

Abstract
Differentiation‐inducing factor‐1 (DIF‐1) has been reported to inhibit the proliferation of various mammalian cells by unknown means, although some possible mechanisms of its action have been proposed, including the activation of glycogen synthase kinase‐3 (GSK‐3). Here, we report an alternative mechanism underlying the action of DIF‐1 in human breast cancer cell line MCF‐7, on which the effects of DIF‐1 have not been examined previously. Intragastric administration of DIF‐1 reduced the tumor growth from MCF‐7 cells injected into a mammary fat pad of nude mice, without causing adverse effects. In cultured MCF‐7, DIF‐1 arrested the cell cycle in G0/G1 phase and suppressed cyclin D1 expression, consistent with our previous results obtained in other cell species. However, DIF‐1 did not inhibit the phosphorylation of GSK‐3. Investigating an alternative mechanism for the reduction of cyclin D1, we found that DIF‐1 reduced the protein levels of signal transducer and activator of transcription 3 (STAT3). The STAT3 inhibitor S3I‐201 suppressed cyclin D1 expression and cell proliferation and the overexpression of STAT3 enhanced cyclin D1 expression and accelerated proliferation. Differentiation‐inducing factor‐1 did not reduce STAT3 mRNA or reduce STAT3 protein in the presence of cycloheximide, suggesting that DIF‐1 inhibited STAT3 protein synthesis. Seeking its mechanism, we revealed that DIF‐1 inhibited the activation of 70 kDa and/or 85 kDa ribosomal protein S6 kinase (p70S6K/p85S6K). Inhibition of p70S6K/p85S6K by rapamycin also reduced the expressions of STAT3 and cyclin D1. Therefore, DIF‐1 suppresses MCF‐7 proliferation by inhibiting p70S6K/p85S6K activity and STAT3 protein synthesis followed by reduction of cyclin D1 expression.
Keywords: cyclin D1, differentiation‐inducing factor‐1, protein synthesis, ribosomal protein S6 kinase, signal transducer and activator of transcription 3
In the present study, we showed that differentiation‐inducing factor‐1 inhibits the growth of MCF‐7‐derived tumors in vivo and the proliferation of cultured MCF‐7 cells in vitro, in agreement with our previous studies on the differentiation‐inducing factors’ action. By contrast, the underlying mechanism here is different from that in the cells examined previously. In MCF‐7, DIF‐1 appeared to suppress cyclin D1 expression in two ways: (i) acceleration of this protein’s degradation by an unidentified, glycogen synthase kinase‐3‐independent mechanism; and (ii) inhibition of cyclin D1 gene expression by downregulation of signal transducer and activator of transcription 3 (STAT3), the mechanisms for which could involve the inhibition of STAT3 mRNA translation to the protein by the reduction of phosphorylated p70S6K/p85S6K.
Abbreviations
- 4E‐BP1
eukaryotic translation initiation factor 4E (eIF4E)‐binding protein 1
- CHX
cycloheximide
- DIF‐1
differentiation‐inducing factor‐1
- GSK‐3
glycogen synthase kinase‐3
- HER2
human epidermal growth factor receptor 2
- p‐STAT3
phosphorylated STAT3
- qRT‐PCR
quantitative RT‐PCR
- S6K
ribosomal protein S6 kinase
- TSS
transcription start site
- t‐STAT3
total STAT3
1. INTRODUCTION
Breast cancer is the most common cancer and the leading cause of cancer‐related deaths among women worldwide. According to the American Cancer Society, approximately 12% of American women develop breast cancer in their lifetime. In 2012, approximately 14.1 million women were diagnosed with breast cancer and more than 8 million women died of this disease all over the world.1, 2 Although anti‐estrogen and anti‐HER2 therapies have significantly improved the prognosis of patients with advanced breast cancer, cancer cells often acquire resistance to these treatments, which allows the cancer to recur.3 Therefore, it is still necessary to develop novel therapeutic strategies against breast cancer.
Differentiation‐inducing factors are low‐molecular‐weight compounds identified in Dictyostelium discoideum as inducers of differentiation of Dictyostelium cells (in a migrating slug) into stalk cells of a fruiting body.4 Nonetheless, the action of DIFs is not limited to Dictyostelium. Differentiation‐inducing factors (particularly DIF‐1 and DIF‐3) have been shown to strongly inhibit the proliferation of various mammalian cells, including vascular smooth muscle cells,5 endothelial cells,6 osteoblast‐like cells,7 and a variety of cancer cells, for example, cells from cervical cancer, squamous cell carcinoma, colon cancer, osteosarcoma, and malignant melanoma8, 9, 10, 11, 12, 13, 14, 15, 16 without cytotoxicity.12, 13, 14 The antiproliferative effect of DIFs has also been revealed in in vivo experiments on murine models of cancer with a colon cancer xenograft (DIF‐1), cervical‐cancer xenograft (DIF‐1), and oxidative‐stress‐induced intestinal cancers (DIF‐1 and DIF‐3).13, 15 Moreover, it was recently reported that DIF‐1 inhibits not only cell proliferation but also in vitro cell migration and in vivo lung colony formation of malignant melanoma cells.14
The mechanisms of DIFs’ action on mammalian cells remain to be elucidated. Some of the reported possible mechanisms of the antiproliferative action on mammalian cancer cells include inhibition of the Wnt‐β‐catenin signaling pathway by the activation of GSK‐3,8, 10, 11, 14 upregulation of cAMP by the inhibition of phosphodiesterase 1,17 and downregulation of TCF7L2 by the inhibition of early growth response protein‐1 induction.11
Because the influence of DIF‐1 on breast cancer cells has not been investigated previously, in the present study, we tested whether DIF‐1 has an antiproliferative effect on breast cancer using human breast cancer cell line MCF‐7, which harbors a mutation in the phosphatidylinositol 3‐kinase gene (PI3K), the most frequently mutated oncogene in breast cancer.18 In this study, we found that DIF‐1 inhibited MCF‐7 cell proliferation through an alternative mechanism: the inhibition of ribosomal protein S6 kinase (p70S6K/p85S6K)‐mediated STAT3 protein synthesis.
2. MATERIALS AND METHODS
2.1. Chemicals and Abs
Differentiation‐inducing factor‐1 was synthesized as described previously.4 We purchased actinomycin D from Sigma‐Aldrich, CHX from Wako, MG132 from Peptide Institute, S3I‐201 from Santa Cruz Biotechnology, rapamycin from Cell Signaling Technology. We purchased an anti‐cyclin D1 mAb (cat. # SC‐20044) from Santa Cruz Biotechnology, mAb to GSK‐3α (cat. #9338S), p‐GSK‐3α (Ser21) (9316S), p‐GSK‐3β (Ser9) (9336S), t‐STAT3 (9139S), p‐STAT3 (Tyr705) (9131S), mTOR (cat. #2983S), p‐mTOR (cat. #5536S), p70S6K (cat. #2708S), p‐p70S6K (Thr389) (cat. # 9234S), p‐p70S6K (Ser371) (cat. #9208S), 4EBP1 (cat. #9644S), and p‐4EBP1 (Thr37/46) (cat. #2855S) from Cell Signaling Technology, a mAb to GSK‐3β (610201) from BD Transduction Laboratories, a mAb to α‐tubulin (CP06) from Calbiochem, and a mAb to GAPDH (ab8245) from Abcam.
2.2. Cell culture
Human breast cancer MCF‐7 cells purchased from the RIKEN cell bank (Tsukuba, Japan) were cultured at 37°C in an atmosphere of 95% air and 5% CO2 in DMEM (Sigma) supplemented with 10% FBS, 100 U/mL penicillin G, and 0.1 μg/mL streptomycin. Human breast cancer SK‐BR3 cells purchased from the ATCC were cultured at 37°C in an atmosphere of 95% air and 5% CO2 in RPMI‐1640 (Sigma) supplemented with 10% FBS, 100 U/mL penicillin G, and 0.1 μg/mL streptomycin. Cells in passages 3‐15 since their receipt were used for the experiments.
2.3. Plasmid transfection
We acquired STAT3‐C Flag pRc/CMV (Plasmid #8722), which expresses a constitutively activated STAT3, from Addgene. Wild‐type human cyclin D1 cDNA was provided by Dr K. Tamai (Medical and Biological Laboratories Co.) and subcloned into pcDNA3 (Invitrogen). Cells were transfected with these plasmids or their empty vectors using the Lipofectamine Plus Reagent (100022052; Invitrogen) according to the manufacturer’s protocol.
2.4. Cell proliferation assay
MCF‐7 cells (2.0 × 104 cells/well) and SK‐BR3 cells (104 cells/well) were seeded in 24‐well plates and cultured either with or without various concentrations of DIF‐1 for various periods. To examine the reversibility of DIF‐1’s effect, MCF‐7 cells (2.0 × 104 cells/well) seeded in 24‐well plates were cultured with or without 30 μmol/L DIF‐1. After 24 hours, cells were washed with PBS and further cultured with or without DIF‐1 for various periods. Cells were harvested by the trypsin‐ EDTA treatment and counted on a Coulter Counter (Beckman Coulter).
2.5. Flow cytometry
Cells (1.0 × 104) suspended in a hypotonic solution containing 50 μg/mL propidium iodide, 0.1% sodium citrate, and 0.1% Triton X‐100 were analyzed for fluorescence on a FACSCalibur flow cytometer (Becton Dickinson).
2.6. Western blot analysis
Protein samples (10 μg/lane) were separated by SDS‐12% PAGE then transferred to a PVDF membrane using a semidry transfer system (1 hour at 12 V). Western blotting was carried out as described previously.15, 19 Protein bands were quantified by optical densitometry and analyzed with ImageJ software (version 1.47; NIH). Data are presented as percentages of the control levels at time 0.
2.7. Quantitative RT‐PCR
Total RNA was isolated with the Fast Gene RNA Basic Kit (NIPPON Genetics). Purity and quantity of the RNA samples were determined on an ND‐1000 spectrophotometer (NanoDrop Technologies). Reverse transcription was undertaken with the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Polymerase chain reaction was carried out with 10 μg of the resultant cDNA and primers specific for CCND1 (assay ID: Hs00765553_m1) encoding cyclin D1, STAT3 (assay ID: Hs00374280_m1), or GAPDH (assay ID: Hs99999905_m1) using the TaqMan Gene Expression Assays (Applied Biosystems). The reactions were carried out on an Applied Biosystems 7500 Real‐Time PCR System (Applied Biosystems) programmed to run 40 cycles of 95°C for 15 seconds and 60°C for 1 minute, after incubation at 95°C for 10 minutes. The data were analyzed by the 2−∆∆CT method.
2.8. In vivo experiments
All mice were housed in a temperature‐controlled environment on a 12:12‐hour light : dark cycle and had ad libitum access to feed and water. MCF‐7 cells were trypsinized and resuspended in 50% Matrigel in PBS at a concentration of 2 × 107 cells/mL. The suspension (0.1 mL) was injected into the left #4 mammary fat pad of 6‐week‐old BALB/c nu/nu female mice (Kyudo, Tosu, Japan) anesthetized with 1.0%‐2.0% isoflurane. Preliminary experiments with this method revealed that 100% of mice developed a visible tumor (data not shown).
Mice were randomly subdivided into 2 groups (each group consisted of 6 mice). Mice in the DIF‐1 treatment group (ID No. 7‐12) orally (intragastrically) received DIF‐1 resuspended in soybean oil by gastric gavage, and those in the control group (ID No. 1‐6) received only soybean oil. Differentiation‐inducing factor‐1 was given every 12 hours (150 mg/kg in the morning and 150 mg/kg in the evening, 10 mL/kg per day) 5 days a week. We were able to carry out this dosing method without complications, such as tracheal dosing or esophageal rupture.13 Body weight of the mice was measured every time DIF‐1 was given and just before the animals were killed. The mice were killed at 14 days after the injection of MCF‐7 cells, and the breast tumors that had grown were excised for analysis. The tumors were photographed and weighed. Blood samples were collected and analyzed for blood cell counts using a Celltac α (MEK‐6450; Nihon Kohden, Tokyo, Japan).
2.9. 5′‐ and 3′‐RACE PCR to determine STAT3 mRNA sequence
Total RNA was isolated from MCF‐7 cells treated with DIF‐1 (30 μmol/L) for 24 hours using Nucleospin RNA (TaKaRa). The primers specific for human Stat3 used for RACE PCR were as follows. Primers used for 5′‐RACE‐PCR: STAT3#10, GATTACGCCAAGCTTAGCATCTGCTGCTTCTCCGTCACCACG; and STAT3#2, GATTACGCCAAGCTTTGAGG GGTGGCAGAATGCAGGTAGGC
Primers used for 3′‐RACE: STAT3#1, GATTACGCCAAGCTTACCTCCCCCATGTGAGGAGCTGAGAACG; and STAT3#3, GATTACGCCAAGCTTCCACCAAGCGAGGACTGAGCATCGAGC. The 5′‐ and 3′‐RACE PCR followed by subcloning the PCR products into pRACE vector were carried out using the SMARTer RACE 5′/3′ Kit (TaKaRa) according to the manufacturer’s instructions. At least 2 clones derived from each RACE PCR product were subjected to sequence analysis (Macrogen Japan).
2.10. Statistical analysis
All experiments were carried out on 3 or more independent samples (biological replicates). The results are expressed as the mean ± SD. Differences between means were analyzed by Student’s t test, one‐way ANOVA with the Bonferroni post hoc test (GraphPad Prism 5.0; GraphPad Software), or 2‐way ANOVA with Tukey’s post hoc test (JMP 13; SAS Institute). Differences were considered statistically significant at P < .05.
2.11. Ethics
The study protocol was approved by the Committee on Ethics of Animal Experiments of Kyushu University (Fukuoka, Japan). Animal handling and procedures were carried out in compliance with the Guidelines for Animal Experiments, Kyushu University, and the Law (No. 105) and Notification (No. 6) of the Japanese Government. All surgical procedures were carried out under inhaled‐isoflurane anesthesia, and every effort was made to minimize the suffering of the experimental animals.
3. RESULTS
3.1. Differentiation‐inducing factor‐1 prevented MCF‐7 tumor growth in vivo
To examine the effect of DIF‐1 on breast cancer tumor growth in vivo, we set up a cancer xenograft model by injecting MCF‐7 cells into a mammary fat pad of immunodeficient mice. After the injection, DIF‐1 suspended in soybean oil (150 mg/kg) was given orally (intragastrically) to the mice every 12 hours for 2 weeks (5 days a week as indicated in Figure 1A), and the tumors that developed from the injected cells were excised on day 14. The dose of DIF‐1 was decided based on our previous pharmacokinetic study.13
Figure 1.
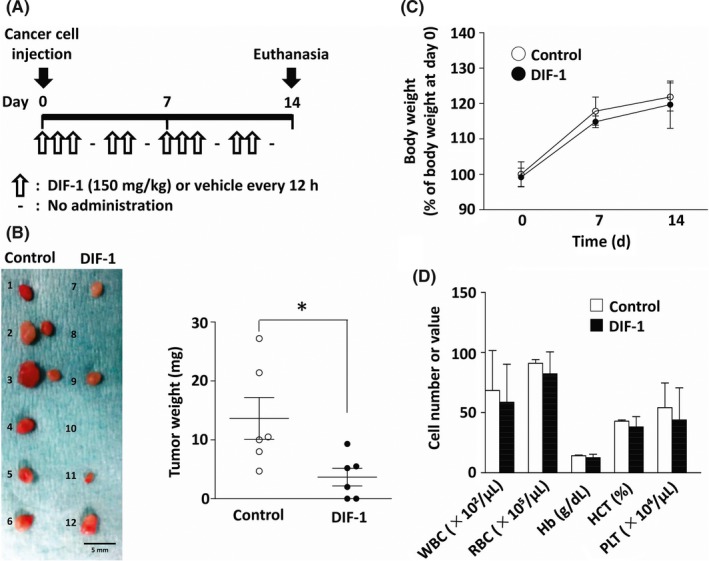
Differentiation‐inducing factor‐1 (DIF‐1) inhibited MCF‐7 tumor growth in vivo. A, Experimental protocol. DIF‐1 or vehicle was given intragastrically to mice every 12 h (5 days a week as indicated) after injection of MCF‐7 cells. The mice were killed, and the grown tumors were collected on day 14. B, Left panel: photograph of excised tumors (1‐6, vehicle‐treated group; 7‐12, DIF‐1‐treated group). Right panel: comparison of tumor weights between the vehicle‐treated group and DIF‐1‐treated group. C, Influence of DIF‐1 on the body weight of mice. Values are shown as percentages of those measured at day 0. D, Effects of DIF‐1 on blood cell counts. Results are shown as mean ± SD of 6 independent experiments. * P < .05. Hb, hemoglobin; HCT, hematocrit; PLT, platelets; RBC, red blood cells; WBC, white blood cells
The treatment with DIF‐1 significantly decreased the weight of tumors (Figure 1B, right panel). We could not find a visible tumor in 2 of the 6 mice treated with DIF‐1 (see Figure 1B, left panel).
We also monitored the general condition of the mice by measuring body weight and counting peripheral blood cells to detect potential adverse effects of DIF‐1. As depicted in Figure 1C,D, DIF‐1 did not affect either body weight or blood cell counts, suggesting that the systemic administration of DIF‐1 apparently was not toxic, in agreement with our previous reports.12, 13, 14
3.2. Differentiation‐inducing factor‐1 reduced expression of cyclin D1 without inhibiting phosphorylation of GSK‐3 in MCF‐7 cells
To identify the mechanism behind the antitumor effect of DIF‐1, we first determined whether DIF‐1 inhibits proliferation of cultured MCF‐7 cells. Differentiation‐inducing factor‐1 significantly inhibited their proliferation in a concentration‐ and time‐dependent manner (Figure 2A, left panel). The proliferation of MCF‐7 cells suppressed by DIF‐1 resumed after DIF‐1 was removed from the medium (Figure 2A, right panel), suggesting that the antiproliferative effect of DIF‐1 was not cytotoxic but reversible, as we reported in a previous study using vascular smooth muscle cells.5 Cell cycle analysis by flow cytometry showed that the treatment with DIF‐1 increased the number of cells in G0/G1 phase and decreased that in S phase (Figure 2B).
Figure 2.

Differentiation‐inducing factor‐1 (DIF‐1) inhibited proliferation and suppressed expression levels of cyclin D1 in MCF‐7 cells. A, Cell proliferation assay. Left panel: MCF‐7 cells were seeded in a 24‐well plate and incubated with various concentrations of DIF‐1 for the indicated periods. Right panel: MCF‐7 cells cultured with DIF‐1 (30 µmol/L) for 24 h were washed with PBS and further cultured with or without DIF‐1 for the indicated periods. B, Cell cycle analyses. Cells were treated with DIF‐1 (20 μmol/L) for 24 h and then harvested with trypsin‐EDTA. Cells were stained with propidium iodide (PI) and nuclear fluorescence was measured by flow cytometry. Percentages of the cells in different cell cycle phases are shown. C, Influence of DIF‐1 on cyclin D1 protein expression. Left panel: time course after addition of DIF‐1 (30 μmol/L). Right panel: concentration dependence. D, Influence of a proteasome inhibitor on the DIF‐1‐induced reduction of cyclin D1 measured in the presence of cycloheximide (CHX). MCF‐7 cells pretreated with or without MG132 (5 μmol/L) for 1 h were incubated with CHX (10 μg/mL) and with or without DIF‐1 (30 μmol/L) for 3 h. Percentages of degraded cyclin D1 were determined by comparing lane 2 with lane 1 (without MG132) and lane 4 with lane 3 (with MG132). E, Effect of DIF‐1 on cyclin D1 mRNA expression. MCF‐7 cells were incubated with or without DIF‐1 (30 μmol/L) for 24 h and then quantitative RT‐PCR was carried out. F, Effect of DIF‐1 on the proliferation of cells overexpressing cyclin D1. Left panel: representative western blot of cyclin D1 and its quantification in pcDNA3‐ and pcDNA3/cyclin D1‐transfected cells. Middle panel: effect of DIF‐1 on cyclin D1 expression and cell proliferation in pcDNA3 (empty vector)‐transfected cells. Right panel: effect of DIF‐1 on cyclin D1 expression and cell proliferation in pcDNA3/cyclin D1‐transfected cells. G, Phosphorylation levels of glycogen synthase kinase‐3α (GSK‐3α) (left) and GSK‐3β (right) after incubation with or without DIF‐1 (30 μmol/L). The results are presented as the mean ± SD of 3 independent experiments. *P < .05; **P < .01; ***P < .001
Subsequently, we examined the effect of DIF‐1 on the expression of cyclin D1 because cyclin D1 plays a crucial role in the progression of G1 phase,20 and we have previously reported that DIF‐1 induces cell cycle arrest in the G0‐G1 transition by suppressing cyclin D1 expression in several other cell types.6, 8, 13, 14, 16 In line with other reports,8, 9, 13, 14 Differentiation‐inducing factor‐1 markedly downregulated the cyclin D1 protein in a time‐ and concentration‐dependent manner (Figure 2C).
We also examined the effect of DIF‐1 on cell proliferation in human breast cancer cell line SK‐BR3, which harbors a mutation in TP53R175H.21, 22 Consist with the results in MCF‐7 cells, DIF‐1 significantly inhibited their proliferation (Figure S1A), and markedly downregulated the cyclin D1 protein (Figure S1B).
Differentiation‐inducing factor‐1 reduced the cyclin D1 protein amount in the presence of CHX, a protein synthesis inhibitor, but the proteasome inhibitor MG132 significantly attenuated this reduction (Figure 2D). These data suggested that DIF‐1 promoted cyclin D1 degradation by the ubiquitin‐proteasome system. Moreover, qRT‐PCR showed that DIF‐1 markedly reduced the expression levels of cyclin D1 mRNA (Figure 2E), again consistent with our previous reports.12, 16 To determine whether the reduction of cyclin D1 was involved in the antiproliferative effect of DIF‐1, we undertook cell proliferation assays using MCF‐7 cells transfected with a cyclin D1 expression plasmid (pcDNA3/cyclin D1) (Figure 2F, left panel). Although DIF‐1 inhibited proliferation of cells transfected with the empty vector (pcDNA3) (Figure 2F, middle panel), the effect of DIF‐1 was attenuated in cells overexpressing cyclin D1 (Figure 2F, right panel), indicating that the reduction of cyclin D1 mediated the effect of DIF‐1.
Our original hypothesis was that the downregulation of cyclin D1 was evoked by DIF‐1‐induced activation of GSK‐3 through the dephosphorylation of Ser21 in GSK‐3α or Ser9 in GSK‐3β, as we have reported in previous studies.6, 8, 9, 10, 12, 13, 14, 15, 16 However, our supposition was wrong because DIF‐1 did not inhibit the phosphorylation of either GSK‐3α or GSK‐3β in MCF‐7 cells (Figure 2G). This result raised the following question: what factors are involved in the DIF‐1‐induced cyclin D1 downregulation in MCF‐7 cells?
3.3. Differentiation‐inducing factor‐1 suppressed expression of STAT3 in MCF‐7 cells
Signal transducer and activator of transcription 3 is constitutively activated in many cancers, including more than 40% of breast cancer cases.23, 24, 25 This transcription factor is activated on the phosphorylation of Tyr705 by cytoplasmic nonreceptor tyrosine kinases.26 Signal transducer and activator of transcription 3 has been shown to mediate gene expression of cyclin D1 in a variety of cell types.27, 28, 29 Therefore, we investigated the effect of DIF‐1 on STAT3 expression and phosphorylation in MCF‐7 cells.
Treatment with DIF‐1 for 24 hours significantly reduced the amounts of p‐STAT3 (Figure 3A,B) and of t‐STAT3 (Figure 3A,C). Because there was no significant difference in p‐STAT3/t‐STAT3 ratios between in the cell groups with and without DIF‐1 (Figure 3D), the downregulation of p‐STAT3 apparently took place in parallel with the decrease in the t‐STAT3 amount. Therefore, the DIF‐1‐induced downregulation of p‐STAT3 seemed to be secondary to the reduction in the t‐STAT3 level. Similar results were obtained in SK‐BR3 cells (Figure S1C‐F).
Figure 3.
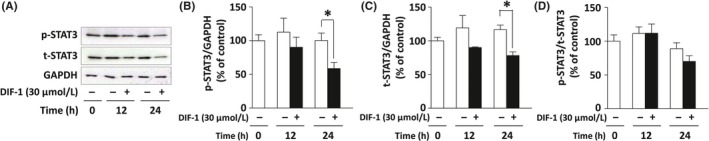
Differentiation‐inducing factor‐1 (DIF‐1) suppressed the expression of signal transducer and activator of transcription 3 (STAT3) in MCF‐7 cells. Cells were incubated with or without DIF‐1 (30 μmol/L) for indicated periods and then western blotting was carried out. A, Representative blots of t‐STAT3 and p‐STAT3. B‐D, Quantification of the western blots by densitometry. Protein bands were quantified and are presented as percentages of the control level at time 0. Results are indicated as the mean ± SD of 5 independent experiments. *P < .05
3.4. Signal transducer and activator of transcription 3 mediates cyclin D1 expression and proliferation in MCF‐7 cells
Next, we verified whether STAT3 mediates cyclin D1 expression and proliferation in MCF‐7 cells using S3I‐201 (a STAT3 inhibitor) and STAT3‐C Flag pRc/CMV (a plasmid coding for constitutively active STAT3).
S3I‐201 binds to the SH2 domain of STAT3 and disrupts formation of a complex of STAT3 with other proteins such as JAK, SRC, and STAT family members.30 As illustrated in Figure 4A, S3I‐201 strongly reduced both t‐STAT3 and p‐STAT3 protein levels. S3I‐201 also strongly suppressed the protein expression of cyclin D1 (Figure 4B) and cell proliferation (Figure 4C). The reduction of t‐STAT3 by the STAT3 inhibitor might have been caused by a positive autoregulatory loop in which STAT3 accelerates transcription of the Jak‐STAT pathway components, including STAT3 itself.31 In contrast, when the constitutively active STAT3 mutant (STAT3‐C) was overexpressed (Figure 4D), the expression levels of cyclin D1 were approximately twice those in the control (Figure 4E). Overexpression of STAT3‐C significantly accelerated cell proliferation too (Figure 4F). These results suggest that STAT3 mediates cyclin D1 expression and proliferation in MCF‐7 cells.
Figure 4.
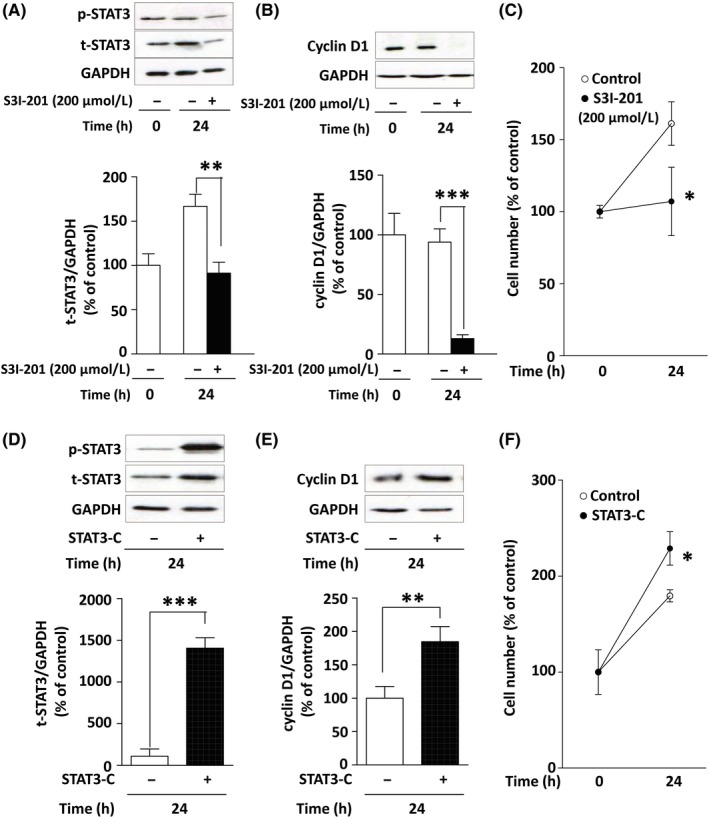
Involvement of signal transducer and activator of transcription 3 (STAT3) in cyclin D1 expression and proliferation in MCF‐7 cells. A, Effect of S3I‐201 (200 μmol/L) on the expression level of STAT3 protein. B, Effect of S3I‐201 on the expression level of cyclin D1 protein. C, Effect of S3I‐201 on the cell proliferation. D, Constitutively active STAT3 was overexpressed after transfection of STAT3‐C Flag pRc/CMV for 24 h. E, Effect of the overexpression of STAT3‐C on the expression level of cyclin D1. F, Effect of the overexpression of STAT3‐C on the cell proliferation. Blots were quantified by densitometry, and the data are shown as percentages of the control level at time 0. Results are presented as the mean ± SD of 3 independent experiments. *P < .05; **P < .01; ***P < .001
3.5. Differentiation‐inducing factor‐1 inhibited STAT3 protein synthesis by reducing phosphorylation levels of p70S6K/p85S6K
Subsequently, we attempted to identify the mechanism underlying the DIF‐1‐induced reduction in the STAT3 protein amount. Unexpectedly, qRT‐PCR revealed that DIF‐1 enhanced the expression of STAT3 mRNA (Figures 5A, left panel, and S1G). This enhancement disappeared when actinomycin D, an inhibitor of transcription, was added (Figure 5A, right panel), suggesting that DIF‐1 activated transcription of the STAT3 gene.
Figure 5.
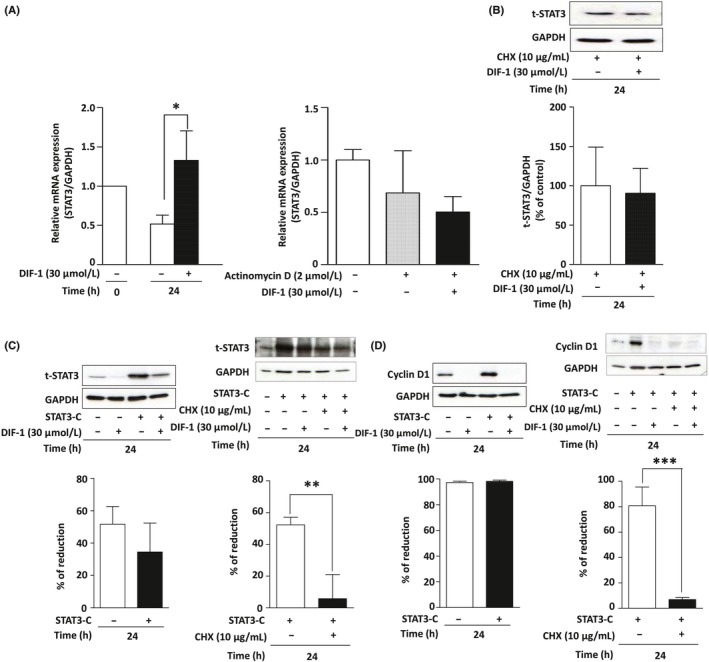
Differentiation‐inducing factor‐1 (DIF‐1) inhibited signal transducer and activator of transcription 3 (STAT3) protein synthesis in MCF‐7 cells. A, Effect of DIF‐1 on the mRNA levels of STAT3. Left panel: MCF‐7 cells were incubated with or without DIF‐1 (30 μmol/L) for 24 h, and the mRNA was quantified by quantitative RT‐PCR. Right panel: MCF‐7 cells were incubated with or without DIF‐1 (30 μmol/L) in the presence or absence of actinomycin D (2 μmol/L) for 24 h, and the mRNA was quantified by quantitative RT‐PCR. B, Effect of DIF‐1 on the stability of STAT3 protein. MCF‐7 cells were incubated with or without DIF‐1 (30 μmol/L) in the presence of cycloheximide (CHX; 10 μg/mL) for 24 h and then western blotting for STAT3 was carried out. C, Effect of DIF‐1 on the expression levels of STAT3 in MCF‐7 cells overexpressing STAT3‐C. Left panel: Influence of overexpression of STAT3‐C on the DIF‐1‐induced reduction of STAT3 protein. Percentages of the reduction of STAT3 by DIF‐1 were determined by comparing lane 2 with lane 1 (STAT3‐C −) and lane 4 with lane 3 (STAT3‐C +). Right panel: influence of CHX on the DIF‐1‐induced reduction of STAT3 protein in cells overexpressing STAT3‐C. Percentages of the reduction in the levels of STAT3 by DIF‐1 were determined by comparing lane 3 with lane 2 (CHX −) and lane 5 with lane 4 (CHX +). D, Effect of DIF‐1 on the expression levels of cyclin D1 in MCF‐7 cells overexpressing STAT3‐C. Left: Influence of overexpression of active STAT3 on DIF‐1‐induced reduction of cyclin D1 protein. Percentages of the reduction of STAT3 by DIF‐1 were determined by comparing lane 2 with lane 1 (STAT3‐C −) and lane 4 with lane 3 (STAT3‐C +). Right panel: Influence of CHX on the DIF‐1‐induced reduction of cyclin D1 protein in cells overexpressing STAT3‐C. Percentages of the reduction in the levels of STAT3 by DIF‐1 were determined by comparing lane 3 with lane 2 (CHX −) and lane 5 with lane 4 (CHX +). Results are presented as the mean ± SD of 3 independent experiments. *P < .05
To test the possibility that DIF‐1 could affect transcription or processing of STAT3 mRNA, we undertook 5′‐ and 3′‐RACE PCR using total RNA purified from MCF‐7 cells treated with DIF‐1 for 24 hours. As shown in Figure S2, we successfully obtained PCR products using 4 different gene‐specific primers for STAT3 (Figure S2A) and determined the DNA sequences of the RACE PCR products (Figure S2B,C). The DNA sequencing revealed that the coding region and 3′‐UTR amplified by RACE PCR completely matched to the consensus sequence of STAT3 variants 1, 2, X1, X2, X3, and X9 (data not shown). Five‐prime UTR regions determined by 5′‐RACE showed high similarity to variant X6 or variant X8. According to DataBase of Transcription Start Sites (DBTSS, https://dbtss.hgc.jp), most of the transcription start sites (TSS) of STAT3 gene locates from position 42 388 520 to 42 388 390 in human chromosome 17. The 5′‐end of the 5′‐RACE PCR products locate within the region corresponding to the TSS of STAT3 gene (Figure S2D), suggesting that transcription of STAT3 mRNA starts from correct sites in the presence of DIF‐1. There were no intronic sequences in any of the RACE PCR products, indicating that DIF‐1 does not affect the normal splicing and processing of STAT3 mRNA. Therefore, it is most likely that DIF‐1 reduces STAT3 protein level by inhibiting its translation or a later process.
Differentiation‐inducing factor‐1 reduced STAT3 protein levels as illustrated in Figure 3. However, DIF‐1 failed to downregulate STAT3 when CHX, a protein synthesis inhibitor, was added to the cell culture medium (Figures 5B and S1H). Therefore, we concluded that DIF‐1 inhibited the translation of STAT3 mRNA into the protein. This conclusion was reinforced by the finding that DIF‐1 decreased the STAT3 protein level and CHX negated this effect of DIF‐1 even in the cells overexpressing STAT3‐C (Figures 5C and S1I,J). Accordingly, DIF‐1 also downregulated cyclin D1 and CHX negated this effect of DIF‐1 in MCF‐7 cells (Figure 5D).
To identify the mechanism for DIF‐1‐induced inhibition of STAT3 translation, we examined the effect of DIF‐1 on the mTOR signaling pathway which regulates protein translation through mTOR‐induced activation of ribosomal protein S6 kinase (p70S6K/p85S6K) and inactivation of 4E‐BP1.32, 33 As shown in Figure 6A, DIF‐1 did not affect the expression or phosphorylation of mTOR. However, DIF‐1 strongly attenuated the phosphorylation levels of p70S6K (Thr389) and p85S6K (Thr412) within 30 minutes (Figure 6B) without affecting Ser371 of p70S6K (Figure 6C) or Thr37/46 of 4E‐BP1 (Figure 6D). Then we examined whether the inhibition of p70S6K/p85S6K is involved in the inhibition of STAT3 translation using rapamycin, an mTOR inhibitor. Similar to DIF‐1, rapamycin reduced the phosphorylation levels of p70S6K/p85S6K (Figure 6E) and the expression levels of STAT3 (Figure 6F) and cyclin D1 (Figure 6G).
Figure 6.
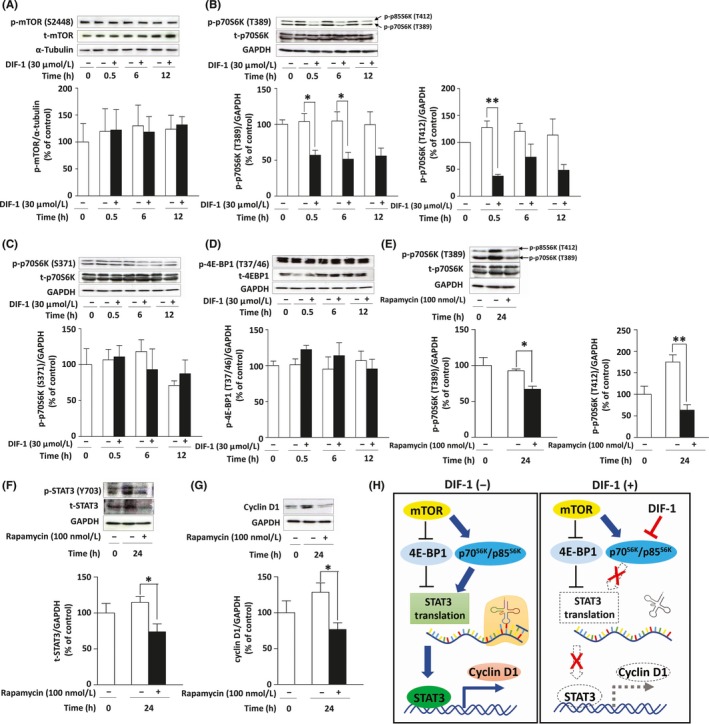
Involvement of the mTOR signaling pathway in differentiation‐inducing factor‐1 (DIF‐1)‐induced inhibition of signal transducer and activator of transcription 3 (STAT3) translation in MCF‐7 cells. A, Effects of DIF‐1 (30 μmol/L) on the expression levels of total (t‐)mTOR and phosphorylated (p‐)mTOR (Ser2448). B‐D, Effects of DIF‐1 (30 μmol/L) on expression levels of t‐p70S6K, p‐p70S6K/p85S6K (Thr389/Thr412), p‐p70S6K (Ser371) and t‐4E‐BP1 and p‐4E‐BP1 (Thr37/46). E‐G, Effects of rapamycin (100 nmol/L) on expression levels of t‐p70S6K, p‐p70S6K/p85S6K (Thr389/Thr412), STAT3, and cyclin D1. Results are presented as the mean ± SD of 3 independent experiments. *P < .05. H, Schematic diagram of the mechanism of DIF‐1’s action on cyclin D1 gene expression
4. DISCUSSION
In the present study, we showed that DIF‐1 inhibits the growth of MCF‐7‐derived tumors in vivo and the proliferation of cultured MCF‐7 cells in vitro, in agreement with our previous studies on the DIFs’ action. By contrast, the underlying mechanism here is different from that in the cells examined previously. In MCF‐7 cells, DIF‐1 appeared to suppress cyclin D1 expression in 2 ways: (i) acceleration of this protein’s degradation by an unidentified, GSK‐3‐independent mechanism; and (ii) inhibition of cyclin D1 gene expression by downregulation of STAT3, the mechanisms for which could involve the inhibition of STAT3 mRNA translation to the protein by the reduction of phosphorylated p70S6K/p85S6K (Figure 6H).
Oral (intragastric) administration of DIF‐1 markedly suppressed tumor growth of MCF‐7 cells injected into a mammary fat pad, in agreement with our previous study using s.c. injected HeLa cervical cancer cells and HCT‐116 colon cancer cells.13 In the current study, we reduced the dose of DIF‐1 from the previous 450 mg/kg/day to 300 mg/kg/day because we have noticed that ingestion of large amounts of soybean oil causes a loss of appetite in mice. Nevertheless, DIF‐1 strongly inhibited MCF‐7 tumor formation, suggesting that DIF‐1 can be administered at low concentrations. It is noteworthy that we could not find a visible tumor in 2 out of the 6 mice treated with DIF‐1. Readers should take into account that in our preliminary experiments, the rate of visible tumor formation was 100% when DIF‐1 was not administered (data not shown).
Consistent with our previous studies, DIF‐1 strongly reduced the expression levels of cyclin D1 by accelerating this protein’s degradation6, 8, 13, 14, 16 and by inhibiting its mRNA expression.12, 16 Initially, we thought that this phenomenon was caused by the activation (dephosphorylation) of GSK‐3, because activated GSK‐3 phosphorylates cyclin D1 to accelerate its degradation and inhibits the Wnt‐β‐catenin signaling pathway which transactivates cyclin D1 gene, as we have uncovered in previous studies.6, 8, 9, 12, 14, 16 Unexpectedly, DIF‐1 did not induce dephosphorylation of GSK‐3 in MCF‐7 cells. Mutated PIK3CA (PI3K P110α catalytic subunit) in MCF‐7 cells constitutively activates AKT, which phosphorylates GSK‐3 to inactivate this kinase.34 This mechanism could be the reason why DIF‐1 failed to activate GSK‐3 in MCF‐7 cells, in contrast to other cell types. In the present study, however, we could not identify the kinase responsible for DIF‐1‐induced cyclin D1 degradation in MCF‐7 cells.
While seeking an alternative mechanism of DIF‐1‐induced downregulation of the cyclin D1 gene, we paid attention to STAT3, because STAT3 induces cyclin D128 and often participates in cancer cell proliferation.35 Moreover, STAT3 promotes malignant tumor formation by altering the expression of protumorigenic gene‐regulatory networks 36, 37, 38, 39, 40 and thereby sustains oxidative‐phosphorylation activities of cancer cells.41, 42, 43 Therefore, STAT3 has been considered to be a promising target for cancer therapy. In fact, a phase I clinical trial was recently undertaken to validate a novel anticancer drug derived from a STAT3 inhibitor.44
We found that DIF‐1 strongly decreased the amounts of p‐STAT3 and t‐STAT3. Therefore, we speculated that the mechanism by which DIF‐1 suppressed cyclin D1 expression could be related to the suppression of STAT3 activity. The STAT3 protein binds directly to the cyclin D1 gene promoter in several cell types including fibroblasts and embryonic kidney cells, and squamous cell carcinoma, fibrosarcoma, glioma, and malignant melanoma cells.27, 28, 29, 45 It has remained unknown, however, whether STAT3 takes part in cyclin D1 expression and cell proliferation in MCF‐7 cells. Our experiments with the STAT3 inhibitor and overexpression of constitutively active STAT3 clearly showed the participation of STAT3 in cyclin D1 expression and in proliferation of MCF‐7 cells.
We initially expected that DIF‐1 would inhibit the phosphorylation of STAT3 because the activity of STAT3 is usually controlled by the phosphorylation of its Tyr705.26 In fact, in gastric cancer cell lines, DIF‐1 has been reported to suppress STAT3 phosphorylation by activating the MEK‐ERK pathway.46 In contrast, here, DIF‐1 did not seem to inhibit STAT3 phosphorylation but suppressed its protein expression. The downregulation of p‐STAT3 was likely to be secondary to the reduction in the t‐STAT3 amount because the p‐STAT3/t‐STAT3 ratio was not changed by DIF‐1. Quantitative RT‐PCR analysis of STAT3 mRNA showed that DIF‐1 did not suppress STAT3 mRNA expression but instead elevated the mRNA levels. These data indicate that the cause of the decrease in STAT3 protein levels was neither the inhibition of STAT3 transcription nor destabilization of STAT3 mRNA. The maintenance of protein homeostasis is essential for healthy cells and therefore, in general, proteins need to be always produced and maintained in an appropriate quantity.47 Therefore, as results show in Figure 5A (left panel), we speculated that cells would have tried to compensate for DIF‐1‐induced reduction of STAT3 protein by enhancing STAT3 mRNA expression. Differentiation‐inducing factor‐1 did not affect the normal splicing and processing of STAT3 mRNA. Therefore, it was presumed that DIF‐1 inhibited STAT3 protein synthesis or accelerated the protein degradation. Of note, DIF‐1 seemed to inhibit the translation of STAT3 mRNA into the protein because DIF‐1 failed to decrease STAT3 protein levels in the presence of CHX. This interpretation was reinforced by the finding that DIF‐1 strongly reduced the expression levels of STAT3 even when constitutively active STAT3 was overexpressed.
To identify the mechanism for DIF‐1‐induced reduction of STAT3 protein level, we investigated possible involvement of mTOR signaling pathway, because this pathway plays an important role in promotion of protein translation32, 33 and is involved in STAT3‐mediated cell survival and proliferation of breast cancer stem‐like cells.48 Intriguingly, DIF‐1 rapidly reduced the phosphorylation levels of Thr389/412 of p70S6K/p85S6K, which is a major downstream of mTOR, phosphorylates S6 protein of ribosomal subunit 40S and induces selective translation of mRNAs.49 The experiments using rapamycin indicated that this pathway is in fact essential for STAT3 translation in MCF‐7 cells.
MCF‐7 cells express estrogen receptor and progesterone receptor but do not overexpress HER2. Approximately 25%‐30% of breast cancers harbor HER2 protooncogene amplification and they show poor prognosis with a lower survival rate and a shorter time to recur.50, 51 Therefore, we additionally examined the effect of DIF‐1 on SK‐BR3 cells that do not express female hormone receptors but overexpress HER2. Additionally, DIF‐1 inhibited cell proliferation and STAT3 expression in SK‐BR3 cells (Figure S1), indicating that DIF‐1’s effect does not depend on the expression levels of HER2 or female hormone receptors.
Against HER2‐overexpressing breast cancers, trastuzumab has been used as a first‐line drug. However, approximately 50% of patients fail to respond to the initial trastuzumab therapy or develop resistance to this Ab therapy.40, 52, 53, 54 In addition, disadvantages of trastuzumab have been reported: trastuzumab treatment increased the risks of cardiotoxicity and brain metastasis by impairment of the blood‐brain barrier in patients with HER2‐overexpressing metastatic breast cancer.55, 56 Therefore, development of novel therapeutic agents against this type of breast cancer is still required. Furthermore, it has been reported that the activation of STAT3 by HER2‐overexpression increased the risk of metastasis through prompting epithelial‐mesenchymal transition in human breast cancer.52 Thus, the effect of DIF‐1 on breast cancer metastasis needs to be investigated. We are attempting to investigate the effect of DIF‐1 on the processes of cancer metastasis, that is, epithelial‐mesenchymal transition, cell infiltration, migration, extravasation, and engraftment of breast cancer cells.
Including ours, there have been several reports on the intracellular signals that mediate the effects of DIFs (mainly DIF‐1 and DIF‐3); DIF‐1 and DIF‐3 interrupted mitochondrial activity.57, 58, 59 Differentiation‐inducing factor‐3 increased the production of reactive oxygen species and intracellular calcium concentration.58, 59 Differentiation‐inducing factor‐1 suppressed the activity of calmodulin‐dependent phosphodiesterase.17 A derivative of DIF‐3 inhibited PAK‐1.60 Differentiation‐inducing factor‐1 activated the MEK‐ERK‐pathway to inhibit STAT3 phosphorylation.46 Differentiation‐inducing factor‐1 activated PI3K and Akt.46 Both DIF‐1 and DIF‐3 activated GSK‐3.9, 61 Here, we added an alternative mechanism: inhibition of S6K‐mediatd STAT3 translation. However, the molecular target of DIFs to which DIFs directly bind has not been identified. If we could identify it, then the development of novel anticancer drugs based on DIF‐1 as the lead compound could be greatly accelerated. Further studies are needed to identify the target molecule by utilizing new approaches such as phosphoproteomic analysis or microRNA expression analysis.
DISCLOSURE
There are no potential conflicts of interest to disclose.
Supporting information
ACKNOWLEDGEMENTS
We appreciate the technical support provided by the Research Support Center at the Graduate School of Medical Sciences, Kyushu University (Fukuoka, Japan). We are grateful for the assistance given by Hideya Ohnishi (Department of Surgery and Oncology, Graduate School of Medical Sciences, Kyushu University). This study was supported by a KAKENHI grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan to MA (Grant No. 17K15581), by Fukuoka Foundation for Sound Health Cancer Research Fund to MA and by the Society for Women’s Health Science Research to FTY.
Tetsuo F, Arioka M, Miura K, et al. Differentiation‐inducing factor‐1 suppresses cyclin D1‐induced cell proliferation of MCF‐7 breast cancer cells by inhibiting S6K‐mediated signal transducer and activator of transcription 3 synthesis. Cancer Sci. 2019;110:3761–3772. 10.1111/cas.14204
Contributor Information
Masaki Arioka, Email: arioka@med.kyushu-u.ac.jp.
Toshiyuki Sasaguri, Email: sasaguri@med.kyushu-u.ac.jp.
REFERENCES
- 1. DeSantis CE, Bray F, Ferlay J, Lortet‐Tieulent J, Anderson BO, Jemal A. International variation in female breast cancer incidence and mortality rates. Cancer Epidemiol Biomark Prev. 2015;24:1495‐1506. [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 3. Tryfonidis K, Zardavas D, Katzenellenbogen BS, Piccart M. Endocrine treatment in breast cancer: cure, resistance and beyond. Cancer Treat Rev. 2016;50:68‐81. [DOI] [PubMed] [Google Scholar]
- 4. Morris HR, Taylor GW, Masento MS, Jermyn KA, Kay RR. Chemical structure of the morphogen differentiation inducing factor from Dictyostelium discoideum. Nature. 1987;328:811‐814. [DOI] [PubMed] [Google Scholar]
- 5. Miwa Y, Sasaguri T, Kosaka C, et al. Differentiation‐inducing factor‐1, a morphogen of dictyostelium, induces G(1) arrest and differentiation of vascular smooth muscle cells. Circ Res. 2000;86:68‐75. [DOI] [PubMed] [Google Scholar]
- 6. Yoshihara T, Takahashi‐Yanaga F, Shiraishi F, et al. Anti‐angiogenic effects of differentiation‐inducing factor‐1 involving VEGFR‐2 expression inhibition independent of the Wnt/beta‐catenin signaling pathway. Mol Cancer. 2010;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsuzaki E, Takahashi‐Yanaga F, Miwa Y, et al. Differentiation‐inducing factor‐1 alters canonical Wnt signaling and suppresses alkaline phosphatase expression in osteoblast‐like cell lines. J Bone Miner Res. 2006;21:1307‐1316. [DOI] [PubMed] [Google Scholar]
- 8. Mori J, Takahashi‐Yanaga F, Miwa Y, et al. Differentiation‐inducing factor‐1 induces cyclin D1 degradation through the phosphorylation of Thr(286) in squamous cell carcinoma. Exp Cell Res. 2005;310:426‐433. [DOI] [PubMed] [Google Scholar]
- 9. Yasmin T, Takahashi‐Yanaga F, Mori J, et al. Differentiation‐inducing factor‐1 suppresses gene expression of cyclin D1 in tumor cells. Biochem Biophys Res Commun. 2005;338:903‐909. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi‐Yanaga F, Mori J, Matsuzaki E, et al. Involvement of GSK‐3beta and DYRK1B in differentiation‐inducing factor‐3‐induced phosphorylation of cyclin D1 in HeLa cells. J Biol Chem. 2006;281:38489‐38497. [DOI] [PubMed] [Google Scholar]
- 11. Jingushi K, Nakamura T, Takahashi‐Yanaga F, et al. Differentiation‐inducing factor‐1 suppresses the expression of c‐Myc in the human cancer cell lines. J Pharmacol Sci. 2013;121:103‐109. [DOI] [PubMed] [Google Scholar]
- 12. Jingushi K, Takahashi‐Yanaga F, Yoshihara T, et al. DIF‐1 inhibits the Wnt/beta‐catenin signaling pathway by inhibiting TCF7L2 expression in colon cancer cell lines. Biochem Pharmacol. 2012;83:47‐56. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi‐Yanaga F, Yoshihara T, Jingushi K, et al. DIF‐1 inhibits tumor growth in vivo reducing phosphorylation of GSK‐3 beta and expressions of cyclin D1 and TCF7L2 in cancer model mice. Biochem Pharmacol. 2014;89:340‐348. [DOI] [PubMed] [Google Scholar]
- 14. Arioka M, Takahashi‐Yanaga F, Kubo M, Igawa K, Tomooka K, Sasaguri T. Anti‐tumor effects of differentiation‐inducing factor‐1 in malignant melanoma: GSK‐3‐mediated inhibition of cell proliferation and GSK‐3‐independent suppression of cell migration and invasion. Biochem Pharmacol. 2017;138:31‐48. [DOI] [PubMed] [Google Scholar]
- 15. Kubokura N, Takahashi‐Yanaga F, Arioka M, et al. Differentiation‐inducing factor‐3 inhibits intestinal tumor growth in vitro and in vivo. J Pharmacol Sci. 2015;127:446‐455. [DOI] [PubMed] [Google Scholar]
- 16. Takahashi‐Yanaga F, Taba Y, Miwa Y, et al. Dictyostelium differentiation‐inducing factor‐3 activates glycogen synthase kinase‐3 beta and degrades cyclin D1 in mammalian cells. J Biol Chem. 2003;278:9663‐9670. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu K, Murata T, Tagawa T, et al. Calmodulin‐dependent cyclic nucleotide phosphodiesterase (PDE1) is a pharmacological target of differentiation‐inducing factor‐1, an antitumor agent isolated from Dictyostelium. Cancer Res. 2004;64:2568‐2571. [DOI] [PubMed] [Google Scholar]
- 18. Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772‐775. [DOI] [PubMed] [Google Scholar]
- 19. Egashira I, Takahashi‐Yanaga F, Nishida R, et al. Celecoxib and 2,5‐dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β‐catenin signaling pathway. Cancer Sci. 2017;108:108‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sherr CJ. Cancer cell cycles. Science. 1996;274:1672‐1677. [DOI] [PubMed] [Google Scholar]
- 21. Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13:293‐325. [DOI] [PubMed] [Google Scholar]
- 22. Strano S, Munarriz E, Rossi M, et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem. 2000;275:29503‐29512. [DOI] [PubMed] [Google Scholar]
- 23. Beguelin W, Flaque M, Proietti CJ, et al. Progesterone receptor induces ErbB‐2 nuclear translocation to promote breast cancer growth via a novel transcriptional effect: ErbB‐2 function as a coactivator of Stat3. Mol Cell Biol. 2010;30:5456‐5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Julien S, Puig I, Caretti E, et al. Activation of NF‐kappa B by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445‐7456. [DOI] [PubMed] [Google Scholar]
- 25. Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer. 2016;138:2570‐2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong Z, Wen Z, Darnell JE. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin‐6. Science. 1994;264:95‐98. [DOI] [PubMed] [Google Scholar]
- 27. Masuda M, Suzui M, Yasumatu R, et al. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Can Res. 2002;62:3351‐3355. [PubMed] [Google Scholar]
- 28. Leslie K, Lang C, Devgan G, et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Can Res. 2006;66:2544‐2552. [DOI] [PubMed] [Google Scholar]
- 29. Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295‐303. [DOI] [PubMed] [Google Scholar]
- 30. Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure‐based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391‐7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He F, Ge W, Martinowich K, et al. A positive autoregulatory loop of Jak‐STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8:616‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holz MK. The role of S6K1 in ER‐positive breast cancer. Cell Cycle. 2012;11:3159‐3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254‐266. [DOI] [PubMed] [Google Scholar]
- 34. Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050‐3060. [DOI] [PubMed] [Google Scholar]
- 35. Zhang L, Li J, Wang Q, et al. The relationship between microRNAs and the STAT3‐related signaling pathway in cancer. Tumour Biol. 2017;39:1010428317719869. [DOI] [PubMed] [Google Scholar]
- 36. Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D. STAT inhibitors for cancer therapy. J Hematol Oncol. 2013;6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schroeder A, Herrmann A, Cherryholmes G, et al. Loss of androgen receptor expression promotes a stem‐like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014;74:1227‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007;251:199‐210. [DOI] [PubMed] [Google Scholar]
- 40. Wang L, Wang Q, Gao M, et al. STAT3 activation confers trastuzumab‐emtansine (T‐DM1) resistance in HER2‐positive breast cancer. Cancer Sci. 2018;109:3305‐3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Genini D, Brambilla L, Laurini E, et al. Mitochondrial dysfunction induced by a SH2 domain‐targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc Natl Acad Sci USA. 2017;114:E4924‐E4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras‐dependent oncogenic transformation. Science. 2009;324:1713‐1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wegrzyn J, Potla R, Chwae YJ, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tolcher A, Flaherty K, Shapiro GI, et al. A first‐in‐human phase I study of OPB‐111077, a small‐molecule STAT3 and oxidative phosphorylation inhibitor, in patients with advanced cancers. Oncologist. 2018;23:658‐e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS. Role of MMP‐2 in the regulation of IL‐6/Stat3 survival signaling via interaction with α5β1 integrin in glioma. Oncogene. 2013;32:327‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kanai M, Konda Y, Nakajima T, et al. Differentiation‐inducing factor‐1 (DIF‐1) inhibits STAT3 activity involved in gastric cancer cell proliferation via MEK‐ERK‐dependent pathway. Oncogene. 2003;22:548‐54. [DOI] [PubMed] [Google Scholar]
- 47. Yalcin A, Hotamisligil GS. Impact of ER protein homeostasis on metabolism. Diabetes. 2013;62:691‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou J, Wulfkuhle J, Zhang H, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem‐like cells is required for viability and maintenance. Proc Natl Acad Sci USA. 2007;104:16158‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bahrami‐B F, Ataie‐Kachoie P, Pourgholami MH, Morris DL. p70 Ribosomal protein S6 kinase (Rps6kb1): an update. J Clin Pathol. 2014;67:1019‐25. [DOI] [PubMed] [Google Scholar]
- 50. Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. 2001;61(Suppl 2):1‐13. [DOI] [PubMed] [Google Scholar]
- 51. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science. 1987;235:177‐82. [DOI] [PubMed] [Google Scholar]
- 52. Chung SS, Giehl N, Wu Y, Vadgama JV. STAT3 activation in HER2‐overexpressing breast cancer promotes epithelial‐mesenchymal transition and cancer stem cell traits. Int J Oncol. 2014;44:403‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hubalek M, Brunner C, Matthä K, Marth C. Resistance to HER2‐targeted therapy: mechanisms of trastuzumab resistance and possible strategies to overcome unresponsiveness to treatment. Wien Med Wochenschr. 2010;160:506‐12. [DOI] [PubMed] [Google Scholar]
- 54. Suzuki E, Toi M. Improving the efficacy of trastuzumab in breast cancer. Cancer Sci. 2007;98:767‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spigel DR, Burstein HJ. HER2 overexpressing metastatic breast cancer. Curr Treat Options Oncol. 2002;3:163‐74. [DOI] [PubMed] [Google Scholar]
- 56. Stemmler HJ, Schmitt M, Willems A, Bernhard H, Harbeck N, Heinemann V. Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2‐positive breast cancer patients with brain metastases and impairment of blood‐brain barrier. Anticancer Drugs. 2007;18:23‐8. [DOI] [PubMed] [Google Scholar]
- 57. Matsuda T, Takahashi‐Yanaga F, Yoshihara T, et al. Dictyostelium differentiation‐inducing factor‐1 binds to mitochondrial malate dehydrogenase and inhibits its activity. J Pharmacol Sci. 2010;112:320‐6. [DOI] [PubMed] [Google Scholar]
- 58. Kubohara Y, Kikuchi H, Matsuo Y, Oshima Y, Homma Y. Mitochondria are the target organelle of differentiation‐inducing factor‐3, an anti‐tumor agent isolated from Dictyostelium discoideum [corrected]. PLoS ONE. 2013;8:e72118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dubois A, Ginet C, Furstoss N, et al. Differentiation inducing factor 3 mediates its anti‐leukemic effect through ROS‐dependent DRP1‐mediated mitochondrial fission and induction of caspase‐independent cell death. Oncotarget. 2016;7:26120‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Oladimeji P, Kubohara Y, Kikuchi H, et al. A derivative of differentiation‐inducing factor‐3 inhibits PAK1 activity and breast cancer cell proliferation. Int J Cancer Clin Res. 2015;2:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takahashi‐Yanaga F, Taba Y, Miwa Y, et al. Dictyostelium differentiation‐inducing factor‐3 activates glycogen synthase kinase‐3beta and degrades cyclin D1 in mammalian cells. J Biol Chem. 2003;278:9663‐70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials