

Abstract
5‐Fluorouracil (5‐FU) is a chemotherapeutic agent commonly used to treat esophageal squamous cell carcinoma (ESCC), but acquisition of chemoresistance frequently occurs and the underlying mechanisms are not fully understood. We found that microRNA (miR)‐338‐5p was underexpressed in ESCC cells with acquired 5‐FU chemoresistance. Forced expression of miR‐338‐5p in these cells resulted in downregulation of Id‐1, and restoration of both in vitro and in vivo sensitivity to 5‐FU treatment. The effects were abolished by reexpression of Id‐1. In contrast, miR‐338‐5p knockdown induced 5‐FU resistance in chemosensitive esophageal cell lines, and knockdown of both miR‐338‐5p and Id‐1 resensitized the cells to 5‐FU. In addition, miR‐338‐5p had suppressive effects on migration and invasion of ESCC cells. Luciferase reporter assay confirmed a direct interaction between miR‐338‐5p and the 3′‐UTR of Id‐1. We also found that miR‐338‐5p was significantly downregulated in tumor tissue and serum samples of patients with ESCC. Notably, low serum miR‐338‐5p expression level was associated with poorer survival and poor response to 5‐FU/cisplatin‐based neoadjuvant chemoradiotherapy. In summary, we found that miR‐338‐5p can modulate 5‐FU chemoresistance and inhibit invasion‐related functions in ESCC by negatively regulating Id‐1, and that serum miR‐338‐5p could be a novel noninvasive prognostic and predictive biomarker in ESCC.
Keywords: chemoresistance, circulating miRNA, esophageal cancer, Id‐1, miR‐338‐5p
We found that microRNA (miR)‐338‐5p was underexpressed in esophageal squamous cell carcinoma cells with acquired 5‐fluorouracil (5‐FU) chemoresistance, and that reexpression of miR‐338‐5p could resensitize them to 5‐FU treatment through targeting Id‐1. MicroRNA‐338‐5p was significantly downregulated in tumor tissue and serum of patients with esophageal squamous cell carcinoma. Low serum miR‐338‐5p was predictive of poor response to 5‐FU/cisplatin‐based neoadjuvant chemoradiotherapy.

Abbreviations
- 5‐FU
5‐fluorouracil
- ESCA
esophageal cancer
- ESCC
esophageal squamous cell carcinoma
- FR
5‐FU resistance
- GEO
Gene Expression Omnibus
- Id‐1
inhibitor of DNA binding 1
- miRNA
microRNA
- Mut
mutant
- PARP
poly(ADP‐ribose) polymerase
- qRT‐PCR
quantitative real‐time PCR
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Esophageal squamous cell carcinoma constitutes more than 80% of esophageal cancer cases worldwide.1, 2 Preoperative chemoradiotherapy with 5‐FU and cisplatin has become a standard treatment for advanced resectable ESCC. However, resistance to therapy is a key factor responsible for the overall low survival rate.3 The underlying mechanisms of chemoresistance in ESCC are still not fully understood.
Recent studies highlight the involvement of miRNAs as tumor suppressors or oncogenes in tumor progression.4 MicroRNAs form a double‐stranded complex with target mRNAs by binding to the 3′‐UTR, and consequently reduce the target protein expression either by suppressing gene translation or by degrading the mRNA.5 MicroRNAs are increasingly recognized as useful diagnostic and prognostic biomarkers. Circulating miRNAs, in particular, are ideal biomarkers for continuous monitoring of cancer progression and cancer surveillance because they are present in a cell‐free form associated with lipids or lipoproteins in the blood, and are therefore protected from degradation.6, 7 Increasing evidence supports the involvement of miRNAs in chemoresistance. For example, gain of miR‐241‐3p expression enhanced sensitivity of ESCC cells to cisplatin,8 whereas loss of miR‐445‐3p had a similar effect.9 It was also reported that modulation of miR‐130a‐3p and miR‐148a‐3p in both directions increased the sensitivity of ESCC cells to 5‐FU and cisplatin.10 We found that ectopic overexpression of miR‐377, which targeted CD133 and suppressed cancer stemness, had a chemosensitizing effect on ESCC cells.11 More recently, we identified the miR‐29c‐3p/FBXO31‐p38 axis as a key regulator of chemoresistance in ESCC.12 Our previous studies also showed that Id‐1 and its downstream PI3K/Akt pathway play an essential role in promoting chemoresistance in ESCC.13, 14, 15 The significance of Id‐1 in ESCC is evident from its frequent aberrant overexpression in ESCC tumors, and the association between its expression level and poor prognosis.15, 16 As little is known about the mechanisms that regulate Id‐1 expression and the strategies by which Id‐1 can be inhibited, this study aimed to identify novel and clinically relevant miRNA(s) that can suppress Id‐1 and regulate chemoresistance in ESCC. Herein, we present data based on in vitro and in vivo functional experiments, as well as analyses of clinical samples, to show that miR‐338‐5p is downregulated in ESCC, and that it regulates chemoresistance and invasion‐related functions in ESCC cells by negatively regulating Id‐1. The potential of circulating miR‐338‐5p as a noninvasive biomarker was also explored.
2. MATERIALS AND METHODS
2.1. Cell lines and transfections
Esophageal squamous cell carcinoma cell lines KYSE410, KYSE150, and KYSE270,17 obtained from DSMZ, were maintained in RPMI‐1640 (Sigma) supplemented with 10% FBS (Invitrogen). The 293T cell line, and the ESCC cell line T.Tn18 obtained from Dr Hitoshi Kawamata (Dokkyo University School of Medicine, Tochigi, Japan), were maintained in DMEM/F12 (Invitrogen) supplemented with 10% FBS. All cell lines were authenticated using short tandem repeat analysis. The FR sublines KYSE410FR and KYSE150FR were generated from parental cell lines (KYSE410 and KYSE150 cells, respectively) by exposing them gradually to increasing concentration of 5‐FU (up to 80 μmol/L) over a period of 18 months.15 Transfection and transduction were carried out as described previously.15 Vectors for transient expression of miR‐338‐5p (ie, pcDNA6.2‐GW/EmGFP‐miR‐338‐5p) and scrambled miRNA (ie, pcDNA6.2‐GW/EmGFP‐Ctrl) were constructed using the Block‐iT Pol II miR RNAi expressing vector kit (Invitrogen). The pLenti‐CMV‐miR‐338‐5p and pLenti‐CMV‐miR‐Ctrl constructs for stable miRNA overexpression were established using the Gateway system (Invitrogen). The pLenti‐CMV‐puro‐DEST empty vector was obtained from Addgene (#17452). Vectors for stable miR‐338‐5p knockdown (pmiRZip‐338‐5p) and scrambled control (pmiRZip‐Ctrl) were purchased from System Biosciences. For transfection, Lipofectamine 2000 reagent (Invitrogen) was used according to the manufacturer’s instructions. Stable overexpressing and knockdown cell lines were established using lentivirus infection. The virus was packaged using 293T, which was transfected with lentiviral plasmids and packaging mix. The stable cell lines were selected with puromycin.
2.2. Clinical samples
Two sets of tissue samples, each consisting of paired ESCC tissue and nonneoplastic mucosa, were used for analysis of miR‐338‐5p expression. Set A included 30 cases of ESCC patients treated at Queen Mary Hospital, Hong Kong, from 2003 to 2007. Set B consisted of an additional 42 pairs of samples from patients who underwent surgical resection of primary ESCC at Queen Mary Hospital between 2011 and 2012, or at the First Affiliated Hospital, Zhengzhou University in Zhengzhou. In addition, pretreatment serum samples of an independent cohort of 104 patients with ESCC who underwent neoadjuvant chemoradiotherapy (with 5‐FU and cisplatin) followed by esophagectomy, and serum samples of 50 healthy individuals, collected from Queen Mary Hospital and Shantou University Medical College (Guangdong, China) were used for determination of circulating miR‐338‐5p. The pathological response to chemoradiotherapy was assessed based on the percentage of viable residual tumor cells in the resected esophagus, categorized into 4 groups according to the guidelines of the Japanese Society for Esophageal Disease.19 All human samples were obtained with informed consent and approval from the Institutional Review Boards of the respective institutions (Queen Mary Hospital, IRB# UW13‐300; Zhengzhou University, IRB# 2017‐09‐142; Shantou University Medical College, IRB# 2015042419).
2.3. Quantitative real‐time PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer’s protocol (Invitrogen). For serum and Set A tissue samples, cDNA was generated using the miScript II RT Kit (Qiagen) based on the oligo(dT) primer method. Quantitative PCR was undertaken using the miScript SYBR Green PCR Kit (Qiagen). The forward primers used were miR‐338‐5p‐Fwd (5′‐gccgaacaatatcctggtgctgagt‐3′), RNU6B‐Fwd (5′‐tgcgggtgctcgcttcggcagc‐3′), and miR‐16‐Fwd (5′‐tagcagcacgtaaatattggcg‐3′). The universal downstream primer was 5′‐ccagtgcagggtccgaggt‐3′. The amounts of miRNAs in ESCC cell lines and Set B tissue samples were quantified using TaqMan miRNA assays (stem‐loop primer method). Reverse transcription reaction was carried out with the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) and qRT‐PCR was carried out by amplifying the cDNA with TaqMan Universal PCR Master Mix II (2×), no UNG (Applied Biosystems), and miRNA‐specific PCR probes. Quantitative PCR was run on an iQ5 Real‐Time PCR Detection System (Bio‐Rad). For in vitro experiments, the mean Ct values of miR‐338‐5p were normalized to that of the internal reference gene or endogenous control to obtain ΔCt, and then to that of the corresponding control or parental cell line to obtain the 2−ΔΔCt value as fold change. The absolute expression value of miR‐338‐5p in clinical samples was expressed as 2−∆Ct.
2.4. Western blot analyses
Preparation of whole‐cell lysates and immunoblotting were undertaken as described previously.20 Primary Abs against Id‐1, β‐actin (Santa Cruz Biotechnology), caspase‐3 and cleaved caspase‐3 (Abcam), and PARP and cleaved PARP (Cell Signaling Technology) were used at 1:1000 dilution. Chemiluminescent signals were developed using the ECL Plus Western blotting detection system (Amersham Biosciences) and visualized by exposure to BioMax Light 64 Film (Kodak) or using myECL Imager (Thermo Fisher Scientific).
2.5. Dual luciferase reporter assays
The 3′‐UTRs of Id‐1 containing WT and Mut miR‐338‐5p binding sites were synthesized by Integrated DNA Technologies. The WT‐ or Mut‐3′‐UTR was then ligated into psiCHECK‐2 vector between XhoI and NotI to generate luciferase reporter vector (psiCHECK‐Id‐1‐3′‐UTR‐WT/Mut). The luciferase reporter assay was carried out using KYSE150 cells. Briefly, cells were seeded in 24‐well plates, and then cotransfected with pcDNA‐6.2‐miR‐338‐5p or pcDNA‐6.2‐miR‐Ctrl and psiCHECK‐Id‐1‐3’UTR‐WT or ‐Mut vector using Lipofectamine 2000 after 24 hours. The activities of firefly and Renilla luciferases were determined using Dual‐Luciferase Reporter Assay System (Promega). The luciferase signals were detected using Victor3 Multilabel Counter (Perkin Elmer), and the values were normalized to that of cells transfected with nontargeting control miRNA and calculated as the means of 3 independent experiments.
2.6. Cell viability assay
Parental ESCC cells and FR cells with manipulated miR‐338‐5p expression were treated with 20 and 40 μmol/L 5‐FU (Calbiochem), respectively, for 48 hours. Cell viability was measured using MTT assay as previously described.21 Relative proliferation was calculated by normalizing to the corresponding miR‐Ctrl or miRZip‐Ctrl cells.
2.7. Migration and invasion assays
Wound healing assay was used to monitor migration of ESCC cells.20 Invasion assay was carried out using Transwell Matrigel‐coated invasion chambers with 8‐μm pore size polycarbonate filters (BD Biosciences) as described previously.20
2.8. Apoptosis assay
Cells were incubated with 5‐FU (40 μmol/L for FR cells and 20 μmol/L for parental ESCC cells). Approximately 1 × 106 cells were harvested 48 hours later and stained with propidium iodide (50 μg/mL)/RNase solution (10 μg/mL RNase in PBS) at 37°C for 30 minutes for flow cytometry analysis (BD FACS Canto II Analyzer; BD Biosciences). The percentage of sub‐G1 population, indicative of cell death, was analyzed with FlowJo.22
2.9. Animal experiments
Approximately 1 × 106 modified ESCC cancer cells (KYSE150FR‐miR‐338‐5p, KYSE150FR‐miR‐338‐5p‐Id‐1, and KYSE150FR‐miR‐Ctrl) were suspended in 50 μL PBS and mixed with 50 μL Matrigel (BD Biosciences). The mixtures (100 μL/animal) were then s.c. injected into the flanks of 3 different groups of nude mice (12 mice per group) to establish tumor xenografts. When the tumors reached approximately 5 mm in diameter, each group of animals was randomly divided into 2 subgroups (n = 6/group) which were either given an i.p. injection of 5‐FU (20 mg/kg, every 3 days) or DMSO as control for 60 days. The tumor volume, calculated according to the equation Volume = (length × width2)/2, was determined at the end of the experiment. All the animal experiments were carried out in accordance with the relevant guidelines and regulations of the Committee on the Use of Live Animals in Teaching and Research of the University of Hong Kong.
2.10. Analysis of public datasets
The expression values of miR‐338‐5p in the ESCA data cohort were downloaded from the Genomic Data Commons Data Portal, NCI (https://portal.gdc.cancer.gov/). Kaplan‐Meier plots were used to compare overall survival using the University of California Santa Cruz’s Xena browser (https://xenabrowser.net). The expression values of miR‐338‐5p in colon cancer and rectal cancer (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115513) and gastric cancer (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE93415 and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63121) were downloaded from NCBI’s GEO.
2.11. Statistical analysis
The data were analyzed using PRISM 5.0 software (GraphPad Software). All the quantitative values were expressed as mean ± SEM. For the in vitro and in vivo experiments, the statistical significance between 2 groups was determined using the unpaired t test. All in vitro experiments were repeated at least 3 times. For quantitative miR‐338‐5p expression in our clinical samples and in public datasets, differences between 2 groups with nonparametric data (as determined using one‐sample Kolmogorov‐Smirnov test for normality) were compared using the Mann‐Whitney U test. The χ2 test was used to analyze the association between miR‐338‐5p expression levels in serum samples and clinicopathological parameters. Pearson’s correlation analysis was used to analyze the relationship between miR‐338‐5p and Id‐1 expression levels in tumor samples. The association between miR‐338‐5p expression level and patient survival was plotted using the Kaplan‐Meier method, and statistical differences were compared using the log‐rank test. Statistical significance level was set at P < .05.
3. RESULTS
3.1. MicroRNA‐338‐5p directly targets 3′‐UTR of Id‐1 and modulates Id‐1 expression in 5‐FU‐resistant ESCC cell lines
TargetScanHuman 6.2 and miRanda algorithms were used to look for novel miRNAs that might target the 3′‐UTR of Id‐1. The prediction results showed that there were 2 putative binding sites for miR‐338‐5p within the 3′‐UTR of Id‐1 (Figure 1A). The luciferase reporter vectors containing WT and Mut Id‐1 3′‐UTR were then constructed accordingly (Figure 1A), and dual‐luciferase reporter assay was carried out to confirm the direct binding of miR‐338‐5p to the Id‐1 3′‐UTR. Cotransfection of miR‐338‐5p and the WT psiCHECK‐Id‐1 3′‐UTR into KYSE150 cells led to a significant decrease (P = .004) of the luciferase signal when compared with scrambled miRNA control (Figure 1B). This inhibitory effect was rescued by mutating the 2 predicted miR‐338‐5p binding sites (Figure 1B). These data indicated that Id‐1 3′‐UTR is a direct target of miR‐338‐5p. We determined the miR‐338‐5p expression level in 42 pairs of esophageal tumor and nonneoplastic tissues (Set B samples), which were previously analyzed for Id‐1 protein expression.15 Pearson’s correlation analysis showed an inverse relationship (r = −0.214, P = .050) between miR‐338‐5p and Id‐1 (Figure 1C).
Figure 1.

MicroRNA (miR)‐338‐5p is a novel suppressor of Id‐1. A, Schematic representation of seed region matching between miR‐338‐5p and the WT and mutant (Mut) sequences of Id‐1 3′‐UTR. B, miR‐338‐5p repressed the expression of a luciferase reporter containing WT Id‐1 3′‐UTR but not the Mut sequence. C, Correlation between miR‐338‐5p and Id‐1 in esophageal squamous cell carcinomas and nonneoplastic esophageal mucosae as determined with linear regression. Ctrl, control
Our group previously reported that Id‐1 can induce chemoresistance in ESCC.14, 15 We therefore speculated that targeting Id‐1 using miR‐338‐5p could help ESCC cancer cells overcome 5‐FU resistance. We found that miR‐338‐5p was downregulated, whereas Id‐1 was upregulated, in KYSE410FR and KYSE150FR cells in comparison with their parental ESCC cell lines (Figure 2A,B). We then undertook miR‐338‐5p knockdown in parental ESCC cell lines using miRZip‐338‐5p (Figure 2C) and found elevation of Id‐1 protein expression levels in both cell lines (Figure 2D). In contrast, stable miR‐338‐5p overexpression decreased Id‐1 protein levels in KYSE410FR and KYSE150FR cells when compared with their corresponding control cell lines (Figure 2E,F), thus indicating that miR‐338‐5p can modulate Id‐1 function in 5‐FU‐resistant ESCC cells.
Figure 2.
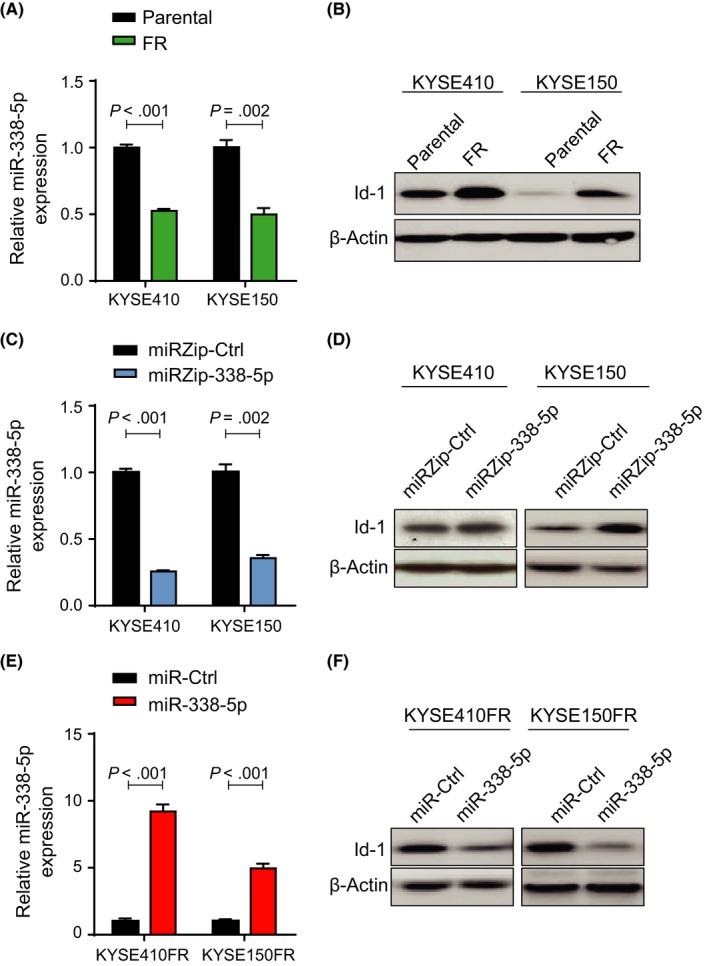
Effects of microRNA (miR)‐338‐5p manipulation on Id‐1 expression in parental and 5‐fluorouracil‐resistant (FR) esophageal cancer cell lines. A, Downregulation of miR‐338‐5p in KYSE410FR and KYSE150FR cells compared with respective parental cell lines, as determined using quantitative real‐time (qRT)‐PCR. B, Overexpression of Id‐1 was detected in KYSE410FR and KYSE150FR cells when compared with respective parental cell lines using western blot analysis. C, Knockdown of miR‐338‐5p by miRZip‐338‐5p in esophageal squamous cell carcinoma (ESCC) parental cell lines was confirmed by qRT‐PCR. Results are shown as fold change relative to the expression level in the control (miR‐Ctrl) transfected cells. D, Knockdown of miR‐338‐5p upregulated Id‐1 in parental ESCC cells. E, qRT‐PCR showing stable overexpression of miR‐338‐5p in KYSE410FR and KYSE150FR cells. F, Stable expression of miR‐338‐5p led to downregulation of Id‐1 in FR ESCC cells
3.2. MicroRNA‐338‐5p overexpression sensitizes 5‐FU‐resistant ESCC cells to 5‐FU treatment in vitro and in vivo
Gain‐of‐function experiments were carried out to investigate the role of miR‐338‐5p in 5‐FU resistance. The results of MTT cell viability assays showed that overexpression of miR‐338‐5p sensitized KYSE410FR and KYSE150FR cells to 5‐FU treatment, and that this effect was abolished by overexpression of Id‐1 (Figure 3A). Western blot analyses showed that miR‐338‐5p expression increased the sensitivity of KYSE410FR and KYSE150FR cells to 5‐FU‐induced apoptosis, as indicated by the increase in cleaved caspase‐3 and cleaved PARP; overexpression of Id‐1 reversed this effect (Figure 3B). Flow cytometry of the sub‐G1 apoptotic cell population confirmed increased apoptosis in the miR‐338‐5p‐overexpressing FR cells, which was abolished by ectopic overexpression of Id‐1 (Figure 3C). In contrast, data from MTT assays showed that knockdown of miR‐338‐5p increased the viability of the 5‐FU‐sensitive parental esophageal cell lines under 5‐FU treatment, and that this effect could be abolished by Id‐1 knockdown (Figure 3D). Western blot analysis of cleaved caspase‐3 and cleaved PARP, and flow cytometric analysis of the sub‐G1 apoptotic cell population, indicated that knockdown of miR‐338‐5p made the parental esophageal cells more resistant to 5‐FU‐induced apoptosis, whereas knockdown of Id‐1 could restore 5‐FU sensitivity (Figure 3E,F). Collectively, these data showed that miR‐338‐5p can modulate 5‐FU chemoresistance by downregulating Id‐1.
Figure 3.
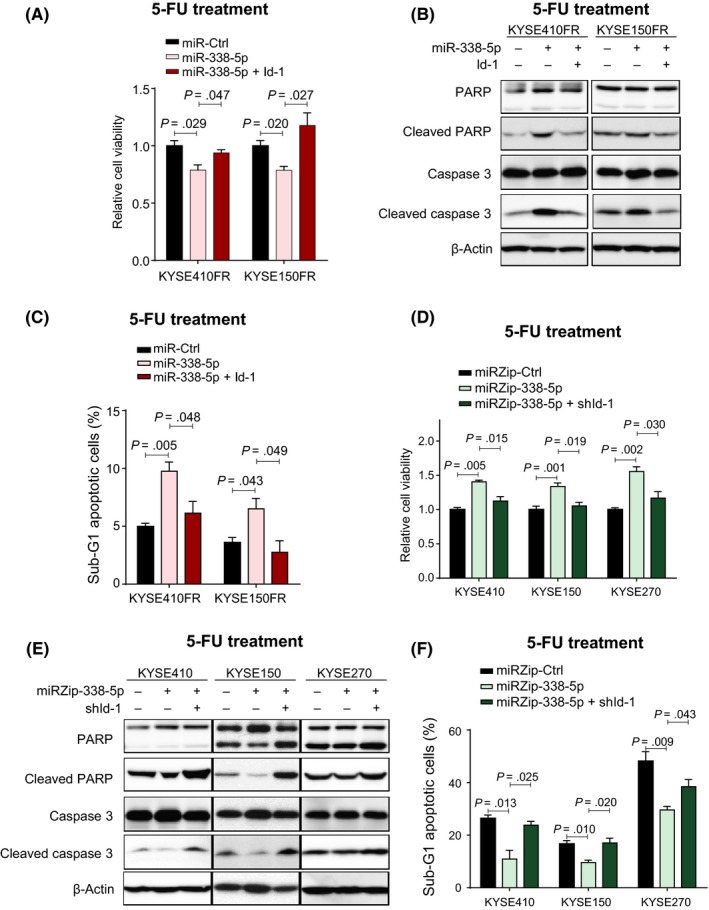
MicroRNA (miR)‐338‐5p increased sensitivity of 5‐fluorouracil (5‐FU)‐resistant (FR) cells to 5‐FU in vitro by inhibiting Id‐1. A, Overexpression of miR‐338‐5p sensitized KYSE150FR and KYSE410FR to 5‐FU treatment, and Id‐1 overexpression rescued the cells. B, C, Western blot analysis of apoptotic markers (B) and flow cytometry analysis of sub‐G1 cell populations (C) indicated that overexpression of miR‐338‐5p induced FR cell lines to undergo apoptosis following 5‐FU treatment, and that this effect was abolished by overexpression of Id‐1. D, MTT assay showed that inhibition of miR‐338‐5p by miRZip‐338‐5p increased the resistance of chemosensitive parental esophageal squamous cell carcinoma cells to 5‐FU, and concurrent suppression of Id‐1 abolished this effect. E, F, Western blot analysis of apoptotic markers (E) and flow cytometry analysis of sub‐G1 cell populations (F) showed that inhibition of miR‐338‐5p could prevent 5‐FU‐induced apoptosis in 5‐FU‐sensitive cell lines, and that this effect was abolished by knockdown of Id‐1
Tumor xenograft experiments were undertaken to determine whether miR‐338‐5p overexpression could render ESCC tumors more sensitive to 5‐FU treatment in vivo (Figure 4A). Comparison of tumor size showed that miR‐338‐5p overexpression in KYSE150FR cells significantly increased the sensitivity of the tumors to 5‐FU therapy, and that concomitant overexpression of Id‐1 significantly restored the 5‐FU resistance (Figure 4B,C).
Figure 4.
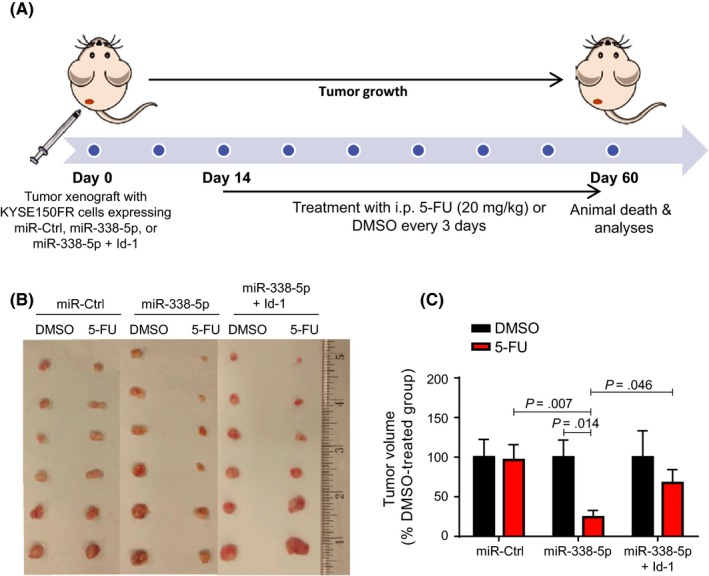
MicroRNA (miR)‐338‐5p increased sensitivity of 5‐fluorouracil (5‐FU)‐resistant (FR) tumors to 5‐FU treatment by inhibiting Id‐1. A, Experimental scheme of 5‐FU treatment in tumor xenograft model established with KYSE150FR cells. B, Photograph of tumors harvested from the 6 experimental groups (n = 6). C, Measurement of tumor volume of 5‐FU‐treated nude mice (normalized to their corresponding DMSO control [Ctrl] groups) showed that overexpression of miR‐338‐5p sensitized the FR tumors to 5‐FU treatment and resulted in smaller tumor volume; such effect was partially abolished by overexpression of Id‐1
3.3. MicroRNA‐338‐5p inhibits ESCC cell invasion and migration by downregulating Id‐1
Several studies reported that overexpression of Id‐1 is associated with tumor invasion and metastasis in different cancer types, including ESCC.20, 23, 24 As our data showed that miR‐338‐5p could downregulate Id‐1, we further speculated that miR‐338‐5p might play a role in regulating invasion and migration of ESCC cells. The results of the Transwell cell invasion assay indicated that forced expression of miR‐338‐5p significantly inhibited the invasive activity of ESCC cells in vitro (Figure 5A). MicroRNA‐338‐5p also reduced the migration activity of ESCC cells, as shown by the wound healing assay (Figure 5B). Reoverexpression of Id‐1 attenuated these effects (Figure 5A,B). Conversely, silencing miR‐338‐5p increased the abilities of KYSE270 and T.Tn cells to migrate and invade, and the effects were ameliorated by Id‐1 knockdown (Figure 5C,D). These findings suggest that miR‐338‐5p has metastasis‐inhibiting functions that are mediated by downregulating Id‐1.
Figure 5.

MicroRNA (miR)‐338‐5p inhibited invasion and migration of esophageal squamous cell carcinoma (ESCC) cells in vitro by inhibiting Id‐1. A, Representative images and quantification of Transwell cell migration assay showing inhibition of invasion of ESCC (KYSE270 and T.Tn) cells by miR‐338‐5p, and the attenuation of such effects by reexpression of Id‐1. B, Wound healing assay showing the effects of miR‐338‐5p overexpression, with and without ectopic expression of Id‐1, on migration of ESCC cells. C, Transwell cell invasion assay of KYSE270 and T.Tn cells transfected with miRZip‐338‐5p, miRZip‐338‐5p in combination with shId‐1, or scrambled control (Ctrl). D, Wound healing assay comparing the migration potential of KYSE270 and T.Tn cells following miR‐338‐5p silencing with and without Id‐1 knockdown
3.4. MicroRNA‐338‐5p is downregulated in ESCC and other gastrointestinal cancers
The expression level of miR‐338‐5p in ESCC tumors and adjacent nonneoplastic esophageal mucosae were examined using real‐time PCR. Among the 30 ESCC samples in Set A, 19 (approximately 63%) had lower expression of miR‐338‐5p relative to their corresponding nonneoplastic tissues (Figure 6A, upper panel). Similarly, analysis of Set B samples showed that 29 of 42 cases (approximately 69%) had lower expression of miR‐338‐5p in tumors than in their corresponding nontumor tissues (Figure 6A, lower panel). In both sets of samples, there was a significant downregulation of miR‐338‐5p expression level in ESCC tumors compared with the nonneoplastic esophageal tissues (Figure 6B). We also analyzed miR‐338‐5p expression in other cancers of the gastrointestinal tract using public sources including TCGA database and GEO datasets. In TCGA data, the expression level of miR‐338‐5p in the ESCA data cohort was significantly lower than that in nontumor tissues (Figure S1A). Furthermore, comparison of data from paired ESCA tumor and nontumor tissues showed downregulation of miR‐338‐5p in the majority (10 of 13) of ESCA samples (Figure S1B). Taken together, these results suggest an association between miR‐338‐5p dysregulation and esophageal cancer. In the GEO datasets, lower expression of miR‐338‐5p was also found in other gastrointestinal cancers (Figure S1C‐F).
Figure 6.
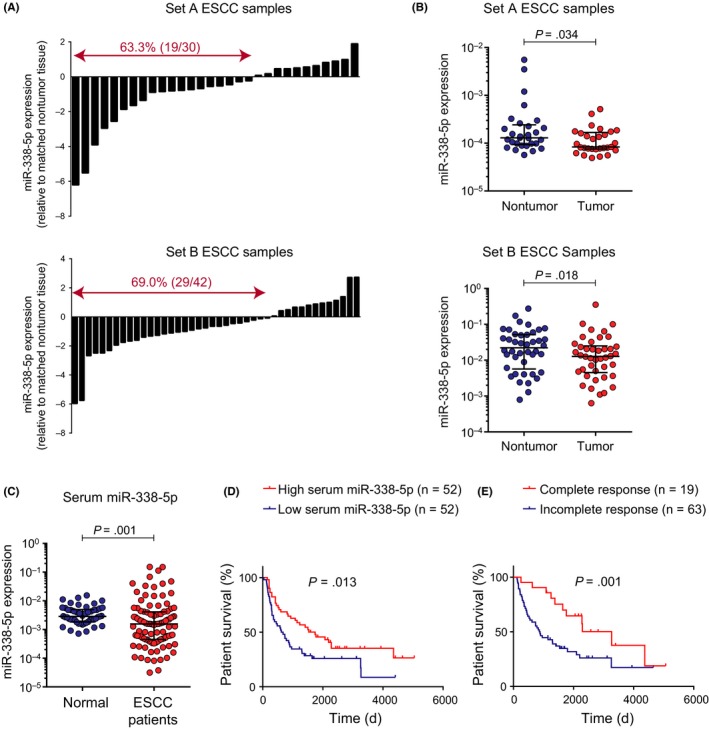
Aberrant expression of microRNA (miR)‐338‐5p in esophageal squamous cell carcinoma (ESCC) tumor and serum samples. A, Results from real‐time PCR showed that the majority of ESCC tumor samples had lower expression of miR‐338‐5p in comparison with corresponding nonneoplastic esophageal mucosae. B, Expression levels of miR‐338‐5p were significantly lower in the ESCC tumors than in nonneoplastic esophageal mucosae. C, Expression of miR‐338‐5p was downregulated in admission/pretreatment sera of patients with ESCC (n = 104) in comparison with serum samples of healthy controls (n = 50). D, Kaplan‐Meier overall survival curves of 104 patients with ESCC who were dichotomized according to serum miR‐338‐5p expression level. E, Survival outcomes of patients stratified by pathologic response (complete vs incomplete) to neoadjuvant chemoradiotherapy
3.5. High serum miR‐338‐5p predicts better survival and response to neoadjuvant chemoradiotherapy in patients with ESCC
Analysis of TCGA‐ESCA data showed that ESCA patients with high miR‐338‐5p expression in their tumors had longer overall survival time (Figure S1G). To determine whether circulating miR‐338‐5p could serve as a noninvasive serum biomarker, we analyzed the expression level of miR‐338‐5p in admission/pretreatment serum samples of 104 patients with ESCC and 50 healthy individuals. The validity of 2 commonly used endogenous controls, namely RNU6B and miR‐16,25 was first examined in order to minimize experimental variation and ensure accurate detection of biological differences among samples. The results showed that there was no significant difference in the serum miR‐16 expression level between patients with ESCC and normal subjects (Figure S2A). However, the serum RNU6B level was significantly lower in patients with ESCC (Figure S2B). Therefore, miR‐16 was selected as the endogenous control in the analysis of serum miR‐338‐5p expression. We found that the expression level of serum miR‐338‐5p in patients with ESCC was significantly lower than that in healthy controls (Figure 6C). The cohort of 104 patients with ESCC was then divided into 2 dichotomous groups of high and low serum miR‐338‐5p expressions using the median as the cut‐off value. The patients with high serum miR‐338‐5p expression had significantly (P = .013) better survival rates (median survival = 58.00 months) than those with low miR‐338‐5p expression (median survival = 21.57 months) (Figure 6D). The advantage of 5‐FU and cisplatin‐based neoadjuvant therapy in improving the overall survival of ESCC patients is evident from Figure 6E, which shows that patients with pathologic complete response had significantly (P = .001) longer survival (median survival = 108.93 months) than those who did not achieve a complete response (median survival = 30.06 months). Correlation with clinicopathologic parameters showed that expression levels of miR‐338‐5p in pretreatment serum were inversely correlated with posttherapy pathologic ypT‐stage (P = .034), ypM‐stage (P = .014), overall pathologic stage (ypTNM; P = .017), and histologic grade (P = .026) (Table 1). Furthermore, high serum miR‐338‐5p expression was associated with lower percentage of residual viable cells in the primary tumor collected during surgery, and with complete response to therapy (Table 1), which indicated that higher pretreatment serum miR‐338‐5p level could predict a better response to chemoradiotherapy.
Table 1.
Correlation between serum microRNA (miR)‐338‐5p expression levels and clinicopathologic parameters in 104 patients with esophageal squamous cell carcinomaa
| Parameters | No. cases | Serum miR‐338‐5p expression | P value | ||
|---|---|---|---|---|---|
| High | Low | ||||
| Age (years) | |||||
| ≤55 | 21 | 9 | 12 | .464 | |
| >55 | 83 | 43 | 40 | ||
| Gender | |||||
| Male | 93 | 45 | 48 | .339 | |
| Female | 11 | 7 | 4 | ||
| Pathologic stage | |||||
| ypT‐stage | |||||
| 0 | 30 | 21 | 9 | .034 | |
| 1/2 | 29 | 14 | 15 | ||
| 3/4 | 23 | 8 | 15 | ||
| ypN‐stage | |||||
| 0 | 47 | 29 | 18 | .090 | |
| 1/2/3 | 35 | 15 | 20 | ||
| ypM stage | |||||
| 0 | 76 | 43 | 33 | .014 | |
| 1 | 5 | 0 | 5 | ||
| ypTNM | |||||
| pCRb | 19 | 15 | 4 | .017 | |
| Othersc | 11 | 6 | 5 | ||
| I/II | 30 | 16 | 14 | ||
| III/IV | 21 | 6 | 15 | ||
| Grade | |||||
| G1 | 38 | 25 | 13 | .026 | |
| G2 | 25 | 12 | 13 | ||
| G3 | 18 | 5 | 13 | ||
| Residual tumor | |||||
| yPV0 (0%) | 38 | 25 | 13 | .007 | |
| yPV1 (1%‐33%) | 42 | 21 | 21 | ||
| yPV2 (34%‐66%) | 8 | 2 | 6 | ||
| yPV3 (67%‐100%) | 16 | 3 | 13 | ||
| Response to chemoradiotherapy | |||||
| Complete | 19 | 15 | 4 | .008 | |
| Incomplete | 63 | 28 | 35 | ||
| Recurrence | |||||
| Yes | 53 | 22 | 31 | .077 | |
| No | 51 | 30 | 21 | ||
Pathologic staging not available for some of the cases.
pCR, pathologic complete response (ie, ypT0N0M0).
ypT0 with lymph node and/or distant metastasis.
4. DISCUSSION
Development of drug resistance is a major obstacle to the effective treatment of human cancer. Despite 5‐FU being an antimetabolic agent widely used in treating gastrointestinal cancers, the response rate of esophageal carcinoma to 5‐FU is only 15%.26, 27, 28 Emerging evidence indicates that miRNAs are associated with chemoresistance, but relatively few miRNAs were experimentally validated to have the ability to regulate the sensitivity of ESCC cells to 5‐FU treatment.10, 11, 12, 29, 30, 31 Our present study found that miR‐338‐5p was underexpressed in 5‐FU‐resistant ESCC cells, and that ectopic overexpression of miR‐338‐5p in these cells could increase their sensitivity to 5‐FU treatment both in vitro and in vivo. We further established that miR‐338‐5p inhibits 5‐FU chemoresistance as well as decreases the invasive and migratory potential of ESCC cells through targeting Id‐1. Upregulation of Id‐1 is a frequent event in chemoresistant and metastatic tumors.32, 33, 34 The role of Id‐1 in cisplatin chemoresistance in esophageal cancer has not been elucidated, but previous studies showed that targeting Id‐1 can sensitize human colon cancer‐initiating cells to oxaliplatin and suppress the metastatic ability of cancer cells in vivo.34, 35 Thus, the miR‐338‐5p/Id‐1 regulatory axis could be worth exploring in developing new treatment strategies for Id‐1 overexpressing or 5‐FU/cisplatin‐resistant ESCC.
Although miR‐338‐5p had been reported to be downregulated in non‐small‐cell lung carcinoma36 and glioblastoma,37, 38 we have found that its expression was reduced in the majority of ESCC. Analysis of GEO datasets showed the same phenomenon in gastric, colon, and rectal cancers, thus suggesting that miR‐338‐5p has tumor suppressor functions in the gastrointestinal tract. In addition to Id‐1, 2 other genes, namely survivin and fermitin family homolog 2 (FERMT2), had been reported to be direct targets of miR‐338‐5p in ESCC.39, 40 Intratumoral injection of miR‐338‐5p mimic could downregulate survivin expression and increase the sensitivity of ESCC tumor xenografts to radiation therapy.40 Esophageal squamous cell carcinoma cells transfected with miR‐338‐5p mimic showed reduced cisplatin resistance and cell proliferation and migration.39 These findings together suggest that miR‐338‐5p replacement therapy could be a potentially useful approach in esophageal cancer therapy because miR‐338‐5p can concurrently target multiple effectors that are implicated in chemoradiotherapy resistance.
Circulating miRNAs are potentially useful minimally invasive biomarkers for prognosis. Our results showed that low miR‐338‐5p levels in serum on admission was significantly associated with poorer survival and advanced posttherapy pathologic staging. Notably, the majority of patients who achieved pathologic complete response to neoadjuvant chemoradiotherapy had high pretreatment serum levels of miR‐338‐5p. Taken together, serum miR‐338‐5p could be a useful predictive marker for response to 5‐FU/cisplatin‐based neoadjuvant chemoradiation. Further confirmation in a larger scale study is warranted.
In summary, by combining clinicopathologic and experimental approaches to study the role of miR‐338‐5p in ESCC, we have provided evidence of its functions and mechanism in reversing 5‐FU chemoresistance as well as suppressing cancer cell invasion and migration.
DISCLOSURE
The authors have no conflicting of interest.
Supporting information
ACKNOWLEDGMENTS
The study was supported by the Research Grants Council of the Hong Kong SAR, China (GRF Projects Nos. 17103814 and 17111917) and the University of Hong Kong CRCG Seed Funding Program for Basic Research (Project No. 201511159123). We acknowledge the University of Hong Kong Li Ka Shing Faculty of Medicine Core Facility for assistance in flow cytometry. We thank Dr Stella Chim, School of Biomedical Sciences, University of Hong Kong, for technical advice. We also thank Professor Liyan Xu, Professor Enmin Li, Hua Zhen Tan, Shan Shan Li, and Yi Wei Xu (Shantou University Medical College, China) for their kind assistance in providing human serum samples. We thank Professor Joan Massague for providing pBabe‐puro‐Id‐1 plasmids, and Professor Kwanghee Baek (Kyung Hee University, Korea) for psiCHECK‐2 vectors. We also thank Paul D. Kaufman and Eric Campeau for the plasmids obtained from Addgene.
Han L, Cui D, Li B, et al. MicroRNA‐338‐5p reverses chemoresistance and inhibits invasion of esophageal squamous cell carcinoma cells by targeting Id‐1. Cancer Sci. 2019;110:3677–3688. 10.1111/cas.14220
REFERENCES
- 1. Lin DC, Wang MR, Koeffler HP. Genomic and epigenomic aberrations in esophageal squamous cell carcinoma and implications for patients. Gastroenterology. 2017;154:374‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241‐2252. [DOI] [PubMed] [Google Scholar]
- 3. Ancona E, Ruol A, Santi S, et al. Only pathologic complete response to neoadjuvant chemotherapy improves significantly the long term survival of patients with resectable esophageal squamous cell carcinoma: final report of a randomized, controlled trial of preoperative chemotherapy versus surgery alone. Cancer. 2001;91:2165‐2174. [PubMed] [Google Scholar]
- 4. Esquela‐Kerscher A, Slack FJ. Oncomirs ‐ microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259‐269. [DOI] [PubMed] [Google Scholar]
- 5. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer‐genetics in the blood. Nat Rev Clin Oncol. 2013;10:472‐484. [DOI] [PubMed] [Google Scholar]
- 7. Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood‐based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513‐10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phatak P, Byrnes KA, Mansour D, et al. Overexpression of miR‐214‐3p in esophageal squamous cancer cells enhances sensitivity to cisplatin by targeting survivin directly and indirectly through CUG‐BP1. Oncogene. 2016;35:2087‐2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu A, Zhu J, Wu G, et al. Antagonizing miR‐455‐3p inhibits chemoresistance and aggressiveness in esophageal squamous cell carcinoma. Mol Cancer. 2017;16:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eichelmann AK, Matuszcak C, Lindner K, et al. Complex role of miR‐130a‐3p and miR‐148a‐3p balance on drug resistance and tumor biology in esophageal squamous cell carcinoma. Sci Rep. 2018;8:17553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li B, Xu WW, Han L, et al. MicroRNA‐377 suppresses initiation and progression of esophageal cancer by inhibiting CD133 and VEGF. Oncogene. 2017;36:3986‐4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li B, Hong P, Zheng C‐C, et al. Identification of miR‐29c and its target FBXO31 as a key regulatory mechanism in esophageal cancer chemoresistance: functional validation and clinical significance. Theranostics. 2019;9:1599‐1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li B, Li J, Xu WW, et al. Suppression of esophageal tumor growth and chemoresistance by directly targeting the PI3K/AKT pathway. Oncotarget. 2014;5:11576‐11587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li B, Tsao SW, Chan KW, et al. Id1‐induced IGF‐II and its autocrine/endocrine promotion of esophageal cancer progression and chemoresistance–implications for IGF‐II and IGF‐IR‐targeted therapy. Clin. Cancer Res. 2014;20:2651‐2662. [DOI] [PubMed] [Google Scholar]
- 15. Li B, Xu WW, Guan XY, et al. Competitive binding between Id1 and E2F1 to Cdc20 regulates E2F1 degradation and thymidylate synthase expression to promote esophageal cancer chemoresistance. Clin. Cancer Res. 2016;22:1243‐1255. [DOI] [PubMed] [Google Scholar]
- 16. Yuen HF, Chan Y‐P, Chan K‐K, et al. Id‐1 and Id‐2 are markers for metastasis and prognosis in oesophageal squamous cell carcinoma. Br J Cancer. 2007;97:1409‐1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69:277‐284. [DOI] [PubMed] [Google Scholar]
- 18. Kawamata H, Furihata T, Omotehara F, ,et al. Identification of genes differentially expressed in a newly isolated human metastasizing esophageal cancer cell line, T.Tn‐AT1, by cDNA microarray. Cancer Sci. 2003;94(8):699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tong DK, Law S, Kwong DLW, et al. Histological regression of squamous esophageal carcinoma assessed by percentage of residual viable cells after neoadjuvant chemoradiation is an important prognostic factor. Ann Surg Oncol. 2010;17:2184‐2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li B, Tsao SW, Li YY, et al. Id‐1 promotes tumorigenicity and metastasis of human esophageal cancer cells through activation of PI3K/AKT signaling pathway. Int J Cancer. 2009;125:2576‐2585. [DOI] [PubMed] [Google Scholar]
- 21. Li B, Li YY, Tsao SW, Cheung AL. Targeting NFkB signaling pathway suppresses tumor growth, angiogenesis, and metastasis of human esophageal cancer. Mol Cancer Ther. 2009;8:2635‐2644. [DOI] [PubMed] [Google Scholar]
- 22. Liu J, Han L, Li B, et al. F‐box only protein 31 (FBXO31) negatively regulates p38 mitogen‐activated protein kinase (MAPK) signaling by mediating lysine 48‐linked ubiquitination and degradation of mitogen‐activated protein kinase kinase 6 (MKK6). J Biol Chem. 2014;289:21508‐21518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo KJ, Wen J, Xie X, et al. Prognostic relevance of Id‐1 expression in patients with resectable esophageal squamous cell carcinoma. Ann Thorac Surg. 2012;93:1682‐1688. [DOI] [PubMed] [Google Scholar]
- 25. Kok MG, Halliani A, Moerland PD, et al. Normalization panels for the reliable quantification of circulating microRNAs by RT‐qPCR. Faseb J. 2015;29:3853‐3862. [DOI] [PubMed] [Google Scholar]
- 26. Enzinger PC, Ilson DH, Kelsen DP. Chemotherapy in esophageal cancer. Semin Oncol. 1999;26:12‐20. [PubMed] [Google Scholar]
- 27. Sumpter K, Harper‐Wynne C, Cunningham D, et al. Report of two protocol planned interim analyses in a randomised multicentre phase III study comparing capecitabine with fluorouracil and oxaliplatin with cisplatin in patients with advanced oesophagogastric cancer receiving ECF. Br J Cancer. 2005;92:1976‐1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ku GY. Systemic therapy for esophageal cancer: chemotherapy. Chin Clin Oncol. 2017;6:49. [DOI] [PubMed] [Google Scholar]
- 29. Nyhan MJ, O’Donovan TR, Boersma AWM, et al. MiR‐193b promotes autophagy and non‐apoptotic cell death in oesophageal cancer cells. BMC Cancer. 2016;16:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin YY, Chen Q‐J, Xu K, et al. Involvement of microRNA‐141‐3p in 5‐fluorouracil and oxaliplatin chemo‐resistance in esophageal cancer cells via regulation of PTEN. Mol Cell Biochem. 2016;422:161‐170. [DOI] [PubMed] [Google Scholar]
- 31. Lindner K, Eichelmann A‐K, Matuszcak C, et al. Complex epigenetic regulation of chemotherapy resistance and biology in esophageal squamous cell carcinoma via microRNAs. Int J Mol Sci. 2018;19:499 10.3390/ijms19020499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang X, Ling MT, Wong YC, Wang X. Evidence of a novel antiapoptotic factor: Role of inhibitor of differentiation or DNA binding (Id‐1) in anticancer drug induced apoptosis. Cancer Sci. 2007;98:308‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun W, Guo M‐M, Han P, et al. Id‐1 and the p65 subunit of NFkB promote migration of nasopharyngeal carcinoma cells and are correlated with poor prognosis. Carcinogenesis. 2012;33:810‐817. [DOI] [PubMed] [Google Scholar]
- 34. Gupta GP, Perk J, Acharyya S, et al. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci USA. 2007;104:19506‐19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fong S, Itahana Y, Sumida T, et al. Id‐1 as a molecular target in therapy for breast cancer cell invasion and metastasis. Proc Natl Acad Sci USA. 2003;100:13543‐13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu N, Yong S, Kim HK, et al. Identification of tumor suppressor microRNAs by integrative microRNA and mRNA sequencing of matched tumor‐normal pairs in lung adenocarcinoma. Mol Oncol. 2019;13:1356-1368. 10.1002/1878-0261.12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Besse A, Sana J, Lakomy R, et al. MiR‐338‐5p sensitizes glioblastoma cells to radiation through regulation of genes involved in DNA damage response. Tumour Biol. 2016;37:7719‐7727. [DOI] [PubMed] [Google Scholar]
- 38. Lei D, Zhang F, Yao D, Xiong N, Jiang X, Zhao H. MiR‐338‐5p suppresses proliferation, migration, invasion, and promote apoptosis of glioblastoma cells by directly targeting EFEMP1. Biomed Pharmacother. 2017;89:957‐965. [DOI] [PubMed] [Google Scholar]
- 39. Lin WC, Chen L‐H, Hsieh Y‐C, et al. miR‐338‐5p inhibits cell proliferation, colony formation, migration, and cisplatin resistance in esophageal squamous cancer cells by targeting FERMT2. Carcinogenesis. 2018;40(7):883–892. [DOI] [PubMed] [Google Scholar]
- 40. Park M, Yoon H‐J, Kang MC, Kwon J, Lee HW. MiR‐338‐5p enhances the radiosensitivity of esophageal squamous cell carcinoma by inducing apoptosis through targeting survivin. Sci Rep. 2017;7:10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials