

Abstract
ARPC2 is a subunit of the Arp2/3 complex, which is essential for lamellipodia, invadopodia and filopodia, and ARPC2 has been identified as a migrastatic target molecule. To identify ARPC2 inhibitors, we generated an ARPC2 knockout DLD‐1 human colon cancer cell line using the clustered regularly interspaced short palindromic repeats/CRISPR‐associated protein 9 (CRISPR/Cas9) system and explored gene signature‐based strategies, such as a connectivity map (CMap) using the gene expression profiling data of ARPC2 knockout and knockdown cells. From the CMap‐based drug discovery strategy, we identified pimozide (a clinically used antipsychotic drug) as a migrastatic drug and ARPC2 inhibitor. Pimozide inhibited the migration and invasion of various cancer cells. Through drug affinity responsive target stability (DARTS) analysis and cellular thermal shift assay (CETSA), it was confirmed that pimozide directly binds to ARPC2. Pimozide increased the lag phase of Arp2/3 complex‐dependent actin polymerization and inhibited the vinculin‐mediated recruitment of ARPC2 to focal adhesions in cancer cells. To validate the likely binding of pimozide to ARPC2, mutant cells, including ARPC2F225A, ARPC2F247A and ARPC2Y250F cells, were prepared using ARPC2 knockout cells prepared by gene‐editing technology. Pimozide strongly inhibited the migration of mutant cells because the mutated ARPC2 likely has a larger binding pocket than the wild‐type ARPC2. Therefore, pimozide is a potential ARPC2 inhibitor, and ARPC2 is a new molecular target. Taken together, the results of the present study provide new insights into the molecular mechanism and target that are responsible for the antitumor and antimetastatic activity of pimozide.
Keywords: actin‐related protein 2/3 complex, actin‐related protein 2/3 complex subunit 2, drug repurposing, metastasis, pimozide
Pimozide is identified as a migrastatic drug and ARPC2 inhibitor from connectivity map‐based drug discovery strategy. Pimozide inhibits migration and invasion in various cancer cell lines, and suppresses metastasis in an in vivo antimetastatic assay. Through drug affinity responsive target stability (DARTS) analysis and cellular thermal shift assay (CETSA), it was confirmed that pimozide directly binds to ARPC2.
Abbreviations
- Arp2/3 complex
actin‐related protein 2/3 complex
- ARPC2
actin‐related protein 2/3 complex subunit 2
- CETSA
cellular thermal shift assay
- DARTS
drug affinity responsive target stability
1. INTRODUCTION
Metastasis is the foremost cause of cancer‐related death, and an antimetastatic drug would be beneficial therapy for cancer patients.1, 2 However, most standard‐of‐care treatments and targeted therapies focus on tumorigenesis and/or primary tumor growth, but not on metastatic activity.3 Tumor cell migration and invasion are critical steps during tumor metastasis, which require actin cytoskeletal reorganization. Cytoskeletal components could provide novel therapeutic approaches to prevent cancer cell migration and metastasis (so called migrastatics). However, there are currently no FDA‐approved anticancer agents due to the poor effective efficacy and toxicity of these agents in clinical trials.4, 5, 6
Actin‐related protein 2/3 complex (Arp2, Arp3, and ARPC1‐5) is a key regulator of actin nucleation and branching for actin cytoskeletal reorganization. Arp2/3 complex, which is activated by nucleation‐promoting factors, regulates actin‐related functions,7, 8 such as migration, membrane trafficking, cell division, endocytosis, phagocytosis, and infection.9, 10 Arp2/3 complex inhibitors, such as CK666 and CK869, inhibit cell migration including cancer and normal cells by targeting Arp2 or Arp3.11, 12 ARPC2 is one of the subunits of the Arp2/3 complex and is required for maintaining the structural integrity of the entire complex. ARPC2 expression is associated with metastasis and poor prognosis of patients with melanoma or gastric tumors.13, 14 Recently, we reported that ARPC2 inhibitor (benproperine, Benp) suppressed the migration and invasion of cancer cells and tumor metastasis in animal models.11 Thus, the discovery of ARPC2 inhibitors is a novel therapeutic approach to prevent cancer cell migration and metastasis.
Connectivity map, which has generated a large collection of transcriptional responses to drug perturbation in human cancer cell lines, primarily aims to understand the pathways modulated by small molecules.15 CMap has become a powerful tool for the identification of biologically active compounds without the need for biological assays because the current version (build 02; https://portals.broadinstitute.org/cmap) of CMap contains more than 7000 gene‐expression profiles representing 1309 compounds.16, 17
For CMap database‐based discovery of ARPC2 inhibitors, ARPC2‐knockout (ARPC2−/−) cancer cells were prepared using gene editing tools, such as the CRISPR/Cas9 system.18 Genome‐wide gene‐expression data of ARPC2‐knockout (ARPC2−/−) or ARPC2‐knockdown cells using siRNA or shRNA have been generated by next‐generation sequencing methods. From the CMap approach, we identified pimozide as an ARPC2 inhibitor through the accompanying analysis of candidate drugs using a migration assay.
Pimozide belongs to the diphenylbutylpiperidine class of drugs that has been approved as an antipsychotic drug by the FDA and targets dopamine receptor D2 (DRD2) by reducing dopamine activity. DRD2 has been identified as a key functional protein for proliferation and survival in pancreatic cancer with high expression of DRD2, suggesting that pimozide is an effective anticancer drug in pancreatic cancer.19 In addition, pimozide has shown antitumor activity in various cancer cells, such as prostate cancer, hepatocellular carcinoma, colorectal cancer, breast cancer, and leukemia, which can occur through the modulation of STAT, Wnt/β‐catenin, USP1, or AKT signaling pathways.20, 21, 22, 23, 24, 25, 26
In the present study, we conducted a CMap approach as a new screening method and found a strong migration inhibitor, pimozide, that binds to ARPC2. Direct interactions between ARPC2 and pimozide have been validated by computational docking studies and label‐free biochemical assays. In addition, pimozide inhibits the vinculin‐ARPC2 interaction that is required for focal adhesion and consequently blocks cell migration in human cancer cells. CMap‐based drug discovery provides an effective method to discover a potential ARPC2 inhibitor, pimozide, and these data suggest that pimozide could be a lead molecule for the development of antimetastatic drugs.
2. MATERIALS AND METHODS
2.1. Cell culture
All cancer cell lines were obtained from ATCC. DLD‐1 (human colon cancer cell), AsPC‐1 (human pancreas cancer cell) and A549 (human lung cancer) were maintained in RPMI‐1640 medium. A375P (human skin cancer cell), CFPAC‐1, MIA PaCa‐2 and PANC‐1 (human pancreas cancer cell) were cultured in DMEM. All culture media were supplemented with 10% heat‐inactivated FBS (Gibco), 100 U/mL penicillin and 100 μg/mL streptomycin. Cell cultures were maintained at 37°C under 5% CO2 in an incubator.
2.2. Generation of ARPC2 knockout or overexpressed DLD‐1 cells
ARPC2 knockout DLD‐1 cells were generated using the CRISPR/Cas‐9 system according to the previously described protocol.18 Suitable sgRNA sequences targeting ARPC2 were selected in the CRISPR design web tool (http://tools.genome-engineering.org): 5′‐TATTTCTTTGAAATTCTACA‐3′ (exon 3 in ARPC2). pX459 (pSpCas9‐2A‐Puro) cut with the BbsI enzyme and guide oligos (5′‐CACCGTATTTCTTTGAAATTCTACA‐3′ and 5′‐AAACTGTAGAATTTCAAAGAAATAC‐3′) were ligated and transfected into DLD‐1 cells. Transfected cells were selected with puromycin (2 μg/mL), and the generation of ARPC2 knockout cells was confirmed by genomic DNA sequencing and western blot assay.
To overexpress ARPC2 or ARPC2 mutant, full‐length ARPC2 in DLD‐1 was obtained by PCR using forward primer (5′‐GGGGTACCATGATCCTGCTGGAGGTGA) and reverse primer (5′‐CCGGAATTCGCGGGATGAAAACGTCTTC‐3′). To eliminate the PAM site (Cas‐9 enzyme recognizes the PAM site for digestion) and convert F225A, F247A and Y250F, we carried out PCR with oligonucleotides (primer). Primers (5′‐GAAATTCTACAAAGAACTTCAGGC‐3′ and 5′‐GCCTGAAGTTCTTTGTAGAATTTC‐3′) were used to mutate lysine65 (AAG) to lysine65 (AAA). The oligo sequence for the ARPC2 mutation is as follows: F225A (5′‐TACATTACCGCTGTGCTGTT‐3′ and 5′‐AACAGCA CAGCGGTAATGTA‐3′), F247A (5′‐GATCCACACGGCCCGGGACTACCT‐3′ and 5′‐AGGTAGTCCCGGGCCGTGTGGATC‐3′) and Y250F (5′‐TTCCGGGACTTCCTGC ACTA‐3′ and 5′‐TAGTGCAGGAAGTCCCGGAA‐3′). All primers were obtained from Bioneer. Mutated ARPC2 PCR products were ligated to the pCDNA3.1(+) vector and transfected into ARPC2 knockout DLD‐1 cells. For transfection, we used Lipofectamine 2000 (Invitrogen) and Plus reagent (Invitrogen) according to the manufacturer's protocol.
2.3. Western blotting
Cell lysates were prepared in RIPA lysis buffer containing a protease inhibitor cocktail (Roche Diagnostics). Proteins (20‐50 μg) were resolved by 8%‐15% SDS‐PAGE and transferred to PVDF membranes (EMD Millipore). The membranes were blocked with 5% nonfat dry milk or 5% BSA in TBST and incubated with primary and secondary antibodies according to the manufacturer's protocol. Antibodies against ARPC2 (ab133315), Arp2 (ab47654), Arp3 (ab56817), ARPC1A (ab135572), ARPC5 (ab51243) and ARPC5L (ab169763) were purchased from Abcam; ARPC2 (SC‐515754), ARPC1B (SC‐271342), ARPC3 (SC‐166630) and GAPDH (SC‐47724) were from Santa Cruz Biotechnology. Antibodies against Myc‐tag (#2278) were purchased from Cell Signaling Technology, ARPC4 (sab100901) was purchased from Sigma‐Aldrich, and vinculin (#05‐386) was purchased from EMD Millipore.
2.4. Immunofluorescence and immunoprecipitation assays
DLD‐1 and ARPC2 knockout DLD‐1 cells were seeded onto 35‐mm μ‐Dish (Ibidi) at a density of 1.0 × 105. After 24 hours, DMSO or pimozide (10 μmol/L) were treated for 4 hours. Cells were washed with PBS and fixed with 4% paraformaldehyde in PBS for 10 minutes at room temperature. After washing with PBS, fixed cells were permeabilized with 0.1% Triton X‐100 for 10 minutes and blocked with 1.0% BSA in PBS for 1 hour. The cells were incubated with anti‐cortactin (Abcam, ab13333) antibody followed by an Alexa Fluor 647‐conjugated donkey antimouse IgG antibody (Thermo Fisher Scientific Inc., A‐31571). For actin staining, FITC‐phalloidin (Cell Signaling Technology, #8878) was also added with secondary antibody. The nuclei were counterstained with DAPI (Santa Cruz Biotechnology, sc‐3579). All images were obtained from a laser scanning confocal microscope (LSM 510 META; Carl Zeiss Vision) and captured with a 40× objective lens.
Cells were lysed in IP‐lysis buffer (20 mmol/L Tris‐HCl pH 7.4, 137 mmol/L NaCl, 1.0% NP‐40). Lysates were centrifuged at 12 000 g and quantified using the Bradford reagent. A total of 500 μg protein was incubated with vinculin antibody overnight at 4°C with rotation, and then 50 μL protein G magnetic beads (Bio‐Rad) was added. After incubation at room temperature for 1 hour, the lysates were removed, and the beads were washed three times with PBS containing 0.1% Tween‐20. Proteins that bound vinculin antibody were gathered with 5× protein loading dye and analyzed by western blotting.
2.5. Next‐generating sequencing and connectivity map
RNAs were isolated from DLD‐1 and ARPC2 knockout DLD‐1 cells using an RNase mini kit (Qiagen). Isolated RNAs were quantitated, and quality was measured in an agarose gel. For RNA‐seq, RNA libraries were generated with TruSeq RNA Sample Prep Kit v2 (Illumina), and size of the RNA library (250‐650 bp) was confirmed in 2% agarose gel. To analyze sequencing, samples that were prepared to 10 nmol/L were assayed using Hi‐Seq 2000 for 100 cycles and paired‐end sequencing (Illumina). Four RNA libraries were pooled in each lane for sequencing, and an average of approximately 11 Gb was obtained for each sample. After mapping using a reference database, gene set analysis and pathway analysis were carried out through the RPKM normalization process and DEG selection.
2.6. Proliferation assay
DLD‐1 cells were seeded onto 96‐well plates at a density of 8000 cells/well in RPMI‐1640 with 10% FBS. After 24 hours, the cells were replenished with fresh complete medium containing the indicated concentrations of compounds or 0.1% DMSO. After incubation for 24‐96 hours, cell proliferation reagent WST‐1 (Dojindo Laboratories) was added to each well. Amount of WST‐1 formazan produced was measured at 450 nm using an ELISA reader (Bio‐Rad).
2.7. Transwell migration and invasion assay
Assay was carried out using 24‐well chambers with Transwell inserts with of 8.0 μm (BD Biosciences). For the invasion assay, the Matrigel basement membrane matrix (Corning) was diluted to 4/1 with serum‐free medium using a cooled pipette and coated at a volume of 200 μL inside the inserts. After incubation on a clean bench for 1 hour, the unbound materials were aspirated. The inside of the inserts was rinsed gently using serum‐free medium and used for assays.
Cells were harvested with trypsin/EDTA (Gibco) and washed twice with serum‐free medium. A total of 80 000 cells in 0.2 mL serum‐free medium was added to the upper chamber, and chemoattractant at the indicated concentrations in 0.5 mL of medium with 10% FBS were placed in the lower chamber. At the end of the incubation period, cells invading the membrane or Matrigel were stained with crystal violet (5 mg/mL in methanol) and imaged using a microscope.
2.8. In vivo antimetastatic assay
All animal works were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee. Six‐week‐old female BALB/c nude mice (Nara Biotech) were used for the lung metastasis assay. AsPC‐1 cells (1 × 106 cells/mouse) that stably expressed luciferase were injected into the lateral tail vein of mice. Mice were imaged for luciferase activity immediately after the tail vein injection to confirm that the cancer cells were successfully xenografted. Pimozide was orally given at a dosage of 30 mg/kg every other day for 28 days. Bioluminescence of cancer cells in lungs was monitored every 7 days using a Photon Imager (Biospace Lab). On the 28th day, mice were killed by CO2 asphyxiation, and their lungs were dissected. Number of metastatic colonies in the lung was counted.
2.9. Drug affinity responsive target stability
DLD‐1 cells were harvested by scraping into ice‐cold M‐PER lysis buffer (Thermo Fisher Scientific Inc.) supplemented with 1 mmol/L NaF, 1× protease inhibitor cocktail and 1 mmol/L Na3VO4. After quantitation, lysates were diluted to 2 mg/mL, and 10× TNC buffer (500 mmol/L Tris‐HCl, 500 mmol/L NaCl and 100 mmol/L CaCl2) was added. After incubation with pimozide or DMSO for 1 hour with rotation at room temperature, lysates were divided into 50‐μL aliquots in Eppendorf tubes and digested with various doses of pronase at room temperature for 10 minutes. After stopping the reaction with 20× protease inhibitor, 5× sample dye was added to each tube, and the proteins were resolved by SDS‐PAGE.
2.10. Intact cellular thermal shift assay
For intact cellular thermal shift assay (CETSA), cells were pretreated with pimozide for 12 hours. Lysates were prepared using lysis buffer (50 mmol/L Tris‐HCl, 100 mmol/L NaCl, 0.2% NP‐40, 5% glycerol, 1.5 mmol/L MgCl2, 25 mmol/L NaF, 1 mmol/L Na3VO4 and 1× protease inhibitor cocktail solution). Lysates that were incubated in ice for 10 minutes were centrifuged at 12 000 g for 10 minutes at 4°C. Cells were quantitated to 2 mg/mL, and 50‐μL aliquots were taken and heated for 5 minutes at a specific temperature using a PCR machine. After incubation in ice for 10 minutes or longer, soluble proteins were separated by centrifugation at 12 000 g for 20 minutes at 4°C. After adding 5× loading dye to each tube, western blot analysis was carried out on the same amount.
2.11. Pull‐down assay
DLD‐1 cells were washed with PBS and homogenized with a 26‐gauge syringe in binding buffer (10 mmol/L Tris‐HCl pH 7.4, 50 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L Na3VO4 and 1× protease inhibitor cocktail solution). The cell lysate was centrifuged, and the supernatant was collected. The cell lysate was precleared by incubation with Neutravidin beads (Thermo Fisher Scientific Inc.) for 1 hour at 4°C. The cleared lysate was incubated with biotinyl‐benproperine (biotinyl‐Benp) for 1 hour at room temperature in the absence or presence of competitor (pimozide or N‐methyl‐pimozide). N‐Methyl‐pimozide was synthesized by reaction of pimozide and methyl iodide in the presence of sodium hydride (Document S1). Proteins associated with biotinyl‐Benp were precipitated with Neutravidin beads and washed three times with washing buffer (50 mmol/L HEPES pH 7.5, 50 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.1% Tween‐20, 10% (v/v) glycerol, 1 mmol/L NaF, 1 mmol/L Na3VO4 and 1× protease inhibitor cocktail solution). Samples were boiled and separated by 10% PAGE.
2.12. In vitro actin polymerization assay
Assays were carried out using an Actin Polymerization Biochem Kit (Cytoskeleton Inc.). Briefly, to convert actin to monomer, actin stock that was diluted in g‐buffer was incubated at 4°C for 1 hour and centrifuged. A total of 200 μL of g‐actin (2 μmol/L) was added to a 96‐well black plate (Corning) with an Arp2/3 complex (Cytoskeleton Inc., 10 nmol/L), WASP‐VCA Domain (Cytoskeleton Inc., 400 nmol/L) or compounds, and polymerization was started with ATP‐added p‐buffer. Excitation and emission were measured at 365 nm and 405 nm, respectively, and the plate was read every 1 minute for 1 hour.
2.13. Statistical analysis
Data are expressed as means ± standard deviation (SD), and the degree of significance was analyzed using Student's t test. Values of P < .05, P < .01 and P < .001 are denoted by *, ** and ***, respectively.
3. RESULTS
3.1. Generation and characterization of ARPC2 knockout DLD‐1 cells
Previously, we found that Benp, which targets ARPC2, strongly inhibited the migration of many different cancer cell types but not normal cells.11 To identify ARPC2 inhibitors by the CMap approach, we established ARPC2 knockout (ARPC2−/−, KO) colon cancer cells (DLD‐1) using the CRISPR/Cas9 system.18 We targeted exon 3 in ARPC2 and obtained two indel sequencing types (Figure 1A). The resulting cells completely lacked ARPC2/p34 and downregulated multiple other subunits of the Arp2/3 complex (Figure 1B). Next, we observed morphological changes in both wild‐type DLD‐1 (WT) and ARPC2−/− cells. ARPC2−/− cells lost their original shape and changed their static type in comparison with WT cells (Figure 1C). To examine the migration activity of WT and ARPC2−/− cells, we carried out a migration assay using a Transwell system. ARPC2−/− cells showed significantly suppressed migration compared with WT cells (Figure 1D). Cell migration requires membrane protrusion at the cell front, known as lamellipodia, which drives cell migration in many normal and cancer cells,27 and lamellipodial protrusion is driven by Arp2/3 complex‐mediated actin polymerization. To investigate whether the migration inhibition results are associated with lamellipodium formation in ARPC2−/− cells, the cells were stained with lamellipodia markers, such as cortactin and F‐actin. As shown in Figure 1E, WT cells generated lamellipodia structures at the leading edge which colocalized with F‐actin. In ARPC2−/− cells, cortactin disappeared at the leading edge of the cell. These confocal results are consistent with a previous result indicating that ARPC2−/− fibroblast cells lack lamellipodia and migrate more slowly than WT cells.28
Figure 1.
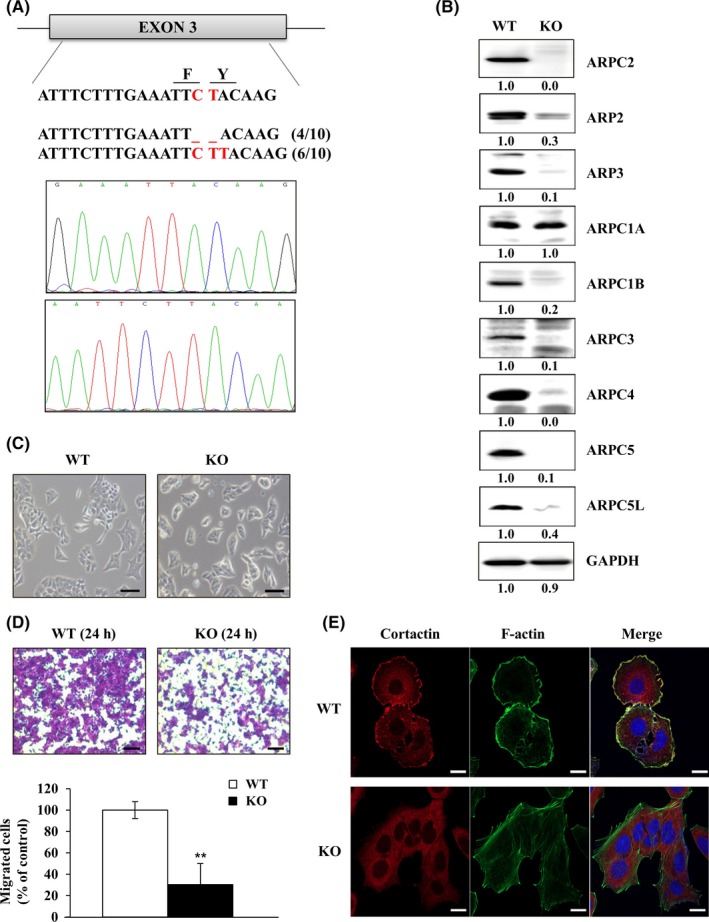
Generation and characterization of ARPC2‐knockout (KO) DLD‐1 colon cancer cells. A, Using the clustered regularly interspaced short palindromic repeats/CRISPR‐associated protein 9 (CRISPR/Cas9) system, we established ARPC2‐KO cell lines in DLD‐1 cells. Cell lines were confirmed by genomic DNA sequencing. B, Disappearance of the Arp2/3 complex was detected by western blot analysis (n = 3). C, The shape of DLD‐1 and DLD‐1‐ARPC2 KO cells was visualized through a microscope (n = 2). Scale bars, 200 μm. D, Cell migration assay was done with DLD‐1 and DLD‐1‐ARPC2 KO cells (n = 3). Scale bars, 20 μm. E, Lamellipodium formation was analyzed by confocal microscopy. Staining of cortactin (lamellipodia marker) and F‐actin shows reduction of lamellipodium formation in DLD‐1‐ARPC2‐KO cell lines (n = 2). Data represent means ± SD compared with the corresponding control, **P < .01
3.2. CMap identifies antipsychotic or antitussive drugs as potential ARPC2 inhibitors
To identify new ARPC2 inhibitors, we used CMap, a biomedical software package produced by the Broad Institute.15, 16 We believe that CMap is able to link the gene expression patterns derived from genetically modified cancer cells to corresponding patterns derived from drug‐treated cancer cell lines, leading to the identification of new ARPC2 inhibitors. Therefore, we built whole‐genome gene expression profiles from siRNA‐ or shRNA‐mediated ARPC2 knockdown, ARPC2−/−, and wild‐type DLD‐1 (WT) cells using a next‐generation sequencing tool. We first determined the fold changes in the levels of gene expression in each type of genetically modified DLD‐1 cell versus WT cells. From each of the above three experimental groups, we selected genes that changed twofold and then converted these to a heatmap, which represents the selected upregulated (red) and downregulated (green) genes WT cells versus ARPC2 knockdown or ARPC2‐knockout cells (Figure 2A). The whole‐genome gene expression profiling patterns are very similar (Figure 2A). Therefore, we selected 416 genes, representing 295 upregulated and 121 downregulated genes, in‐WT cells versus ARPC2−/− cells‐for the CMap study.
Figure 2.
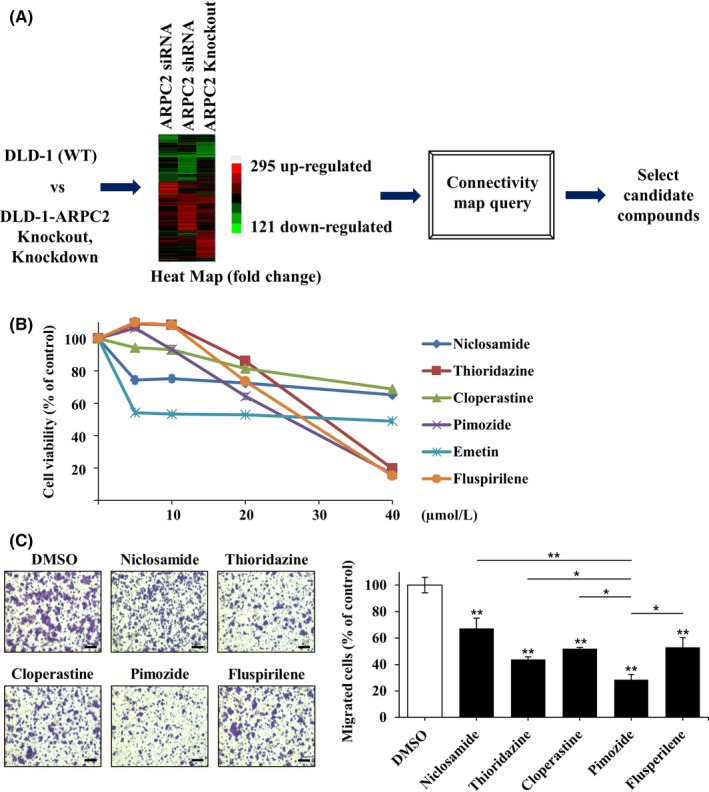
Pimozide was identified as a migration inhibitor using gene expression data and connectivity map (CMap) analysis. A, Diagram of the CMap process. B, Viability of DLD‐1 cells at various concentrations (0, 5, 10, 20 or 40 μmol/L) of selected compounds from the CMap analysis was measured using a WST‐1 assay. Incubation time of compounds was 24 h (n = 3). C, Migration assay with DLD‐1 cells was carried out in the presence of DMSO (0.1%) or 10 μmol/L compounds (Niclosamide, Thioridazine, Cloperastine, Pimozide or Fluspirilene). The cells attached to the lower surface of the membrane were stained with crystal violet and visualized on an optical microscope (n = 3). The migrated cells were counted by the Image‐ProPlus 5.0 program. Scale bars, 200 μm. Data represent means ± SD; *P < .05; **P < .01 compared with DMSO group
The query contained 416 genes that had been submitted to the CMap database for analysis (Figure 2A). The top 10 correlated drugs with lower p‐values and a positive enrichment score were selected by the CMap database (Table 1). From this analysis, we found that many antipsychotic drugs such as thioridazine, trifluoperazine, prochlorperazine, fluspirilene, and pimozide were potential ARPC2 inhibitors. It is very interesting that cloperastine, an antitussive drug, was also selected as an ARPC2 inhibitor because we previously reported that the antitussive drug, Benp is an ARPC2 inhibitor that suppresses cancer cell migration and tumor metastasis.11 Among the drugs positively associated with ARPC2 knockout, we excluded cytotoxic compounds, such as niclosamide, valinomycin, and emetine. We examined a few compounds using proliferation and migration assay and selected pimozide as a potential ARPC2 inhibitor because pimozide suppressed migration by approximately 65% in a Transwell migration assay at 10 μmol/L without cytotoxicity (Figure 2B, C), and various psychiatric drugs, including pimozide, are under investigation for repurposing as antitumor therapeutics.29
Table 1.
Results of connectivity map (CMap) analysis
| Rank | CMap name | Function | Mean | n | Enrichment | P | Specificity | Percent non‐null |
|---|---|---|---|---|---|---|---|---|
| 1 | Niclosamide | Anthelmintic | 0.774 | 5 | 0.963 | 0 | 0 | 100 |
| 2 | Valinomycin | Antibiotic | 0.794 | 4 | 0.943 | 0 | 0.0116 | 100 |
| 3 | Thioridazine | Antipsychotic | 0.618 | 20 | 0.695 | 0 | 0.0274 | 95 |
| 4 | Trifluoperazine | Antipsychotic | 0.553 | 16 | 0.638 | 0 | 0.0481 | 93 |
| 5 | LY‐294002 | PI3K inhibitor | 0.381 | 61 | 0.458 | 0 | 0.0872 | 77 |
| 6 | Cloperastine | Antitussive | 0.609 | 6 | 0.867 | .00002 | 0 | 100 |
| 7 | Prochlorperazine | Antipsychotic | 0.476 | 16 | 0.592 | .00002 | 0.0485 | 87 |
| 8 | Emetine | Amebicide | 0.652 | 4 | 0.925 | .00004 | 0.0211 | 100 |
| 9 | Pimozide | Antipsychotic | 0.694 | 4 | 0.918 | .00004 | 0.0101 | 100 |
| 10 | Fluspirilene | Antipsychotic | 0.604 | 4 | 0.865 | .00046 | 0.005 | 100 |
3.3. Pimozide inhibits migration and invasion in various cancer cell lines, and suppresses metastasis in an in vivo antimetastatic assay
To investigate the growth‐suppressive effect on DLD‐1 human colon cancer cells with very low levels of DRD2 expression, we treated cells with up to 40 µmol/L pimozide for 24, 48, 72, or 96 hours and measured the antiproliferative effects using a WST‐1 assay. Although pimozide inhibited the growth of DLD‐1 cells in a dose‐ and time‐dependent method, interestingly, more than 80% of cell viability was maintained after treatment with pimozide up to a concentration of 10 μmol/L (Figure 3B). These results indicate that pimozide has very weak cytotoxic effects against DLD‐1 cells and strongly inhibits migration of the cells without cytotoxicity. To determine migrastatic activity against various cancer cells, we carried out a Transwell migration assay in various cell lines with 10 μmol/L pimozide treatment. Pimozide inhibited the migration of various types of cancer cells, including PANC‐1 cells with high levels of DRD2 expression and AsPC‐1 cells with very low levels of DRD2 expression (Figure 3C, D).19 Pimozide inhibited the migration and invasion of DLD‐1 and AsPC‐1 cells with IC50 values of 5‐8 μmol/L (Figure 3E, F). These results suggest that pimozide is an effective inhibitor of the migration and invasion of cancer cells in a DRD2‐independent and an ARPC2‐dependent method, because ARPC2 downregulation strongly inhibited AsPC‐1 cell migration.11
Figure 3.
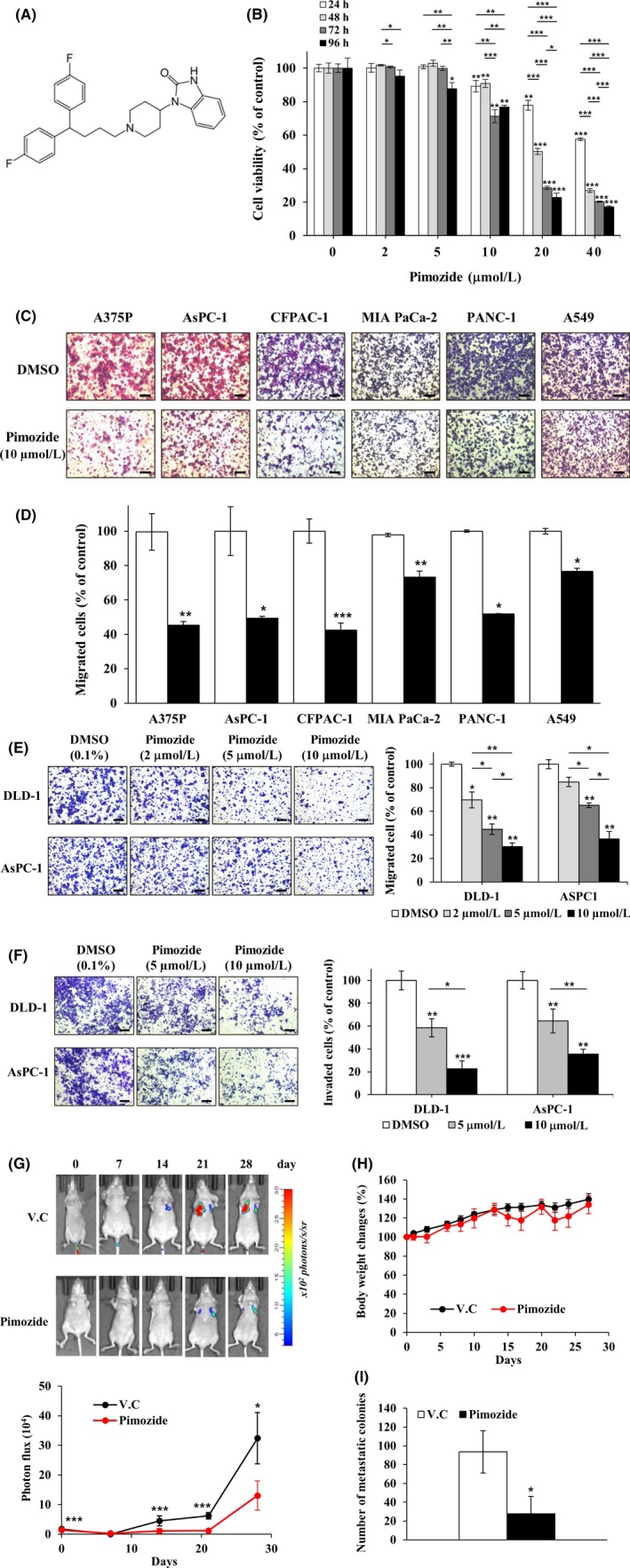
Inhibition effect of pimozide on growth, migration, invasion, and metastasis. A, Chemical structure of pimozide. B, DLD‐1 cells were treated with pimozide (0, 2, 5, 10, 20 or 40 μM) for 24, 48, 72 or 96 h. Cell proliferation was measured by a WST‐1 assay (n = 3). C‐E, Cell migration assays were carried out with a Transwell system in different types of cancer cell lines. Migrated cells were stained with crystal violet and counted using the Image‐ProPlus 5.0 program (n = 3). Scale bars, 200 μm. F, Cell invasion assays were done using Matrigel‐coated Transwell inserts in DLD‐1 and AsPC‐1 cell lines and the migrating cells were quantified (n = 3). Scale bars, 200 μm. G, Inhibition of pancreatic cancer cell metastasis to the lungs by pimozide in the lung metastatic mouse model. Top, Representative images from luciferase‐expressing AsPC‐1 cells in the whole body (n = 6 per group). Bottom, Quantification of photon flux in the lungs at the indicated time points. V.C., vehicle control. H, Body weight was measured on each indicated day. I, On the 28th day, mice were killed and their lungs were dissected. Number of metastatic colonies in the lung was counted and averaged. (n = 6 per group). Data represent mean ± SD compared with the corresponding control, *P < .05, **P < .01, ***P < .001
To examine the antimetastatic activity of pimozide, a lung metastatic assay was carried out using AsPC‐1 cells that stably expressed luciferase. AsPC‐1 cells were directly injected into the tail vein of female BALB/c nude mice (6 weeks old). The pimozide‐treated group showed a marked decrease in the number of lung metastases and mouse body weight was not affected by pimozide treatment (Figure 3G, H). In addition, pimozide inhibited the formation of metastatic colonies in the lung by 62.8% compared to vehicle control (Figure 3I and Figure S1).
3.4. Pimozide directly binds to ARPC2
In previous studies of pimozide in breast cancer cell lines, USP1 downregulation significantly inhibited the abilities of cell migration and invasion.24 We examined whether knockdown of USP1 in the DLD‐1 colon cancer cell line used in this article influences migration. The mRNA level of USP1 was decreased by siRNA of USP1, and migration was not affected (data not shown). These results suggest that USP1 is not affected, at least in DLD‐1 cell lines.
Drug‐target engagement is a critical factor for the pharmacological effects of drugs and therapeutic target validation at the cellular level. There is a wide variety of methods to examine drug‐target engagement, including CETSA and DARTS analysis. CETSA uses drug‐induced changes in the thermal stability of a target protein.30 DARTS relies on the idea that drug‐target engagement protects against proteolysis of the target protein.31 The greatest advantage of these methods is their ability to use native small molecules without chemical modification, such as biotin or fluorescent tags, or photoaffinity labels.
From CMap analysis, we identified pimozide as an ARPC2 inhibitor, and confirmed its migrastatic activity against various cancer cells. To determine whether pimozide directly binds to ARPC2, we used antibody‐based CETSA in intact cells and DARTS in cell lysates. DLD‐1 cell lysates were incubated with DMSO or pimozide (100 or 200 µmol/L, respectively), and the lysates were exposed to increasing doses of pronase. As shown in Figure 4A, we found that pronase‐induced digestion of ARPC2 was prevented by pimozide at a concentration of 0.05% pronase. The protective effect of pimozide against pronase increased in a dose‐dependent way at 0.05% pronase treatment (Figure 4A). GAPDH, as a control protein, was not affected by pimozide treatment in this experiment. To confirm direct engagement between pimozide and ARPC2 in intact cells, we carried out an intact‐CETSA experiment. DLD‐1 cells were treated with 10 μmol/L pimozide for 12 hours and then incubated at different temperatures. Pimozide strongly induced the thermal stability of ARPC2 at a variety of temperatures (Figure 4B).
Figure 4.
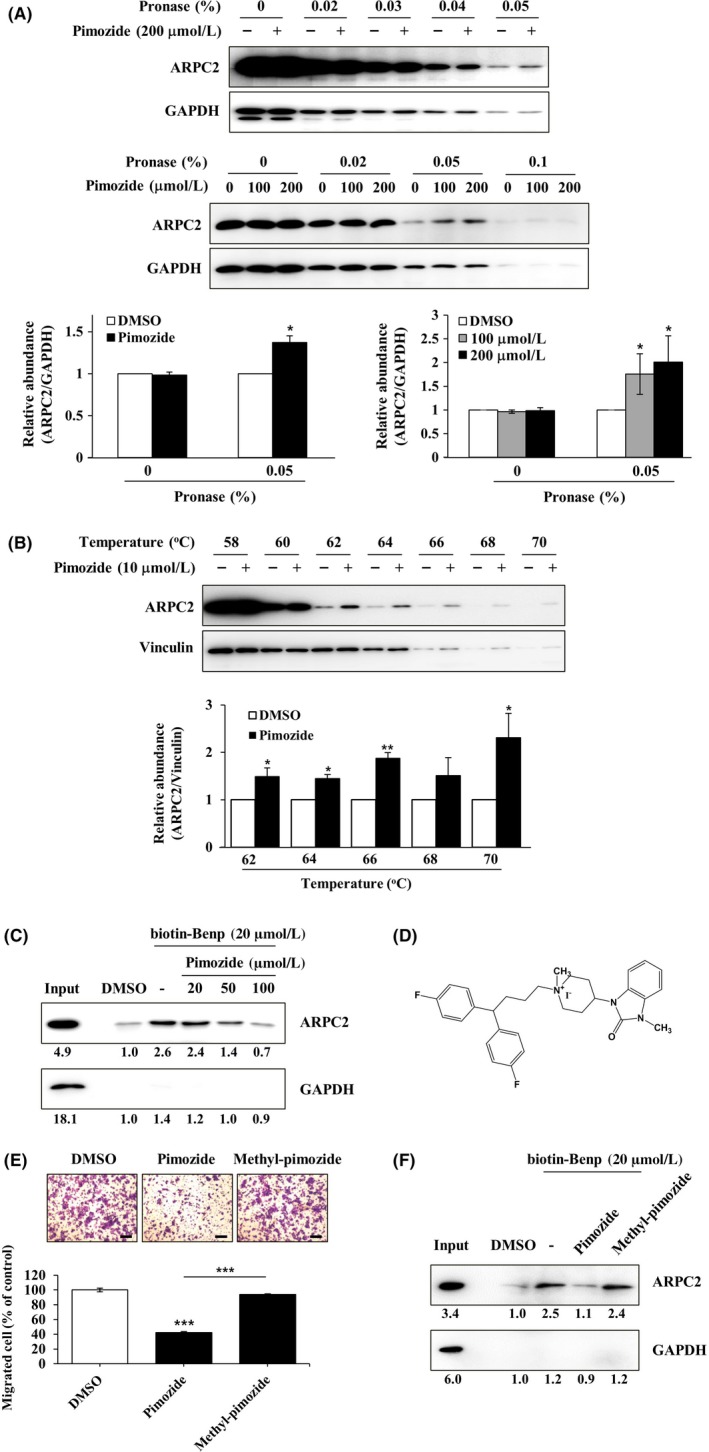
Direct binding of pimozide with ARPC2. A, DLD‐1 cell lysates were incubated in the presence or absence of pimozide (100 or 200 μmol/L) for 1 h at room temperature, followed by proteolysis with various pronases in a dose‐dependent way. GAPDH, which served as the loading control, is relatively resistant to proteolysis (n = 3). B, DLD‐1 cells were treated for 12 h with 10 μmol/L pimozide, and then CETSA was carried out to measure binding ability. Pimozide increased the thermal stability of ARPC2 compared with DMSO. Vinculin is a nontarget protein of pimozide (n = 3). Band intensity was quantified using the MultiGauge program. C, DLD‐1 cell lysates was incubated with 20 μmol/L biotinyl benproperine (biotinyl‐Benp) and competed with pimozide at the indicated concentration. Proteins were captured with NeutrAvidin‐Agarose resin (ThermoFisher Scientific Inc., Waltham, MA, USA) and eluted proteins were analyzed by western blotting (n = 2). D, Chemical structure of N‐methyl‐pimozide. E, Cell migration assay of DLD‐1 cells that were treated with DMSO, pimozide, or N‐methyl‐pimozide for 18 h and quantification of the migrated cells (n = 3). Scale bars, 200 μm. F, Pull‐down assay with biotinyl‐Benp was done in the absence or presence of compounds as competitor (100 μM). Bound proteins on the beads were separated by SDS‐PAGE, and western blot was carried out using anti‐ARPC2 and anti‐GAPDH antibodies (n = 2). Data represent the means ± SD; *P < .05, **P < .01, ***P < .001 compared with the DMSO group
In a previous article, we reported the binding activity of Benp to ARPC2 using biotinyl‐Benp.11 We thought that it was impossible to prepare biotinylpimozide because it does have not an active site for biotinylation. Therefore, we carried out experiments that showed direct binding activity of pimozide using biotinyl‐Benp. Pimozide inhibits binding between ARPC2 and biotinyl‐Benp in a dose‐dependent way (Figure 4C). To further confirm direct binding of ARPC2 and pimozide, we synthesized a compound with a methyl group on pimozide (N‐methyl‐pimozide) (Figure 4D and Document S1). As shown in Figure 4E, N‐methyl‐pimozide did not inhibit migration of DLD‐1 cells. N‐methyl‐pimozide does not block the interaction between ARPC2 and biotinyl‐Benp (Figure 4F). These results indicated that N‐methyl‐pimozide did not bind to ARPC2, resulting in a failure to inhibit the migration of DLD‐1 cells. Our data suggest that pimozide directly interacts with ARPC2 in DLD‐1 cells, which results in the inhibition of the migration of DLD‐1 cells.
3.5. Inhibition of cancer cell migration by blocking the functions of ARPC2
The Arp2/3 complex has an important role in several cellular processes, including cell migration and adhesion.32 Actin polymerization in the form of branched networks in lamellipodia is the major mechanism driving leading‐edge protrusion in migrating cells.33, 34 Therefore, we carried out an actin polymerization assay using the Actin Polymerization Biochem Kit, supplied by Cytoskeleton, Inc. Pyrene‐labeled monomeric actin was incubated with the Arp2/3 complex, and the VCA (Verprolin, Central, Acidic) domain of WASP stimulated the rate of actin polymerization. Addition of the Arp2/3 complex and VCA into the actin polymerization mixture enhanced the actin polymerization rate; further addition of pimozide or CK869 to the actin polymerization mixture induced a delay in actin polymerization initiation (Figure 5A). Increase in the lag time at the outset of actin polymerization implied that pimozide inhibited actin polymerization at the nucleation step.
Figure 5.
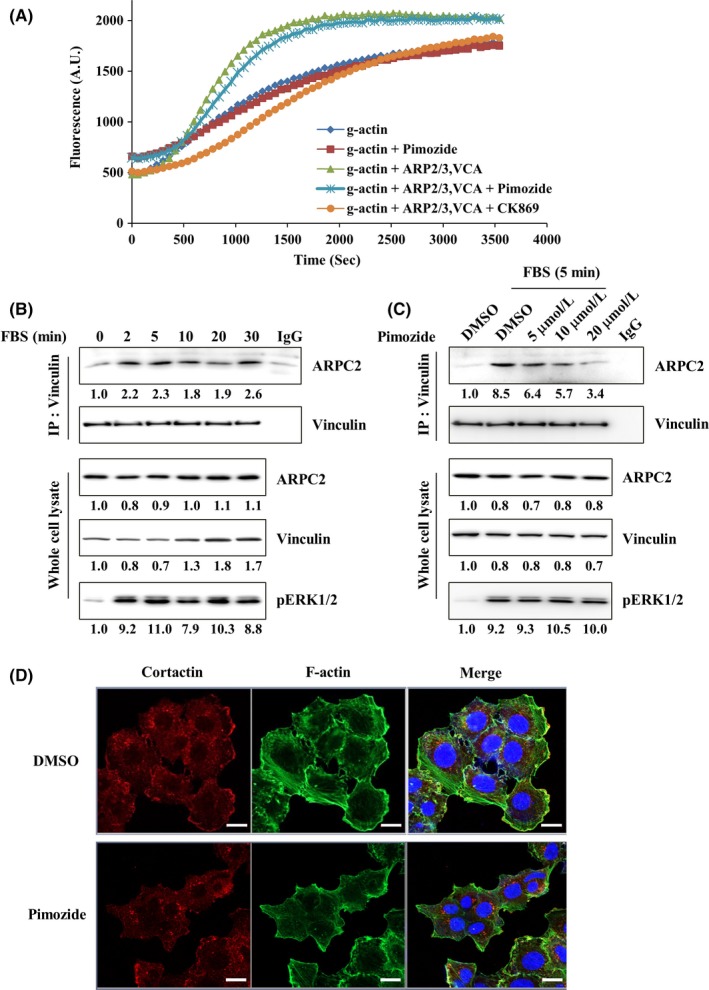
Pimozide binding to ARPC2 delays the outset of Arp2/3 complex‐mediated actin polymerization and inhibits vinculin‐dependent Arp2/3 complex localization to focal adhesion. A, Assay was done using an actin polymerization biochemical kit. Actin polymerization was measured by pyrene fluorescence at 365 nm/405 nm (n = 3). B, Starved DLD‐1 cells were activated with FBS for the indicated times and immunoprecipitated with a vinculin antibody. Expression level of p‐ERK1/2 shows that cells are activated sufficiently by FBS. (n = 3) C, Immunoprecipitation of vinculin antibody from starved DLD‐1 cells that were treated with DMSO or pimozide (5, 10 or 20 μmol/L) for 30 min. Activation time of FBS was 5 min (n = 3). D, Confocal images of DLD‐1 cells that were treated with DMSO or pimozide (10 μmol/L) for 4 h (n = 2)
Pimozide strongly inhibited the migration of cancer cells (Figure 3C); however, as shown in Figure 5A, pimozide did not strongly inhibit actin polymerization in comparison with CK869. Therefore, we thought that pimozide might inhibit the migration of cancer cells through modulation of interactions between ARPC2 and the proteins existing at the lamellipodia, because we observed the disappearance of lamellipodia in ARPC2‐/‐ DLD‐1 cells (Figure 1E). It was also reported that phenotypic loss of lamellipodia is due to a specific loss of Arp2/3 complex activity.11, 35 One of the proteins is vinculin, which plays a role in the mechanical coupling of integrin to the actin cytoskeleton, and binding of the Arp2/3 complex to vinculin promotes the extension of lamellipodia and cell spreading.36, 37
To determine whether ARPC2 and vinculin interact, we treated FBS as an activator for the indicated time in DLD‐1 cells that were starved for 24 hours. Interaction of ARPC2 and vinculin occurred within 5 minutes (Figure 5B), and the interaction between ARPC2 and vinculin lasted up to 30 minutes and then decreased within 60 minutes (data not shown). Because the interaction between ARPC2 and vinculin is the strongest at 5 minutes, we incubated pimozide and FBS for 5 minutes. Pimozide suppressed the interaction between ARPC2 and vinculin in a dose dependent way (Figure 5C). Next, we carried out immunocytochemistry to determine what changes occurred at the leading edge of the cells by blocking the engagement of pimozide with vinculin. In the cells treated with pimozide for 4 hours, cortactin, which is a marker protein of lamellipodia, was not located at the edge of the cell and led to destruction of lamellipodia (Figure 5D). Collectively, these findings indicated that pimozide strongly disrupted the ability of ARPC2 to bind to vinculin, resulting in the inhibition of cell migration.
3.6. Validation of the binding sites of pimozide in ARPC2 using gene‐knockout methods
We confirmed the binding of pimozide to ARPC2 using label‐free methods, such as CETSA and DARTS, which led to inhibition of migration of a variety of cancer cells through blocking the interaction between ARPC2 and vinculin at the leading edge of the cells. Through computer prediction modeling based on our published data,11 we found that pimozide fit into the binding pocket of Benp through hydrophobic interactions with F225 and F247 in ARPC2 (Figure 6A). To confirm the binding pocket, we prepared DLD‐1 cells with the F225A, F247A, or Y250F mutant forms of ARPC2. To exclude the effect of endogenous ARPC2 WT, each ARPC2 mutant gene was transfected into ARPC2−/− DLD‐1 (KO) cells. We observed the expression of Arp2/3 complex proteins in the mutated cells, which disappeared in KO cells (Figure 6B). We validated the binding sites through the change in the inhibitory effect of pimozide on the migration of these mutated cells. Interestingly, pimozide strongly inhibited the migration of ARPC2F247A and ARPC2Y250F cells compared to WT cells, and there was no difference in the migration of ARPC2F225A cells (Figure 6C). These results indicate that pimozide binds to the pocket sites of Benp and requires a larger binding pocket than that of Benp. We confirmed the results using a computational docking model and calculation of a theoretical binding affinity with the generalized Born and surface area continuum solvation (MM/GBSA).38 From the structural model for pimozide in ARPC2, we found that the bis‐(4‐fluorophenyl)‐group of pimozide moves to the deep pocket, leading to an increase in activity in ARPC2F247A and ARPC2Y250F mutant cells. Theoretical binding affinity of pimozide and ARPC2 shows that ARPC2F247A and ARPC2Y250F more strongly bind with pimozide than WT ARPC2 (Figure 2S). Furthermore, we prepared other ARPC2 mutated cells such as E184A and F185A in ARPC2 and found that these mutated cells were resistant by approximately 15% relative to WT cells (Figure 3S). These experimental and theoretical data confirmed that pimozide inhibited the migration of DLD‐1 cells through direct binding with ARPC2.
Figure 6.
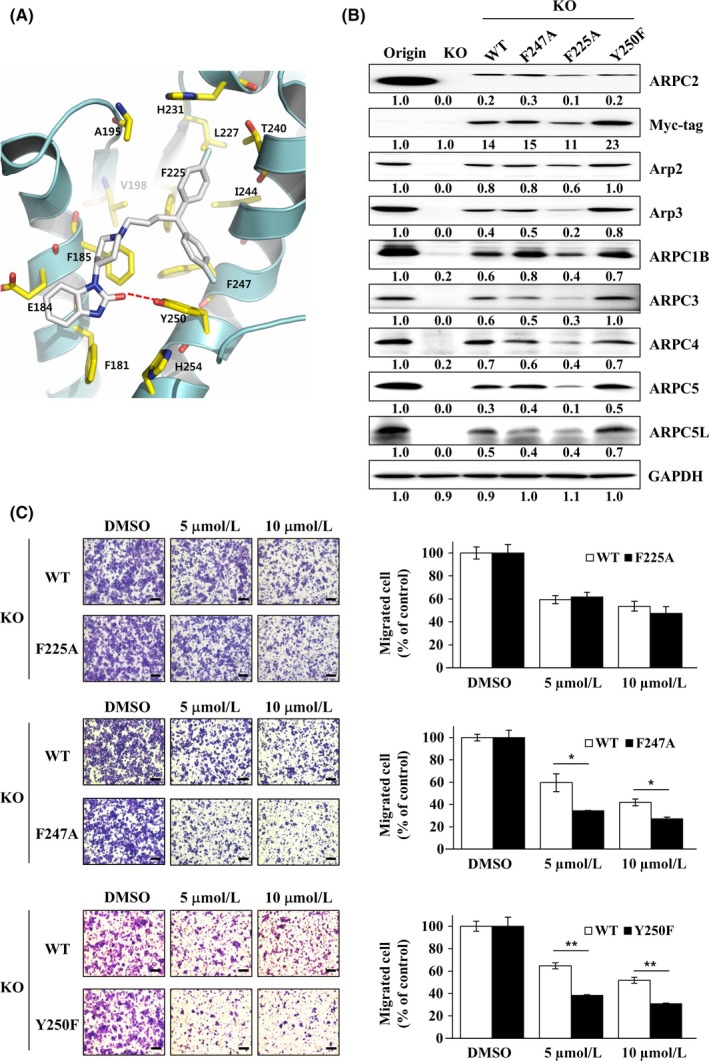
Identification of pimozide binding sites and confirmation of the prediction using ARPC2 mutated cells. A, Expected computational structure model for pimozide in complex with ARPC2. B, Immunoblotting of the Arp2/3 complex, tagging protein (Myc) and loading control protein (GAPDH) in DLD‐1 ARPC2−/− cells stably transfected with ARPC2 WT or ARPC2 mutants (F225A, F247A and Y250F) vectors (n = 3). C, Migration assay of ARPC2−/− cells stably transfected with ARPC2 WT and mutant at various concentrations (5 or 10 μmol/L) of pimozide for 18 h (n = 3). The migrated cells were counted by the Image‐ProPlus 5.0 program. Scale bars, 200 μm. Data represent means ± SD compared with the corresponding control, *P < .05, **P < .01
4. DISCUSSION
Metastasis is associated with poor patient prognosis and is the main cause of cancer motility. To control metastasis effectively, we must inhibit fundamental metastatic processes and develop specific antimetastatic drugs that do not rely on primary tumor responses.3 The early steps of migration and invasion are the breaking away of cancer cells from the primary tumor followed by entry into the circulatory system. Therefore, migration and invasion of cancer cells are critical steps during tumor metastasis, and inhibition of cancer cell migration and invasion (so‐called migrastatics) could be a strategy for the development of specific antimetastatic drugs.4, 5, 6 In general, migrating cancer cells form actin‐based protrusions, and the Arp2/3 complex mediates actin polymerization during lamellipodium formation and migration in cells. Several studies have shown that the Arp2/3 complex or Arp2/3‐stimulating factors, such as cortactin, are upregulated in malignant gliomas, and inhibition of Arp2/3 activity reduces lamellipodium formation, migration, and invasion.39, 40 Arp2/3 inhibitors block the migration of both normal and cancer cells; however, ARPC2 inhibitors selectively suppress migration, invasion, and cancer metastasis.11 siRNA‐mediated silencing of Arp2/3 complex subunits is sufficient to maintain a functional Arp2/3 complex, thereby leading only to a reduction in the migration of subunit‐specific pancreatic cells.41 Subunits of the Arp2/3 complex might be a good therapeutic target for the discovery of migrastatic drugs. In the present study, we found that pimozide, as a functional modulator of ARPC2, inhibited the migration and invasion of a variety of cancer cells (Figure 3C‐F).
Exploring antitumor compounds from noncancer drugs provides an opportunity for rapid advancement of therapeutic strategies into clinical trials. This approach, alternatively called ‘new uses for old drugs’ or ‘drug repurposing’ has gained considerable attention over the past decade.42 Several strategies have been used to identify noncancer drugs for cancer‐related treatment. One successful strategy is the CMap approach, which is a web‐based tool that establishes genetic expression profiles observed in cancer cells.17 To identify new migrastatic agents through blocking ARPC2 functions, we used CMap. For this study, we prepared ARPC2−/− colon cancer cells (DLD‐1) using the CRISPR/Cas9 system. Fortunately, the ARPC2−/− cells did not induce lethality, but their growth and migration rates were relatively slow in comparison with those of WT DLD‐1. The phenotype of the ARPC2−/− cells is very similar to Benp‐treated DLD‐1 cells, such as the loss of lamellipodium formation (Figure 1E).11 Therefore, we carried out CMap analysis using genome expression profiling data from ARPC2−/− cells and selected pimozide as a migrastatic drug and ARPC2 modulator.
Interestingly, previous studies have already shown antiproliferative and migrastatic effects of pimozide in pancreatic cancer cells with high expression levels of DRD2,19 MDA‐MB‐231 breast cancer cells, and A549 lung cancer cells.43 Consequently, many antipsychotic dopamine receptor antagonists have recently been tested in a variety of cancer cells, and these drugs inhibit the growth of cancer cells through the modulation of different targets, such as DRD2, STAT, and AKT.43, 44 In this study, we found that a few antipsychotic drugs, including pimozide, thioridazine and fluspirilene inhibited cancer cell migration and might be ARPC2 functional modulators. These results could provide new therapeutic opportunities for antipsychotic drugs as migrastatic agents.
Lamellipodium formation is one of the key steps for cancer cell migration. To investigate whether migration inhibition results are associated with lamellipodium formation in ARPC2−/− and pimozide‐treated DLD‐1 cells, the cells were stained with lamellipodia markers. As shown in Figures 1E and 5D, WT cells generated small and punctuated cortactin‐rich lamellipodia at cell edges, which colocalized with F‐actin, showing disruption of the structure of the lamellipodial actin networks in ARPC2−/− and pimozide‐treated DLD‐1 cells. Pimozide inhibited the formation of lamellipodia at the leading edge of cancer cells; however, it did not strongly inhibit the Arp2/3 complex mediating actin polymerization. Understanding the mechanism by which pimozide disrupts lamellipodium formation is necessary for validation of ARPC2 as a target of pimozide. It was reported that a vinculin‐Arp2/3 hybrid complex induced focal adhesion and lamellipodium formation.36, 37 Pimozide dissociates the interaction between ARPC2 and vinculin, which means that pimozide inhibits cancer cell migration by impairing ARPC2 function (Figure 5C).
Validation of target engagement of candidate compounds is highly important in early drug discovery to improve the overall efficiency of the compounds. Label‐free methods, such as CETSA and DARTS, are novel methods to identify drug engagement with target proteins in intact cells or cell lysates. We applied these methods to confirm the interaction between ARPC2 and pimozide in cells. Pimozide induced stability against pronase in a dose‐dependent way and thermal stability of ARPC2 in a temperature‐dependent way (Figure 4A, B), which might be due to pimozide directly binding to ARPC2. As shown in Figure 4C and D, the interaction between ARPC2 and biotinyl‐Benp was effectively inhibited by treatment with pimozide, which means that pimozide and biotinyl‐Benp competitively bind with ARPC2. From the experiment using the inactive compound, N‐methyl‐pimozide (Document S1), it was also confirmed that pimozide inhibited the migration of DLD‐1 cells by binding to ARPC2 (Figure 4E, F).
Molecular docking and cell‐based assays are also valuable tools for the validation of candidate compounds. Based on this strategy, the present study predicted the binding pocket of pimozide in ARPC2, and we proved the prediction using DLD‐1 cells with F225A, F247A, or Y250F mutant forms of ARPC2 (Figure 6A, B). From these investigations, F247A or Y250F mutant cells provide a larger pocket than WT DLD‐1 cells, which leads the bis‐(4‐fluorophenyl)‐group of pimozide to move to the deep pocket leading to an increase in activity in the mutant cells. Furthermore, the binding affinity of pimozide was calculated in each mutant cell line using MM/GBSA (Figure S2), which is consistent with the results in Figure 6C. Based on these results, we are currently refining the drug design to achieve stronger candidates.
In the present study, we found that pimozide inhibited cancer cell migration and invasion without cytotoxicity through the modulation of ARPC2 functions (Figure 7). These results will provide valuable information to help to elucidate the antitumor and antimetastatic effects of pimozide, which could be a useful lead molecule for the development of migrastatic agents.
Figure 7.
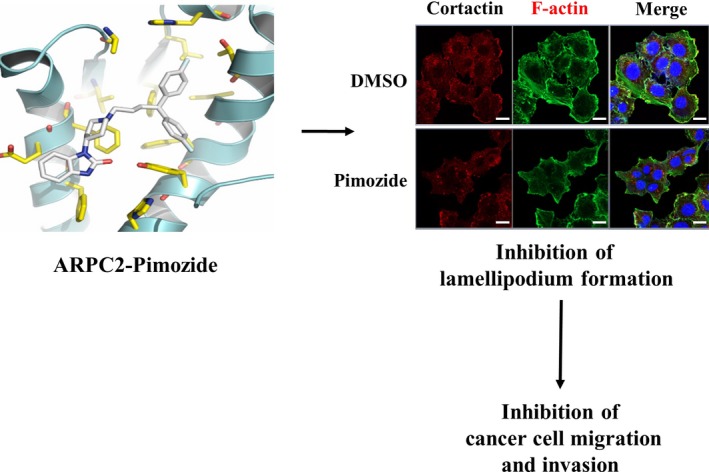
Proposed mechanism for anti‐migrastatic and ‐metastatic activity of pimozide by ARPC2 inhibition
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the KRIBB Research Initiative Program, the Bio‐Synergy Research Project, and the Bio & Medical Technology Development Program of the National Research Foundation and funded by the Korean government (NRF‐2012M3A9C4048777, NRF‐2015M3A9B5030311 and NRF‐2019M3E5D5066600).
Choi J, Lee Y‐J, Yoon YJ, et al. Pimozide suppresses cancer cell migration and tumor metastasis through binding to ARPC2, a subunit of the Arp2/3 complex. Cancer Sci. 2019;110:3788–3801. 10.1111/cas.14205
REFERENCES
- 1. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559‐1564. [DOI] [PubMed] [Google Scholar]
- 2. Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson RL, Balasas T, Callaghan J, et al. A framework for the development of effective anti‐metastatic agents. Nat Rev Clin Oncol. 2019;16:185‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gandalovicova A, Rosel D, Fernandes M, et al. Migrastatics‐anti‐metastatic and anti‐invasion drugs: promises and challenges. Trends Cancer. 2017;3:391‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br J Pharmacol. 2014;171:5507‐5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dumontet C, Jordan MA. Microtubule‐binding agents: A dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rohatgi R, Ma L, Miki H, et al. The interaction between N‐WASP and the Arp2/3 complex links Cdc42‐dependent signals to actin assembly. Cell. 1999;97:221‐231. [DOI] [PubMed] [Google Scholar]
- 8. Machesky LM, Mullins RD, Higgs HN, et al. Scar, a WASp‐related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci U S A. 1999;96:3739‐3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nurnberg A, Kitzing T, Grosse R. Nucleating actin for invasion. Nat Rev Cancer. 2011;11:177‐187. [DOI] [PubMed] [Google Scholar]
- 10. Gross SR. Actin binding proteins: their ups and downs in metastatic life. Cell Adh Migr. 2013;7:199‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoon YJ, Han YM, Choi J, et al. Benproperine, an ARPC2 inhibitor, suppresses cancer cell migration and tumor metastasis. Biochem Pharmacol. 2019;163:46‐59. [DOI] [PubMed] [Google Scholar]
- 12. Mlinie N, Gautreau XA. The Arp2/3 regulatory system and its deregulation in cancer. Physiol Rev. 2018;98:215‐238. [DOI] [PubMed] [Google Scholar]
- 13. Hanniford D, Segura MF, Zhong J, et al. Identification of Metastasis‐Suppressive microRNAs in Primary Melanoma. J Natl Cancer Inst. 2015;107:dju494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Liu Y, Yu CJ, et al. Role of ARPC2 in human gastric cancer. Mediators Inflamm. 2017;2017:5432818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamb J, Crawford ED, Peck D, et al. The connectivity map: using gene‐expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929‐1935. [DOI] [PubMed] [Google Scholar]
- 16. Liu TP, Hsieh YY, Chou CJ, Yang P‐M. Systematic polypharmacology and drug repurposing via an integrated L1000‐based Connectivity Map database mining. R Soc Open Sci. 2018;5:181321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Subramanian A, Narayan R, Corsello SM, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171:1437‐1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8:2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jandaghi P, Najafabadi HS, Bauer AS, et al. Expression of DRD2 Is Increased in Human Pancreatic Ductal Adenocarcinoma and Inhibitors Slow Tumor Growth in Mice. Gastroenterology. 2016;151:1218‐1231. [DOI] [PubMed] [Google Scholar]
- 20. Zhou W, Chen MK, Yu HT, et al. The antipsychotic drug pimozide inhibits cell growth in prostate cancer through suppression of STAT3 activation. Int J Oncol. 2016;48:322‐328. [DOI] [PubMed] [Google Scholar]
- 21. Fako V, Yu Z, Henrich CJ, et al. Inhibition of wnt/beta‐catenin signaling in hepatocellular carcinoma by an antipsychotic drug pimozide. Int J Biol Sci. 2016;12:768‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen JJ, Cai N, Chen GZ, et al. The neuroleptic drug pimozide inhibits stem‐like cell maintenance and tumorigenicity in hepatocellular carcinoma. Oncotarget. 2017;8:17593‐17609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ren Y, Tao J, Jiang Z, et al. Pimozide suppresses colorectal cancer via inhibition of Wnt/beta‐catenin signaling pathway. Life Sci. 2018;209:267‐273. [DOI] [PubMed] [Google Scholar]
- 24. Ma A, Tang M, Zhang L, et al. USP1 inhibition destabilizes KPNA2 and suppresses breast cancer metastasis. Oncogene. 2019;38:2405‐2419. [DOI] [PubMed] [Google Scholar]
- 25. Strobl JS, Kirkwood KI, Lantz TK, et al. Inhibition of human breast cancer cell proliferation in tissue culture by the neuroleptic agents pimozide and thioridazine. Cancer Res. 1990;50:5399‐5405. [PubMed] [Google Scholar]
- 26. Mistry H, Hsieh G, Buhrlage JJ, et al. Small‐molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12:2651‐2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol. 2014;15:577‐590. [DOI] [PubMed] [Google Scholar]
- 28. Rotty JD, Wu C, Haynes EM, et al. Profilin‐1 serves as a gatekeeper for actin assembly by Arp2/3‐dependent and ‐independent pathways. Dev Cell. 2015;32:54‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang J, Zhao D, Liu Z. Repurposing psychiatric drugs as anti‐cancer agents. Cancer Lett. 2018;419:257‐265. [DOI] [PubMed] [Google Scholar]
- 30. Martinez Molina D, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84‐87. [DOI] [PubMed] [Google Scholar]
- 31. Lomenick B, Hao R, Jonai N, et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci USA. 2009;106:21984‐21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol. 2006;7:713‐726. [DOI] [PubMed] [Google Scholar]
- 33. Svitkina TM. Ultrastructure of protrusive actin filament arrays. Curr Opin Cell Biol. 2013;25:574‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beltzner CC, Pollard TD. Pathway of actin filament branch formation by Arp2/3 complex. J Biol Chem. 2008;283:7135‐7144. [DOI] [PubMed] [Google Scholar]
- 35. Wu C, Asokan SB, Berginski ME, et al. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DeMali KA, Barlow CA, Burridge K. Recruitment of the Arp2/3 complex to vinculin: coupling membrane protrusion to matrix adhesion. J Cell Biol. 2002;159:881‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chorev DS, Moscovitz O, Geiger B, et al. Regulation of focal adhesion formation by a vinculin‐Arp2/3 hybrid complex. Nat Commun. 2014;5:3758. [DOI] [PubMed] [Google Scholar]
- 38. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand‐binding affinities. Expert Opin Drug Discov. 2015;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Z, Yang X, Chen C, et al. Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol Rep. 2013;30:2127‐2136. [DOI] [PubMed] [Google Scholar]
- 40. Swaney KF, Li R. Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr Opin Cell Biol. 2016;42:63‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rauhala HE, Teppo S, Niemela S, et al. Silencing of the ARP2/3 complex disturbs pancreatic cancer cell migration. Anticancer Res. 2013;33:45‐52. [PubMed] [Google Scholar]
- 42. Gupta SC, Sung B, Prasad S, et al. Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends Pharmacol Sci. 2013;34:508‐517. [DOI] [PubMed] [Google Scholar]
- 43. Roney SI, Park SK. Antipsychotic dopamine receptor antagonists, cancer, and cancer stem cells. Arch Pharm Res. 2018;41:384‐408. [DOI] [PubMed] [Google Scholar]
- 44. Dakir EH, Pickard A, Srivastava K, et al. The anti‐psychotic drug pimozide is a novel chemotherapeutic for breast cancer. Oncotarget. 2018;9:34889‐34910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials