

Abstract
As for the lack of simple and effective diagnostic methods at the early of the nasopharyngeal carcinoma (NPC), the mortality rate of NPC still remains high. Therefore, it is meaningful to explore the precise molecular mechanisms involved in the proliferation, carcinogenesis, and recurrence of NPC and thus find an effective diagnostic way and make a better therapeutic strategy.
Three gene expression data sets (GSE64634, GSE53819, and GSE12452) were downloaded from Gene Expression Omnibus (GEO) and analyzed using the online tool GEO2R to identify differentially expressed genes (DEGs). Gene ontology functional analysis and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of the DEGs were performed in Database for Annotation, Visualization and Integrated Discovery. The Search Tool for the Retrieval of Interacting Genes database was used to evaluate the interactions of DEGs and to construct a protein–protein interaction network using Cytoscape software. Hub genes were validated with the cBioPortal database.
The overlap among the 3 data sets contained 306 genes were identified to be differentially expressed between NPC and non-NPC samples. A total of 13 genes (DNAAF1, PARPBP, TTC18, GSTA3, RCN1, MUC5AC, POU2AF1, FAM83B, SLC22A16, SPEF2, ERICH3, CCDC81, and IL33) were identified as hub genes with degrees ≥10.
The present study was attempted to identify and functionally analyze the DEGs that may be involved in the carcinogenesis or progression of NPC by using comprehensive bioinformatics analyses and unveiled a series of hub genes and pathways. A total of 306 DEGs and 13 hub genes were identified and may be regarded as diagnostic biomarkers for NPC. However, more experimental studies are needed to carried out elucidate the biologic function of these genes results for NPC.
Keywords: nasopharyngeal carcinoma, bioinformatic analysis, gene
1. Introduction
Nasopharyngeal carcinoma (NPC) has remained high in endemic regions and is the most common malignant tumor in southern China and South East Asia.[1] It is primarily a malignant tumor derived from nasopharyngeal epithelium located in the upper part of the nasopharyngeal cavity and on the side wall, with a strong tendency to metastasize.[2] Its incidence very high in head and neck tumors approximately 0.2 to 0.5 cases per 100,000 people.[3] The main clinical manifestations of the patient are nasal congestion, blood stasis, ear blockage, hearing loss, and vision. Things like ghosts and headaches and other symptoms.[3] Diagnosing the disease in the early needs a high index of clinical acumen and confirmation is only dependent on histology.[2] The potential highly risk factors for NPC include Epstein–Barr virus (EBV) infection,[4] alcohol consumption, exposure to dust, formaldehyde, the function of genetic factors, and cigarette smoking.[5–8] EBV infection is found in 90% to 100% of NPC cases in endemic regions.[9] EBV is associated with multiple types of human cancer, such as Burkitt lymphoma and Hodgkin disease, while in Asia it is closely association with NPC. Accumulating evidence has demonstrated that abnormal expression and mutation of genes are involved in the carcinogenesis and progression of NPC, including glutathione S-transferase A1 (GSTA1), NGX6, COX-2, as well as mutations of tumor-suppressor genes. Recent genomic study of NPC has found dysregulated nuclear factor kappa B (NF-κB) signaling in NPC as well as multiple somatic mutations in the upstream negative regulators of NF-κB signaling.[5] Ye et al[10] reported that RASSF1A promoter methylation may be used for clinical diagnosis of nasopharyngeal carcinoma. Chen et al[11] used immunohistochemistry to detect the expression of p53R2 in 201 patients with NPC and find p53R2 was positively expressed in 92.5% (186/201) of NPC tissue with a high expression rate of 38.3% (77/201). Multivariate analysis of Cox model showed that IGFBP6 is an independent prognostic biomarker for recurrence and distant metastasis.[12] Peng et al[13] reported that the chronic stimulation of COX-2 plays a key role in the neoplastic conversion and development of NPC. However, as for the lack of simple and effective diagnostic methods at the early of the disease, the mortality rate of NPC still remains high. Therefore, it is meaningful to explore the precise molecular mechanisms involved in the proliferation, carcinogenesis, and recurrence of NPC and thus find an effective diagnostic way and make a better therapeutic strategy.
In this larger data age, microarray technology and bioinformatics analysis have been widely used to throughput and simultaneously detects thousands of genes at the genome level. Microarray gene expression dazzling features are integrated, automated, miniaturized[14] which have helped us identify the differentially expressed genes (DEGs) and functional pathways involved in the carcinogenesis and progression of NPC. A single microarray analysis cannot obtain reliable results. Thus, we downloaded 3 mRNA microarray data sets from Gene Expression Omnibus (GEO) and analyzed to get DEGs between nasopharyngeal carcinoma tissues and noncancerous tissues. Subsequently, gene ontology (GO), protein–protein interaction (PPI) network analyses and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed to make us clearness the molecular mechanisms underlying carcinogenesis and progression. In conclusion, a total of 306 DEGs and 13 hub genes were identified the 13 hub genes present in front of us were DNAAF1, PARPBP, TTC18, GSTA3, RCN1, MUC5AC, POU2AF1, FAM83B, SLC22A16, SPEF2, ERICH3, CCDC81, and IL33.
2. Materials and methods
2.1. Data resources
The GEO (http://www.ncbi.nlm.nih.gov/geo)[15] is a public functional genomics data repository which included throughout gene expression data, chips, and microarrays. Three gene expression data sets (GSE64634,[16] GSE53819, and GSE12452[17]) were downloaded from GEO (GPL6480, Agilent-014850 Whole Human Genome Microarray 4x44K G4112F and Affymetrix GPL570 platform, Affymetrix Human Genome U133 Plus 2.0 Array). Observing the download of GSE64634 database included 14 NPC samples and 4 normal samples, GSE53819 data set contained 18 NPC tissue samples and 18 noncancerous samples, GSE12452 contained 31 NPC samples and 10 noncancerous samples. Ethical approval was not necessary for this study because our study is bioinformatic analysis.
2.2. Identification of DEGs
The identification of DEGs between NPC and noncancerous samples was performed using GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r). GEO2R is an online tool designed that allows users to compare different data sets in a GEO series for identify DEGs across experimental conditions. To correct the discovery of statistically significant genes and limitations of false-positives, we using the adjusted P-value and Benjamini and Hochberg false discovery rate method. A |log2FC| > 2 and P-value < .01 were used as considered statistically significant.
2.3. KEGG and GO enrichment analyses of DEGs
The Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.ncifcrf.gov) (version 6.8)[18] which is a useful online platform database that integrates biologic data and provides a comprehensive set of functional annotation information of genes as well as proteins for users to analyze the functions or signaling pathways. The Kyoto Encyclopedia of Genes and Genomes (KEGG)[19] is a database resource for understanding high-level gene functions and linking genomic information from large-scale molecular data sets. GO[20] function analysis (biologic processes [BPs], cellular components [CCs], and molecular functions [MFs]) is a powerful bioinformatics tool to analyze BP and annotate genes. To analyze the function of the identified DEGs, biologic analyses were performed using GO enrichment and KEGG pathway analysis via DAVID online database. P < .05 as the cutoff criterion considered statistically significant.
2.4. PPI network construction and module analysis
Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org)[21] online database was using to predicted the PPI network information. Analyzing the interactions and functions between DEGs may provide information about the mechanisms of generation and development of disease (PPI score > 0.4). Cytoscape (version 3.7.1) is a bioinformatics platform for constructing and visualizing molecular interaction networks.[22] The plug-in Molecular Complex Detection (MCODE) of Cytoscape was applied to detect densely connected regions in PPI networks. The PPI networks were constructed using Cytoscape and the most significant module in the PPI networks was selected using MCODE. The criteria for selection were set as follows: Max depth = 100, degree cut-off = 2, Node score cut-off = 0.2, MCODE scores >5, and K-score = 2.
2.5. Hub genes selection and analysis
A network of the integrative relationships of the hub genes and their coexpression genes clinical characteristics in NPC was analyzed using cBioPortal for Cancer Genomics (http://www.cbioportal.org/),[23] which is an open-access resource for analyzing and exploring genetic alterations from multidimensional studies samples. The analyses of genomic mutations in the selected TCGA data sets could be analyzed in the cBioPortal online according to the instructions.
3. Results
3.1. Identification of DEGs in NPC
After standardization of the microarray results, DEGs (1099 in GSE64634, 2377 in GSE5381, and 1344 in GSE12452) were identified. The overlap among the 3 data sets contained 306 genes were identified to be differentially expressed between NPC and non-NPC samples with the threshold of P < .01 and a minimal 2-fold change of expression as shown in the Venn diagram (Fig. 1A), consisting of 280 downregulated genes and 26 upregulated genes between nasopharyngeal carcinoma tissues and noncancerous tissues. The volcano plots and the heatmap of top 50 DEGs are shown in Figures 2 and 3.
Figure 1.

Venn diagram and the most significant module of differentially expressed genes (DEGs). (A) DEGs were selected with a fold change >2 and P-value < .01 among the mRNA expression profiling sets GSE64634, GSE53819, and GSE12452. The 3 data sets showed an overlap of 306 genes. (B) The most significant module was obtained from protein–protein interaction network with 21 nodes and 153 edges.
Figure 2.
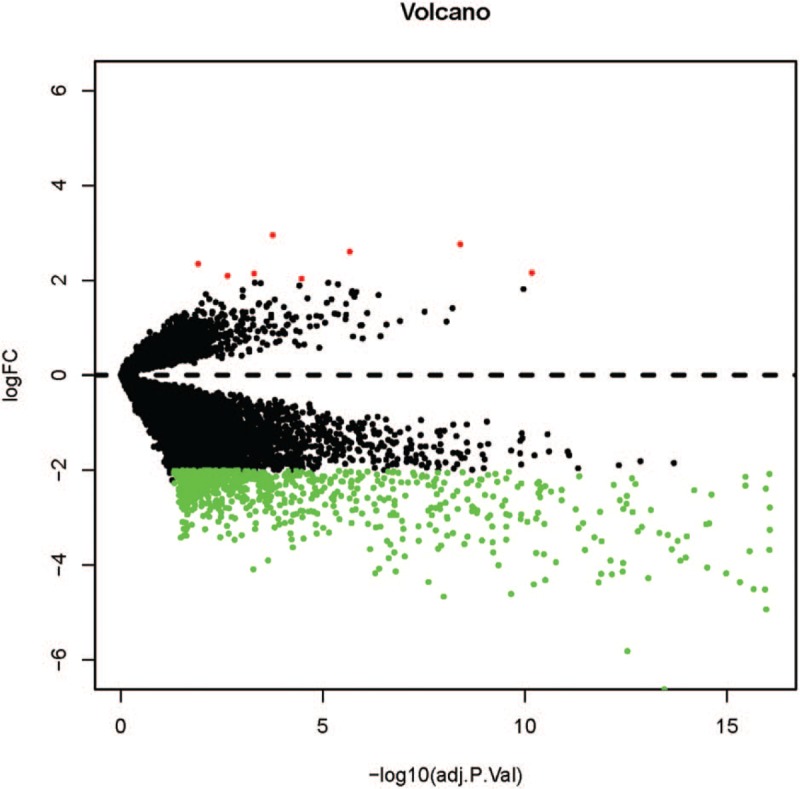
The volcano plots of differentially expressed genes in GSE64634, GSE53819, and GSE12452.
Figure 3.
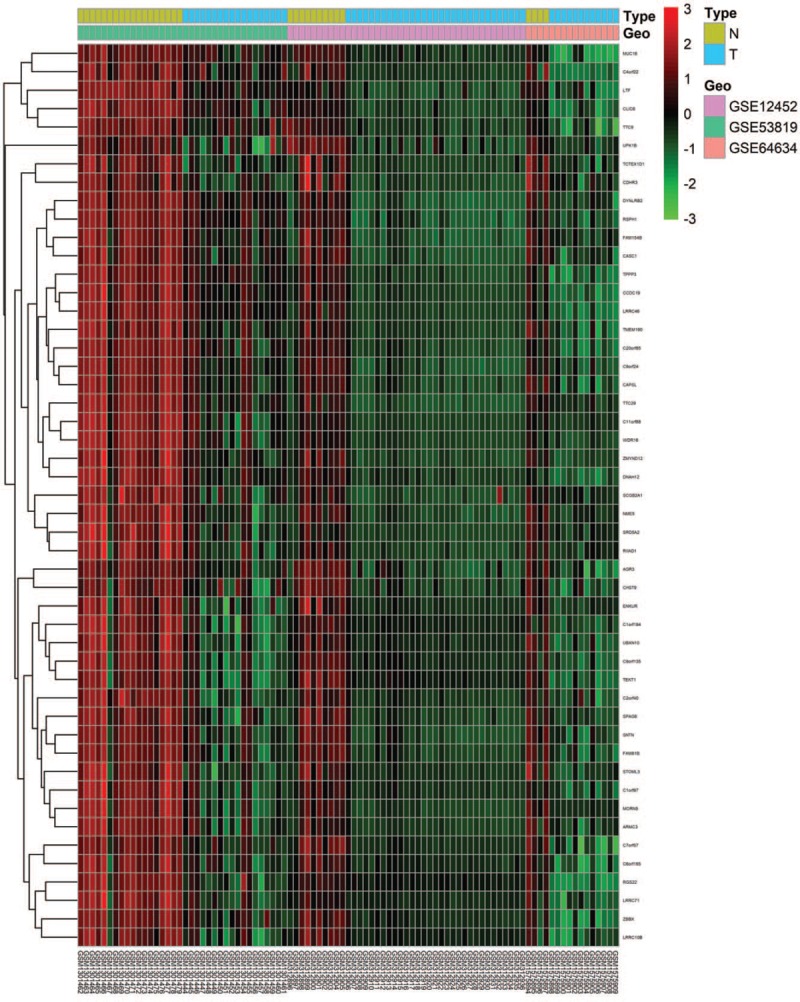
The heatmap of top 50 differentially expressed differentially expressed genes in GSE64634, GSE53819, and GSE12452.
3.2. GO enrichment and KEGG analyses of DEGs
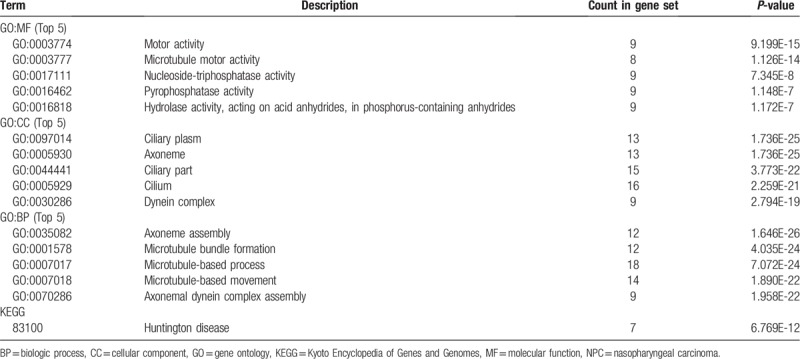
To further investigate the biologic functions and mechanisms of the DEGs, functional and pathway enrichment analyses were performed using DAVID tool. GO analysis results showed that changes in BPs of DEGs were significantly enriched in axoneme assembly, cilium organization, microtubule bundle formation, cilium assembly, and cilium movement (Table 1). Changes in MF were mainly enriched in microtubule motor activity, motor activity, alcohol dehydrogenase activity, zinc-dependent, alcohol dehydrogenase (NAD) activity, and epidermal growth factor receptor binding (Table 1). Changes in CC of DEGs were mainly enriched in the cilium, ciliary part, ciliary plasm, axoneme, and axoneme part (Table 1). KEGG pathway analysis revealed that the DEGs were mainly enriched in drug metabolism, metabolism of xenobiotic by cytochrome P450, chemical carcinogenesis, tyrosine metabolism, Huntington disease, fatty acid degradation, and adenine ribonucleotide biosynthesis.
Table 1.
GO and KEGG pathway enrichment analysis of differentially expressed genes in NPC samples.

3.3. PPI network construction and module analysis
To further explore the connection between DEGs at the protein level, the PPI networks were constructed based on the interactions of DEGs (Fig. 4) and the most significant module was obtained using Cytoscape (Fig. 1B). A total of 678 interactions and 225 nodes were screened to establish the PPI network and the biologic functional analyses of genes involved in this most significant module were analyzed using DAVID. KEGG results showed that genes in this module were mainly enriched in Huntington disease, and GO analysis results are summarized in Table 2.
Figure 4.
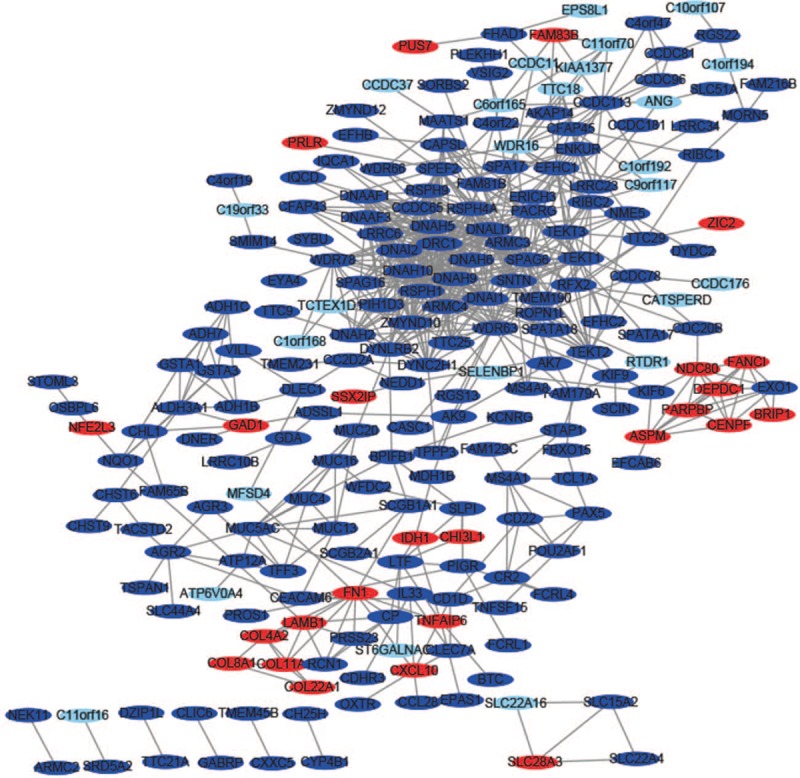
The protein–protein interaction network of differentially expressed genes was constructed using Cytoscape. Upregulated genes are marked in light red; downregulated genes are marked in light blue.
Table 2.
GO and KEGG pathway enrichment analysis of differentially expressed genes in the most significant module.
3.4. Hub gene selection and analysis
A total of 13 genes (DNAAF1, PARPBP, TTC18, GSTA3, RCN1, MUC5AC, POU2AF1, FAM83B, SLC22A16, SPEF2, ERICH3, CCDC81, and IL33) were identified as hub genes with degrees ≥10. The full names, abbreviations, also known as, and functions for these 13 hub genes are shown in Table 3. A network of the hub genes and their coexpression genes were performed via cBioPortal online platform (Fig. 5).
Table 3.
Functional roles of 13 hub genes with degree ≥10.
Figure 5.
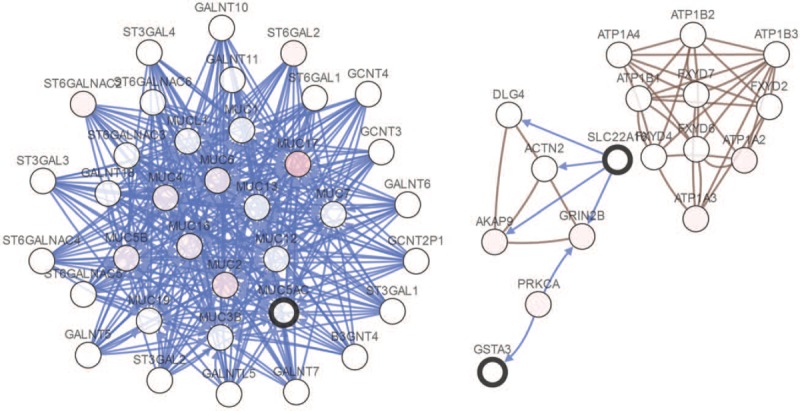
Interaction network and biologic process analysis of the hub genes. Hub genes and their coexpression genes were analyzed using cBioPortal. Nodes with bold black outline represent hub genes. Nodes with thin black outline represent the coexpression genes.
4. Discussion
More than 95% of nasopharyngeal carcinomas belong to poorly differentiated and undifferentiated cancer types. They have high malignancy and fast growth and are prone to lymph nodes or blood metastasis. There are currently no clinically specific molecular markers for nasopharyngeal carcinoma. Therefore, it is necessary to elucidate the detailed molecular mechanisms that are independently associated with tumor prognosis and invasiveness. With the development of high-throughput sequencing technology and microarrays technology, the profiling of DEGs closely link to NPC has become more common worldwide, providing a novel and effective way to predict the promising potential diagnostic and therapeutic targets in preventing and treating NPC. In this study, the 3 gene expression data sets were extracted from GEO, and a total of 306 DEGs between NPC and normal tissues were screened. GO enrichment and KEGG analyses showed that these DEGs were robustly related to various biologic functions most of which are closely to the progression and genesis of cancer. Moreover, coexpression genes was performed via cBioPortal platform and a PPI network with the DEGs was constructed, and 13 hub genes, including DNAAF1, PARPBP, TTC18, GSTA3, RCN1, MUC5AC, POU2AF1, FAM83B, SLC22A16, SPEF2, ERICH3, CCDC81, and IL33, were identified as the key genes in NPC.
The protein encoded by DNAAF1 gene is cilium specific and involved in the regulation of microtubule-based cilia and actin-based brush border microvilli, DNAAF1 mutations can cause primary ciliary dyskinesia.[24] DNAAF1 was initially identified as a dynein assembly factor, with mutations causing reduced ciliary beat frequency and a block in outer dynein arm assembly.[25] Mutations in this gene may suggest a disorder in nasal ciliary motor function in patients with nasopharyngeal carcinoma. Thus, DNAAF1 may be a potential biomarker may be used to predict cancer prognosis and sensitivity to cancer treatment. PARPBP which is highly expressed in nonsmall cell lung cancer.[26] PARPBP can encode DNA repair enzymes to protect DNA against anticancer drug damage. Therefore, PARP-1 inhibitors have become a promising approach for developing chemosensitizers in cancers.[27] GSTA3 gene encodes a glutathione S-tranferase (GST) belonging to the alpha class genes that are located in a cluster mapped to chromosome 6. Genes of the alpha class are highly related and encode enzymes with glutathione peroxidase activity. The enzyme encoded by this gene catalyzes the double bond isomerization of precursors for progesterone and testosterone during the biosynthesis of steroid hormones.
The GSTs are phase II detoxification enzymes that may have evolved in response to changes of environmental substrates.[28] Thus GSTA3 may be plays an important role in the occurrence and development of NPC. RCN1 is a calcium-binding protein located in the lumen of the ER. This protein localizes to the plasma membrane in human prostate cancer cell lines. It is speculated that the RCN1 has played the function in Ca2+-dependent cell adhesion,[29] as dysregulation of RCN1 protein resulting in multifarious diseases, such as cancer. Reports indicate that RCN1 was found in multiple tumorous types, including kidney cancer, breast cancer, liver cancer, colorectal cancer, and so on.[29–32] These findings remind us that overexpression of RCN1 might contribute to the tumorigenesis and tumor progression. MUC5AC expressed in human normal airways and is significantly upregulated in the sinus mucosa of patients with chronic rhinosinusitis.[33,34] Ye et al demonstrates that autophagy is essential for activation of the JNK-AP-1 signaling pathway, and this subsequently promotes MUC5AC production.[35] POU2AF1 a known B-cell transcriptional coactivator[36] and is regulated by CD40-L, a continuous stimulation may resulting in an overexpression of this gene on B cells.[37] FAM83B has also been reported significantly upregulated in breast cancer,[38] gastric cancer,[39] pancreatic ductal adenocarcinoma,[40] and lung squamous cell carcinoma.[41] SLC22A16 belongs to transporter protein family which transports carnitine and successful treatment has been related to the level of activity of this transporter in tumor cells.[42] SPEF2 is essential for motile cilia, and lack of SPEF2 function causes primary ciliary dyskinesia.[43] ERICH3 are related to plasma serotonin concentrations and dysfunction may lead to selective serotonin reuptake inhibitor.[44] CCDC81 may as a potential cargo-binding protein in conjunction with Dynein-VII.[45] IL33 is involved in the maturation of Th2 cells and the activation of basophils, mast cells, eosinophils, and natural killer cells.[46] TTC18 also known as CFAP70, a novel cilia-related gene and may play important in NPC.[47]
Although similar studies have been explored before,[48] but we 1st used a more stringent screening criteria and to obtain more reliable results, we 1st downloaded 3 mRNA microarray data sets from GEO database. While our study may exit some limitations, such as due to insufficient data, we cannot evaluate the correlations between the hub genes and survival prognosis. But a network of the hub genes and their coexpression genes was performed using cBioPortal platform.
5. Conclusion
The present study was attempted to identify and functionally analyze the DEGs that may be involved in the carcinogenesis or progression of NPC by using comprehensive bioinformatics analyses and unveiled a series of hub genes and pathways. A total of 306 DEGs and 13 hub genes were identified and may be regarded as diagnostic biomarkers for NPC. However, more experimental studies are needed to carried out elucidate the biologic function of these genes results for NPC.
Author contributions
Data curation: Ji-Zhou Zhang.
Methodology: Zeng-Hong Wu.
Validation: Qing Cheng.
Writing – original draft: Zeng-Hong Wu, Qing Cheng.
Writing – review & editing: Zeng-Hong Wu.
Footnotes
Abbreviations: BPs = biologic processes, CC = cellular component, CCDC81 = coiled-coil domain containing 81, DAVID = Database for Annotation, Visualization and Integrated Discovery, DEG = differentially expressed genes, DNAAF1 = dynein axonemal assembly factor 1, ERICH3 = glutamate rich 3, FAM83B = family with sequence similarity 83 member B, GEO = gene expression omnibus, GO = gene ontology, GSTA3 = glutathione S-transferase alpha 3, IL33 = interleukin 33, KEGG = Kyoto Encyclopedia of Genes and Genomes, MCODE = Molecular Complex Detection, MF = molecular function, MUC5AC = mucin 5AC oligomeric mucus/gel-forming, NPC = nasopharyngeal carcinoma, PARPBP = PARP1-binding protein, POU2AF1 = POU class 2 homeobox associating factor 1, PPI = protein–protein interaction, RCN1 = reticulocalbin 1, SLC22A16 = solute carrier family 22 member 16, SPEF2 = sperm flagellar 2, STRING = search tool for the retrieval of interacting genes/proteins, TTC18 = tetratricopeptide repeat domain 18.
How to cite this article: Zhang JZ, Wu ZH, Cheng Q. Screening and identification of key biomarkers in nasopharyngeal carcinoma: evidence from bioinformatic analysis. Medicine. 2019;98:48(e17997).
As the work is a bioinformatic analysis, ethical approval was not necessary.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Chua MLK, Wee JTS, Hui EP, et al. Nasopharyngeal carcinoma 2016;387:1012–24. [DOI] [PubMed] [Google Scholar]
- [2].Kamran SC, Riaz N, Lee N. Nasopharyngeal carcinoma. Surg Oncol Clin N Am 2015;24:547–61. [DOI] [PubMed] [Google Scholar]
- [3].Yu MC. Nasopharyngeal carcinoma: epidemiology and dietary factors. IARC Sci Publ 1991;39–47. [PubMed] [Google Scholar]
- [4].Scott RS. Epstein–Barr virus: a master epigenetic manipulator. Curr Opin Virol 2017;26:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tsao SW, Tsang CM, To KF, et al. The role of Epstein-Barr virus in epithelial malignancies. J Pathol 2015;235:323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nakanishi Y, Wakisaka N, Kondo S, et al. Progression of understanding for the role of Epstein–Barr virus and management of nasopharyngeal carcinoma. Cancer Metastasis Rev 2017;36:435–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Henderson BE, Louie E, SooHoo Jing J, et al. Risk factors associated with nasopharyngeal carcinoma. N Engl J Med 1976;295:1101–6. [DOI] [PubMed] [Google Scholar]
- [8].Nam JM, McLaughlin JK, Blot WJ. Cigarette smoking, alcohol, and nasopharyngeal carcinoma: a case-control study among U.S. whites. J Natl Cancer Inst 1992;84:619–22. [DOI] [PubMed] [Google Scholar]
- [9].Ho Y, Tsao SW, Zeng M, et al. STAT3 as a therapeutic target for Epstein-Barr virus (EBV): associated nasopharyngeal carcinoma. Cancer Lett 2013;330:141–9. [DOI] [PubMed] [Google Scholar]
- [10].Ye M, Huang T, Ni C, et al. Diagnostic capacity of RASSF1A promoter methylation as a biomarker in tissue, brushing, and blood samples of nasopharyngeal carcinoma. EBioMedicine 2017;18:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen J, Li S, Xiao Y, et al. p53R2 as a novel prognostic biomarker in nasopharyngeal carcinoma. BMC Cancer 2017;17:846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen Q, Qin S, Liu Y, et al. IGFBP6 is a novel nasopha-ryngeal carcinoma prognostic biomarker. Oncotarget 2016;7:68140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Peng JP, Chang HC, Hwang CF, et al. Overexpression of cyclooxygenase-2 in nasopharyngeal carcinoma and association with lymph node metastasis. Oral Oncol 2005;41:903–8. [DOI] [PubMed] [Google Scholar]
- [14].Ge Y, He Z, Xiang Y, et al. The identification of key genes in nasopharyngeal carcinoma by bioinformatics analysis of high-throughput data. ??? 2019;46:2829–40. [DOI] [PubMed] [Google Scholar]
- [15].Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bo H, Gong Z, Zhang W, et al. Upregulated long non-coding RNA AFAP1-AS1 expression is associated with progression and poor prognosis of nasopharyngeal carcinoma. Oncotarget 2015;6:20404–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dodd LE, Sengupta S, Chen IH, et al. Genes involved in DNA repair and nitrosamine metabolism and those located on chromosome 14q32 are dysregulated in nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev 2006;15:2216–25. [DOI] [PubMed] [Google Scholar]
- [18].Huang DW, Sherman BT, Tan Q, et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007;8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Altermann E, Klaenhammer TR. PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics 2005;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: proteinprotein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jianjiong G, Bülent Arman A, Ugur D, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:l1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Loges NT, Olbrich H, Becker-Heck A, et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am J Hum Genet 2009;85:883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Freshour J, Yokoyama R, Mitchell DR. Chlamydomonas flagellar outer row dynein assembly protein ODA7 interacts with both outer row and I1 inner row dyneins. J Biol Chem 2007;282:5404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen K, Li YJ, Xu H, et al. An analysis of the gene interaction networks identifying the role of PARP1 in metastasis of nonsmall cell lung cancer. Oncotarget 2017;8:87263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cepeda V, Fuertes MA, Castilla J, et al. Poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors in cancer chemotherapy. Recent Pat Anti-Cancer 2006;1:39–53. [DOI] [PubMed] [Google Scholar]
- [28].Tan HM, Low WY. Rapid birth-death evolution and positive selection in detoxification-type glutathione S-transferases in mammals. PLoS One 2018;13:e0209336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nimmrich I, Erdmann S, Melchers U, et al. Seven genes that are differentially transcribed in colorectal tumor cell lines. Cancer Lett 2000;160:37–43. [DOI] [PubMed] [Google Scholar]
- [30].Liu Z, Brattain MG, Appert H. Differential display of reticulocalbin in the highly invasive cell line, MDA-MB-435, versus the poorly invasive cell line, MCF-7. Biochem Biophys Res Commun 1997;231:283–9. [DOI] [PubMed] [Google Scholar]
- [31].Yu LR, Zeng R, Shao XX, et al. Identification of differentially expressed proteins between human hepatoma and normal liver cell lines by two-dimensional electrophoresis and liquid chromatography-ion trap mass spectrometry. Electrophoresis 2000;21:3058–68. [DOI] [PubMed] [Google Scholar]
- [32].Giribaldi G, Barbero G, Mandili G, et al. Proteomic identification of reticulocalbin 1 as potential tumor marker in renal cell carcinoma. J Proteomics 2013;91:385–92. [DOI] [PubMed] [Google Scholar]
- [33].Huang X, Feng K, Liu D, et al. Expression and relationship of pendrin and MUC5AC in patients with chronic rhinosinusitis. Zhonghua Yi Xue Za Zhi 2015;95:1078–82. [PubMed] [Google Scholar]
- [34].Luo Q, Zhang J, Wang HT, et al. Expression and regulation of transcription factor foxa2 in chronic rhinosinusitis with and without nasal polyps. Allergy Asthma Immunol Res 2015;7:458–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ye Y, Zhao J, Ye J, et al. The role of autophagy in the overexpression of MUC5AC in patients with chronic rhinosinusitis. Int Immunopharmacol 2019;71:169–80. [DOI] [PubMed] [Google Scholar]
- [36].Auer RL, Starczynski J, McElwaine S, et al. Identification of a potential role forPOU2AF1 andBTG4 in the deletion of 11q23 in chronic lympho-cytic leukemia. Genes Chromosomes Cancer 2005;43:1–0. [DOI] [PubMed] [Google Scholar]
- [37].Teitell MA. OCA-B regulation of B-cell development and function. Trends Immunol 2003;24:546–53. [DOI] [PubMed] [Google Scholar]
- [38].Cipriano R, Miskimen KL, Bryson BL, et al. FAM83B-mediated activation of PI3K/AKT and MAPK signaling cooperates to promote epithelial cell transformation and resistance to targeted therapies. Oncotarget 2013;4:729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zigui Z, Tianshi M, Xuezhi H, et al. Long intergenic non-coding RNA 00324 promotes gastric cancer cell proliferation via binding with HuR and stabilizing FAM83B expression. Cell Death Dis 2018;9:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shen CQ, Yan TT, Liu W, et al. High expression of FAM83B predicts poor prognosis in patients with pancreatic ductal adenocarcinoma and correlates with cell cycle and cell proliferation. J Cancer 2017;8:3154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Okabe N, Ezaki J, Yamaura T, et al. FAM83B is a novel biomarker for diagnosis and prognosis of lung squamous cell carcinoma. Int J Oncol 2015;46:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aouida M, Poulin R, Ramotar D. The human carnitine transporter SLC22A16 mediates high affinity uptake of the anticancer polyamine analogue bleomycin-A5. J Biologic Chem 2010;285:6275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lehti MS, Henriksson H, Rummukainen P, et al. Cilia-related protein SPEF2 regulates osteoblast differentiation. Sci Rep 2018;8:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gupta M, Neavin D, Liu D, et al. TSPAN5, ERICH3 and selective serotonin reuptake inhibitors in major depressive disorder: pharmacometabolomics-informed pharmacogenomics. Mol Psychiatry 2016;21:1717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Burroughs AM, Kaur G, Zhang D, et al. Novel clades of the HU/IHF superfamily point to unexpected roles in the eukaryotic centrosome, chromosome partitioning, and biologic conflicts. Cell Cycle 2017;16:1093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Buzzelli JN, Chalinor HV, Pavlic DI, et al. IL33 is a stomach alarmin that initiates a skewed Th2 response to injury and infection. Cell Mol Gastroenterol Hepatol 2015;1:203–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shamoto N, Narita K, Kubo T, et al. CFAP70 is a novel axoneme-binding protein that localizes at the base of the outer dynein arm and regulates ciliary motility. Cells 2018;7: E124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ge Y, He Z, Xiang Y, et al. The identification of key genes in nasopharyngeal carcinoma by bioinformatics analysis of high-throughput data. Mol Biol Rep 2019;46:2829–40. [DOI] [PubMed] [Google Scholar]