmonary function test showing isolated moderate reduction in diffusion lung capacity (DLCO) with normal volumes. (H) Right heart catheterization data showing severely elevated pulmonary artery (PA) pressures and pulmonary vascular resistance (PVR) with normal pulmonary capillary wedge pressure (PCWP) typical of PAH. Cardiac output (CO) is reduced with normal right atrial (RA) pressure. FEV1=forced expiratory volume in 1 sec; FVC=forced vital capacity; TLC=total lung capacity
Many patients with PAH have more than one condition contributing to their total pulmonary hypertension burden. Endothelial dysfunction and hypoxia, although not causal, often exacerbate PAH. The clinician is often challenged to determine whether a patient has PAH that is somewhat exacerbated by concomitant mild lung disease, left heart disease, or pulmonary embolism. Conversely, many group 2 patients, such as those with mitral stenosis, and group 3 patients with chronic obstructive pulmonary disease or interstitial lung disease have a disproportionate pulmonary hypertensive response, which reflects underlying pulmonary arterial vasoconstriction, adverse remodeling, or both. Such group 2 and 3 pulmonary hypertension patients have physiology that is reminiscent of PAH, notably elevated PVR26 27
Differentiating PAH from pulmonary hypertension due to HFpEF
Among the most common challenges in clinical practice is distinguishing between PAH and group 2 pulmonary hypertension secondary to HFpEF.27 Both syndromes have normal left ventricular systolic function and abnormal left ventricular diastolic parameters. Correct case classification is crucial, as HFpEF accounts for a considerable proportion of referrals to pulmonary hypertension centers,28 and PAH specific therapies are not effective in group 2 pulmonary hypertension.29 30 Two metrics that differentiate the HFpEF group are increased left atrial size on echocardiography and increased PCWP on RHC. In normal older patients the mean PCWP is 9 (SD 3) mm Hg in men and 11 (3) mm Hg in women.31 Including patients with PCWP values above 15 mm Hg in the group 1 category is thus inappropriate. Skilled measurement of PCWP is critical to distinguish between group 2 pulmonary hypertension and PAH (fig 4).32 End expiratory PCWP as opposed to mean PCWP correlates better with left ventricular end diastolic pressure (LVEDP).32 33 If an accurate PCWP cannot be obtained, left heart catheterization should be done to measure LVEDP.
Fig 4.

Accurate method to measure pulmonary capillary wedge pressure (PCWP) tracing to differentiate pulmonary arterial hypertension from pulmonary hypertension due to heart failure with preserved ejection fraction. Pressure measurement should be made at end expiration. Computer generated (digital) mean pressures should be avoided as it underestimates pressures. Reproduced with permission from Ryan JJ, et al. Am Heart J 2012;163:589-9432
Clinical and echocardiographic models can differentiate between group 2 pulmonary hypertension and PAH (fig 5).27 34 Older patients with more cardiovascular comorbidities are more likely to have pulmonary hypertension due to HFpEF than PAH. Whereas the clinical model has greater than 90% accuracy in differentiating pulmonary hypertension secondary to HFpEF and PAH, the echocardiographic model has only 59% specificity for identifying PAH. Exercise hemodynamic assessment and volume challenge during RHC have also been proposed to distinguish PAH from pulmonary hypertension due to HFpEF.35 36 However, these techniques have not been standardized and should be used only in experienced centers.
Fig 5.
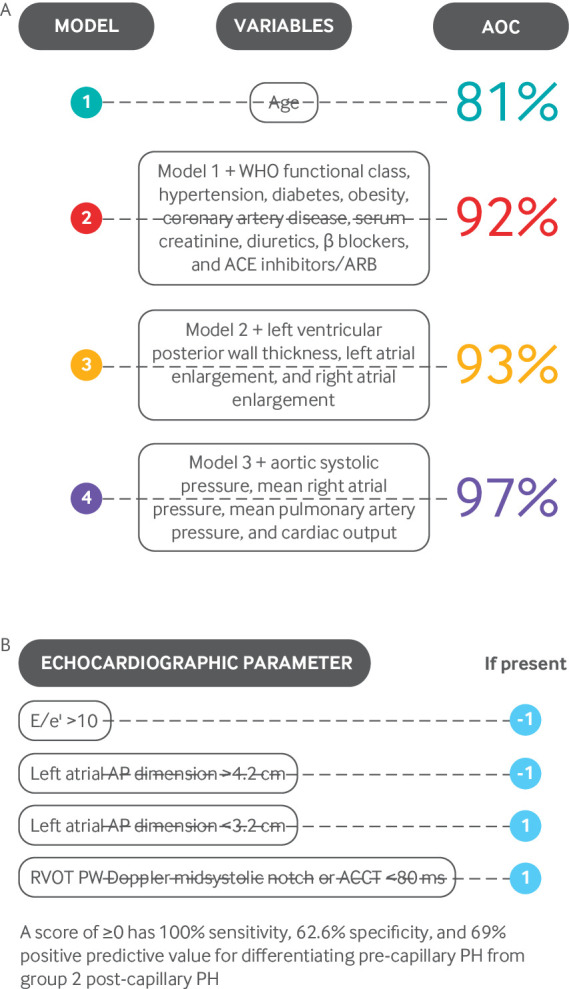
Clinical and echocardiographic models for differentiating pulmonary arterial hypertension from pulmonary hypertension (PH) due to heart failure with preserved ejection fraction. ACCT=pulmonary artery acceleration time; ACE=angiotensin converting enzyme; AOC=area under curve; AP=anteroposterior; ARB=angiotensin receptor blocker; PW=pulse wave; RVOT=right ventricular outflow tract. A: Adapted from Thenappan T, et al. Circulation Heart Fail 2011;4:257-65.27 B): Adapted with permission from Opotowsky AR, et al. Circ Cardiovasc Imaging 2012;5:765-7534
Differentiating PAH from pulmonary hypertension due to chronic lung disease or hypoxia
Table 2 lists the characteristics that can help physicians in differentiating PAH from group 3 pulmonary hypertension due to chronic lung disease. This is important, as PAH specific therapies have not been shown to be effective in pulmonary hypertension due to lung disease.38
Table 2.
Characteristics differentiating pulmonary arterial hypertension from group 3 pulmonary hypertension37
| Characteristics | Pulmonary arterial hypertension | Group 3 pulmonary hypertension |
|---|---|---|
| Ventilatory function: | ||
| FEV1 (in COPD) | >60% predicted | ≤60% predicted |
| FVC (in ILD) | >70% predicted | ≤70% predicted |
| TLC (in ILD) | >60% predicted | ≤60% predicted |
| High resolution CT scan of chest | Absence of or only modest airway or parenchymal abnormalities | Characteristic airway and/or parenchymal abnormalities |
| Cardiopulmonary exercise testing | Circulatory limitations | Ventilatory limitations |
| Preserved breathing reserve | Reduced breathing reserve | |
| Reduced oxygen pulse | Normal oxygen pulse | |
| Low CO/VO2 slope | Normal CO/VO2 slope | |
| No change or decrease in PaCO2 during exercise | Increase in PaCO2 during exercise | |
| Mixed venous oxygen saturation at lower limit | Mixed venous oxygen saturation above lower limit |
CO=cardiac output; COPD=chronic obstructive pulmonary disease; CT=computed tomography; FEV1=forced expiratory volume in 1 sec; FVC=forced vital capacity; ILD=interstitial lung disease; PaCO2=partial pressure of carbon dioxide in arterial blood; TLC=total lung capacity; VO2=oxygen consumption.
Assessing right ventricular function
Right ventricular function is the major determinant of long term outcomes in PAH.39 Several echocardiographic parameters that reflect increased right ventricular and right atrial size or decreased right ventricular contractility are better prognostic indicators in PAH than RVSP.39 Increased mortality risk is predicted by reduced right ventricular systolic and diastolic function, the presence of pericardial effusion, increased right ventricular or right atrial size, decreased tricuspid annular plane systolic excursion (TAPSE) (fig 6A), decreased Tei index (fig 6B),40 reduced peak systolic tricuspid lateral annular velocity (S’) (fig 6C),41 42 decreased isovolumic contraction velocity (fig 6C),43 44 45 46 47 48 and abnormal right ventricular free wall strain.
Fig 6.
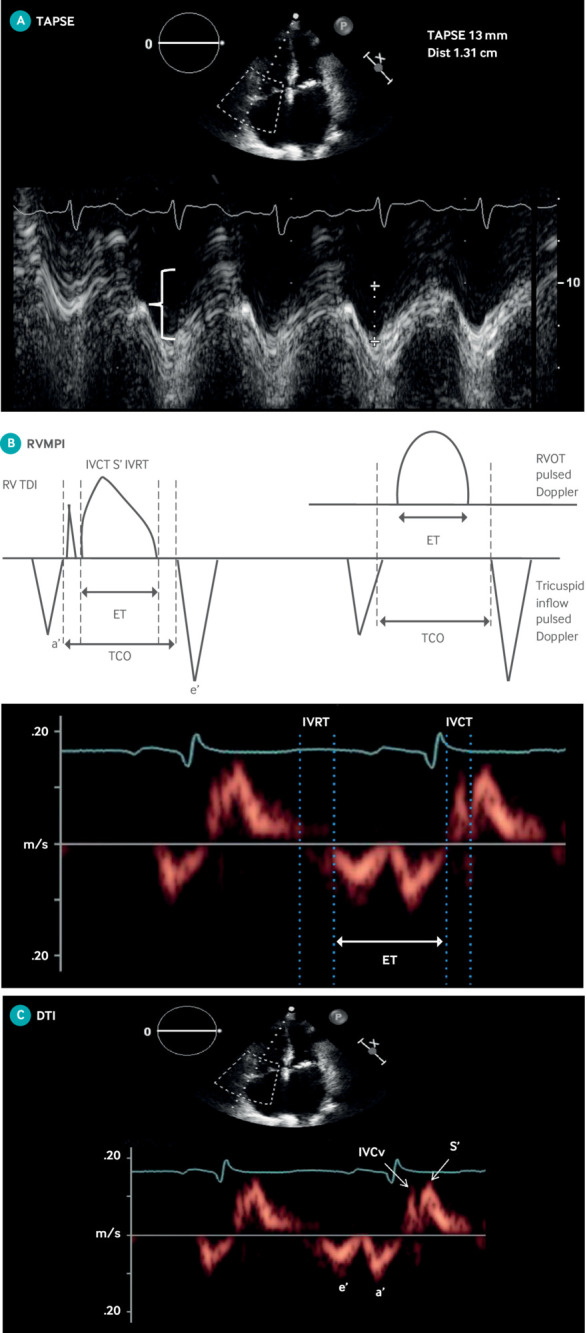
Echocardiographic measures of right ventricular function. (A) TAPSE. M mode cursor placed through right ventricular (RV) apex to lateral tricuspid annulus in apical four chamber view for purpose of measuring distance traveled by annulus in centimeters from end diastole to end systole. Abnormal TAPSE of 1.3 cm is noted by crosshatches. (B) Right ventricular myocardial performance index (RVMPI—Tei index) is defined as sum of isovolumic contraction (IVCT) and isovolumic relaxation (IVRT) time divided by ejection time (ET). Above is representation of two ways to calculate RVMPI, on tissue Doppler and on pulsed wave Doppler. Below are IVCT, IVRT, and ET where RVMPI=(IVCT+IVRT)/ET. (C) Doppler tissue imaging (DTI) of tricuspid annulus. S’ is highest systolic velocity measured by pulsed DTI of tricuspid annulus. Isovolumic contraction velocity (IVCv) is defined as peak velocity by DTI measurement at level of tricuspid annulus in early systole when right ventricle contracts and pressures acutely rise without any change in ventricular volume. These measurements can be done after high frame rate acquisition with color coded Doppler offline (not shown)
Unlike the left ventricle, the right ventricle shortens from apex to base with contraction. TAPSE assesses longitudinal right ventricular shortening by measuring annular excursion in the apical four chamber view, using M mode ultrasound (fig 6A). A TAPSE below 18 mm predicts increased mortality in PAH.44 Although convenient and reproducible, TAPSE measures only basal right ventricular systolic function and is not as accurate a measure of global right ventricular function as cardiac magnetic resonance imaging (MRI) or three dimensional echocardiography.49 TAPSE can be influenced by left ventricular function.50 51 In addition, the timing of the deterioration in TAPSE relative to the onset of RVF is poorly defined.52
The geometry of the right ventricle and its heavy trabeculation make two dimensional transthoracic estimation of right ventricular function and volume difficult. Three dimensional echocardiography has improved the ability to accurately assess the right ventricle.53 Right ventricular volume acquisitions and right ventricular ejection fraction (RVEF) measurements using three dimensional echocardiography are accurate and reproducible with minimal inter-observer variability (approximately 4%).54 Changes in right ventricular volumes and function measured by three dimensional echocardiography correlate with clinical outcomes in PAH.55 However, because of its low measurement variability, cardiac MRI is more accurate and reproducible than either the two or three dimensional transthoracic technique. For example, to detect a change of 5% in RVEF in a clinical trial population, two and three dimensional echocardiography would require twice and 2.5 times the patient sample size needed for cardiac MRI.49
Current treatments for PAH
The current treatment strategy for PAH can be broadly divided into general measures, supportive therapies (box 1), and PAH specific therapies.
Box 1. General supportive measures and background therapies in PAH management.
General supportive measures
Restrict salt and fluid intake to reduce volume overload in light of their limited right ventricular reserve
Supplemental oxygen therapy should be used if needed to maintain systemic oxygen saturation above 90% as it improves exercise capacity56
Exercise training is advocated in PAH. Exercise training increases six minute walk distance (+52 m), peak oxygen uptake, and quality of life in PAH.57 Low level, symptom limited, aerobic exercises are preferred, and intense isometric exercises can lead to syncope
Vaccination against influenza and pneumococcal pneumonia is recommended
Women in the reproductive age group are strongly counseled to use contraceptive measures. Pregnancy is associated with substantial mortality in PAH.58 Hysteroscopic sterilization is the preferred method of contraception, but other options include progesterone-only intrauterine devices and pills and tubal ligation.59 Estrogen containing contraceptives and injectable progesterone are contraindicated because they increase the risk of thrombosis59
Background therapies for PAH
Diuretics—Diuretics are used to treat venous congestion resulting from RVF
Digoxin—Digoxin improves right ventricular contractility and cardiac output in acute hemodynamic studies60; however, no data support its long term use in PAH. The benefits and risks should be weighed
Anticoagulation—In situ thrombosis occurs in the small resistance pulmonary arteries of PAH patients.61 Retrospective and prospective studies have reported improved survival with long term warfarin therapy in PAH.61 62 63 Registry data have questioned the benefits of anticoagulation, especially in patients with APAH.64 65 In the COMPERA registry, the use of anticoagulation was associated with a 21% improvement in survival in patients with IPAH but no benefit in those with APAH.64 The REVEAL registry found no survival benefit with anticoagulation in IPAH and reduced survival in APAH.65 Long term warfarin therapy is recommended only in IPAH, heritable PAH, or anorexigen associated PAH patients with an international normalized ratio goal of 1.5-2.5.24 66 The role of novel oral anticoagulants is unknown
PAH specific therapies
Fourteen PAH specific therapies are available for PAH. They target components of four PAH relevant molecular pathways: voltage gated, L type calcium channels, nitric oxide cyclic guanosine monophosphate (cGMP), endothelin, and prostacyclin.2 These drugs do not directly target the adverse vascular remodeling that obliterates, obstructs, and stiffens the pulmonary vasculature, and most do not improve the function of the right ventricle. The vasodilator therapies may indirectly inhibit cell proliferation and regress adverse vascular remodeling when given chronically, particularly if they reduce mPAP. For example, pulmonary artery banding, which reduces pulmonary arterial pressure distal to the band, reduces downstream adverse vascular remodeling in the Sugen 5416/hypoxia rat model of PAH.67 This suggests that pulmonary hypertension begets pulmonary hypertension and conversely implies that improving hemodynamics, however achieved, may regress vascular remodeling.
Calcium channel blockers
Calcium channel blockers (CCBs) are effective in the 5-10% of PAH patients who respond to acute vasodilatory challenge during RHC with a drop in mPAP by between 10 and 40 mm Hg, with no drop in cardiac output.68 Inhaled nitric oxide, intravenous epoprostenol, or intravenous adenosine are used for acute vasodilator testing. Vasoreactivity may be due to genetic predisposition, resulting from enrichment in vascular smooth muscle cell contraction gene variants. Early studies suggest that vasoreactivity can potentially be identified on the basis of a simple peripheral blood mRNA expression profile (reduced levels of mRNA for DSG2, a desmosomal cadherin, and RHOQ, a cytoskeletal protein).69 70
CCBs are indicated only in PAH patients with a documented, positive vasodilator test. These patients are uncommon (5-10% of all cases) and have a different natural history with a five year survival rate of 90% with CCB monotherapy.62 Eligible PAH patients generally need higher than usual doses of CCB: amlodipine 20 mg, nifedipine 120-240 mg, or diltiazem 240-720 mg daily. Verapamil is not used because of its negative inotropic effects. Although diltiazem can also have a negative inotropic effect, it is preferred over amlodipine or nifedipine in patients with sinus or atrial tachycardia. Patients treated with CCBs should be closely monitored for adequate response and transitioned to PAH specific therapies if symptoms progress. Adequate long term response to CCBs in patients with APAH is rare.71
Drugs targeting the nitric oxide pathway
Nitric oxide is a potent pulmonary vasodilator that activates soluble guanylate cyclase (sGC) to generate cGMP. cGMP causes pulmonary artery smooth muscle cell (PASMC) relaxation through cGMP dependent protein kinases, which activate downstream targets, including the large conductance, calcium sensitive potassium channel BKca.72 Patients with PAH have reduced lung expression of endothelial nitric oxide synthase (eNOS), which synthesizes nitric oxide, and increased expression of phosphodiesterase 5 (PDE5), which degrades cGMP to 5′-GMP. The resulting decrease in cGMP is implicated in adverse pulmonary vascular remodeling in PAH.72 PDE5 inhibitors and sGC stimulators augment the nitric oxide-cGMP pathway and are approved for treating PAH. Although inhaled nitric oxide is useful for acute treatment in the intensive care unit and for acute vasodilator testing,66 73 it is not used for chronic ambulatory therapy owing to the logistical challenges of delivering this short lived gas. A phase III trial is evaluating the safety and efficacy of long term inhaled nitric oxide in PAH (NCT02725372).
PDE5 inhibitors—Sildenafil and tadalafil are approved for treatment of PAH.72 Sildenafil improves six minute walk distance (6MWD), World Health Organization functional class, and mPAP at all three doses studied (20 mg, 40 mg, and 80 mg three times a day) (table 3).77 It had no effect on clinical worsening. Sildenafil is approved at 20 mg three times a day for treatment of PAH. However, long term extension studies showed significant improvement in hemodynamics with higher doses.77 Tadalafil (40 mg daily) increases 6MWD, time to clinical worsening, and health related quality of life (table 3).79 The major side effects of PDE5 inhibitors include headache, flushing, dyspepsia, and epistaxis.77 79
Table 3.
Landmark clinical trials in pulmonary hypertension
| Date | Interventions | Characteristics | Study cohort | Primary outcome | Results |
|---|---|---|---|---|---|
| 199674 | IV epoprostenol v placebo | 12 week, prospective, randomized, open trial | 81 patients: IPAH, heritable, drug induced PAH | 6MWD; survival | 6MWD improved by 32 m (P=0.002); no deaths with epoprostenol but deaths in control group (P=0.003) |
| 200075 | IV epoprostenol v placebo | 12 week, prospective, randomized, open trial | 111 patients: scleroderma related PAH | 6MWD | Difference in 6MWD between treatment groups: 108 (95% CI 55 to 180) m (P<0.001) |
| 2002 (BREATH)76 | Bosentan 125 mg BID v placebo | 12 week, prospective, randomized, double blind trial | 213 patients: IPAH, heritable, drug induced PAH, CTD-PAH (scleroderma and lupus) | 6MWD | Difference in 6MWD between treatment groups: 44 (21 to 67) m (P<0.001) |
| 200275 | SC treprostinil v placebo | 12 week, prospective, randomized, double blind trial | 470 patients: IPAH, heritable, drug induced PAH, CTD-PAH, CHD-PAH | 6MWD | Difference in 6MWD between treatment groups: 16 (4.4 to 27.6) m (P=0.006) |
| 2005 (SUPER)77 | Sildenafil (20 mg, 40 mg, and 80 mg) v placebo | 12 week, prospective, randomized, double blind trial | 278 patients: IPAH, CTD-PAH, CHD-PAH | 6MWD | Difference in 6MWD between treatment groups v placebo: 20 mg—45 (21 to 70) m (P<0.01); 40 mg—46 (20 to 72) m (P<0.01); 80 mg—50 (23 to 70) m (P<0.01) |
| 2008 ARIES 178 | Ambrisentan (5 mg and 10 mg) v placebo | 12 week, prospective, randomized, double blind trial | 202 patients: IPAH, drug induced PAH, CTD-PAH | 6MWD | Difference in 6MWD between treatment groups v placebo: 5 mg—31 (3 to 59) m (P=0.008); 10 mg—51 (21 to 76) m (P<0.001) |
| 2008 ARIES 278 | Ambrisentan (2.5 mg and 5 mg) v placebo | 12 week, prospective, randomized, double blind trial | 192 patients: IPAH, drug induced PAH, CTD-PAH | 6MWD | Difference in 6MWD between treatment groups v placebo: 2.5 mg—32 (2 to 63) m (P=0.008); 5 mg—59 (30 to 89) m (P<0.001) |
| 2009 (PHIRST)79 | Tadalafil (2.5 mg, 10 mg, 20 mg, and 40 mg) v placebo | 16 week, prospective, randomized, double blind trial | 405 patients: IPAH, heritable, drug induced PAH, CTD-PAH, CHD-PAH | 6MWD | Difference in 6MWD between treatment groups v placebo: 2.5 mg—14 (6 to 33) m (P=NS); 10 mg—20 (1 to 39) m (P=NS); 20 mg—27 (11 to 44) m (P=NS); 40 mg—33 (15 to 50) m (P<0.01) |
| 201080 | Inhaled treprostinil 9 breaths QID v placebo | 12 week, prospective, randomized, double blind trial | 225 patients on background bosentan or sildenafil: IPAH, heritable, CTD-PAH, HIV-PAH, drug induced PAH | 6MWD | Difference in 6MWD between treatment groups: 20 (8 to 32.8) m (P<0.001) |
| 2013 (PATENT)81 | Riociguat 2.5 mg v placebo | 12 week, prospective, randomized, double blind trial | 443 patients: IPAH, heritable, CTD-PAH, CHD-PAH, PoPH, drug induced PAH | 6MWD | Difference in 6MWD between treatment groups: 36 m (95% CI, 20 m to 52 m, p<0.001) |
| 2012 (FREEDOM M)82 | Oral treprostinil v placebo | 16 week, prospective, randomized, double blind trial | 350 patients on ERA, PDE5 inhibitor, or both: IPAH, heritable, drug induced PAH, CTD-PAH, CHD-PAH, HIV-PAH | 6MWD | Difference in 6MWD between treatment groups: 11 (0 to 22) m (P=0.07) |
| 2013 (FREEDOM C)83 | Oral treprostinil v placebo | 12 week, prospective, randomized, double blind trial | 349 treatment-naive patients: IPAH, heritable, drug induced PAH, CTD-PAH, CHD-PAH, HIV-PAH | 6MWD | Difference in 6MWD between treatment groups: 23.0 (4 to 41) m (P=0.0125) |
| 2013 (SERAPHIN)84 | Macitentan (3 mg and 10 mg) v placebo | Event driven, prospective, randomized, double blind trial | 750 patients: IPAH, heritable, drug induced PAH, CTD-PAH, CHD-PAH, HIV-PAH | Time to composite endpoint of death, worsening of PAH, initiation of IV/SC prostanoids, lung transplant, atrial septostomy | HR: 3 mg dose—0.70 (97.5% CI 0.52 to 0.96) (P=0.01); 10 mg dose—0.55 (0.39 to 0.76) (P<0.001) |
| 2015 (GRIPHON)85 | Selexipag v placebo | Event driven, prospective, randomized, double blind trial | 1156 patients: IPAH, heritable, drug induced PAH, CTD-PAH, CHD-PAH, HIV-PAH | Time to composite endpoint of death, PAH hospital admission, initiation of IV/SC prostanoid therapy or long term oxygen therapy, lung transplantation, atrial septostomy | HR: 0.60 (99% CI 0.46 to 0.78) (P<0.001) |
| 2015 (AMBITION)86 | Ambrisentan + tadalafil v ambrisentan or tadalafil monotherapy | Event driven, prospective, randomized, double blind trial | 605 treatment-naive patients: IPAH, heritable, drug induced PAH, CTD-PAH, CHD-PAH, HIV-PAH | Time to composite endpoint of death, PAH hospital admission, PAH worsening, unsatisfactory long term clinical response | HR for combination therapy v pooled monotherapy: 0.50 (95% CI 0.35 to 0.72) (P<0.001) |
6MWD=six minute walk distance; BID=twice daily; CHD=congenital heart disease; CTD=connective tissue disease; ERA=endothelin receptor blockers; HR=hazard ratio; IPAH=idiopathic pulmonary arterial hypertension; IV=intravenous; PAH=pulmonary arterial hypertension; PDE5=phosphodiesterase; QID=four times a day; SC=subcutaneous.
sGC stimulators—sGC is a heterodimeric enzyme that generates cGMP. In PAH, sGC is often dysfunctional because it is oxidized or has lost its heme group.87 Riociguat directly stimulates sGC independent of nitric oxide, resulting in increased cGMP and pulmonary vasodilation. Riociguat 2.5 mg three times a day improves 6MWD, PVR, serum N terminal pro B type natriuretic peptide (NT-proBNP) concentrations, time to clinical worsening, and WHO functional class (table 3).81 The improvement in 6MWD is observed both in treatment-naive patients and those taking other PAH specific therapies.81 sGC and PDE5 inhibitors should not be given concurrently owing to the risk of hypotension. However, transition to sGC from PDE5 inhibitors improves exercise capacity and hemodynamics in patients who have inadequate responses to PDE5 inhibitors.88 Headache, dizziness, hypotension, dyspepsia, and gastroesophageal reflux are the most common adverse effects of riociguat.
Drugs targeting the endothelin pathway: endothelin receptors antagonists
Endothelin is a potent vasoconstrictor and smooth muscle mitogen. It acts through endothelin A and endothelin B receptors. Endothelin is overexpressed in the lungs and plasma of patients with PAH. The endothelial receptor antagonists (ERAs) bosentan, ambrisentan, and macitentan are beneficial in PAH.
Bosentan and macitentan are dual ERAs, blocking both endothelin A and endothelin B receptors. Bosentan 125 mg twice a day improves 6MWD, WHO functional class, Borg dyspnea score, and time to clinical worsening (table 3).76 Bosentan also improves PVR and 6MWD in patients with mild symptoms (WHO functional class II).89 Compared with bosentan, macitentan has greater tissue penetration and produces more sustained receptor blockade. Macitentan 3 mg and 10 mg daily reduce a composite endpoint of long term morbidity and mortality by 30% and 45%, respectively (table 3).84 Importantly, this is driven exclusively by the reduction in worsening of PAH, and no reduction is seen in either all cause or PAH related mortality.84 Ambrisentan (10 mg daily), a selective ERA antagonist, improves 6MWD, time to clinical worsening, WHO functional class, quality of life, and NT-proBNP when given as monotherapy.78
The major adverse effects of ERAs include hepatotoxicity, peripheral edema, anemia, and nasal congestion.90 In a meta-analysis, hepatotoxicity was more commonly observed with bosentan, anemia with bosentan and macitentan, and peripheral edema with bosentan and ambrisentan.90 Monthly liver function testing is warranted in patients taking bosentan. Although not mandated, serial liver function testing is advisable in patients taking macitentan and ambrisentan. ERAs should be discontinued if liver aminotransferases are more than five times the upper limit of normal, if aminotransferase elevations are accompanied by an increase in serum bilirubin concentrations (more than twice the upper limit of normal), or if patients develop signs of hepatic failure. Bosentan has several relevant drug interactions, notably decreasing serum sildenafil concentrations, which can impair the clinical benefits of combining these drugs.91 92 Bosentan decreases warfarin concentrations, necessitating close monitoring of the international normalized ratio.93
Drugs targeting the prostacyclin pathway
Prostacyclin and prostanoids bind IP receptors, which increases cyclic adenosine monophosphate concentrations causing non-selective pulmonary vasodilatation.94 They also have antiplatelet, antithrombotic, antiproliferative, and anti-inflammatory properties.94 Prostacyclin expression is reduced in the lungs of patients with PAH.2 Depending on the preparation and specific molecule, prostanoids can be given as a continuous infusion (intravenously (epoprostenol and treprostinil) or subcutaneously (treprostinil)), via inhalation (iloprost and treprostinil), or orally (treprostinil and beraprost).
Epoprostenol is a synthetic prostacyclin analog. It has a very short half life (less than five minutes) and is unstable at room temperature. Therefore, it must be refrigerated and administered by continuous intravenous infusion. Epoprostenol improves exercise capacity, mPAP, PVR, cardiac output, quality of life, and survival (table 3).74 75 The administration of epoprostenol requires a complex delivery system including a continuous infusion pump, central venous catheter, and ice packs to maintain temperature. It must be administered through a central venous catheter, as it is an irritant to the peripheral veins. A newer formulation (Veletri) is stable at room temperature, simplifying administration. Epoprostenol is usually started at 2 ng/kg/min, and the dose is gradually increased. The average dose of epoprostenol used for PAH ranges from 25 to 40 ng/kg/min. The most frequent adverse effects include gastrointestinal symptoms (nausea, vomiting, and diarrhea), headache, flushing, and jaw pain.
Treprostinil is a prostacyclin analog that has several advantages, including a longer half life (three hours) and stability at room temperature. Various formulations of treprostinil are approved to treat PAH (subcutaneous, intravenous, inhalational, and oral). Subcutaneous treprostinil improves 6MWD, hemodynamics, and quality of life (table 3).95 The major limitation of subcutaneous administration is infusion site pain, which occurs in 85% of patients.95 Although intravenous treprostinil has not been studied in a randomized trial, the US Food and Drug Administration approved its use for PAH on the basis of bioequivalence. Open label, long term extension studies have shown improvement in exercise capacity and hemodynamics and delay in time to clinical worsening with intravenous treprostinil.96 Subcutaneous or intravenous treprostinil is started at 2 ng/kg/min and increased gradually to an average target dose of 60-80 ng/kg/min. The adverse effects of treprostinil and epoprostenol are similar.
Catheter related bloodstream infection in patients receiving intravenous prostanoids can be life threatening. Patients must apply a meticulously sterile technique to reduce infection when refilling the pump at home. A fully implantable pump decreases catheter related side effects and increases patients’ satisfaction97; however, this pump is not approved for clinical use.
Inhaled treprostinil improves 6MWD and quality of life as an add-on therapy for patients who have symptoms despite taking sildenafil or bosentan (table 3).80 Inhaled treprostinil is started at three inhalations four times a day and increased gradually to a maximum of nine to 12 inhalations four times a day. It has minimal systemic effects owing to reduced systemic absorption and also lacks catheter related side effects. The common adverse effects include dry cough and headache.80
Treprostinil diolamine is an oral, salt form of treprostinil. It improves 6MWD in treatment-naive PAH patients with no improvement in WHO functional class or time to clinical worsening.14 82 However, no significant benefit is seen when oral treprostinil is used as an add-on therapy in patients who have symptoms when taking an ERA or a PDE5 inhibitor.83 Thus, oral treprostinil is approved only as a monotherapy for improving exercise capacity in treatment-naive PAH patients. Oral treprostinil is started at 0.125 mg three times a day and increased by 0.125 mg every three to four days. Common adverse effects include nausea, diarrhea, headache, and jaw pain.82
Selexipag is an orally available, non-prostanoid activator of IP receptors.98 Both the parent drug and its metabolite have a high affinity for IP receptors. This selectivity minimizes adverse effects and facilitates dose escalation. In a long term, event driven trial, selexipag reduced the composite endpoint of death, lung transplantation, atrial septostomy, hospital admission for worsening PAH, or worsening of PAH by 40% (table 3).85 The benefits were mainly driven by reduction in hospital admissions for PAH, with no improvement in mortality. The reduction in morbidity was not dose dependent. Selexipag is started at 200 μg twice daily and increased weekly to a maximum of 1600 μg twice daily. The common adverse effects include nausea, vomiting, diarrhea, headache, and jaw pain.
Treatment approach
Patients with positive acute vasodilator testing should be treated initially with CCBs and monitored closely. Non-responders to vasodilator are stratified on the basis of clinical, echocardiographic, and hemodynamic evaluations that assess right ventricular function as high or low risk (table 4). Patients with a high risk profile have worse survival.99 100 101 Thus, patients at high risk should be considered for initial parenteral prostanoid therapy. Epoprostenol is preferred in these patients given its survival benefit. In a retrospective review of 19 patients at high risk presenting with cardiac index less than 2 L/min/m2 and PVR greater than 20 WU, starting triple combination therapy with epoprostenol, a PDE5 inhibitor, and an ERA at diagnosis was associated with improvement in exercise capacity and hemodynamics and 100% survival at three years.102 The efficacy, safety, and cost effectiveness of this approach needs further assessment in a randomized controlled trial.
Table 4.
Risk assessment in pulmonary arterial hypertension patients for treatment approach24
| Determinants of risk | Low risk | High risk |
|---|---|---|
| Evidence of RV failure | No | Yes |
| Progression | Gradual | Rapid |
| WHO class | II, III | IV |
| Six minute walk distance | Longer (>400-500 m) | Shorter (<300 m) |
| Cardiopulmonary exercise testing | VO2max>14.5 mL/min/kg | VO2max<12 mL/min/kg |
| BNP/NT-proBNP | Minimally elevated and stable | Very elevated and/or rising |
| Blood gases | PaCO2>34 mm Hg | PaCO2<32 mm Hg |
| Echocardiographic findings | Minimal RV dysfunction TAPSE>2.0 cm | Pericardial effusion RV dysfunction TAPSE<1.5 cm |
| Hemodynamics | Normal/near normal RAP and CI | High RAP, low CI |
BNP=brain natriuretic peptide; CI=cardiac index; NT-proBNP=N terminal pro B type natriuretic peptide; PaCO2=partial pressure of carbon dioxide; RAP=right atrial pressure; RV=right ventricular; TAPSE=tricuspid annular plane systolic excursion; VO2max=maximum oxygen consumption; WHO=World Health Organization.
Patients with a low risk profile are treated by either sequential combination therapy or initial combination therapy. In the first approach, patients are started on oral monotherapy. A second drug, targeting a different pathway, is added if the response to monotherapy is inadequate. In the initial combination therapy approach, patients are started on an oral combination of PDE5 inhibitor and ERA simultaneously at diagnosis.66 Compared with initial monotherapy, initial combination therapy with ambrisentan and tadalafil improved a composite endpoint of death, lung transplantation, hospital admission for worsening PAH, worsening PAH, and persistent worsening after six months by 50% (table 3)86; however, no mortality advantage was seen.86 On the basis of these results, initial combination therapy has been preferred over sequential combination therapy in most patients. Sequential combination therapy is considered primarily for patients with mild disease and preserved right ventricular function.
Patients should be routinely monitored to ensure an adequate response; in those with inadequate responses to combination therapy, a third agent should be added.66 Patients who have high risk features despite receiving combination therapy should receive parenteral prostacyclin therapy. For patients who are not at treatment goal but who lack high risk features, addition of inhaled treprostinil or selexipag or starting a transition from PDE5 inhibitor to sGC stimulators can improve outcomes. The role of oral treprostinil therapy in patients who are already receiving combination therapy is limited, as it is known to improve exercise capacity only in treatment-naive patients.
Patients who continue to have WHO functional class III or IV symptoms on maximal medical therapy should be referred as early as possible for lung transplantation.24 Atrial septostomy is considered as a bridge to transplant in patients who are unstable while awaiting transplant or as a palliative procedure in patients who are not candidates for transplant.24 Veno-arterial extracorporeal membrane oxygenators and pumpless oxygenators (Novo lung) have also been used successfully as a bridge to transplant in patients with PAH and RVF.103
Pathogenesis of PAH
Despite the benefits of current therapies to the quality of life and time to clinical worsening, these treatments do not decrease mortality, apart from epoprostenol which improved survival in PAH patients with WHO functional class IV.74 The average improvement in functional capacity (6MWD) and hemodynamics (mPAP) for each of the four classes of PAH specific therapies is modest. In randomized clinical trials, the average fall in mPAP with an optimal dose of sGC, PDE5 inhibitor, ERA, or prostanoid is less than 6 mm Hg (table 5).87 The primary criticism of current classes of PAH specific therapies, beyond their limited hemodynamic efficacy, is that they primarily target excessive vasoconstriction, which is a dominant pathophysiologic feature in less than 10% of patients with PAH.4 Better understanding of disease pathogenesis is thus needed to identify new targets for therapy. Ongoing research has provided new understanding of the cellular, genetic, and epigenetic changes that drive pathological vascular remodeling in the lungs of patients with PAH (fig 7). Box 2 describes the five emerging concepts in the pathogenesis of PAH.
Table 5.
Change in hemodynamics with approved pulmonary arterial hypertension specific therapies87
| Drug | MPAP (mm Hg) | PVR (Wood units) | Cardiac index (L/min/m2) |
|---|---|---|---|
| Intravenous epoprostenol | 6.7 (2.6 to 10.7) | 4.9 (2.3 to 7.6) | 0.5 (0.2 to 0.9) |
| Subcutaneous treprostinil | 2.3 (0.5) | 3.5 (0.6) | 0.12 (0.04) |
| Sildenafil | 4.7 (6.7 to 2.8) | 3.3 (1.9 to 4.6) | 0.37 (0.20 to 0.55) |
| Tadalafil | 8.5 (4 to 13) | 3.2 (1.5 to 4.9) | 0.6 (0.1 to 1.6) |
| Riociguat | 4 (2-6) | 2.8 (2.1 to 3.5) | 0.9 (0.7 to 1.2)* |
| Macitentan | 6.4 (2.5 to 10.2) | 0.5 (0.3 to 0.6) | 0.63 (0.33 to 0.93) |
All values represent placebo corrected change as mean (SD) or mean (95% CI).
MPAP=mean pulmonary artery pressure; PVR=pulmonary vascular resistance.
Change in cardiac output rather than change in cardiac index.
Fig 7.

Mechanisms implicated in pathogenesis of pulmonary arterial hypertension (PAH). PAH is a panvasculopathy, meaning that all layers of the vascular wall are involved. PAH is also reflective of gene environment interactions and has important genetic and epigenetic mechanisms. This figure shows abnormalities in the gene and environment, blood, and each layer of the pulmonary artery, from intima (endothelial cells), to media (pulmonary arterial smooth muscle cells—PASMCs) to adventitia (fibroblasts). Because of the many reports that inform this composite figure, individual sources for the information are not referenced. The normal state is shown on the left side, the abnormalities that occur in PAH are highlighted in the middle section, and the consequences of these abnormalities are shown on the right. The net effect of these abnormalities is a state of vasoconstriction, inflammation, thrombosis with a hyperproliferative, apoptosis resistant PASMC population, which promotes vasoconstriction and vascular obstruction, and excessive fibrosis, which reduces vascular compliance. These vascular changes ultimately increase right ventricular (RV) afterload and impair RV-pulmonary artery coupling, leading to RV failure. 5-HHT=5 hydroxytryptamine; ADMA=asymmetric dimethylarginine; APN=adiponectin; BMPR2=bone morphogenetic protein receptor 2; BNP=brain natriuretic peptide; Ca2+=calcium; DNAMT=DNA methyltransferase; Drp-1=dynamin related protein 1; ET1=endothelin; HDAC=histone deacetylases; HIF=hypoxia inducible factor; IL=interleukin; MCP-1=monocyte chemoattractant protein-1; MCUC=mitochondrial calcium uniporter complex; miRNA=micro RNA; MMP=matrix metalloproteinase; NFAT=nuclear factor of activated T cells; NF-kB=nuclear factor kappa light chain enhancer of activated B cells; NK=natural killer cells; NO=nitric oxide; PDGFR=platelet derived growth factor receptor; PDGR=platelet derived growth factor; PDH=pyruvate dehydrogenase; PDK=pyruvate dehydrogenase kinase; PGI2=prostacyclin; PKM-2=pyruvate kinase M2; PPAR=peroxisome proliferator activated receptor; SERCA=sarco-endoplasmic reticulum Ca2+ ATPase; SERT=serotonin transporter; SNP=single nucleotide polymorphism; SOD=superoxide dismutase; Th2=T helper cells; TNF=tumor necrosis factor; TRPC=transient receptor potential cation channel; T-reg=regulatory T cells; TxA2=thromboxane A2; VIP=vasoactive intestinal peptide
Box 2. Concepts in PAH.
Characterization of a cancer-like phenotype in PAH that accounts for the obstructive pulmonary vasculopathy. This is defined by altered mitochondrial metabolic function (Warburg metabolism) and mitochondrial dynamic function (fragmentation). These changes drive cell proliferation and impair apoptosis. They are downstream from genetic mechanisms, such as BMPR2 mutation, and epigenetic mechanisms
PAH is promoted by genetic and epigenetic factors that contribute to disease pathogenesis. The most common genetic mechanism is mutation of BMPR2
PAH is in part a disease of altered immunity and increased inflammation. Improved understanding of the role of chronic inflammation, fibrosis, and immune mediated mechanisms offers potential therapies to reverse adverse vascular remodeling
PAH is predominantly a disease of women, although afflicted men have worse prognosis. There is an emerging appreciation for the importance of sex differences in the incidence, therapeutic responsiveness, and outcomes of PAH
Patients with PAH die from right ventricular failure. There is an increasing recognition of the unique embryologic origins and response to increased afterload of the right ventricle, and the crucial role that right ventricular adaptation plays, both in determining prognosis and as a target for therapy. It seems that the ischemic, metabolically remodeled right ventricle in PAH is an example of hibernating myocardium
Cancer-like metabolic and mitochondrial cellular phenotype of PAH
In 1891 Romberg described medial thickening and occlusive vascular lesions in the lungs of patients with PAH.104 Controversies remain regarding the cellular and molecular origins of this pathological remodeling, including some uncertainly as to which cells proliferate (PASMC, clonal endothelial cells, fibroblasts, inflammatory cells, or some combination of these). There is also debate about the relative importance of an early excess of endothelial apoptosis (at the time of an initial vascular insult) versus a later resistance to apoptosis that promotes vascular obstruction. However, strong evidence from multiple laboratories, using both patient derived primary cell lines and animal models, shows that a proliferative, apoptosis resistant, cancer-like phenotype occurs in PASMCs, endothelial cells, and fibroblasts in established PAH.105 106 107
Cytosolic calcium and ion channels
Increased cytosolic calcium ([Ca2+]cyt) contributes to the contractile, hyperproliferative, and anti-apoptotic phenotype of PAH PASMCs. [Ca2+]cyt is regulated by several ion channels that control calcium influx, as well as Ca2+ sequestration within the sarcoplasmic reticulum and mitochondria. Elevated [Ca2+]cyt in PASMCs from patients with PAH has been linked to the activation of store operated Ca2+ channels, including the transient receptor potential channel TrpC6,108 and downregulation of voltage gated potassium channels, such as Kv1.5.109 Decreased expression of Kv1.5 channels, which normally maintain PASMC membrane potential,110 leads to membrane depolarization and influx of Ca2+ through voltage dependent calcium channels (CaL). In rodent models of pulmonary hypertension, restoring Kv1.5 expression through adenoviral gene transfer improves hemodynamics.111 In addition to promoting smooth muscle cell contractility, prolonged increases in [Ca2+]cyt can increase proliferation by driving cells into the cell cycle.109 Activation of the Ca2+/calcineurin sensitive transcription factor nuclear factor of activated T cells (NFAT) in PAH PASMCs perpetuates the elevation in [Ca2+]cyt by suppressing Kv1.5 expression.112 NFAT activation also promotes apoptosis resistance by increasing expression of bcl-2.112 Failure of mitochondrial calcium uptake, caused by downregulation of the mitochondrial calcium uniporter (MCU), further contributes to the cytosolic calcium overload in PAH.113 Rho kinase activation in PAH causes calcium sensitization, leading to greater vasoconstriction for a given level of [Ca2+]cyt. Moreover, the vasoconstriction caused by rho kinase activation is not reversed by conventional vasodilators and likely contributes to vascular stiffening.114 Rho kinase inhibitors, such as fasudil, have been studied in limited human PAH cohorts.115 They seem to be safe and effective and may reduce mortality in PAH patients with right ventricular failure.116
Although there is little debate that calcium concentrations are elevated in PAH PASMCs, the relative importance of excessive calcium entry via store operated channels and/or CaL channels (which are therapeutically targeted by CCBs) versus impaired organelle uptake of calcium (in mitochondria and endoplasmic reticulum) versus calcium sensitization (by rho kinase) remains uncertain.
Mitochondrial metabolic dysfunction
Mitochondrial dysfunction in PAH PASMCs includes a metabolic shift from glucose oxidation toward uncoupled aerobic glycolysis, a metabolic pattern first described by Otto Warburg in cancer cells. In the presence of oxygen, rates of glycolysis in normal vascular cells are closely coupled to rates of glucose oxidation. In Warburg metabolism, uncoupled glycolysis is increased because, whereas mitochondrial respiration (glucose oxidation) is actively suppressed, glycolysis is disproportionately elevated providing the abnormal cell with sufficient energy to thrive.
Although this disease signature in PAH was initially identified in PASMCs,117 118 similar metabolic and mitochondrial changes occur in pulmonary artery endothelial cells (PAECs)104 119 120 and adventitial fibroblasts of PAH patients.121 More recently, examination of the diseased right ventricle in PAH patients and animal models has identified a similar Warburg phenotype in cardiomyocytes, where it reduces contractility.122
In healthy cells, the pyruvate produced by glycolysis enters the mitochondria via the mitochondrial pyruvate transporter. There it is converted by pyruvate dehydrogenase (PDH) into acetyl coenzyme A, which fuels the Krebs cycle. In PAH, oxidative phosphorylation is actively suppressed by upregulated expression of pyruvate dehydrogenase kinase (PDK). PDK phosphorylates and inhibits PDH.123 124 This shifts the cell to rely on glycolysis, which is energetically inefficient. However, in PAH, glycolysis is upregulated, both by an increase in glucose influx mediated by increased glucose transporter (glut) expression and by alterations in splice variant expression of the terminal glycolytic enzyme pyruvate kinase.106 107 This metabolic shift supports rapid proliferation while avoiding mitochondrial apoptosis.106 107 124
Warburg metabolism accounts for the increase in fluorodeoxyglucose uptake in the lungs and right ventricle of PAH patients and preclinical PAH models observed using positron emission tomography.125 In patients with PAH, a lactate gradient exists from superior vena cava to pulmonary artery.126 Whether this pre-pulmonary lactate gradient reflects right ventricular ischemia or Warburg metabolism is unknown.
Emerging metabolic therapies that exploit this pathway include the small molecule PDK inhibitor dichloroacetate. Dichloroacetate reverses the Warburg phenotype in PASMCs and right ventricular cardiomyocytes and regresses PAH in preclinical models.124 127 Dichloroacetate has been safely used to chronically treat children with mitochondrial disease and lactic acidosis.128 A four month, open label study of dichloroacetate (3 to 6.25 mg/kg twice daily) in patients with IPAH taking approved PAH therapies showed that dichloroacetate reduced mPAP and PVR while improving 6MWTD; however, some patients did not respond to dichloroacetate, and these patients had functional variants of SIRT3 and UCP2.129
Vascular cells isolated from patients with PAH also show additional signs of mitochondrial dysfunction, including fragmentation and membrane hyperpolarization. Hyperpolarization of mitochondrial membranes contributes to apoptosis resistance by blocking the release of pro-apoptotic mediators, such as cytochrome C(103). The mitochondria in vascular cells exist in dynamic networks that are continuously dividing (fission) and joining together (fusion).130 For nuclear division to occur, mitosis must be coordinated with mitochondrial division. Increased rates of mitotic fission in PAH cause PAH PASMCs to show fragmented mitochondria (fig 8), and this can be therapeutically targeted.117 A fission/fusion imbalance in PAH results from increased activation of the mitochondrial fission mediator dynamin related protein 1 (Drp1) and reduced expression of the fusion mediator, mitofusin 2.117 130 Drp1 activation and mitofusin 2 downregulation are secondary to other changes in the PAH milieu, including elevated [Ca2+]cyt, increased mitogen concentrations (platelet derived growth factor, endothelin 1), and impaired expression of the mitochondrial biogenesis promoter peroxisome proliferator activated receptor γ coactivator 1-α.117 131
Fig 8.

Mitochondrial fragmentation in pulmonary arterial hypertension (PAH). Confocal imaging of mitochondria in human pulmonary arterial smooth muscle cells (PASMCs). Left side: Mitochondria are stained red with potentiometric dye tetramethylrhodamine methyl ester. Nuclei are blue (stained with ‘6-diamidino-2-phenylindole). Note fused network in normal mitochondria versus fragmented network in PAH PASMC. This fragmentation reflects increase in mitotic fission in PAH that results from increased expression of activated dynamin related protein 1 and reduced expression of fission mediator mitofusin 2. Right side: To directly measure fission, PASMC were transfected with mitochondrial matrix targeted, photoactivatable green fluorescent protein (mito-PA-GFP) and mitochondrial targeted red fluorescent protein (mito-Ds-red). Mito-Ds-red is tonically fluorescent whereas mito-PA-GFP does not fluoresce until photoactivated. See supplemental movies for dynamic images of these files. In these movies, mito-PA-GFP is selectively activated in a few mitochondria (using a focused 488 nm laser) and serial observations allow measurement of spread of green protein within adjoined mitochondria. More fissioned network in PAH PASMC has less spread of matrix GFP green signal outside activation zone (white box) than does control PASMC imaged at same time interval. Reproduced with permission from Marsboom G, et al. Circ Res 2012;110:1484-97117
Reduced expression of the MCU in PASMCs from PAH patients and rodent models provides a potential unifying mechanism linking dysregulated mitochondrial metabolism and dynamics and partially explains the observed increase in PASMC proliferation and apoptosis resistance in PAH.113 MCU is the major functional subunit of the MCU complex that allows for the influx of Ca2+ into the mitochondrial matrix. Reduced expression of MCU (and increased expression of MICU1, a negative regulator of MCU) increases [Ca2+]cyt in PAH while lowering intramitochondrial calcium. Increased [Ca2+]cyt promotes vasoconstriction and enhances mitochondrial fission and proliferation. Low intramitochondrial calcium inhibits calcium sensitive dehydrogenases in the mitochondrial matrix (including PDH) and thereby inhibits glucose oxidation.113 MCU downregulation in PAH is epigenetically mediated by increases in the micro-RNAs miR25 and miR138. Anti-miRs or MCU gene transfer reverse the mitochondrial phenotype in PAH PASMCs and regress PAH in the monocrotaline model.113
Genetic contributions to PAH
Heritable forms of PAH account for 6-10% of all PAH.24 Heterozygous, germline mutations in BMPR2, the gene encoding the type II bone morphogenetic protein (BMP) receptor (BMPR-II), account for 70-80% of heritable PAH cases,132 133 134 135 as well as 15-25% of IPAH cases.132 134 135 136 137 Box 3 describes the discovery of BMPR2 gene mutations in PAH. At risk siblings from families with PAH carrying a BMPR2 mutation have a disease penetrance in mutation carriers of only 27%. Disease penetrance is greater in females (42%) than in males (14%).138 Thus, a BMPR2 mutation increases an individual’s chance of developing PAH 25 000-fold, from roughly 1 in 100 000 to 1 in 4.8 138 A meta-analysis of PAH patients also showed that, compared with patients without BMPR2 mutations, mutation carriers are younger at diagnosis, develop more hemodynamically severe disease, are less likely to respond to vasodilators, and are at an increased risk of death.139 However, BMPR2 mutations are considered to be permissive of disease, requiring additional genetic, epigenetic, or environmental factors for the development of PAH in people with mutations.
Box 3. Discovery of BMPR2 mutations in PAH.
1951—First description of “primary pulmonary hypertension” by Dresdale includes recognition of heritable PAH
1997—Microsatellite markers and linkage analysis are used to map the “PAH gene” to a region on chromosome 2q31-32 128 129
2000—Two independent groups identify the affected gene as BMPR2, a receptor of the transforming growth factor-β superfamily 130 131
BMPR-II protein concentrations are reduced by about 75% in lung tissue and endothelial cells from patients with PAH.140 141 This reduction is greater than expected from haploinsufficiency alone and occurs in patients lacking BMPR2 mutations, as well as in non-genetic rodent models.140 141 This suggests that factors associated with disease, independent of mutation status, suppress BMPR-II expression and provides biologic plausibility that targeting BMPR-II deficiency might be beneficial even in PAH patients lacking BMPR2 mutations.
Impaired BMPR-II signaling has been shown to promote accelerated cell proliferation,142 while potentially contributing to disease initiation by enhancing the susceptibility of PAECs to apoptosis.143 Loss of BMPR2 in the endothelium also causes mitochondrial dysfunction and inflammation,119 providing a potential mechanism linking BMPR2 mutations to mitochondrial dysfunction in PAH. Emerging BMPR-II related therapies for PAH include strategies to rescue the functionality of mutated BMPR2 alleles or enhance BMPR-II signaling through the functional receptors that are produced by the non-mutated allele. Rescue strategies include the use of read through compounds such as Ataluren (PTC-124), which promote the transcriptional read through of premature termination codons and the production of functional, full length BMPR-II protein from mutated alleles.144 Missense mutations can also be rescued in preclinical studies by using chemical chaperones, such as 4-phenylbutyrate, which increase trafficking of misfolded, but otherwise functional, BMPR-II protein from the endoplasmic reticulum to the cell surface.145 Preclinical trials of lung targeted BMPR-II gene therapy have had mixed results.146 147 Examples of enhancing signaling via non-mutated BMPR-II protein include the delivery of recombinant BMP ligands, such as BMP9,143 or small molecule agonists of canonical BMP signaling, such as FK506.148 These approaches enhance BMPR-II mediated signaling in the endothelium and can reverse established disease in the Sugen 5416/hypoxia rat model.143 148 Inhibition of BMPR-II degradation, through lysosomal inhibitors such as hydroxychloroquine, also enhances BMPR-II mediated signaling in human PAECs and prevents disease in the monocrotaline model.149
Mutations in other components of the BMP signaling pathway including ACVRL1, which encodes the type I BMP receptor ALK1, ENG, the gene encoding the accessory receptor endoglin, and SMAD9, which encodes the BMP transcriptional mediator, Smad 8, contribute to a small percentage of PAH cases.150 151 152 153 Whole exome sequencing studies of families with PAH have also identified mutations in genes unrelated to canonical BMP signaling pathways in 1-3% of all cases of PAH. These include mutations in KCNK3, which encodes the pH sensitive potassium channel TASK-1,154 and CAV1, which encodes caveolin 1, a membrane protein that is essential for the formation of lipid rafts, known as caveolae.155 Whole exome sequencing approaches have also been used in the examination of pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH), which are classified as group 1 pulmonary hypertension. Independent studies examining families with autosomal recessive forms of either PVOD or PCH identified bi-allelic mutations of EIF2AK4 (or GCN2), the gene encoding eukaryotic translation initiation factor 2 α kinase, as the mutation underlying the hereditary forms of these conditions.156 157 Thus, genotyping shows that PVOD and PCH are phenotypic variants of the same disease. Bi-allelic EIF2AK4 mutations were also identified in 20-25% of sporadic cases of both conditions.156 158
Preliminary data from the PAH Biobank sponsored by the National Heart, Lung, and Blood Institute at Cincinnati Children’s Hospital Medical Center have been provided by study principal investigator William Nichols (CCHMC/University of Cincinnati). The incidence of pathogenic/suspected pathogenic gene variants identified, using panel sequencing of 12 genes in the 2251 PAH patients, is 10.8%. Pathogenic/suspected pathogenic variants are less frequent in APAH (5.8%) and more common in IPAH (13.2%) (personal communication, W Nichols). Nicholas Morrell (University of Cambridge) is conducting a study of IPAH and heritable PAH patients (n=1250), using whole genome sequencing as part of the National Institutes for Health Research Bioresource for Rare Diseases Study. These two studies will ultimately define the genetic contribution to various PAH patient populations and help to determine the value of routine genetic testing in patients with PAH.
Involvement of epigenetic factors in PAH
The expression of genes in PAH is also influenced through epigenetic processes, defined as mechanisms that alter gene expression without changing the sequence of genomic DNA. Epigenetic mechanisms in PAH include DNA methylation, histone modification, and RNA interference via micro-RNAs (box 4).
Box 4. Epigenetic mechanisms in PAH.
DNA methylation—DNA methylation involves the covalent attachment of a methyl group to cytosine residues in CpG dinucleotide sequences. Methylation occurs in CpG rich regions of the genome known as CpG islands. These CpG islands are usually near the gene promoters, and their hypermethylation interferes with gene transcription
Histone acetylation—The modification of histones influences the transcriptional activity of genes through the regulation of their accessibility to transcription factors. Histone acetylation, which is associated with increased transcriptional activity, is carried out by histone acetyltransferases and reversed by histone deacetylases
Production of micro-RNA—miRs are encoded by intronic DNA and regulate gene expression through RNA interference. Short (20-22 nucleotide) segments of RNA bind to complementary sequences in the 3′-untranslated region of mRNA, leading to mRNA degradation or the repression of translation. As a single miR holds the potential to bind and regulate multiple gene targets, these molecules can serve as “master regulators” for programs of targeted gene expression
DNA methylation
Epigenetic regulation of a pulmonary hypertension phenotype was first identified in the assessment of spontaneous PAH in the fawn hooded rat model.159 Hypermethylation and silencing of the gene encoding superoxide dismutase 2 in this model caused a 50% reduction in pulmonary artery superoxide dismutase 2 protein expression (also seen in PAH patients). Superoxide dismutase 2 is a mitochondrial enzyme that converts superoxide to hydrogen peroxide. Hydrogen peroxide serves as a diffusible redox signaling molecule. Reduced hydrogen peroxide in the fawn hooded rat model causes normoxic activation of hypoxia inducible factor (HIF-1α), which triggers the Warburg effect by upregulating PDK and glut expression and increased PASMC proliferation,160 and also inhibits Kv1.5 channel function/expression, causing vasoconstriction.118 This epigenetic mechanism results from lung specific increases in the expression of DNA methyltransferases 1 and 3B (DNMT). A similar mechanism of reduced superoxide dismutase 2 and elevated HIF-1α occurs in endothelial cells from patients with IPAH.120 Increased DNA methylation is implicated in the exaggerated pulmonary hypertensive response of mice born to mothers subjected to a restrictive diet during pregnancy.161 Future therapies may involve inhibiting DNA methyltransferases,162 as is currently done in some hematologic malignancies (using decitabine),163 modulating the enzymes that demethylate DNA (ten eleven dioxygenases),164 and/or manipulating methyl binding proteins, which alter the transcription of methylated genes.
Histone modification
An examination of lung tissue from patients with IPAH, as well as lungs and right ventricles from rats with chronic hypoxic pulmonary hypertension, identified increased expression of histone deacetylases (HDAC1 and HDAC5) in remodeled pulmonary vessels.165 Additional work in PAECs from PAH patients identified increased nuclear accumulation of HDAC4 and HDAC5, leading to downregulation of the transcription factor myocyte enhancer factor 2 and a reduction of several genes involved in pulmonary vascular integrity and homeostasis, including Krüppel-like factors 2 and 4 and connexins 37 and 40.166 Interestingly some metabolic enzymes, such as PDH, act within the nucleus to generate the acetyl coenzyme A required for histone regulation.167 Increased histone acetylation has also been reported in a rat model of persistent pulmonary hypertension in the newborn,168 where a compensatory sixfold increase in eNOS expression was attributed to enhanced acetylation of the eNOS promoter and a modest decrease in gene methylation.
The HDAC inhibitors valproic acid and suberoylanilide hydroxamic acid reduced established hypoxia induced pulmonary hypertension in rodent models.165 Similar results were also obtained with the HDAC inhibitor MGCD0103, which prevented hypoxic pulmonary vascular remodeling in rats.169 Although valproic acid improves RVH in both the monocrotaline rat model and the pulmonary artery banding model of RVF,170 another HDAC inhibitor, trichostatin A, worsened RVH and fibrosis in the rat pulmonary artery banding model.171 These differences may reflect the need to target specific subtypes of HDAC in PAH and/or differences in the role of HDACs in various forms of RVH.
Micro-RNAs
Micro-RNA dysregulation occurs in whole lung tissue in PAH, as well as in cultured PAECs, PASMCs, and fibroblasts. Table 6 summarizes the miRs identified in PAH and their effect on in vitro or in vivo processes.172 173 174 175 176 177 178 180 181 183 184 185 186 Many of these dysregulated miRs influence pathways that are critical to creating the cancer-like, mitochondrial-metabolic phenotype of PAH.
Table 6.
Micro-RNAs related to pathogenesis of pulmonary arterial hypertension (PAH)
| Micro-RNA | Tissue/model | Effect |
|---|---|---|
| miR-17-92 cluster: | ||
| miR-17172 | Increased in chronic hypoxia mouse model | Increased PASMC proliferation |
| miR-17-5p, miR-20a173 | HEK293 cells | Induced by IL-6; overexpression downregulates BMPR-II |
| miR-21174-177 | Increased in PAH patient lungs, hypoxic PASMCs, interleukin 6 overexpression, and hypoxic PH models | Decreased NOS expression in hypoxic PAECs, increased PASMC proliferation; miR-21 deletion enhances PH in mice; miR-21 overexpression prevents PH in mice |
| Decreased in PAH patient lungs, serum, monocrotaline rats | ||
| miR-21 and miR-27a178 | Decreased in PAH PAECs, PASMCs | Both miRs suppress PAEC and PASMC proliferation |
| miR-26a179 | Decreased in circulation of PAH patients and MCT rats | |
| miR-124180 181 | Reduced in hypoxic PASMCs and PAH adventitial fibroblasts | Increased NFAT activity and proliferation |
| miR-138 and miR-25113 | Increased in PAH PASMCs and monocrotaline rat model | Downregulation of MCU, increased PASMC proliferation, apoptosis resistance; inhibition of miRs prevents PH in monocrotaline model |
| miR-140-5p182 | Reduced in PAH whole blood, monocrotaline and Sugen-hypoxia rat model | Increased PASMC proliferation via increased SMURF1 |
| miR-143/145183 | Increased in PAH patients and hypoxic mouse model | miR-145 inhibition blocks hypoxia induced PH |
| miR-150184 | Reduced in circulation of PAH patients | Associated with poor survival |
| miR-204185 | Reduced in PAH PASMCs, hypoxic and monocrotaline rat models | Increased NFAT, PASMC proliferation; miR-204 mimics prevent PH in monocrotaline model |
| miR-424/503186 | Reduced in PAH PAECs | Reduced endothelial proliferation, decreased expression of FGF-2 and FGF receptor-1; restoration of miRs prevents monocrotaline and Sugen-hypoxia induced PH |
BMPR-II=type II bone morphogenetic protein receptor; FGF=fibroblast growth factor; MCT=monocrotaline; MCU=mitochondrial calcium uniporter; NFAT=nuclear factor of activated T cells; NOS=nitric oxide synthase; PAEC=pulmonary artery endothelial cells; PASMC=pulmonary artery smooth muscle cells; PH=pulmonary hypertension.
Therapeutic modulation of disease associated miRs, using miR-mimics or antago-miRs, has shown potential to prevent or reverse PAH in rodent models.182 Although these findings are promising, the ability of miRs to regulate a broad array of genes, which is the very basis of their value, also raises the potential for undesirable off-target effects that could limit their translation into the treatment of human disease. miRs, particularly those present at altered concentrations in the circulation, represent useful biomarkers that can predict survival.179 184
Two recent studies provide an example of the intersection of genetics, epigenetics, and metabolism in the pathogenesis of PAH. Caruso et al and Zhang et al showed that altered activity of the enzyme pyruvate kinase contributes to the Warburg phenomenon in endothelial cells and fibroblasts in PAH, respectively.106 107 Pyruvate kinase is the final step in glycolysis, catalyzing phosphate transfer from phosphoenolpyruvate to ADP, producing pyruvate and ATP. Both groups showed that this pro-glycolytic mechanism is stimulated by upregulation of a heterogeneous nuclear ribonucleoprotein, polypyrimidine tract binding protein (PTBP1), in response to an epigenetic mechanism—namely, downregulation of miR124. The increase in PTBP1 changes the splice variant expression of pyruvate kinase, favoring the proglycolytic variant pyruvate kinase M2. The mitochondrial supply of pyruvate is further compromised by downregulation of the mitochondrial pyruvate transporter. Remarkably, BMPR2 mutation/downregulation leads to the same Warburg phenotype and causes proliferation of endothelial cells. This work shows coherence between apparently disparate theories, illustrating how seemingly unrelated gene mutation or epigenetic changes can promote the Warburg mitochondrial metabolic phenotype, a final common pathway of the proliferative PAH phenotype in many vascular cells.
Inflammation and fibrosis in PAH
Chronic inflammation and maladaptive fibrosis play an essential role in PAH. Immune dysfunction is a central feature of APAH, especially in patients with connective tissue diseases (for example, systemic lupus erythematosus and scleroderma) and infections (for example, HIV and schistosomiasis).187 The prevalence of schistosomiasis in the developing world makes it the single largest cause of PAH, with an estimated 2 000 000 cases worldwide.188
Chronic inflammation is also observed in IPAH. Histologic examinations of lung tissue from IPAH patients have identified the presence of immune cell infiltrates, composed of lymphocytes, macrophages, dendritic cells, and mast cells in pulmonary vascular lesions.189 190 191 Circulating concentrations of inflammatory cytokines including interleukins 1β, 2, 4, 6, 8, 10, and 12p70,192 193 tumor necrosis factor α,192 193 and the chemokines CXC3L1 (or fractalkine),194 CCL2 (or monocyte chemotactic protein 1),195 and CCL5 (or RANTES)196 are elevated in IPAH. Elevated concentrations of interleukins 6, 8, 10, and 12 predicted survival in a cohort of 21 PAH patients better than traditional prognostic markers, such as 6MWD or cardiopulmonary hemodynamics.193
Fibroblasts isolated from the adventitia of pulmonary arteries from both PAH patients and chronic hypoxia models of pulmonary hypertension show a hyperproliferative, apoptosis resistant, pro-inflammatory phenotype, marked by the production of inflammatory molecules and expression of myofibroblast markers.181 197 198 These changes are associated with epigenetic modifications including increased HDAC activity and decreased miR124 expression.181 Interestingly, the depletion of circulating monocytic cell populations blocks adventitial remodeling in a rat model of hypoxic pulmonary hypertension, suggesting that fibrotic remodeling is at least partially dependent on the recruitment of circulating monocytic mesenchymal precursors.199 The interplay between adventitial fibroblasts and monocytic cells is further supported by the demonstration that human and experimental PAH fibroblasts activate a STAT3 mediated, pro-inflammatory phenotype in macrophages by means of increased interleukin 6 secretion.197
Recent mechanistic studies examining the contribution of cellular immunity to the pathogenesis of pulmonary hypertension have also focused on the role of specific lymphocyte populations, including T cell, B cell, and natural killer cell subsets. The role of T lymphocytes in disease pathogenesis is controversial. Athymic nude rats lack mature T cells and develop more severe pulmonary hypertension in response to either monocrotaline or the vascular endothelial growth factor receptor blocker SU-5416,200 201 suggesting a protective role for regulatory T cells.202 In Sugen rats, PAH is attenuated by blockade of leukotriene B4 activity with leukotriene A4 hydrolase inhibitors. More recently, blockade of interleukin 6, using the anti-interleukin 6 receptor antibody MR16-1, was shown to prevent chronic hypoxia induced pulmonary hypertension in mice by blocking the accumulation of Th17 cell derived interleukin 21 and M2 macrophages in the lungs of these mice.203 Thus the role of T cell subsets in disease is context specific and likely varies among species.
The contribution of humoral immunity and excessive B cell activation to the pathogenesis of PAH is supported by multiple studies identifying circulating autoantibodies and the ectopic expansion of pulmonary lymphoid tissues in patients with PAH.204 Increased bronchus associated lymphoid tissues and autoantibody production has also been reported in the monocrotaline rat model, with passive autoantibody transfer being sufficient to transmit disease between animals.205
Impairment of innate lymphocytes, such as natural killer cells, is reported in human and experimental PAH.206 Natural killer cells can adopt an angiogenic phenotype and contribute to vascular remodeling in pregnancy and cancer through the production of angiogenic cytokines and matrix degrading enzymes.207 208 In PAH, natural killer cells have an impaired phenotype, marked by increased transforming growth factor β and matrix metalloproteinase 9 production.206 Deficiencies in natural killer cells and CD8+ killer T cells were linked to an increased risk of death in a small cohort of IPAH and APAH patients.209
Right ventricular dysfunction in PAH
The right ventricle’s ability to adapt to pressure overload determines functional status and prognosis in PAH. A dilated right ventricle or a small left ventricle independently predicts poor survival in PAH.210 Adult PAH patients with RVF admitted for inotropic therapy have a 41% acute mortality rate,211 far exceeding the mortality of patients admitted with left ventricular failure.
Chronic pressure overload in PAH stimulates RVH in a compensatory attempt to maintain cardiac output while minimizing wall stress (fig 9A, right panel). However, in some patients, the right ventricle dilates owing to maladaptive remodeling and eventually fails (fig 9A, left panel, and fig 9B). In general, PAH patients with congenital heart disease (Eisenmenger’s syndrome) show adaptive, concentric RVH and are free from RVF for decades215; conversely, APAH patients, particularly those with scleroderma, have maladaptive remodeling with worse right ventricular function and a higher incidence and severity of RVF, despite lower mPAP.216 Adaptive RVH has been associated with a lower risk of clinical worsening compared with maladaptive RVH in patients with IPAH.38 217
Fig 9.

Right ventricular changes in pulmonary arterial hypertension (PAH). (A) Adaptive versus maladaptive right ventricular hypertrophy (RVH). Adaptive RVH on left is concentric, and patient had long survival with group 1 pulmonary hypertension before succumbing to cancer. Maladaptive patient (on right) had associated PAH (scleroderma) and died within three years of diagnosis with eccentric dilatation and thinning of right ventricle and right ventricular failure. Reproduced with permission from Rich S, et al. Chest 2010;138:1234-9.212 (B) cardiac magnetic resonance imaging (MRI) showing severe dilatation right ventricle (RV) with flattening of interventricular septum and small left ventricle (LV). Reproduced with permission from Michelakis E, et al. Circulation 2003;108:2066-9.213 (C) Cardiac positron emission tomography (PET) scan showing increased fluorodeoxyglucose (FDG) uptake in right ventricle in patient with PAH. Reproduced with permission from Oikawa M, et al. JACC 2005;45:1849.214 (D) Cardiac MRI with gadolinium showing late gadolinium enhancement (white areas identified by arrows) in interventricular septum at right ventricular free wall insertion sites. LGE=late gadolinium enhancement
Abnormalities of right ventricular perfusion, angiogenesis, autonomic signaling, metabolism, and fibrosis contribute to the development of maladaptive RVH and RVF. Many of the changes in the right ventricle during RVH, such as activation of the autonomic nervous system and uncoupled glycolysis,218 219 are ultimately maladaptive. Whether these chamber specific, cardiac abnormalities reflect primary insults to the right ventricle or are simply the maladaptive responses to increased right ventricular afterload remains uncertain. Nonetheless, they represent important pathophysiologic abnormalities and offer many therapeutic targets.
Right ventricular ischemia
Patients with PAH often present with evidence of myocardial ischemia, including angina-like chest pain and elevated concentrations of serum troponin.220 Right ventricular ischemia may reflect reduced right coronary artery perfusion pressure. However, right ventricular function is usually preserved as long as coronary perfusion pressure, the difference in pressure between the aorta and the right ventricle in systole or diastole, exceeds 50 mm Hg.221 In PAH, systemic (aortic) hypotension and a rise in right ventricular pressure (in both systole and diastole) combine to reduce the pressure gradient that drives perfusion of the right coronary artery, contributing to right ventricular ischemia. Ischemia in RVH may also reflect loss of small vessels (microvascular rarefaction).222 The observation that RVF is more prevalent in patients with scleroderma, who have circulating autoantibodies toxic to the endothelium, and in PAH models that are characterized by endothelial injury (induced by SU5416) suggests a potential role for primary endothelial damage in the right ventricular microcirculation.223 Capillary rarefaction in experimental PAH is reversible with β-adrenergic blockers.222
Autonomic activation and downregulation of β1 receptors in RVH
The right ventricle is less responsive to inotropes than the left ventricle.224 Circulating catecholamines are elevated in PAH patients with RVF.225 The autonomic activation exceeds that seen in left ventricular failure.226 Like PAH patients, rats with PAH induced maladaptive RVH show selective downregulation and uncoupling of β1-adrenoreceptors, α1-adrenoreceptors, and dopaminergic receptors in right ventricular myocytes. This is due to G protein coupled receptor kinase 2 (GRK2) mediated uncoupling of receptor signaling and impairs responses to inotropes. GRK2 inhibition seems to be therapeutically beneficial in PAH.227 228 In rodent models of PAH and RVH, dobutamine is the optimal right ventricular inotrope; however, this has not been established in PAH patients.227 The observed downregulation of the cardiomyocyte β1-receptors suggests biologic plausibility for using β-adrenergic blockers for RVF in PAH patients. However, in a study of 19 patients, bisoprolol reduced cardiac output and exercise capacity with no improvement in RVEF.229 In contrast, carvedilol reduced glycolytic rate and increased β-adrenergic receptor expression in the right ventricle with improvement in right ventricular fractional area change and no reduction in exercise capacity and cardiac output.230 More definitive studies are needed.
Right ventricular metabolism in RVH
The fetal right ventricle relies on glycolysis and glucose oxidation as the major sources of ATP production.231 In the adult right ventricle, there is a transition to reliance on fatty acid oxidation (FAO), which accounts for 60-90% of ATP production. FAO needs about 12% more oxygen per mole of ATP than does glucose oxidation. The hypertrophied right ventricle depends on glucose metabolism.232 The metabolic fate of glucose in RVH is altered because processes, including PDK mediated inhibition of PDH, suppress mitochondrial metabolism. In this scenario, lactate (the end product of glycolysis) accumulates, causing acidosis and exacerbating right ventricular hypokinesis.228 Activating glucose oxidation by using the PDK inhibitor dichloroacetate improves right ventricular function while regressing RVH. Thus, the right ventricle in PAH can be resuscitated metabolically. Consistent with this, increased right ventricular uptake of fluorodeoxyglucose was observed in a small series of PAH patients studied with positron emission tomography, a consequence of reliance on uncoupled glycolysis, and was reduced by effective PAH targeted therapy (fig 9C).214
In PAH patients, there is evidence of cardiac steatosis and lipotoxicity, which murine data suggest relates to reduced FAO.93 Whether differences in FAO occur in adaptive versus maladaptive RVH remains to be determined. In light of this report of impaired FAO in the right ventricles of patients with PAH, it is uncertain whether FAO inhibitors, such as trimetazidine and ranolazine, would be helpful in PAH, as they are in preclinical models of RVH induced by pulmonary artery banding.223
Right ventricular hibernation
Right ventricular ischemia leads to right ventricular hibernation, defined as a reduction in right ventricular function caused by a chronic decrease in perfusion with persistent myocardial viability.228 Thus, future therapeutic opportunities might include anti-ischemic or angiogenic therapy. Alternatively, it might be empirically easier to treat RVF and ischemia by pharmacologically changing metabolism or adrenergic signaling to “do more with less” (that is, use of drugs that alter cardiac metabolism or anti-adrenergic drugs).228 229 However, the clinical trial data that would be needed to alter practice are lacking.
Right ventricular fibrosis
The role of right ventricular fibrosis in RVF in PAH is unknown. However, late gadolinium enhancement on MRI at right ventricular insertion points may indicate localized fibrosis (fig 9D)233 and is an independent risk factor for poor prognosis.234
Chamber specific responses to RVH
Quantitative differences in the expression of many transporters and pumps in the right versus left ventricle, seen in fetal hearts, persist into adulthood.235 This raises the possibility that chamber selective therapeutic targets may exist in RVH. For example, PDE5 expression, normally absent in the right ventricle, is induced during RVH. Sildenafil has a direct right ventricular inotropic effect that occurs only in RVH.72 236
Sex differences in PAH
Although woman are more likely to acquire PAH, men (particularly those aged over 60 years) have a worse prognosis. This may relate to more maladaptive response of the male right ventricle to PAH. Male patients are less likely to improve their RVEF after starting PAH targeted therapy. Male and female patients showed a similar reduction in PVR one year after starting PAH targeted therapy, but RVEF improved in female patients, whereas it deteriorated in male patients.13 This difference accounted for much of the male survival disadvantage. Even healthy people have differences in right ventricular function, such that men have lower RVEF than women (by about 4%), despite greater right ventricular mass (approximately 8%) and volumes.237 Beyond the adaptive response of the right ventricle, other important sex differences in the pathogenesis of PAH exist in both the development and progression of vascular obstruction and the responsiveness to PAH targeted therapy.238 These are beyond the scope of our review, and the interested reader is referred to the literature.99 100 101 239
Guidelines
Three main guideline statements about the care of adult patients with pulmonary hypertension are available.24 66 240 Differences between guidelines documents are reviewed in box 5.
Box 5. Differences between PAH guideline documents24 66 241 .
Referral to PAH centers
All three documents:
Referral to centers expert in the management of PAH
Warfarin use
CHEST guideline and expert panel report 2007 and ACCF/AHA 2009 expert consensus document:
Warfarin anticoagulation in IPAH with a target international normalized ratio 1.5-2.5
2015 ESC/ERS guidelines:
May be considered in patients with IPAH, heritable PAH, and anorexigen associated PAH (level IIb recommendation)
Vasoreactivity testing
All three documents:
Perform vasoreactivity testing before starting PAH targeted therapy
Use of high dose CCBs in patients with positive vasoreactivity testing
All three documents:
Indicated in patients with IPAH, heritable PAH, and anorexigen related PAH
2015 ESC/ERS guidelines:
Vasodilator responsiveness does not predict favorable long term response to CCB therapy in APAH
Combination PAH targeted therapy
CHEST guideline and expert panel report 2007:
Start monotherapy in patients who are functional class II and III. Add second agent for patients “who remain symptomatic on stable and appropriate doses” of ERAs or PDE5 inhibitors
ACCF/AHA 2009 expert consensus document:
The safety and efficacy of combination therapy in PAH is a subject of active investigation
2015 ESC/ERS guidelines (reflecting results of AMBITION trial published in 2015 86):
Level I recommendation for the initial use of ambrisentan and tadalafil as therapy in patients who are functional class II and III. Other combination therapies are given level IIa and IIIb recommendations
Emerging therapies
Table 7 summarizes emerging therapies for PAH that address novel targets.
Table 7.
Emerging therapies for pulmonary arterial hypertension targeting novel targets implicated in pathogenesis
| Target | Therapy | |
|---|---|---|
| Warburg metabolism and altered mitochondrial dynamics93 117 124 131 223 | Inhibitors of pyruvate dehydrogenase kinase (eg, dichloroacetate); inhibitors of fatty acid oxidation (eg, trimetazidine and ranolazine); anti-fission/pro-fusion therapies | |
| Vasoconstriction115 116 | Rho kinase inhibitors | |
| BMPR-II deficiency: | ||
| With BMPR2 mutation: | ||
| Nonsense mutation (premature termination codon)144 | Ataluren (PTC-124) | |
| Missense mutation145 | Chemical chaperones (eg, 4-PBA) | |
| Independent of BMPR2 mutation status143 148 149 | Recombinant BMP9; FK506 (tacrolimus); hydroxychloroquine | |
| Epigenetic mechanisms of disease161-164 168 177 242 | DNMT inhibitors; HDAC inhibitors (eg, valproic acid); micro-RNA therapies: miR mimics, antago-miRs | |
| Immune dysfunction187 203 | Anti-interleukin 6 receptor antibodies; immunosuppressants; leukotriene-A4 hydrolase inhibitors | |
| Right ventricular dysfunction227 229 230 | Anti-ischemic angiogenic right ventricle therapies; adrenergic modulators (GRK2 inhibitors, β-adrenergic blockers) | |
4-PBA=4-phenylbutyrate; BMPR-II=type II bone morphogenetic protein receptor; BMPR2=bone morphogenic protein receptor; DNMT=DNA methyltransferases; GRK=G protein receptor kinase; HDAC=histone deacetylases; PTC=premature termination codons.
Conclusions
Tremendous advances have been made in the diagnosis and management of PAH. As a result, patients have access to four classes of PAH specific agents. However, none of these agents is curative and all are expensive. A concerning trend in clinical trials is to use composite endpoints that include mortality and imply survival benefits when outcomes are driven by reductions in hospital admission or improved functional capacity. Arguably, the available PAH specific therapies do not target key features of this syndrome, including its neoplastic, mitochondrial-metabolic, inflammatory, and fibrotic phenotypes. Although basic science studies have clarified several key molecular pathways that underlie PAH, the translation of these studies into clinically available biomarkers and new therapeutic agents has been slow, and no clear path exists to move a drug or biomarker from the bench top to the bedside. Moreover, preclinical studies are siloed, and researchers (us included) often fail to consider or integrate opposing or complementary theories of disease. As a result, PAH is variably viewed as a genetic disease, an epigenetic syndrome, an autoimmune inflammatory syndrome, or a disease of disordered mitochondrial dynamics and metabolism. Like the parable of the blind man and the elephant, each reductionist perspective fails to integrate the totality of the data and thus fails to fully portray the broader picture that is the PAH elephant. It is increasingly clear that these mechanisms intersect, offering new therapeutic targets.
Questions for future research.
Clinical
-
How should right ventricular function best be measured in PAH patients?
MRI versus three dimensional echocardiography versus two dimensional echocardiography
What is the role of right ventricle relevant biomarkers?
-
Is right ventricle-pulmonary artery coupling an important metric in PAH?
Can right ventricle-pulmonary artery coupling be modified therapeutically using drugs or mechanical devices?
What is the role of assessing right ventricular contractile reserve in patients with PAH?
Should genetic testing be routinely done in patients with confirmed PAH and their family members?
-
Screening for associated PAH
How often should patients with risk factors for PAH (eg, scleroderma or HIV infection) have screening echocardiograms for diagnosing PAH?
-
Is the initial combination PAH targeted therapy strategy superior to a sequential combination therapy strategy?
What is the health economic implication of initial combination therapy?
-
The use of inotropes in PAH patients with RVF is associated with high mortality rates, and the inotrope of preference varies tremendously between and within centers.
What is the optimal right ventricular inotrope for use in PAH patients?
Are inotropes increasing mortality in PAH?
Is there a role for right ventricular assist devices or oxygenators in supporting PAH patients?
-
Therapeutic application of drugs approved for group 1 pulmonary hypertension in other groups (with the exception of riociguat for group 4 disease) is not supported by evidence
How can therapeutic creep best be avoided?
Are there subsets of group 2 and group 3 patients with elevated PVR that might benefit from a PAH targeted therapeutic agent?
-
How can we accelerate translation of investigator initiated research to advance therapy and diagnosis of PAH?
Can we create a forum to allow industry to examine potential therapeutic targets and biomarkers discovered by investigators to enhance knowledge translation?
-
Is there a need for more advanced and sensitive endpoints to evaluate progression and regression of PAH?
What is the role of routine cardiac MRI, MRI lung perfusion, exercise right heart catheterization, and measurements of right ventricle-pulmonary artery coupling?
-
Patients are not consulted routinely in the design of clinical trials of PAH therapies
What endpoints are most meaningful to PAH patients?
Can PAH patients advocate for the funding of investigator initiated therapies?
-
PAH is a very heterogeneous syndrome
How can we begin to apply a personalized medicine approach to these patients?
Is the best level to personalize PAH therapy identified at the genomic, epigenomic, or proteomic level?
Epidemiology
-
Much of the epidemiology in PAH comes from registries and specialized clinics
What is the epidemiology of PAH in population based studies in adults and children?
Should we routinely compare the epidemiology of group 1 disease with the other pulmonary hypertension groups?
Does the changing epidemiology of group 1 pulmonary hypertension (with more older, obese patients) reflect misclassification of group 2 pulmonary hypertension patients with HFpEF?
What is the basis for the female preponderance in PAH?
How best can regional and national PAH registries be standardized and shared?
Basic and translational science
-
How does the BMPR2 pathway intersect with other important PAH pathways?
Why is BMPR2 downregulated in patients and animals lacking gene mutations?
Does the BMPR2 pathway offer viable therapeutic targets?
-
What classes of drugs should be the focus of phase I clinical trials in PAH?
Candidates include anti-inflammatory agents, immunomodulators, metabolic modulators that target Warburg metabolism, agents targeting fibrosis, agents modulating mitochondrial dynamics, inhibitors of rho kinase, and agents that enhance expression of or function of BMPR2
-
Although animal models of PAH are imperfect, it may be that the experimental design of preclinical studies is more to blame for false positive reports of therapeutic benefits that may not be reproducible in patients.
What is the role for randomization and blinding in preclinical pulmonary hypertension studies?
Should papers assessing preclinical therapies for pulmonary hypertension be required to include measures of pulmonary arterial pressure, cardiac output, and/or PVR?
Is there a standard duration of observation and ethically acceptable mortality surrogate that should be used in preclinical studies to avoid falsely optimistic assessment of drug benefit?
Can we create standardized protocols and models for preclinical assessment of PAH to make findings more robust and representative of the human disease?
-
How can we better share DNA, mRNA, proteins, cells, and tissues from patients with PAH to advance discovery?
Should resource sharing always result in authorship, or would a fee for service repository of biomaterials be beneficial?
-
Are the peripheral vasculature, right ventricular coronary vasculature, or skeletal muscles directly affected in PAH?
Are circulating inflammatory, autoimmune, or epigenetic factors directly targeting tissues beyond the pulmonary vasculature in PAH?
Glossary of abbreviations.
6MWD—six minute walk distance
APAH—associated pulmonary arterial hypertension
BMP—bone morphogenetic protein
BMPR-II—type II bone morphogenetic protein receptor
[Ca2+]cyt—cytosolic calcium
CCB—calcium channels blocker
CTEPH—chronic pulmonary thromboembolic pulmonary hypertension
cGMP—cyclic guanosine monophosphate
eNOS—endothelial nitric oxide synthase
ERA—endothelin receptor antagonists
FAO—fatty acid oxidation (FAO)
GRK2—G protein-coupled receptor kinase-2
HDAC—histone deacetylases
HFpEF—heart failure and preserved ejection fraction
IPAH—idiopathic pulmonary arterial hypertension
Kv—voltage-gated potassium channels
LVEDP—left ventricular end-diastolic pressure
MCU—mitochondrial calcium uniporter
miR—micro RNA
mPAP—mean pulmonary artery pressure
MRI—magnetic resonance imaging
NFAT—nuclear factor of activated T cells
NIH—National Institutes of Health
NT-proBNP—N terminal pro B type natriuretic peptide
PAEC—pulmonary arterial endothelial cells
PAH—pulmonary arterial hypertension
PASMC—pulmonary arterial smooth muscle cells
PCH—pulmonary capillary hemangiomatosis
PCWP—pulmonary capillary wedge pressure
PDE5—phosphodiesterase 5
PDH—pyruvate dehydrogenase
PDK—pyruvate dehydrogenase kinase
PTBP1—polypyrimidine tract-binding protein
PVOD—pulmonary veno-occlusive disease
PVR—pulmonary vascular resistance
RHC—right heart catheterization
RVEF—right ventricular ejection fraction
RVF—right ventricular failure
RVH—right ventricular hypertrophy
RVSP—right ventricular systolic pressure
sGC—soluble guanylate cyclase stimulators
TAPSE—tricuspid annular plane systolic excursion
Web Extra.
Extra material supplied by the author
Supplementary movie 1
Supplementary movie 2
Contributors: All authors defined intellectual content, conducted literature research, acquired data, and participated in manuscript preparation, editing, and critical review.
Funding: SLA is supported by Canada Foundation for Innovation (229252 and 33012), NIH RO1s HL113003 and HL071115, a Tier 1 Canada research chair in mitochondrial dynamics and translational medicine (950-229252), and a grant from the William J Henderson Foundation. TT is funded by AHA scientist development grant 15SDG25560048. MLO is supported by a CIHR project grant (PJT-152916) and a Tier 2 Canada research chair.
Competing interests: We have read and understood the BMJ policy on declaration of interests and declare the following interests: TT received a modest honorarium from Gilead and Actelion for participating in an advisory board.
Provenance and peer review: Commissioned; externally peer reviewed.
Patient involvement: No patients were involved in the creation of this article.
Series explanation: State of the Art Reviews are commissioned on the basis of their relevance to academics and specialists in the US and internationally. For this reason they are written predominantly by US authors
References
- 1. Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(Suppl):D34-41. 10.1016/j.jacc.2013.10.029 [DOI] [PubMed] [Google Scholar]
- 2. Tuder RM, Archer SL, Dorfmüller P, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 2013;62(Suppl):D4-12. 10.1016/j.jacc.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987;107:216-23. 10.7326/0003-4819-107-2-216 [DOI] [PubMed] [Google Scholar]
- 4. Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006 [eng.]. Eur Respir J 2007;30:1103-10. 10.1183/09031936.00042107 [DOI] [PubMed] [Google Scholar]
- 5. Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30:104-9. 10.1183/09031936.00092306 [DOI] [PubMed] [Google Scholar]
- 6. Kane GC, Maradit-Kremers H, Slusser JP, Scott CG, Frantz RP, McGoon MD. Integration of clinical and hemodynamic parameters in the prediction of long-term survival in patients with pulmonary arterial hypertension. Chest 2011;139:1285-93. 10.1378/chest.10-1293 [DOI] [PubMed] [Google Scholar]
- 7. Jing ZC, Xu XQ, Han ZY, et al. Registry and survival study in chinese patients with idiopathic and familial pulmonary arterial hypertension. Chest 2007;132:373-9. 10.1378/chest.06-2913 [DOI] [PubMed] [Google Scholar]
- 8. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173:1023-30. 10.1164/rccm.200510-1668OC [DOI] [PubMed] [Google Scholar]
- 9. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 2010;137:376-87. 10.1378/chest.09-1140 [DOI] [PubMed] [Google Scholar]
- 10. Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186:790-6. 10.1164/rccm.201203-0383OC [DOI] [PubMed] [Google Scholar]
- 11. Zhang R, Dai LZ, Xie WP, et al. Survival of Chinese patients with pulmonary arterial hypertension in the modern treatment era. Chest 2011;140:301-9. 10.1378/chest.10-2327 [DOI] [PubMed] [Google Scholar]
- 12. Hoeper MM, Huscher D, Ghofrani HA, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 2013;168:871-80. 10.1016/j.ijcard.2012.10.026 [DOI] [PubMed] [Google Scholar]
- 13. Jacobs W, van de Veerdonk MC, Trip P, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014;145:1230-6. 10.1378/chest.13-1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hatton N, Ryan JJ. Sex differences in response to pulmonary arterial hypertension therapy: is what’s good for the goose, good for the gander? Chest 2014;145:1184-6. 10.1378/chest.13-3061 [DOI] [PubMed] [Google Scholar]
- 15. Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J 2010;35:1079-87. 10.1183/09031936.00072709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122:156-63. 10.1161/CIRCULATIONAHA.109.911818 [DOI] [PubMed] [Google Scholar]
- 17. Benza RL, Foreman AJ, Prucka WR, et al., eds. Predicting survival in pulmonary arterial hypertension using the REVEAL database. American Thoracic Society, 2009. 10.1164/ajrccm-conference.2009.179.1_MeetingAbstracts.A2651. [DOI] [Google Scholar]
- 18. Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010;122:164-72. 10.1161/CIRCULATIONAHA.109.898122 [DOI] [PubMed] [Google Scholar]
- 19. Anand V, Roy SS, Archer SL, et al. Trends and Outcomes of Pulmonary Arterial Hypertension-Related Hospitalizations in the United States: Analysis of the Nationwide Inpatient Sample Database From 2001 Through 2012. JAMA Cardiol 2016;1:1021-9. 10.1001/jamacardio.2016.3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013;62(Suppl):D42-50. 10.1016/j.jacc.2013.10.032 [DOI] [PubMed] [Google Scholar]
- 21. Strange G, Playford D, Stewart S, et al. Pulmonary hypertension: prevalence and mortality in the Armadale echocardiography cohort. Heart 2012;98:1805-11. 10.1136/heartjnl-2012-301992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee SL, Daimon M, Kawata T, et al. Estimation of right atrial pressure on inferior vena cava ultrasound in Asian patients. Circ J 2014;78:962-6. 10.1253/circj.CJ-13-1234 [DOI] [PubMed] [Google Scholar]
- 23. Rich JD, Shah SJ, Swamy RS, Kamp A, Rich S. Inaccuracy of Doppler echocardiographic estimates of pulmonary artery pressures in patients with pulmonary hypertension: implications for clinical practice. Chest 2011;139:988-93. 10.1378/chest.10-1269 [DOI] [PubMed] [Google Scholar]
- 24. McLaughlin VV, Archer SL, Badesch DB, et al. American College of Cardiology Foundation Task Force on Expert Consensus Documents. American Heart Association. American College of Chest Physicians. American Thoracic Society, Inc. Pulmonary Hypertension Association ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009;53:1573-619. 10.1016/j.jacc.2009.01.004 [DOI] [PubMed] [Google Scholar]
- 25. Tunariu N, Gibbs SJ, Win Z, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med 2007;48:680-4. 10.2967/jnumed.106.039438 [DOI] [PubMed] [Google Scholar]
- 26. Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005;172:189-94. 10.1164/rccm.200401-006OC [DOI] [PubMed] [Google Scholar]
- 27. Thenappan T, Shah SJ, Gomberg-Maitland M, et al. Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circ Heart Fail 2011;4:257-65. 10.1161/CIRCHEARTFAILURE.110.958801 [DOI] [PubMed] [Google Scholar]
- 28. Deaño RC, Glassner-Kolmin C, Rubenfire M, et al. Referral of patients with pulmonary hypertension diagnoses to tertiary pulmonary hypertension centers: the multicenter RePHerral study. JAMA Intern Med 2013;173:887-93. 10.1001/jamainternmed.2013.319 [DOI] [PubMed] [Google Scholar]
- 29. Califf RM, Adams KF, McKenna WJ, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: The Flolan International Randomized Survival Trial (FIRST). Am Heart J 1997;134:44-54. 10.1016/S0002-8703(97)70105-4 [DOI] [PubMed] [Google Scholar]
- 30. Redfield MM, Chen HH, Borlaug BA, et al. RELAX Trial Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013;309:1268-77. 10.1001/jama.2013.2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Esfandiari S, Wright SP, Goodman JM, Sasson Z, Mak S. Pulmonary Artery Wedge Pressure Relative to Exercise Work Rate in Older Men and Women. Med Sci Sports Exerc 2017;49:1297-304. 10.1249/MSS.0000000000001227 [DOI] [PubMed] [Google Scholar]
- 32. Ryan JJ, Rich JD, Thiruvoipati T, Swamy R, Kim GH, Rich S. Current practice for determining pulmonary capillary wedge pressure predisposes to serious errors in the classification of patients with pulmonary hypertension. Am Heart J 2012;163:589-94. 10.1016/j.ahj.2012.01.024 [DOI] [PubMed] [Google Scholar]
- 33. Halpern SD, Taichman DB. Misclassification of pulmonary hypertension due to reliance on pulmonary capillary wedge pressure rather than left ventricular end-diastolic pressure. Chest 2009;136:37-43. 10.1378/chest.08-2784 [DOI] [PubMed] [Google Scholar]
- 34. Opotowsky AR, Ojeda J, Rogers F, et al. A simple echocardiographic prediction rule for hemodynamics in pulmonary hypertension. Circ Cardiovasc Imaging 2012;5:765-75. 10.1161/CIRCIMAGING.112.976654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robbins IM, Hemnes AR, Pugh ME, et al. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circ Heart Fail 2014;7:116-22. 10.1161/CIRCHEARTFAILURE.113.000468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Borlaug BA, Nishimura RA, Sorajja P, Lam CS, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail 2010;3:588-95. 10.1161/CIRCHEARTFAILURE.109.930701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seeger W, Adir Y, Barberà JA, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol 2013;62(Suppl):D109-16. 10.1016/j.jacc.2013.10.036 [DOI] [PubMed] [Google Scholar]
- 38. Prins KW, Duval S, Markowitz J, Pritzker M, Thenappan T. Chronic use of PAH-specific therapy in World Health Organization Group III Pulmonary Hypertension: a systematic review and meta-analysis. Pulm Circ 2017;7:145-55. 10.1086/690017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van de Veerdonk MC, Kind T, Marcus JT, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011;58:2511-9. 10.1016/j.jacc.2011.06.068 [DOI] [PubMed] [Google Scholar]
- 40. Tei C, Dujardin KS, Hodge DO, et al. Doppler echocardiographic index for assessment of global right ventricular function. J Am Soc Echocardiogr 1996;9:838-47. 10.1016/S0894-7317(96)90476-9 [DOI] [PubMed] [Google Scholar]
- 41. Lindqvist P, Waldenström A, Henein M, Mörner S, Kazzam E. Regional and global right ventricular function in healthy individuals aged 20-90 years: a pulsed Doppler tissue imaging study: Umeå General Population Heart Study. Echocardiography 2005;22:305-14. 10.1111/j.1540-8175.2005.04023.x [DOI] [PubMed] [Google Scholar]
- 42. Meluzín J, Spinarová L, Bakala J, et al. Pulsed Doppler tissue imaging of the velocity of tricuspid annular systolic motion; a new, rapid, and non-invasive method of evaluating right ventricular systolic function. Eur Heart J 2001;22:340-8. 10.1053/euhj.2000.2296 [DOI] [PubMed] [Google Scholar]
- 43. Raymond RJ, Hinderliter AL, Willis PW, et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol 2002;39:1214-9. 10.1016/S0735-1097(02)01744-8 [DOI] [PubMed] [Google Scholar]
- 44. Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 2006;174:1034-41. 10.1164/rccm.200604-547OC [DOI] [PubMed] [Google Scholar]
- 45. Ghio S, Pazzano AS, Klersy C, et al. Clinical and prognostic relevance of echocardiographic evaluation of right ventricular geometry in patients with idiopathic pulmonary arterial hypertension. Am J Cardiol 2011;107:628-32. 10.1016/j.amjcard.2010.10.027 [DOI] [PubMed] [Google Scholar]
- 46. Ernande L, Cottin V, Leroux PY, et al. Right isovolumic contraction velocity predicts survival in pulmonary hypertension. J Am Soc Echocardiogr 2013;26:297-306. 10.1016/j.echo.2012.11.011 [DOI] [PubMed] [Google Scholar]
- 47. Sachdev A, Villarraga HR, Frantz RP, et al. Right ventricular strain for prediction of survival in patients with pulmonary arterial hypertension. Chest 2011;139:1299-309. 10.1378/chest.10-2015 [DOI] [PubMed] [Google Scholar]
- 48. Leather HA, Ama’ R, Missant C, Rex S, Rademakers FE, Wouters PF. Longitudinal but not circumferential deformation reflects global contractile function in the right ventricle with open pericardium. Am J Physiol Heart Circ Physiol 2006;290:H2369-75. 10.1152/ajpheart.01211.2004 [DOI] [PubMed] [Google Scholar]
- 49. Addetia K, Bhave NM, Tabit CE, et al. Sample size and cost analysis for pulmonary arterial hypertension drug trials using various imaging modalities to assess right ventricular size and function end points. Circ Cardiovasc Imaging 2014;7:115-24. 10.1161/CIRCIMAGING.113.000932 [DOI] [PubMed] [Google Scholar]
- 50. Anavekar NS, Gerson D, Skali H, Kwong RY, Yucel EK, Solomon SD. Two-dimensional assessment of right ventricular function: an echocardiographic-MRI correlative study. Echocardiography 2007;24:452-6. 10.1111/j.1540-8175.2007.00424.x [DOI] [PubMed] [Google Scholar]
- 51. Gupta S, Khan F, Shapiro M, Weeks SG, Litwin SE, Michaels AD. The associations between tricuspid annular plane systolic excursion (TAPSE), ventricular dyssynchrony, and ventricular interaction in heart failure patients. Eur J Echocardiogr 2008;9:766-71. 10.1093/ejechocard/jen147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hardziyenka M, Campian ME, de Bruin-Bon HA, Michel MC, Tan HL. Sequence of echocardiographic changes during development of right ventricular failure in rat. J Am Soc Echocardiogr 2006;19:1272-9. 10.1016/j.echo.2006.04.036 [DOI] [PubMed] [Google Scholar]
- 53. Chua S, Levine RA, Yosefy C, et al. Assessment of right ventricular function by real-time three-dimensional echocardiography improves accuracy and decreases interobserver variability compared with conventional two-dimensional views. Eur J Echocardiogr 2009;10:619-24. 10.1093/ejechocard/jep013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bhave NM, Patel AR, Weinert L, et al. Three-dimensional modeling of the right ventricle from two-dimensional transthoracic echocardiographic images: utility of knowledge-based reconstruction in pulmonary arterial hypertension. J Am Soc Echocardiogr 2013;26:860-7. 10.1016/j.echo.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 55. Nagata Y, Wu VC, Kado Y, et al. Prognostic Value of Right Ventricular Ejection Fraction Assessed by Transthoracic 3D Echocardiography. Circ Cardiovasc Imaging 2017;10:e005384. 10.1161/CIRCIMAGING.116.005384 [DOI] [PubMed] [Google Scholar]
- 56. Ulrich S, Hasler ED, Saxer S, et al. Effect of breathing oxygen-enriched air on exercise performance in patients with precapillary pulmonary hypertension: randomized, sham-controlled cross-over trial. Eur Heart J 2017;38:1159-68. 10.1093/eurheartj/ehx099 [DOI] [PubMed] [Google Scholar]
- 57. Pandey A, Garg S, Khunger M, et al. Efficacy and Safety of Exercise Training in Chronic Pulmonary Hypertension: Systematic Review and Meta-Analysis. Circ Heart Fail 2015;8:1032-43. [DOI] [PubMed] [Google Scholar]
- 58. Jaïs X, Olsson KM, Barbera JA, et al. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur Respir J 2012;40:881-5. 10.1183/09031936.00141211 [DOI] [PubMed] [Google Scholar]
- 59. Hemnes AR, Kiely DG, Cockrill BA, et al. Statement on pregnancy in pulmonary hypertension from the Pulmonary Vascular Research Institute. Pulm Circ 2015;5:435-65. 10.1086/682230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rich S, Seidlitz M, Dodin E, et al. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest 1998;114:787-92. 10.1378/chest.114.3.787 [DOI] [PubMed] [Google Scholar]
- 61. Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984;70:580-7. 10.1161/01.CIR.70.4.580 [DOI] [PubMed] [Google Scholar]
- 62. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992;327:76-81. 10.1056/NEJM199207093270203 [DOI] [PubMed] [Google Scholar]
- 63. Frank H, Mlczoch J, Huber K, Schuster E, Gurtner HP, Kneussl M. The effect of anticoagulant therapy in primary and anorectic drug-induced pulmonary hypertension. Chest 1997;112:714-21. 10.1378/chest.112.3.714 [DOI] [PubMed] [Google Scholar]
- 64. Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014;129:57-65. 10.1161/CIRCULATIONAHA.113.004526 [DOI] [PubMed] [Google Scholar]
- 65. Preston IR, Roberts KE, Miller DP, et al. Effect of Warfarin Treatment on Survival of Patients With Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation 2015;132:2403-11. 10.1161/CIRCULATIONAHA.115.018435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016;37:67-119. 10.1093/eurheartj/ehv317 [DOI] [PubMed] [Google Scholar]
- 67. Abe K, Shinoda M, Tanaka M, et al. Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc Res 2016;111:16-25. 10.1093/cvr/cvw070 [DOI] [PubMed] [Google Scholar]
- 68. Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111:3105-11. 10.1161/CIRCULATIONAHA.104.488486 [DOI] [PubMed] [Google Scholar]
- 69. Hemnes AR, Trammell AW, Archer SL, et al. Peripheral blood signature of vasodilator-responsive pulmonary arterial hypertension. Circulation 2015;131:401-9, discussion 409. 10.1161/CIRCULATIONAHA.114.013317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hemnes AR, Zhao M, West J, et al. Critical Genomic Networks and Vasoreactive Variants in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2016;194:464-75. 10.1164/rccm.201508-1678OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Montani D, Savale L, Natali D, et al. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J 2010;31:1898-907. 10.1093/eurheartj/ehq170 [DOI] [PubMed] [Google Scholar]
- 72. Archer SL, Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med 2009;361:1864-71. 10.1056/NEJMct0904473 [DOI] [PubMed] [Google Scholar]
- 73. Decoene C, Bourzoufi K, Moreau D, Narducci F, Crepin F, Krivosic-Horber R. Use of inhaled nitric oxide for emergency Cesarean section in a woman with unexpected primary pulmonary hypertension. Can J Anaesth 2001;48:584-7. 10.1007/BF03016836 [DOI] [PubMed] [Google Scholar]
- 74. Barst RJ, Rubin LJ, Long WA, et al. Primary Pulmonary Hypertension Study Group A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296-301. 10.1056/NEJM199602013340504 [DOI] [PubMed] [Google Scholar]
- 75. Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med 2000;132:425-34. 10.7326/0003-4819-132-6-200003210-00002 [DOI] [PubMed] [Google Scholar]
- 76. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896-903. 10.1056/NEJMoa012212 [DOI] [PubMed] [Google Scholar]
- 77. Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148-57. 10.1056/NEJMoa050010 [DOI] [PubMed] [Google Scholar]
- 78. Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES) Group Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117:3010-9. 10.1161/CIRCULATIONAHA.107.742510 [DOI] [PubMed] [Google Scholar]
- 79. Galiè N, Brundage BH, Ghofrani HA, et al. Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119:2894-903. 10.1161/CIRCULATIONAHA.108.839274 [DOI] [PubMed] [Google Scholar]
- 80. McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010;55:1915-22. 10.1016/j.jacc.2010.01.027 [DOI] [PubMed] [Google Scholar]
- 81. Ghofrani HA, Galiè N, Grimminger F, et al. PATENT-1 Study Group Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330-40. 10.1056/NEJMoa1209655 [DOI] [PubMed] [Google Scholar]
- 82. Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation 2013;127:624-33. 10.1161/CIRCULATIONAHA.112.124388 [DOI] [PubMed] [Google Scholar]
- 83. Tapson VF, Jing ZC, Xu KF, et al. FREEDOM-C2 Study Team Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest 2013;144:952-8. 10.1378/chest.12-2875 [DOI] [PubMed] [Google Scholar]
- 84. Pulido T, Adzerikho I, Channick RN, et al. SERAPHIN Investigators Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18. 10.1056/NEJMoa1213917 [DOI] [PubMed] [Google Scholar]
- 85. Sitbon O, Channick R, Chin KM, et al. GRIPHON Investigators Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015;373:2522-33. 10.1056/NEJMoa1503184 [DOI] [PubMed] [Google Scholar]
- 86. Galiè N, Barberà JA, Frost AE, et al. AMBITION Investigators Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med 2015;373:834-44. 10.1056/NEJMoa1413687 [DOI] [PubMed] [Google Scholar]
- 87. Dasgupta A, Bowman L, D’Arsigny CL, Archer SL. Soluble guanylate cyclase: a new therapeutic target for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Clin Pharmacol Ther 2015;97:88-102. 10.1002/cpt.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hoeper MCP, Klinger JR, Langleben D, Naeije R, Simonneau G, Benza RL. RESPITE: Riociguat in pulmonary arterial hypertension patients with an inadequate response to phosphodiesterase type 5 inhibitors. Eur Respir J 2016;48(suppl 60):OA263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Galiè N, Rubin Lj, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008;371:2093-100. 10.1016/S0140-6736(08)60919-8 [DOI] [PubMed] [Google Scholar]
- 90. Wei A, Gu Z, Li J, et al. Clinical Adverse Effects of Endothelin Receptor Antagonists: Insights From the Meta-Analysis of 4894 Patients From 24 Randomized Double-Blind Placebo-Controlled Clinical Trials. J Am Heart Assoc 2016;5:e003896. 10.1161/JAHA.116.003896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Paul GA, Gibbs JS, Boobis AR, Abbas A, Wilkins MR. Bosentan decreases the plasma concentration of sildenafil when coprescribed in pulmonary hypertension. Br J Clin Pharmacol 2005;60:107-12. 10.1111/j.1365-2125.2005.02383.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. McLaughlin V, Channick RN, Ghofrani HA, et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J 2015;46:405-13. 10.1183/13993003.02044-2014 [DOI] [PubMed] [Google Scholar]
- 93. Brittain EL, Talati M, Fessel JP, et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016;133:1936-44. 10.1161/CIRCULATIONAHA.115.019351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Moncada S. Prostacyclin, from discovery to clinical application. J Pharmacol 1985;16(Suppl 1):71-88. [PubMed] [Google Scholar]
- 95. Simonneau G, Barst RJ, Galie N, et al. Treprostinil Study Group Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165:800-4. 10.1164/ajrccm.165.6.2106079 [DOI] [PubMed] [Google Scholar]
- 96. Benza RL, Tapson VF, Gomberg-Maitland M, Poms A, Barst RJ, McLaughlin VV. One-year experience with intravenous treprostinil for pulmonary arterial hypertension. J Heart Lung Transplant 2013;32:889-96. 10.1016/j.healun.2013.06.008 [DOI] [PubMed] [Google Scholar]
- 97. Waxman AB, McElderry HT, Gomberg-Maitland M, et al. Totally Implantable IV Treprostinil Therapy in Pulmonary Hypertension Assessment of the Implantation Procedure. Chest 2017;Jun 3:S0012-3692(17)31021-8. [DOI] [PubMed] [Google Scholar]
- 98. McLaughlin V, Channick R, Chin K, et al. Effect Of Selexipag On Morbidity/Mortality In Pulmonary Arterial Hypertension: Results Of The Griphon Study. J Am Coll Cardiol 2015;65(Suppl):A1538 10.1016/S0735-1097(15)61538-8. [DOI] [Google Scholar]
- 99. Lahm T, Kawut SM. Inhibiting oestrogen signalling in pulmonary arterial hypertension: sex, drugs and research. Eur Respir J 2017;50:1700983. 10.1183/13993003.00983-2017 [DOI] [PubMed] [Google Scholar]
- 100. Mathai SC, Hassoun PM, Puhan MA, Zhou Y, Wise RA. Sex differences in response to tadalafil in pulmonary arterial hypertension. Chest 2015;147:188-97. 10.1378/chest.14-0263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Foderaro A, Ventetuolo CE. Pulmonary Arterial Hypertension and the Sex Hormone Paradox. Curr Hypertens Rep 2016;18:84. 10.1007/s11906-016-0689-7 [DOI] [PubMed] [Google Scholar]
- 102. Sitbon O, Jaïs X, Savale L, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J 2014;43:1691-7. 10.1183/09031936.00116313 [DOI] [PubMed] [Google Scholar]
- 103. Machuca TN, de Perrot M. Mechanical Support for the Failing Right Ventricle in Patients With Precapillary Pulmonary Hypertension. Circulation 2015;132:526-36. 10.1161/CIRCULATIONAHA.114.012593 [DOI] [PubMed] [Google Scholar]
- 104. Romberg E. Ueber Sklerose der Lungen arterie. Dtsch Arch Klin Med 1891;48:197-206. [Google Scholar]
- 105. Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 2008;294:H570-8. 10.1152/ajpheart.01324.2007 [DOI] [PubMed] [Google Scholar]
- 106. Caruso P, Dunmore BJ, Schlosser K, et al. Identification of miR-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 and PKM2. Circulation 2017;CIRCULATIONAHA.117.028034. 10.1161/CIRCULATIONAHA.117.028034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhang H, Wang D, Li M, et al. The Metabolic and Proliferative State of Vascular Adventitial Fibroblasts in Pulmonary Hypertension is Regulated through a MiR-124/PTBP1/PKM Axis. Circulation 2017;CIRCULATIONAHA.117.028069. 10.1161/CIRCULATIONAHA.117.028069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yu Y, Keller SH, Remillard CV, et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009;119:2313-22. 10.1161/CIRCULATIONAHA.108.782458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yuan JX, Aldinger AM, Juhaszova M, et al. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 1998;98:1400-6. 10.1161/01.CIR.98.14.1400 [DOI] [PubMed] [Google Scholar]
- 110. Archer SL, Souil E, Dinh-Xuan AT, et al. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 1998;101:2319-30. 10.1172/JCI333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pozeg ZI, Michelakis ED, McMurtry MS, et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003;107:2037-44. 10.1161/01.CIR.0000062688.76508.B3 [DOI] [PubMed] [Google Scholar]
- 112. Bonnet S, Rochefort G, Sutendra G, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A 2007;104:11418-23. 10.1073/pnas.0610467104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hong Z, Chen KH, DasGupta A, et al. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype. Am J Respir Crit Care Med 2017;195:515-29. 10.1164/rccm.201604-0814OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Oka M, Homma N, Taraseviciene-Stewart L, et al. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 2007;100:923-9. 10.1161/01.RES.0000261658.12024.18 [DOI] [PubMed] [Google Scholar]
- 115. Nahar K, Absar S, Gupta N, et al. Peptide-coated liposomal fasudil enhances site specific vasodilation in pulmonary arterial hypertension. Mol Pharm 2014;11:4374-84. 10.1021/mp500456k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jiang R, Ai ZS, Jiang X, et al. Intravenous fasudil improves in-hospital mortality of patients with right heart failure in severe pulmonary hypertension. Hypertens Res 2015;38:539-44. 10.1038/hr.2015.33 [DOI] [PubMed] [Google Scholar]
- 117. Marsboom G, Toth PT, Ryan JJ, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 2012;110:1484-97. 10.1161/CIRCRESAHA.111.263848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bonnet S, Michelakis ED, Porter CJ, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006;113:2630-41. 10.1161/CIRCULATIONAHA.105.609008 [DOI] [PubMed] [Google Scholar]
- 119. Diebold I, Hennigs JK, Miyagawa K, et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab 2015;21:596-608. 10.1016/j.cmet.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Fijalkowska I, Xu W, Comhair SA, et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 2010;176:1130-8. 10.2353/ajpath.2010.090832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Li M, Riddle S, Zhang H, et al. Metabolic Reprogramming Regulates the Proliferative and Inflammatory Phenotype of Adventitial Fibroblasts in Pulmonary Hypertension Through the Transcriptional Corepressor C-Terminal Binding Protein-1. Circulation 2016;134:1105-21. 10.1161/CIRCULATIONAHA.116.023171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015;131:1691-702. 10.1161/CIRCULATIONAHA.114.006979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. McMurtry MS, Bonnet S, Wu X, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 2004;95:830-40. 10.1161/01.RES.0000145360.16770.9f [DOI] [PubMed] [Google Scholar]
- 124. Michelakis ED, McMurtry MS, Wu XC, et al. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 2002;105:244-50. 10.1161/hc0202.101974 [DOI] [PubMed] [Google Scholar]
- 125. Marsboom G, Wietholt C, Haney CR, et al. Lung ¹⁸F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;185:670-9. 10.1164/rccm.201108-1562OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gutierrez G, Venbrux A, Ignacio E, Reiner J, Chawla L, Desai A. The concentration of oxygen, lactate and glucose in the central veins, right heart, and pulmonary artery: a study in patients with pulmonary hypertension. Crit Care 2007;11:R44. 10.1186/cc5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Guignabert C, Tu L, Izikki M, et al. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22alpha-targeted overexpression of the serotonin transporter. FASEB J 2009;23:4135-47. 10.1096/fj.09-131664 [DOI] [PubMed] [Google Scholar]
- 128. Abdelmalak M, Lew A, Ramezani R, et al. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab 2013;109:139-43. 10.1016/j.ymgme.2013.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Michelakis ED, Gurtu V, Webster L, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med 2017;9:eaao4583. 10.1126/scitranslmed.aao4583 [DOI] [PubMed] [Google Scholar]
- 130. Archer SL. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med 2013;369:2236-51. 10.1056/NEJMra1215233 [DOI] [PubMed] [Google Scholar]
- 131. Ryan JJ, Marsboom G, Fang YH, et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187:865-78. 10.1164/rccm.201209-1687OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Aldred MA, Vijayakrishnan J, James V, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat 2006;27:212-3. 10.1002/humu.9398 [DOI] [PubMed] [Google Scholar]
- 133. Cogan JD, Pauciulo MW, Batchman AP, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174:590-8. 10.1164/rccm.200602-165OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Morisaki H, Nakanishi N, Kyotani S, Takashima A, Tomoike H, Morisaki T. BMPR2 mutations found in Japanese patients with familial and sporadic primary pulmonary hypertension. Hum Mutat 2004;23:632. 10.1002/humu.9251 [DOI] [PubMed] [Google Scholar]
- 135. Machado RD, Pauciulo MW, Thomson JR, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet 2001;68:92-102. 10.1086/316947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Koehler R, Grünig E, Pauciulo MW, et al. Low frequency of BMPR2 mutations in a German cohort of patients with sporadic idiopathic pulmonary arterial hypertension. J Med Genet 2004;41:e127. 10.1136/jmg.2004.023101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cogan JD, Vnencak-Jones CL, Phillips JA, 3rd, et al. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med 2005;7:169-74. 10.1097/01.GIM.0000156525.09595.E9 [DOI] [PubMed] [Google Scholar]
- 138. Larkin EK, Newman JH, Austin ED, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:892-6. 10.1164/rccm.201205-0886OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Evans JD, Girerd B, Montani D, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 2016;4:129-37. 10.1016/S2213-2600(15)00544-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Atkinson C, Stewart S, Upton PD, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002;105:1672-8. 10.1161/01.CIR.0000012754.72951.3D [DOI] [PubMed] [Google Scholar]
- 141. Lavoie JR, Ormiston ML, Perez-Iratxeta C, et al. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation 2014;129:2125-35. 10.1161/CIRCULATIONAHA.114.008777 [DOI] [PubMed] [Google Scholar]
- 142. Hurst LA, Dunmore BJ, Long L, et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat Commun 2017;8:14079. 10.1038/ncomms14079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Long L, Ormiston ML, Yang X, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med 2015;21:777-85. 10.1038/nm.3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Drake KM, Dunmore BJ, McNelly LN, Morrell NW, Aldred MA. Correction of nonsense BMPR2 and SMAD9 mutations by ataluren in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2013;49:403-9. 10.1165/rcmb.2013-0100OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Sobolewski A, Rudarakanchana N, Upton PD, et al. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: potential for rescue. Hum Mol Genet 2008;17:3180-90. 10.1093/hmg/ddn214 [DOI] [PubMed] [Google Scholar]
- 146. McMurtry MS, Moudgil R, Hashimoto K, Bonnet S, Michelakis ED, Archer SL. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2007;292:L872-8. 10.1152/ajplung.00309.2006 [DOI] [PubMed] [Google Scholar]
- 147. Reynolds AM, Xia W, Holmes MD, et al. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2007;292:L1182-92. 10.1152/ajplung.00020.2006 [DOI] [PubMed] [Google Scholar]
- 148. Spiekerkoetter E, Tian X, Cai J, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 2013;123:3600-13. 10.1172/JCI65592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Long L, Yang X, Southwood M, et al. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res 2013;112:1159-70. 10.1161/CIRCRESAHA.111.300483 [DOI] [PubMed] [Google Scholar]
- 150. Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001;345:325-34. 10.1056/NEJM200108023450503 [DOI] [PubMed] [Google Scholar]
- 151. Aldred MA, Comhair SA, Varella-Garcia M, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 2010;182:1153-60. 10.1164/rccm.201003-0491OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chaouat A, Coulet F, Favre C, et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004;59:446-8. 10.1136/thx.2003.11890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Harrison RE, Flanagan JA, Sankelo M, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 2003;40:865-71. 10.1136/jmg.40.12.865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ma L, Roman-Campos D, Austin ED, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013;369:351-61. 10.1056/NEJMoa1211097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Austin ED, Ma L, LeDuc C, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012;5:336-43. 10.1161/CIRCGENETICS.111.961888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Eyries M, Montani D, Girerd B, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 2014;46:65-9. 10.1038/ng.2844 [DOI] [PubMed] [Google Scholar]
- 157. Best DH, Sumner KL, Austin ED, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014;145:231-6. 10.1378/chest.13-2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Montani D, Girerd B, Jaïs X, et al. Clinical phenotypes and outcomes of heritable and sporadic pulmonary veno-occlusive disease: a population-based study. Lancet Respir Med 2017;5:125-34. 10.1016/S2213-2600(16)30438-6 [DOI] [PubMed] [Google Scholar]
- 159. Archer SL, Marsboom G, Kim GH, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 2010;121:2661-71. 10.1161/CIRCULATIONAHA.109.916098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr 2007;39:231-4. 10.1007/s10863-007-9081-2 [DOI] [PubMed] [Google Scholar]
- 161. Rexhaj E, Bloch J, Jayet PY, et al. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol 2011;301:H247-52. 10.1152/ajpheart.01309.2010 [DOI] [PubMed] [Google Scholar]
- 162. Maresca A, Zaffagnini M, Caporali L, Carelli V, Zanna C. DNA methyltransferase 1 mutations and mitochondrial pathology: is mtDNA methylated? Front Genet 2015;6:90. 10.3389/fgene.2015.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Momparler RL, Côté S, Momparler LF, Idaghdour Y. Inhibition of DNA and Histone Methylation by 5-Aza-2′-Deoxycytidine (Decitabine) and 3-Deazaneplanocin-A on Antineoplastic Action and Gene Expression in Myeloid Leukemic Cells. Front Oncol 2017;7:19. 10.3389/fonc.2017.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Scourzic L, Mouly E, Bernard OA. TET proteins and the control of cytosine demethylation in cancer. Genome Med 2015;7:9. 10.1186/s13073-015-0134-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Zhao L, Chen CN, Hajji N, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012;126:455-67. 10.1161/CIRCULATIONAHA.112.103176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Kim J, Hwangbo C, Hu X, et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 2015;131:190-9. 10.1161/CIRCULATIONAHA.114.013339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Sutendra G, Kinnaird A, Dromparis P, et al. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014;158:84-97. 10.1016/j.cell.2014.04.046 [DOI] [PubMed] [Google Scholar]
- 168. Xu XF, Ma XL, Shen Z, Wu XL, Cheng F, Du LZ. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J Hypertens 2010;28:2227-35. 10.1097/HJH.0b013e32833e08f1 [DOI] [PubMed] [Google Scholar]
- 169. Cavasin MA, Demos-Davies K, Horn TR, et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res 2012;110:739-48. 10.1161/CIRCRESAHA.111.258426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cho YK, Eom GH, Kee HJ, et al. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ J 2010;74:760-70. 10.1253/circj.CJ-09-0580 [DOI] [PubMed] [Google Scholar]
- 171. Bogaard HJ, Mizuno S, Hussaini AA, et al. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 2011;183:1402-10. 10.1164/rccm.201007-1106OC [DOI] [PubMed] [Google Scholar]
- 172. Pullamsetti SS, Doebele C, Fischer A, et al. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am J Respir Crit Care Med 2012;185:409-19. 10.1164/rccm.201106-1093OC [DOI] [PubMed] [Google Scholar]
- 173. Brock M, Trenkmann M, Gay RE, et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 2009;104:1184-91. 10.1161/CIRCRESAHA.109.197491 [DOI] [PubMed] [Google Scholar]
- 174. Caruso P, MacLean MR, Khanin R, et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol 2010;30:716-23. 10.1161/ATVBAHA.109.202028 [DOI] [PubMed] [Google Scholar]
- 175. Parikh VN, Jin RC, Rabello S, et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 2012;125:1520-32. 10.1161/CIRCULATIONAHA.111.060269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 2010;299:L861-71. 10.1152/ajplung.00201.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. White K, Dempsie Y, Caruso P, et al. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 2014;64:185-94. 10.1161/HYPERTENSIONAHA.113.03037 [DOI] [PubMed] [Google Scholar]
- 178. Drake KM, Zygmunt D, Mavrakis L, et al. Altered MicroRNA processing in heritable pulmonary arterial hypertension: an important role for Smad-8. Am J Respir Crit Care Med 2011;184:1400-8. 10.1164/rccm.201106-1130OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Schlosser K, White RJ, Stewart DJ. miR-26a linked to pulmonary hypertension by global assessment of circulating extracellular microRNAs. Am J Respir Crit Care Med 2013;188:1472-5. 10.1164/rccm.201308-1403LE [DOI] [PubMed] [Google Scholar]
- 180. Kang K, Peng X, Zhang X, et al. MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem 2013;288:25414-27. 10.1074/jbc.M113.460287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Wang D, Zhang H, Li M, et al. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ Res 2014;114:67-78. 10.1161/CIRCRESAHA.114.301633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Rothman AM, Arnold ND, Pickworth JA, et al. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest 2016;126:2495-508. 10.1172/JCI83361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Caruso P, Dempsie Y, Stevens HC, et al. A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res 2012;111:290-300. 10.1161/CIRCRESAHA.112.267591 [DOI] [PubMed] [Google Scholar]
- 184. Rhodes CJ, Wharton J, Boon RA, et al. Reduced microRNA-150 is associated with poor survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187:294-302. 10.1164/rccm.201205-0839OC [DOI] [PubMed] [Google Scholar]
- 185. Courboulin A, Paulin R, Giguère NJ, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 2011;208:535-48. 10.1084/jem.20101812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kim J, Kang Y, Kojima Y, et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 2013;19:74-82. 10.1038/nm.3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 2014;115:165-75. 10.1161/CIRCRESAHA.113.301141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Graham BB, Bandeira AP, Morrell NW, Butrous G, Tuder RM. Schistosomiasis-associated pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest 2010;137(Suppl):20S-9S. 10.1378/chest.10-0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension [vii.]. Clin Chest Med 2007;28:23-42, vii. 10.1016/j.ccm.2006.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Heath D, Yacoub M. Lung mast cells in plexogenic pulmonary arteriopathy. J Clin Pathol 1991;44:1003-6. 10.1136/jcp.44.12.1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Perros F, Dorfmüller P, Souza R, et al. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur Respir J 2007;29:462-8. 10.1183/09031936.00094706 [DOI] [PubMed] [Google Scholar]
- 192. Humbert M, Monti G, Brenot F, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 1995;151:1628-31. 10.1164/ajrccm.151.5.7735624 [DOI] [PubMed] [Google Scholar]
- 193. Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010;122:920-7. 10.1161/CIRCULATIONAHA.109.933762 [DOI] [PubMed] [Google Scholar]
- 194. Perros F, Dorfmüller P, Souza R, et al. Fractalkine-induced smooth muscle cell proliferation in pulmonary hypertension. Eur Respir J 2007;29:937-43. 10.1183/09031936.00104706 [DOI] [PubMed] [Google Scholar]
- 195. Sanchez O, Marcos E, Perros F, et al. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2007;176:1041-7. 10.1164/rccm.200610-1559OC [DOI] [PubMed] [Google Scholar]
- 196. Dorfmüller P, Zarka V, Durand-Gasselin I, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med 2002;165:534-9. 10.1164/ajrccm.165.4.2012112 [DOI] [PubMed] [Google Scholar]
- 197. El Kasmi KC, Pugliese SC, Riddle SR, et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol 2014;193:597-609. 10.4049/jimmunol.1303048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Li M, Riddle SR, Frid MG, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol 2011;187:2711-22. 10.4049/jimmunol.1100479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Frid MG, Brunetti JA, Burke DL, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol 2006;168:659-69. 10.2353/ajpath.2006.050599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Miyata M, Sakuma F, Ito M, Ohira H, Sato Y, Kasukawa R. Athymic nude rats develop severe pulmonary hypertension following monocrotaline administration. Int Arch Allergy Immunol 2000;121:246-52. 10.1159/000024324 [DOI] [PubMed] [Google Scholar]
- 201. Tamosiuniene R, Tian W, Dhillon G, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 2011;109:867-79. 10.1161/CIRCRESAHA.110.236927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Qian J, Tian W, Jiang X, et al. Leukotriene B4 Activates Pulmonary Artery Adventitial Fibroblasts in Pulmonary Hypertension. Hypertension 2015;66:1227-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Hashimoto-Kataoka T, Hosen N, Sonobe T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci U S A 2015;112:E2677-86. 10.1073/pnas.1424774112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Perros F, Dorfmüller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;185:311-21. 10.1164/rccm.201105-0927OC [DOI] [PubMed] [Google Scholar]
- 205. Colvin KL, Cripe PJ, Ivy DD, Stenmark KR, Yeager ME. Bronchus-associated lymphoid tissue in pulmonary hypertension produces pathologic autoantibodies. Am J Respir Crit Care Med 2013;188:1126-36. 10.1164/rccm.201302-0403OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Ormiston ML, Chang C, Long LL, et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 2012;126:1099-109. 10.1161/CIRCULATIONAHA.112.110619 [DOI] [PubMed] [Google Scholar]
- 207. Gotthardt D, Putz EM, Grundschober E, et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov 2016;6:414-29. 10.1158/2159-8290.CD-15-0732 [DOI] [PubMed] [Google Scholar]
- 208. Hanna J, Goldman-Wohl D, Hamani Y, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med 2006;12:1065-74. 10.1038/nm1452 [DOI] [PubMed] [Google Scholar]
- 209. Edwards AL, Gunningham SP, Clare GC, et al. Professional killer cell deficiencies and decreased survival in pulmonary arterial hypertension. Respirology 2013;18:1271-7. 10.1111/resp.12152 [DOI] [PubMed] [Google Scholar]
- 210. van Wolferen SA, Marcus JT, Boonstra A, et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007;28:1250-7. 10.1093/eurheartj/ehl477 [DOI] [PubMed] [Google Scholar]
- 211. Chaouat A, Savale L, Chouaid C, et al. Role for interleukin-6 in COPD-related pulmonary hypertension. Chest 2009;136:678-87. 10.1378/chest.08-2420 [DOI] [PubMed] [Google Scholar]
- 212. Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 2010;138:1234-9. 10.1378/chest.09-2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Michelakis ED, Tymchak W, Noga M, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation 2003;108:2066-9. 10.1161/01.CIR.0000099502.17776.C2 [DOI] [PubMed] [Google Scholar]
- 214. Oikawa M, Kagaya Y, Otani H, et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol 2005;45:1849-55. 10.1016/j.jacc.2005.02.065 [DOI] [PubMed] [Google Scholar]
- 215. Mukerji B, Alpert MA, Mukerji V. Right ventricular alterations in scuba divers: findings on electrocardiography and echocardiography. South Med J 2000;93:673-6. 10.1097/00007611-200007000-00006 [DOI] [PubMed] [Google Scholar]
- 216. Overbeek MJ, Lankhaar JW, Westerhof N, et al. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur Respir J 2008;31:1160-6. 10.1183/09031936.00135407 [DOI] [PubMed] [Google Scholar]
- 217. Georgianos PI, Sarafidis PA, Liakopoulos V. Arterial Stiffness: A Novel Risk Factor for Kidney Injury Progression? Am J Hypertens 2015;28:958-65. 10.1093/ajh/hpv004 [DOI] [PubMed] [Google Scholar]
- 218. Syrris P, Ward D, Evans A, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet 2006;79:978-84. 10.1086/509122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 2010;121:1533-41. 10.1161/CIRCULATIONAHA.108.840827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Torbicki A, Kurzyna M, Kuca P, et al. Detectable serum cardiac troponin T as a marker of poor prognosis among patients with chronic precapillary pulmonary hypertension. Circulation 2003;108:844-8. 10.1161/01.CIR.0000084544.54513.E2 [DOI] [PubMed] [Google Scholar]
- 221. Bian X, Williams AG, Jr, Gwirtz PA, Downey HF. Right coronary autoregulation in conscious, chronically instrumented dogs. Am J Physiol 1998;275:H169-75. [DOI] [PubMed] [Google Scholar]
- 222. Bogaard HJ, Natarajan R, Mizuno S, et al. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med 2010;182:652-60. 10.1164/rccm.201003-0335OC [DOI] [PubMed] [Google Scholar]
- 223. Fang YH, Piao L, Hong Z, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med (Berl) 2012;90:31-43. 10.1007/s00109-011-0804-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Wang GY, McCloskey DT, Turcato S, Swigart PM, Simpson PC, Baker AJ. Contrasting inotropic responses to alpha1-adrenergic receptor stimulation in left versus right ventricular myocardium. Am J Physiol Heart Circ Physiol 2006;291:H2013-7. 10.1152/ajpheart.00167.2006 [DOI] [PubMed] [Google Scholar]
- 225. Nootens M, Kaufmann E, Rector T, et al. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol 1995;26:1581-5. 10.1016/0735-1097(95)00399-1 [DOI] [PubMed] [Google Scholar]
- 226. Bristow MR, Minobe W, Rasmussen R, et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest 1992;89:803-15. 10.1172/JCI115659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Piao L, Fang YH, Parikh KS, et al. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation 2012;126:2859-69. 10.1161/CIRCULATIONAHA.112.109868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Piao L, Fang YH, Cadete VJ, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl) 2010;88:47-60. 10.1007/s00109-009-0524-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. van Campen JS, de Boer K, van de Veerdonk MC, et al. Bisoprolol in idiopathic pulmonary arterial hypertension: an explorative study. Eur Respir J 2016;48:787-96. 10.1183/13993003.00090-2016 [DOI] [PubMed] [Google Scholar]
- 230. Farha S, Saygin D, Park MM, et al. Pulmonary arterial hypertension treatment with carvedilol for heart failure: a randomized controlled trial. JCI Insight 2017;2:e95240. 10.1172/jci.insight.95240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev 2007;12:331-43. 10.1007/s10741-007-9034-1 [DOI] [PubMed] [Google Scholar]
- 232. Sharma S, Taegtmeyer H, Adrogue J, et al. Dynamic changes of gene expression in hypoxia-induced right ventricular hypertrophy. Am J Physiol Heart Circ Physiol 2004;286:H1185-92. 10.1152/ajpheart.00916.2003 [DOI] [PubMed] [Google Scholar]
- 233. Shehata ML, Lossnitzer D, Skrok J, et al. Myocardial delayed enhancement in pulmonary hypertension: pulmonary hemodynamics, right ventricular function, and remodeling. AJR Am J Roentgenol 2011;196:87-94. 10.2214/AJR.09.4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Freed BH, Gomberg-Maitland M, Chandra S, et al. Late gadolinium enhancement cardiovascular magnetic resonance predicts clinical worsening in patients with pulmonary hypertension. J Cardiovasc Magn Reson 2012;14:11. 10.1186/1532-429X-14-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Chugh SS, Whitesel S, Turner M, Roberts CT, Jr, Nagalla SR. Genetic basis for chamber-specific ventricular phenotypes in the rat infarct model. Cardiovasc Res 2003;57:477-85. 10.1016/S0008-6363(02)00703-4 [DOI] [PubMed] [Google Scholar]
- 236. Nagendran J, Archer SL, Soliman D, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007;116:238-48. 10.1161/CIRCULATIONAHA.106.655266 [DOI] [PubMed] [Google Scholar]
- 237. Kawut SM, Lima JA, Barr RG, et al. Sex and race differences in right ventricular structure and function: the multi-ethnic study of atherosclerosis-right ventricle study. Circulation 2011;123:2542-51. 10.1161/CIRCULATIONAHA.110.985515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest 2012;141:363-73. 10.1378/chest.10-3114 [DOI] [PubMed] [Google Scholar]
- 239. Mair KM, Johansen AK, Wright AF, Wallace E, MacLean MR. Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. Br J Pharmacol 2014;171:567-79. 10.1111/bph.12281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Taichman DB, Ornelas J, Chung L, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST guideline and expert panel report. Chest 2014;146:449-75. 10.1378/chest.14-0793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest 2007;131:1917-28. 10.1378/chest.06-2674 [DOI] [PubMed] [Google Scholar]
- 242. Cavasin MA, Stenmark KR, McKinsey TA. Emerging roles for histone deacetylases in pulmonary hypertension and right ventricular remodeling (2013 Grover Conference series). Pulm Circ 2015;5:63-72. 10.1086/679700 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary movie 1
Supplementary movie 2