

The extensive use of β-lactam antibiotics and the bacterial adaptive capacity have led to the apparently unstoppable increase of antimicrobial resistance, one of the current major global health challenges. In the leading nosocomial pathogen Pseudomonas aeruginosa, the mutation-driven AmpC β-lactamase hyperproduction stands out as the main resistance mechanism, but the molecular cues enabling this system have remained elusive until now. Here, we provide for the first time direct and quantitative information about the soluble cell wall-derived fragments accounting for the different levels and pathways of AmpC hyperproduction. Based on these results, we propose a hierarchical model of signals which ultimately govern ampC hyperexpression and resistance.
KEYWORDS: AmpC β-lactamase, Pseudomonas aeruginosa, muropeptide, peptidoglycan
ABSTRACT
The hyperproduction of the chromosomal AmpC β-lactamase is the main mechanism driving β-lactam resistance in Pseudomonas aeruginosa, one of the leading opportunistic pathogens causing nosocomial acute and chronic infections in patients with underlying respiratory diseases. In the current scenario of the shortage of effective antipseudomonal drugs, understanding the molecular mechanisms mediating AmpC hyperproduction in order to develop new therapeutics against this fearsome pathogen is of great importance. It has been accepted for decades that certain cell wall-derived soluble fragments (muropeptides) modulate AmpC production by complexing with the transcriptional regulator AmpR and acquiring different conformations that activate/repress ampC expression. However, these peptidoglycan-derived signals have never been characterized in the highly prevalent P. aeruginosa stable AmpC hyperproducer mutants. Here, we demonstrate that the previously described fragments enabling the transient ampC hyperexpression during cefoxitin induction (1,6-anhydro-N-acetylmuramyl-pentapeptides) also underlie the dacB (penicillin binding protein 4 [PBP4]) mutation-driven stable hyperproduction but differ from the 1,6-anhydro-N-acetylmuramyl-tripeptides notably overaccumulated in the ampD knockout mutant. In addition, a simultaneous greater accumulation of both activators appears linked to higher levels of AmpC hyperproduction, although our results suggest a much stronger AmpC-activating potency for the 1,6-anhydro-N-acetylmuramyl-pentapeptide. Collectively, our results propose a model of AmpC control where the activator fragments, with qualitative and quantitative particularities depending on the pathways and levels of β-lactamase production, dominate over the repressor (UDP-N-acetylmuramyl-pentapeptide). This study represents a major step in understanding the foundations of AmpC-dependent β-lactam resistance in P. aeruginosa, potentially useful to open new therapeutic conceptions intended to interfere with the abovementioned cell wall-derived signaling.
IMPORTANCE The extensive use of β-lactam antibiotics and the bacterial adaptive capacity have led to the apparently unstoppable increase of antimicrobial resistance, one of the current major global health challenges. In the leading nosocomial pathogen Pseudomonas aeruginosa, the mutation-driven AmpC β-lactamase hyperproduction stands out as the main resistance mechanism, but the molecular cues enabling this system have remained elusive until now. Here, we provide for the first time direct and quantitative information about the soluble cell wall-derived fragments accounting for the different levels and pathways of AmpC hyperproduction. Based on these results, we propose a hierarchical model of signals which ultimately govern ampC hyperexpression and resistance.
INTRODUCTION
Pseudomonas aeruginosa stands out among the human opportunistic pathogens, since it is the primary cause of ventilator-associated pneumonia and burn wound infections (1, 2) along with chronic respiratory infections in patients with chronic underlying diseases (3–5). Among its well-known plethora of resources for antibiotic resistance, the hyperproduction of the intrinsic inducible cephalosporinase AmpC is the main mechanism used by this pathogen to cope with β-lactams (6–8).
The regulation of AmpC β-lactamases was initially studied in other Gram-negative bacteria harboring this type of intrinsic enzyme, such as Citrobacter freundii and Enterobacter cloacae. It was proposed that the expression of these β-lactamases was controlled by the LysR-type AmpR transcriptional regulator complexed with certain cell wall (peptidoglycan)-derived fragments, generically known as muropeptides (9–12). As in other species, P. aeruginosa ampR and ampC genes form a divergent operon with overlapping promoters and with the fragment in between acting as an AmpR binding site (13). It was proposed that upon muropeptide binding, AmpR would change its conformation to modulate RNA polymerase activity and thus ampC transcription (11, 14–16). Further, this mechanism was shown to be intimately linked to the peptidoglycan recycling biology. During growth, the bacterial cell wall needs to expand, and peptidoglycan is cleaved on each generation by hydrolytic enzymes to allow new material insertion and cell division. Although part of the cleaved peptidoglycan fragments are released to the extracellular medium, most of them are transported into the cytoplasm through the AmpG permease for recycling (17–21). Once in the cytosol, the muropeptides (mainly N-acetylglucosamine-1,6-anhydro-N-acetylmuramyl-peptides [NAG-anhNAM-peptides]) are further processed by NagZ, which removes the N-acetylglucosamine residue (NAG) to produce 1,6-anhydro-N-acetylmuramyl-peptides (anhNAM-peptides), and by the amidase AmpD, which cleaves the bond between the anhNAM and the stem peptides (22, 23). The concept of recycling precisely comes from the fact that the resulting monosaccharides (NAG and anhNAM) and peptides are reused to assemble an essential unit for peptidoglycan synthesis: the uridine 5′-pyrophosphoryl-N-acetylmuramic acid-pentapeptide (UDP-NAM-P5) (13, 19–21, 24, 25). Under normal conditions, the UDP-NAM-P5 units bind to AmpR, and this complex is believed to repress ampC expression to basal levels. This is the reason why UDP-NAM-P5 has been known as an AmpC repressor (13, 16, 17). Conversely, when bacteria are exposed to the so-called inducer β-lactams (e.g., cefoxitin), the derived inhibition of certain penicillin binding proteins (PBPs), such as PBP4 (encoded by dacB), alters the peptidoglycan turnover balance in favor of autolysis (16, 17, 26–31). It is believed that this situation boosts the amount of muropeptides reaching the cytosol, which saturate NagZ and AmpD activities, providing an increased quantity of NAG-anhNAM-peptides and/or anhNAM-peptides that displace the repressor UDP-NAM-P5 from AmpR (32, 33). Then, the new AmpR-muropeptide complex promotes ampC hyperexpression entailing resistance to hydrolyzable β-lactams such as cefoxitin (7, 16, 17, 25, 31, 34). Besides this transient induction, the selection of mutations leading to stable hyperproduction is a very common event among clinical strains, obviously due to the pressure exerted by β-lactam treatments. Among the different mutations that enable AmpR-dependent AmpC hyperproduction (7, 13, 24, 25, 34–36), those on ampD and dacB are the most frequently reported in P. aeruginosa (12, 37–41).
As stated before, while certain peptidoglycan-derived fragments (known as activators) were described to enable AmpR-mediated AmpC hyperproduction decades ago in different species (13, 16, 17, 42, 43), the identification of NAG-anhNAM-P5 and its derivative anhNAM-P5 as the drivers for the transient AmpC induction in P. aeruginosa is recent (33). Other works, based on in vitro experiments, have suggested that only muropeptides containing a terminal d-Ala-d-Ala motif (i.e., muropentapeptides) bind AmpR for ampC induction (32, 44). Thus, these results contrast with other work in different Gram-negative bacteria, such as C. freundii (16, 17), E. cloacae (43), Aeromonas hydrophila (45), or Stenotrophomonas maltophilia (46), where different AmpC-activating signals (murotripeptides [anhNAM-P3] and murotetrapeptides [anhNAM-P4]) were reported. However, the signaling for AmpC hyperproduction in P. aeruginosa strains bearing the mentioned mutations in dacB or ampD (8, 37, 47) and thus of extraordinary clinical relevance has never been ascertained. Some clues could be deduced from the work with AmpD mutants in other species (16, 17, 42, 43), but today there are no data regarding the signals sustaining the highly prevalent dacB mutational pathway.
Given these important gaps in the knowledge regarding P. aeruginosa AmpC regulation, and the current scenario of increasing prevalence of its resistance in health care settings (48–51), deciphering the signals enabling stable AmpC hyperproduction in this species becomes crucial. In this work, we have performed a comprehensive and quantitative analysis of soluble muropeptides in diverse mutant backgrounds to identify the signals governing AmpC hyperproduction. Our results revealed a very particular dynamic of AmpC regulation for P. aeruginosa, in which the AmpC production level depends on the nature and quantity of cell wall-derived activators. Thus, this work is a major step toward understanding the basis of P. aeruginosa AmpC-mediated hyperproduction, an essential instrument to conceive future therapies intended to interfere with the involved signaling for combating β-lactam resistance.
RESULTS
AmpC hyperproduction in Pseudomonas aeruginosa: different pathways, different signaling.
The ultrahigh-pressure liquid chromatography–mass spectrometry (UPLC-MS) analyses of soluble intracellular muropeptides in the wild-type strain PAO1 (Fig. 1) showed that the most abundant molecules within the total anhNAM-peptide pool were anhNAM-P3 (ca. 65%) followed by anhNAM-P4 (ca. 35%) and, at a great distance, by anhNAM-P5 (ca. 1%). However, the amount of this last muropeptide increased ca. 10-fold upon cefoxitin treatment (Fig. 1A and B). Interestingly, the levels of accumulated anhNAM-P5 in the dacB mutant (PAΔdB) were similar to those of cefoxitin-induced PAO1 (7.3- ± 2.9-fold compared to PAO1 [Fig. 1A and B]). As can be observed in Fig. 1B, this anhNAM-P5 increase appeared in exchange of a notable reduction in the proportion of anhNAM-P4 within the respective anhNAM-peptide pool (the proportion of anhNAM-P3 remaining stable), which suggests that the total anhNAM-peptide amount both in cefoxitin-induced PAO1 and in PAΔdB is similar to that of PAO1 and that anhNAM-P4 does not play an important role to promote ampC expression in these strains.
FIG 1.

Accumulation of the AmpC activator anhNAM-muropeptides in the cefoxitin-induced PAO1 and in hyperproducer mutants of Pseudomonas aeruginosa. (A and C) The columns represent the mean values of anhNAM-P5 (A) and anhNAM-P3 (C), compared to PAO1, from three independent determinations, and the error bars show the standard deviation (SD). (B) The different-colored sections within the circles represent the proportion of each muropeptide with regard to the total amount of soluble anhNAM-peptides of each strain; the data were extracted from the cited three independent experiments. ND, not detected.
Besides anhNAM-P5, there was a significant increase in the accumulation of its parent molecule, NAG-anhNAM-P5, in the cefoxitin-induced PAO1 and PAΔdB, compared to PAO1 (ca. 5-fold [data not shown]). However, since NagZ allows the cytosolic cleavage of NAG from NAG-anhNAM-peptides, it is understandable that the accumulation of anhNAM-peptides is proportional to the amount of NAG-anhNAM-peptides reaching the cytosol. For these reasons, in this study we have focused on anhNAM-peptide quantification, a conception supported by previous works (42, 43), in which anhNAM-P5 was considered the genuine AmpR-binding signal.
In contrast to PAΔdB, the ampD mutant (PAΔD) showed levels of anhNAM-P5 (Fig. 1A and B) below the wild type and even below the detection limit. Conversely, the intracellular accumulation of anhNAM-P3 increased ca. 10-fold in PAΔD compared to PAO1 (10.9 ± 3.0 [Fig. 1C]). Since anhNAM-P3 is the most abundant chemical species within the anhNAM-peptide pool under normal conditions (ca. 65% [Fig. 1B]), an increase of 10-fold in this muropeptide accumulation necessarily entails a significant increase in the total amount of soluble anhNAM-peptides in PAΔD. Moreover, given that the anhNAM-P5 accumulation (Fig. 1A and B) was under the detection limit in PAΔD, it seems plausible that the amount of this chemical species remained at wild-type levels, thus losing mass within the total pool. These facts suggest that anhNAM-P5 does not significantly sustain AmpC hyperproduction in PAΔD.
Simultaneous accumulation of 1,6-anhydro-N-acetylmuramyl-tripeptides and -pentapeptides drives high-level AmpC hyperproduction.
To better understand the role of anhNAM-P3 and anhNAM-P5 as AmpC activators, we intracellularly quantified them in a panel of mutants displaying increasing levels of AmpC production. This panel included single knockout mutants in the periplasmic amidases AmpDh2 (PAΔDh2) and AmpDh3 (PAΔDh3) (52–55) and different combinations among them and/or with the abovementioned ampD or dacB deletions (Table 1; Fig. 2). As expected, we observed no significant differences in the level of anhNAM-P3 or anhNAM-P5 accumulation between PAΔDh2 or PAΔDh3 and PAO1 (Fig. 2A and B), since these mutants show basal ampC expression (Table 1) (56). However, the anhNAM-P3 (but not anhNAM-P5) level increased ca. 3 times in the double mutant PAΔDh2Dh3, consistent with a slight but significant increase of its ampC expression (ca. 2-fold compared to PAO1 [Table 1]). Interestingly, a simultaneous overaccumulation of both anhNAM-P3 and anhNAM-P5 was observed in the PAΔDDh2, PAΔDDh3, PAΔDDh2Dh3, and PAdacBΔD strains (Fig. 2A and B), all of them displaying high levels of ampC expression (Table 1) (47, 56, 57). Particularly, the two highest AmpC-hyperproducing strains, PAΔDDh2Dh3 and PAdacBΔD (both with ampC mRNA levels over 1,000-fold higher than PAO1), showed an increase of ca. 600-fold (655- ± 141-fold) of anhNAM-P3 and almost 150-fold of anhNAM-P5 and of around 100-fold (105- ± 42-fold) of anhNAM-P3 and 160-fold (160- ± 40-fold) of anhNAM-P5, respectively, compared to PAO1 (Fig. 2A and B). Finally, and similarly to what has been mentioned above for PAΔD, our results suggest that an important increase in the total anhNAM-peptide pool must exist in the high-level AmpC-hyperproducing mutants, since an increase of more than 100-fold with regard to wild type in the most abundant anhNAM-peptide (anhNAM-P3) should necessarily affect the whole (Fig. 2C). Additionally, the fact that the proportion of anhNAM-P5 was barely reduced (or even increased in PAdacBΔD) in this set of mutants within the anhNAM-peptide pool (Fig. 2C) supports the net increase of this species compared to wild-type level (Fig. 2B), whereas the drastic reduction of anhNAM-P4 within the whole indicates that this species remained at wild-type levels, if not below.
TABLE 1.
Strains used in this work, displayed with their features related to the profile of β-lactam resistance (ceftazidime MIC and ampC expression)
| Strain | Genotype/relevant characteristic(s) | Ceftazidime MIC (mg/liter) |
ampC mRNA expression, mean ± SDa |
Source or reference |
|---|---|---|---|---|
| PAO1b | Completely sequenced reference strain | 1 | 1 | Laboratory collection |
| PAΔD | PAO1 ΔampD::lox; AmpD is an N-acetyl-anhydromuramyl-l-alanine amidase involved in peptidoglycan recycling; negative regulator of AmpC expression |
8 | 47 ± 9.5 | 56 |
| PAΔDh2 | PAO1 ΔampDh2::lox; AmpDh2 is an additional AmpD homologue of P. aeruginosa |
0.75 | 1.1 ± 0.2 | 56 |
| PAΔDh3 | PAO1 ΔampDh3::lox; AmpDh3 is an additional AmpD homologue of P. aeruginosa |
1 | 1.2 ± 0.3 | 56 |
| PAΔDh2Dh3 | PAO1 ΔampDh2::lox ΔampDh3::lox | 0.75 | 2.3 ± 0.14 | 56 |
| PAΔDDh2 | PAO1 ΔampD::lox ΔampDh2::lox | 12 | 79.5 ± 12.2 | 56 |
| PAΔDDh3 | PAO1 ΔampD::lox ΔampDh3::lox | 48 | 251.2 ± 51.9 | 56 |
| PAΔDDh2Dh3 | PAO1 ΔampD::lox ΔampDh2::lox ΔampDh3::lox; mutant derepressed for AmpC production |
48 | 1,225 ± 101 | 56 |
| PAΔdB | PAO1 ΔdacB::lox; dacB encodes the nonessential penicillin-binding protein 4 |
24 | 51 ± 16 | 57 |
| PAdacBΔD | 1A1 spontaneous dacB knockout mutant (W273X) of PAO1; ampD::lox; mutant derepressed for AmpC production |
96 | 1,770 ± 401 | 57 |
| PAΔAG | PAO1 ΔampG::lox; ampG encodes the specific permease allowing the entry of certain muropeptides into the cytosol |
1 | 1.1 ± 0.4 | 47 |
| PAΔDDh2Dh3ΔAG | PAO1 ΔampD::lox ΔampDh2::lox ΔampDh3::lox ΔampG::lox | 1 | 0.95 ± 0.2 | 74 |
| PAdacBΔDG | 1A1 spontaneous dacB knockout mutant (W273X) of PAO1; ampD::lox ampG::lox |
0.75 | 1.7 ± 0.6 | 47 |
Data obtained from the indicated references.
The ampC mRNA level in PAO1 under cefoxitin induction was 50 ± 14 with regard to basal conditions.
FIG 2.

Accumulation of the AmpC activator anhNAM-muropeptides in high-level hyperproducer mutants of Pseudomonas aeruginosa. (A and B) The columns represent the mean values of anhNAM-P3 (A) and anhNAM-P5 (B) (both compared to PAO1 values), from three independent determinations, and the error bars show the SD. (C) The different-colored sections within the circles represent the proportion of each muropeptide with regard to the total amount of soluble anhNAM-peptides of each strain; the data were extracted from the cited three independent experiments. ND, not detected.
AmpG disruption reduces the intracellular accumulation of 1,6-anhydro-N-acetylmuramyl-peptides, preventing AmpC hyperproduction.
The results displayed in Fig. 3A and B confirm that ampG deletion drastically decreases the intracellular accumulation of anhNAM-P3 and anhNAM-P5 in P. aeruginosa. This can be observed in the comparison of PAO1 with PAΔAG, in which the amount of these signals was barely quantifiable, but also in PAΔDDh2Dh3ΔAG and PAdacBΔDG. In these two mutants, which proceed from PAΔDDh2Dh3 and PAdacBΔD (high-level AmpC hyperproducers), respectively, the AmpG disruption not only abolished AmpC hyperproduction (Table 1) but also significantly reduced the amount of both activator signals, between 10- and 15-fold with regard to their parent strains.
FIG 3.

Accumulation of the AmpC activator anhNAM-muropeptides in mutants of Pseudomonas aeruginosa with impaired peptidoglycan recycling through AmpG disruption. The columns represent the mean values of anhNAM-P3 (A) and anhNAM-P5 (B), compared to PAO1, from three independent determinations, and the error bars show the SD. ND, not detected.
The levels of UDP-N-acetylmuramic acid-pentapeptide are not fully proportional to AmpC production but respond to peptidoglycan recycling blockade.
The results shown in Fig. 4 indicate that the variations in the amount of the alleged AmpC repressor UDP-NAM-P5 are much smaller (approximately ranging from 0.5- to 5-fold compared to wild type) than those of the AmpC-activating signals among our strains. Furthermore, the accumulation of UDP-NAM-P5 was not consistently inversely proportional to the ampC expression levels (Table 1). In fact, a clear inverse correlation between AmpC production and UDP-NAM-P5 levels was seen only for PAΔDDh2Dh3, and thus, the amount of this repressor was variable in the rest of the strains and even greater than that of PAO1 in some cases, such as PAΔdB, PAdacBΔD, or the cefoxitin-induced PAO1. In contrast, its amount was consistently reduced (even below half the wild-type value) in the strains in which the peptidoglycan recycling was seriously impaired, such as those containing the triple AmpD homologues and/or AmpG disruptions (Fig. 4).
FIG 4.
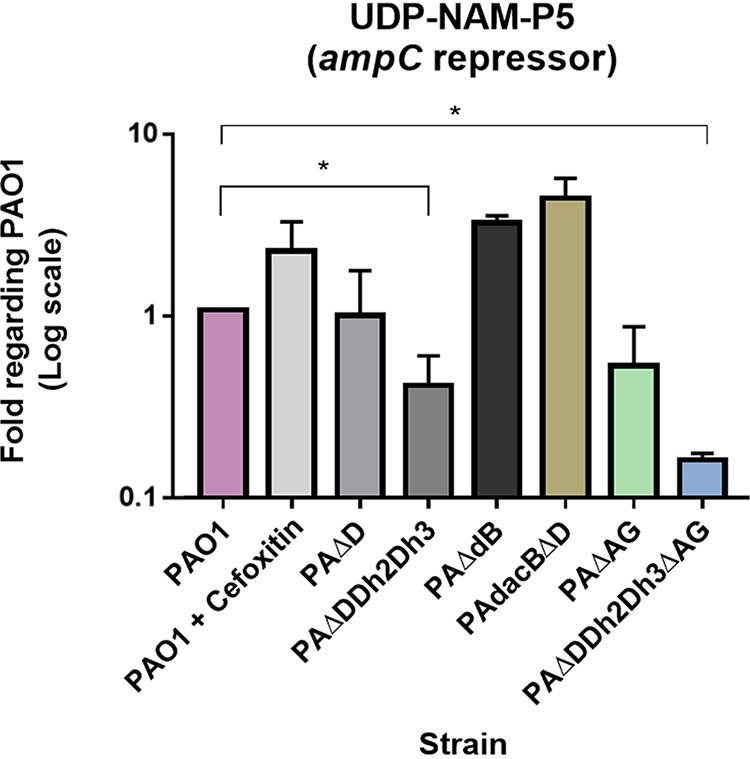
Quantification of the AmpC repressor (UDP-NAM-P5), in different hyperproducer and/or peptidoglycan-recycling-impaired strains of Pseudomonas aeruginosa. The columns represent the mean values of UDP-NAM-P5 with regard to PAO1 (considered 1), obtained from three independent determinations, and the error bars show the SD. *, Student’s t test P value of <0.05.
DISCUSSION
Through the present study, we have identified for the first time the overaccumulation of anhNAM-P5 as the activator signal underlying the PBP4 mutational route of AmpC hyperproduction in P. aeruginosa, the most frequent mechanism causing AmpC-mediated resistance in clinical strains (37). Therefore, this pathway shares the same signal that was previously proposed to mediate the cefoxitin induction (33), which is also supported by our results (Fig. 1). In contrast, our observations indicate that anhNAM-P3 is the main signal accounting for AmpC hyperproduction in the AmpD inactivation-mediated pathway, also very frequently found in P. aeruginosa of clinical origin. This would be in agreement with previous studies that showed increased anhNAM-P3 accumulation in ampD-defective mutants of other Gram-negative species (16, 17, 42, 43). Thus, our results suggest that the mild levels of AmpC hyperproduction (ampC mRNA ca. 50-fold higher than PAO1 [56, 57]), like those shown by the cefoxitin-induced PAO1, PAΔD, or PAΔdB (Table 1), can be achieved through increased levels of one of the two different activators, anhNAM-P3 or anhNAM-P5. However, our results also indicate that anhNAM-P5 has a much stronger AmpC-activating capacity, given that with a substantially smaller amount than that of anhNAM-P3, it triggers a similar ampC hyperexpression. In other words, anhNAM-P3 could play the same activator role but requiring a much larger number of molecules. Therefore, although our results do not support the full need for a terminal d-Ala-d-Ala in the stem peptide of the cell wall-derived signal for ampC expression promotion, they suggest a much higher capacity for AmpR binding if this moiety is present, in agreement with previous works (32).
The mentioned increased accumulation of anhNAM-P3 in PAΔD can be explained by the facts that, under regular conditions, (i) most of the released muropeptides taken up through AmpG have tetrapeptide stems, given that the d,d-transpeptidases (e.g., high-molecular-mass PBPs) and transpeptidation-independent d,d-carboxypeptidases readily cleave the terminal d-Ala from the pentapeptide stems in the sacculus (18, 33, 58, 59); (ii) NAG from NAG-anhNAM-P4 is efficiently cleaved by NagZ (42, 43); and (iii) the terminal d-Ala from anhNAM-P4 is largely trimmed by the cytosolic l,d-carboxypeptidase LdcA (60–62). Finally, the AmpD disruption would disable the cytosolic cleavage to release the ahnNAM from the stem tripeptide, thereby increasing the amount of the activator anhNAM-P3 (Fig. 1C). Accordingly, given that AmpD should not affect the periplasmic turnover and stem peptide length but rather the cytosolic processing of muropeptides, the very low anhNAM-P5 level in PAΔD seems also logical (Fig. 1A and B).
Conversely, cefoxitin causes an increase in the proportion of pentapeptides within the murein sacculus through the inhibition of the d,d-carboxypeptidase activities of the low-molecular-mass PBPs such as PBP4 (as shown in A. hydrophila and P. aeruginosa, for instance [31, 45, 63–66]). This inhibitory effect is believed to move the turnover balance toward an increased autolysis (16, 17, 26, 28, 29, 31), leading to a rise in the released NAG-anhNAM-P5 and anh-NAM-P5 (16, 17, 28, 31, 67), which would ultimately saturate AmpD and bind AmpR to induce ampC expression (33) (Fig. 1A and B). In this regard, our results suggest that dacB inactivation mimics the cefoxitin induction in P. aeruginosa; in fact, an increase in the level of stem pentapeptides in the peptidoglycan is observed when dacB is deleted together with other low-mass PBPs (59). Further, the inactivation of PBP4 would obviously resemble the abovementioned cefoxitin effects, causing a higher peptidoglycan autolysis coresponsible for the increased amount of cytosolic anhNAM-P5. These ideas are supported by the fact that both cefoxitin induction and PBP4 deletion have been shown to cause the activation of the CreBC two-component system in contrast with AmpD disruption, which has no effect on this system (53, 68, 69). CreBC activation has pleiotropic effects, among them the documented increase in the β-lactam resistance output derived from a given AmpC production level, which evidences its relevance (13, 53, 69–71). Thus, our results suggest that the abnormal overaccumulation of soluble anhNAM-P5 would be the driver of the role of PBP4 as cell wall damage sentinel and the signal detected by the membrane-bound CreC sensor leading to the activation of CreBC on one hand and to the promotion of ampC expression (via AmpR binding) on the other (57, 68).
With regard to the high-level AmpC hyperproduction, our results clearly demonstrate a concomitant overaccumulation of anhNAM-P3 and anhNAM-P5 in the strains with this profile (ca. 100- to 1,000-fold of ampC mRNA compared to PAO1), regardless of the pathway: PAΔDDh2, PAΔDDh3, PAΔDDh2Dh3, and PAdacBΔD. Additionally, we have also shown that this simultaneous increase (on the order of hundreds-fold compared to wild type) necessarily entails a significant increase in the total amount of soluble anhNAM-peptide pool (Fig. 2C). This would contrast with what was proposed in the work of Lee et al. (33), in which, although the AmpC inducers NAG-anhNAM-P5 and anhNAM-P5 experienced a net increase, the whole peptidoglycan-derived soluble fragment pool was shown to be reduced ca. 4-fold upon cefoxitin challenge. These facts suppose another interesting difference between the mechanisms underlying cefoxitin induction and the dynamics of ampC hyperexpression in the high-level hyperproducer strains.
Although the two highest hyperproducing mutants, PAΔDDh2Dh3 and PAdacBΔD, display the simultaneous accumulation of anhNAM-P3 and anhNAM-P5 (Fig. 2), the notable quantitative variations of these signals among the two strains did not significantly affect the final outcome of ampC expression level (Table 1). This is probably due to the AmpR saturation, supported by the fact that these two strains are not further inducible (57). As stated before, the AmpC-activating power (via AmpR binding) of anhNAM-P5 seems much stronger than that of anhNAM-P3, but in this high-level hyperproduction context, in which the amount of both activators is highly increased (but the number of anhNAM-P3 molecules continues to be much greater than that of anhNAM-P5), it is difficult to predict which species outcompetes the other for AmpR binding, or even if an indistinct binding does occur.
Further analysis of the PAΔDDh2Dh3 mutant helps to explain this simultaneous accumulation of anhNAM-P3 and anhNAM-P5. Since AmpDh2 and AmpDh3 are periplasmic amidases cleaving stem peptides from the peptidoglycan (52–55) and thus initiating the recycling-related role that AmpD exerts within the cytosol, it seems logical that the triple inactivation caused an exaggerated anhNAM-P3 accumulation in comparison with PAΔD (ca. 60-fold higher [Fig. 2A]). A similar conclusion could be deduced with regard to anhNAM-P5: under basal conditions, the amount of this molecule is low (Fig. 1A and B) (33) not only because of the abovementioned d,d-carboxypeptidase activities but also because of stem peptide cleavage by AmpDh2 and AmpDh3. Therefore, the absence of these two proteins should consequently increase the amount of released muropeptides instead of free peptides. In the AmpDh2-AmpDh3 defective background (AmpD still functional), no striking consequences are seen (Fig. 2A and B), suggesting that AmpD is self-sufficient to metabolize this increased amount of cytosolic muropeptides. But when AmpD (the main amidase cleaving the AmpC activators for ulterior anabolism [52, 56, 72]) is absent, the partial or total lack of amidase activity in the periplasm entails a dramatic increase in anhNAM-peptide accumulation and ampC expression (47, 56, 57). Despite the high-level β-lactam resistance of PAΔDDh2Dh3, this kind of triple mutant has never been found in nature, likely because it is dramatically impaired in fitness and virulence (73–75). Conversely, PAdacBΔd-type mutants have been readily found in the clinical setting (47). Furthermore, an interesting question arises with this strain: why the activator muropeptide accumulation is much higher (between 10- and 20-fold for anhNAM-P3 and anhNAM-P5) than in the PAΔD and PAΔdB single mutants? The total absence of AmpD in the double mutant (compared to the mere AmpD saturation likely occurring in PAΔdB) could explain the anhNAM-P5 increase. However, the reason why the amount of anhNAM-P3 also increases in PAdacBΔD but is not in appreciable in PAΔdB is more intriguing and could suggest a potential preferential trimming activity of AmpD over anhNAM-P3 versus anhNAM-P5. Finally, here we show that the amount of anhNAM-P4 does not increase in parallel with the ampC expression levels in any of the pathways (47, 56, 57) but rather the contrary (Fig. 1B and Fig. 2C), and thus, this muropeptide seems to be expendable for AmpC hyperproduction in P. aeruginosa, in contrast to what has been described for S. maltophilia (46).
It has been previously shown that AmpG inactivation prevents the entrance of NAG-anhNAM-peptides and consequently the cytosolic accumulation of AmpC-activator signals, thereby disabling ampC overexpression (47, 76–80). These facts are confirmed by our results, in which ampG deletion drastically decreased the intracellular accumulation of anhNAM-P3 and anhNAM-P5 in PAO1 but also in the high-level hyperproducer mutants in comparison with the respective parent strain, which leads to a block of ampC expression. Since AmpG inactivation disables the bona fide NAG-anhNAM-peptide cytosolic gate, the still-increased (with regard to PAO1) accumulation of anhNAM-P3 and anhNAM-P5 in PAΔDDh2Dh3ΔAG and PAdacBΔDG (Fig. 3) necessarily corresponds to their increased generation in the periplasm driven by the inactivation of dacB and/or ampD homologues. One could argue that instead of anhNAM-P3 and anhNAM-P5, in PAΔDDh2Dh3ΔAG and PAdacBΔDG a periplasmic accumulation of NAG-anhNAM-P3 and NAG-anhNAM-P5 should appear, inasmuch as NagZ, responsible for the NAG cleavage, is cytosolic. Nevertheless, as previously proposed (33, 81), the existence of N-acetylglucosaminidase activity in the P. aeruginosa periplasm seems highly likely, which would enable the NAG cleavage and explain the large amount of anhNAM-peptides. On the other hand, the previously described increased release of muropeptides to the extracellular medium in AmpG-defective mutants (82–84) could contribute to the reduced amount of soluble anhNAM-peptides of PAΔDDh2Dh3ΔAG and PAdacBΔDG compared to the parent strains (Fig. 3A and B).
In this study, we finally sought to analyze if the peptidoglycan biosynthesis precursor UDP-NAM-P5, which has been reported to act as an AmpC repressor through AmpR binding in different species (17, 19, 32), could have a similar role in P. aeruginosa. Nevertheless, our results suggest that the repressor capacity of UDP-NAM-P5 is fairly minor in P. aeruginosa, since the level of ampC expression was generally not inversely proportional to the UDP-NAM-P5 amount in our strains. In fact, in some of our hyperproducer mutants, this precursor was at wild-type (or even higher) levels, a circumstance that has been previously shown for AmpD defective mutants in different species (17). In contrast, our results show a consistent UDP-NAM-P5 reduction in those strains with impaired peptidoglycan recycling, namely, PAΔDDh2Dh3, PAΔDDh2Dh3ΔG, and PAΔAG (74, 75) (Fig. 4). These facts, previously reported in AmpG mutants from other species (17), seem quite logical since recycling disruption blocks a very important pipeline of materials for the anabolism of new UDP-NAM-P5 units. Consistently, PAΔDDh2Dh3ΔG displayed the lowest levels of UDP-NAM-P5 (Fig. 4) given its impairment for both periplasmic turnover (in terms of AmpDh2/AmpDh3-driven stem peptide cleavage that would allow the recycling of the latter) and uptake of NAG-anhNAM-containing fragments via AmpG (25, 34, 36). Therefore, our results do not support a major influence of UDP-NAM-P5 over AmpC regulation in P. aeruginosa. A possible explanation could be that when the activator muropeptides are present, they exert a dominant role, thereby displacing the repressor UDP-NAM-P5 from AmpR (17). In fact, in accordance with this very modest regulatory role that we propose for UDP-NAM-P5, the deletion of AmpR (which eliminates the repression exerted through binding with this precursor) was previously reported to cause only a slight increase of ampC expression in P. aeruginosa (below 5-fold compared to wild type) (57).
The results we show in this study (summarized in Fig. 5A) allow us to draw a novel model for peptidoglycan-derived signaling enabling ampC hyperexpression in P. aeruginosa, which takes into account the different pathways, levels of AmpC production, and the involved activator muropeptides (Fig. 5B). Although with certain quantitative particularities, an excellent correlation (considering Spearman’s coefficients) between the amount of the activator muropeptide anhNAM-P5 and ampC expression does exist and is a little weaker for anhNAM-P3. Meanwhile, no correlation with UDP-NAM-P5 is found, indicating a residual role for this molecule as AmpC regulator. Moreover, when applying combinatorial analysis, i.e., choosing the highest value for each strain (anhNAM-P3 or anhNAM-P5), the correlation with ampC expression is almost perfect, similar to that obtained by summing the two types of muropeptides, which suggests their additive nature for AmpC activation.
FIG 5.

Model for Pseudomonas aeruginosa AmpC derepression, depicting the variations in the pathways (different mutational routes versus cefoxitin induction) and levels of enzyme production, based on the differential accumulation of muropeptides. (A) The gray bars represent the level of ampC expression obtained from previous works by our group (52, 53, 74), whereas the red circles, green squares, and yellowish green triangles represent the accumulation of anhNAM-P3, anhNAM-P5, and UDP-NAM-P5, respectively, in each strain (always in terms of fold with regard to PAO1, considered 1). The error bars represent the SD. The scale on the right is valid for only UDP-NAM-P5, whereas that on the left is for the other parameters (both are log scales). (B) Smooth Loess curves of the linear regressions between ampC expression and (i) the accumulation of the different independent muropeptides, (ii) the sum of both anhNAM-P3 and anhNAM-P5 (SUM), or (iii) the highest muropeptide of each strain (MAX). The Spearman coefficient for each correlation is shown.
Therefore, our model, which displays certain particularities in comparison with other species’ β-lactamase signaling (16, 17, 42, 43, 45, 46), entails a definitive step to understand the basis for P. aeruginosa AmpC regulation, which could be useful to open new therapeutic conceptions oriented to interfere with the involved peptidoglycan-derived signaling.
MATERIALS AND METHODS
Bacterial strains.
A list and description of the strains used in this work are shown in Table 1. P. aeruginosa PAO1 strain-derived single or combined knockout mutants previously constructed according to described procedures (56, 57, 85), based on the Cre-lox system for gene deletion in P. aeruginosa (86), were used. Their data regarding susceptibility to β-lactams, i.e., ceftazidime MICs and ampC expression, were obtained from the previous work of our group indicated in Table 1.
Cefoxitin induction experiments.
For induction experiments, before the realization of each assay, the overnight cultures were diluted 1:50 and were grown in the presence of 50 μg/ml cefoxitin for 3 h (37°C, 180-rpm agitation), as previously described (85).
Generation of soluble peptidoglycan precursor pools and UPLC-MS analysis.
Sample preparation (at least three independent preparations per strain were done, always referring the data of each determination to the wild type) for collection of cytosolic soluble muropeptides was performed following the protocol described previously by Lee et al. (33) with some modifications. Briefly, bacteria were grown until exponential phase and cooled down on ice for 10 min, and then after adjusting the OD600 of all the cultures (to have the same number of bacteria in each sample), normalized volumes of cells were harvested by centrifugation at 4,000 rpm. at 4°C for 20 min. Cell pellets were then gently resuspended, washed with ice-cold 0.9% NaCl solution, and finally resuspended in water and boiled for 15 min. After centrifugation to remove cell debris at 14,000 rpm for 15 min, soluble fractions (containing intracellular soluble muropeptides) were transferred into new tubes and stored at −20°C. Samples were filtered using 0.2-μm-pore-size filters, dried by speed vacuum, resuspended into water, and used for UPLC-MS analyses.
Detection and characterization of soluble muropeptides were performed on a UPLC system interfaced with a Xevo G2/XS quadrupole-time of flight (Q-TOF) mass spectrometer (Waters Corp.). Chromatographic separation was achieved using an Acquity UPLC BEH C18 column, 130 Å, 1.7 μm, 2.1 mm by 150 mm (Waters Corp.), heated at 45°C. Formic acid at 0.1% in Milli-Q water (buffer A) and 0.1% formic acid in acetonitrile (buffer B) were used as eluents. The gradient of buffer B was set as follows: 0 to 3 min, 5%; 3 to 6 min, 5 to 6.8%; 6 to 7.5 min, 6.8 to 9%; 7.5 to 9 min, 9 to 14%; 9 to 11 min, 14 to 20%; 11 to 12 min, hold at 20% with a flow rate of 0.175 ml/min; 12 to 12.10 min, 20 to 90%; 12.1 to 13.5 min, hold at 90%; 13.5 to 13.6 min, 90 to 2%, 13.6 to 16 min, hold at 2% with a flow rate of 0.3 ml/min; then 16 to 18 min, hold at 2% with a flow rate of 0.25 ml/min. The Q-TOF MS instrument was operated in positive ionization mode using MSe (mass spectrometry using elevated collision energies). The following parameters were set for electrospray ionization (ESI): capillary voltage at 3.0 kV, source temperature to 120°C, desolvation temperature to 350°C, sample cone voltage to 40 V, cone gas flow of 100 liters/h, and desolvation gas flow of 500 liters/h. Data acquisition and processing were performed using the UNIFI software package (Waters Corp.).
The molecular structure of the soluble muropeptides expected to be found in the cytosolic samples was obtained by using ChemSketch version 14.01 (ACD/Labs, Toronto, Ontario, Canada) and used to build a compound library in UNIFI. This compound library was used for processing the data, detection, and identification of muropeptides. Subsequent confirmation of the structure of automatically detected muropeptide was performed by further analysis of the MS fragmentation pattern and comparison with data previously obtained from the analysis of standard muropeptides. The area of the extracted ion chromatogram of identified muropeptides was considered the quantitative value.
Data analysis.
GraphPad Prism 5 software was used for graphical representation and statistical analysis. Quantitative variables were compared using two-tailed Student’s t test or Mann-Whitney U test as appropriate. A P value of <0.05 was considered statistically significant. To find correlations between the accumulation of muropeptides and ampC expression, the linear regressions represented by smooth Loess curves were calculated, together with the respective Spearman correlation coefficients, using the R software (version 3.6.0; R Foundation for Statistical Computing, Vienna, Austria).
ACKNOWLEDGMENTS
Work in the Oliver lab is supported by the Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC), the Ministerio de Economía y Competitividad of Spain, and the Instituto de Salud Carlos III, cofinanced by the European Regional Development Fund (ERDF; A way to achieve Europe) through the Spanish Network for the Research in Infectious Diseases (RD12/0015 and RD16/0016), and grants CP12/03324, PI15/00088, PI15/02212, PI18/00681, and PI18/00076. Work in the Cava lab is supported by the Swedish Research Council (VR), the Knut and Alice Wallenberg Foundation (KAW), the Laboratory of Molecular Infection Medicine Sweden (MIMS), and the Kempe Foundation.
C.J., F.C., and A.O. designed research; G.T. and S.B.H. performed research; B.M., F.C., and S.B.H. contributed reagents/analytic tools; G.T., S.B.H., A.O., C.J., F.C., J.A.A., and B.M. analyzed data; C.J., F.C., and A.O. wrote the paper, with contribution and review from J.A.A., B.M., G.T., and S.B.H.
REFERENCES
- 1.Vincent J-L. 2003. Nosocomial infections in adult intensive-care units. Lancet 361:2068–2077. doi: 10.1016/S0140-6736(03)13644-6. [DOI] [PubMed] [Google Scholar]
- 2.Bassetti M, Taramasso L, Giacobbe DR, Pelosi P. 2012. Management of ventilator-associated pneumonia: epidemiology, diagnosis and antimicrobial therapy. Expert Rev Anti Infect Ther 10:585–596. doi: 10.1586/eri.12.36. [DOI] [PubMed] [Google Scholar]
- 3.Doring G, Parameswaran IG, Murphy TF. 2011. Differential adaptation of microbial pathogens to airways of patients with cystic fibrosis and chronic obstructive pulmonary disease. FEMS Microbiol Rev 35:124–146. doi: 10.1111/j.1574-6976.2010.00237.x. [DOI] [PubMed] [Google Scholar]
- 4.Lyczak JB, Cannon CL, Pier GB. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect 2:1051–1060. doi: 10.1016/s1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 5.Gellatly SL, Hancock REW. 2013. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis 67:159–173. doi: 10.1111/2049-632X.12033. [DOI] [PubMed] [Google Scholar]
- 6.Livermore DM. 2009. Has the era of untreatable infections arrived? J Antimicrob Chemother 64(Suppl 1):i29–i36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- 7.Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Del Barrio-Tofiño E, López-Causapé C, Cabot G, Rivera A, Benito N, Segura C, Montero MM, Sorlí L, Tubau F, Gómez-Zorrilla S, Tormo N, Durá-Navarro R, Viedma E, Resino-Foz E, Fernández-Martínez M, González-Rico C, Alejo-Cancho I, Martínez JA, Labayru-Echverria C, Dueñas C, Ayestarán I, Zamorano L, Martinez-Martinez L, Horcajada JP, Oliver A. 2017. Genomics and susceptibility profiles of extensively drug-resistant Pseudomonas aeruginosa isolates from Spain. Antimicrob Agents Chemother 62:e02352-17. doi: 10.1128/AAC.02352-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindberg F, Westman L, Normark S. 1985. Regulatory components in Citrobacter freundii ampC beta-lactamase induction. Proc Natl Acad Sci U S A 82:4620–4624. doi: 10.1073/pnas.82.14.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindberg F, Normark S. 1987. Common mechanism of ampC beta-lactamase induction in enterobacteria: regulation of the cloned Enterobacter cloacae P99 beta-lactamase gene. J Bacteriol 169:758–763. doi: 10.1128/jb.169.2.758-763.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindquist S, Lindberg F, Normark S. 1989. Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC beta-lactamase gene. J Bacteriol 171:3746–3753. doi: 10.1128/jb.171.7.3746-3753.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartowsky E, Normark S. 1993. Interactions of wild-type and mutant AmpR of Citrobacter freundii with target DNA. Mol Microbiol 10:555–565. doi: 10.1111/j.1365-2958.1993.tb00927.x. [DOI] [PubMed] [Google Scholar]
- 13.Juan C, Torrens G, González-Nicolau M, Oliver A. 2017. Diversity and regulation of intrinsic β-lactamases from non-fermenting and other Gram-negative opportunistic pathogens. FEMS Microbiol Rev 41:781–815. doi: 10.1093/femsre/fux043. [DOI] [PubMed] [Google Scholar]
- 14.Honoré N, Nicolas MH, Cole ST. 1986. Inducible cephalosporinase production in clinical isolates of Enterobacter cloacae is controlled by a regulatory gene that has been deleted from Escherichia coli. EMBO J 5:3709–3714. doi: 10.1002/j.1460-2075.1986.tb04704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lodge JM, Minchin SD, Piddock LJ, Busby SJ. 1990. Cloning, sequencing and analysis of the structural gene and regulatory region of the Pseudomonas aeruginosa chromosomal ampC beta-lactamase. Biochem J 272:627–631. doi: 10.1042/bj2720627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. 1994. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for beta-lactamase induction. EMBO J 13:4684–4694. doi: 10.1002/j.1460-2075.1994.tb06792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobs C, Frère JM, Normark S. 1997. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible β-lactam resistance in gram-negative bacteria. Cell 88:823–832. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 18.Höltje JV. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev 62:181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JT, Uehara T. 2008. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol Mol Biol Rev 72:211–227. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Heijenoort J. 2011. Peptidoglycan hydrolases of Escherichia coli. Microbiol Mol Biol Rev 75:636–663. doi: 10.1128/MMBR.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vermassen A, Leroy S, Talon R, Provot C, Popowska M, Desvaux M. 2019. Cell wall hydrolases in bacteria: insight on the diversity of cell wall amidases, glycosidases and peptidases toward peptidoglycan. Front Microbiol 10:331. doi: 10.3389/fmicb.2019.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hesek D, Lee M, Zhang W, Noll BC, Mobashery S. 2009. Total synthesis of N-acetylglucosamine-1,6-anhydro-N-acetylmuramylpentapeptide and evaluation of its turnover by AmpD from Escherichia coli. J Am Chem Soc 131:5187–5193. doi: 10.1021/ja808498m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zamorano L, Reeve TM, Deng L, Juan C, Moyá B, Cabot G, Vocadlo DJ, Mark BL, Oliver A. 2010. NagZ inactivation prevents and reverts β-lactam resistance, driven by AmpD and PBP 4 mutations, in Pseudomonas aeruginosa. Antimicrob Agents Chemother 54:3557–3563. doi: 10.1128/AAC.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langaee TY, Gagnon L, Huletsky A. 2000. Inactivation of the ampD gene in Pseudomonas aeruginosa leads to moderate-basal-level and hyperinducible AmpC β-lactamase expression. Antimicrob Agents Chemother 44:583–589. doi: 10.1128/aac.44.3.583-589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dik DA, Fisher JF, Mobashery S. 2018. Cell-wall recycling of the Gram-negative bacteria and the nexus to antibiotic resistance. Chem Rev 118:5952–5984. doi: 10.1021/acs.chemrev.8b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitano K, Tomasz A. 1979. Triggering of autolytic cell wall degradation in Escherichia coli by beta-lactam antibiotics. Antimicrob Agents Chemother 16:838–848. doi: 10.1128/aac.16.6.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomasz A. 1979. From penicillin-binding proteins to the lysis and death of bacteria: a 1979 view. Rev Infect Dis 1:434–467. doi: 10.1093/clinids/1.3.434. [DOI] [PubMed] [Google Scholar]
- 28.Sanders CC, Bradford PA, Ehrhardt AF, Bush K, Young KD, Henderson TA, Sanders WE. 1997. Penicillin-binding proteins and induction of AmpC beta-lactamase. Antimicrob Agents Chemother 41:2013–2015. doi: 10.1128/AAC.41.9.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho H, Uehara T, Bernhardt TG. 2014. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee M, Hesek D, Blázquez B, Lastochkin E, Boggess B, Fisher JF, Mobashery S. 2015. Catalytic spectrum of the penicillin-binding protein 4 of Pseudomonas aeruginosa, a nexus for the induction of β-lactam antibiotic resistance. J Am Chem Soc 137:190–200. doi: 10.1021/ja5111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vadlamani G, Thomas MD, Patel TR, Donald LJ, Reeve TM, Stetefeld J, Standing KG, Vocadlo DJ, Mark BL. 2015. The β-lactamase gene regulator AmpR is a tetramer that recognizes and binds the D-Ala-D-Ala motif of its repressor UDP-N-acetylmuramic acid (MurNAc)-pentapeptide. J Biol Chem 290:2630–2643. doi: 10.1074/jbc.M114.618199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee M, Dhar S, De Benedetti S, Hesek D, Boggess B, Blázquez B, Mathee K, Mobashery S. 2016. Muropeptides in Pseudomonas aeruginosa and their role as elicitors of β-lactam-antibiotic resistance. Angew Chem Int Ed Engl 55:6882–6886. doi: 10.1002/anie.201601693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fisher JF, Mobashery S. 2014. The sentinel role of peptidoglycan recycling in the β-lactam resistance of the Gram-negative Enterobacteriaceae and Pseudomonas aeruginosa. Bioorg Chem 56:41–48. doi: 10.1016/j.bioorg.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juan C, Torrens G, Barceló IM, Oliver A. 2018. Interplay between peptidoglycan biology and virulence in Gram-negative pathogens. Microbiol Mol Biol Rev 82:e00033-18. doi: 10.1128/MMBR.00033-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhar S, Kumari H, Balasubramanian D, Mathee K. 2018. Cell-wall recycling and synthesis in Escherichia coli and Pseudomonas aeruginosa—their role in the development of resistance. J Med Microbiol 67:1–21. doi: 10.1099/jmm.0.000636. [DOI] [PubMed] [Google Scholar]
- 37.López-Causapé C, Cabot G, del Barrio-Tofiño E, Oliver A. 2018. The versatile mutational resistome of Pseudomonas aeruginosa. Front Microbiol 9:685. doi: 10.3389/fmicb.2018.00685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuga A, Okamoto R, Inoue M. 2000. ampR gene mutations that greatly increase class C β-lactamase activity in Enterobacter cloacae. Antimicrob Agents Chemother 44:561–567. doi: 10.1128/aac.44.3.561-567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bagge N, Ciofu O, Hentzer M, Campbell JIA, Givskov M, Høiby N. 2002. Constitutive high expression of chromosomal β-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS1669) located in ampD. Antimicrob Agents Chemother 46:3406–3411. doi: 10.1128/aac.46.11.3406-3411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balcewich MD, Reeve TM, Orlikow EA, Donald LJ, Vocadlo DJ, Mark BL. 2010. Crystal structure of the AmpR effector binding domain provides insight into the molecular regulation of inducible AmpC β-lactamase. J Mol Biol 400:998–1010. doi: 10.1016/j.jmb.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 41.Cabot G, Ocampo-Sosa AA, Domínguez MA, Gago JF, Juan C, Tubau F, Rodríguez C, Moyà B, Peña C, Martínez-Martínez L, Oliver A. 2012. Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob Agents Chemother 56:6349–6357. doi: 10.1128/AAC.01388-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dietz H, Pfeifle D, Wiedemann B. 1996. Location of N-acetylmuramyl-L-alanyl-D-glutamylmesodiaminopimelic acid, presumed signal molecule for beta-lactamase induction, in the bacterial cell. Antimicrob Agents Chemother 40:2173–2177. doi: 10.1128/AAC.40.9.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dietz H, Pfeifle D, Wiedemann B. 1997. The signal molecule for beta-lactamase induction in Enterobacter cloacae is the anhydromuramyl-pentapeptide. Antimicrob Agents Chemother 41:2113–2120. doi: 10.1128/AAC.41.10.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dik DA, Domínguez-Gil T, Lee M, Hesek D, Byun B, Fishovitz J, Boggess B, Hellman LM, Fisher JF, Hermoso JA, Mobashery S. 2017. Muropeptide binding and the X-ray structure of the effector domain of the transcriptional regulator AmpR of Pseudomonas aeruginosa. J Am Chem Soc 139:1448–1451. doi: 10.1021/jacs.6b12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tayler AE, Ayala JA, Niumsup P, Westphal K, Baker JA, Zhang L, Walsh TR, Wiedemann B, Bennett PM, Avison MB. 2010. Induction of β-lactamase production in Aeromonas hydrophila is responsive to β-lactam-mediated changes in peptidoglycan composition. Microbiology 156:2327–2335. doi: 10.1099/mic.0.035220-0. [DOI] [PubMed] [Google Scholar]
- 46.Huang Y-W, Wang Y, Lin Y, Lin C, Lin Y-T, Hsu C-C, Yang T-C. 2017. Impacts of penicillin binding protein 2 inactivation on β-lactamase expression and muropeptide profile in Stenotrophomonas maltophilia. mSystems 2:e00077-17. doi: 10.1128/mSystems.00077-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zamorano L, Reeve TM, Juan C, Moyá B, Cabot G, Vocadlo DJ, Mark BL, Oliver A. 2011. AmpG inactivation restores susceptibility of pan-β-lactam-resistant Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother 55:1990–1996. doi: 10.1128/AAC.01688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breidenstein EBM, de la Fuente-Núñez C, Hancock R. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Hirsch EB, Tam VH. 2010. Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev Pharmacoecon Outcomes Res 10:441–451. doi: 10.1586/erp.10.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.López-Causapé C, Rojo-Molinero E, MacIà MD, Oliver A. 2015. The problems of antibiotic resistance in cystic fibrosis and solutions. Expert Rev Respir Med 9:73–88. doi: 10.1586/17476348.2015.995640. [DOI] [PubMed] [Google Scholar]
- 51.Oliver A, Mulet X, López-Causapé C, Juan C. 2015. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist Updat 21–22:41–59. doi: 10.1016/j.drup.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Zhang W, Lee M, Hesek D, Lastochkin E, Boggess B, Mobashery S. 2013. Reactions of the three AmpD enzymes of Pseudomonas aeruginosa. J Am Chem Soc 135:4950–4953. doi: 10.1021/ja400970n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee M, Artola-Recolons C, Carrasco-López C, Martínez-Caballero S, Hesek D, Spink E, Lastochkin E, Zhang W, Hellman LM, Boggess B, Hermoso JA, Mobashery S. 2013. Cell-wall remodeling by the zinc-protease AmpDh3 from Pseudomonas aeruginosa. J Am Chem Soc 135:12604–12607. doi: 10.1021/ja407445x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martínez-Caballero S, Lee M, Artola-Recolons C, Carrasco-López C, Hesek D, Spink E, Lastochkin E, Zhang W, Hellman LM, Boggess B, Mobashery S, Hermoso JA. 2013. Reaction products and the X-ray structure of AmpDh2, a virulence determinant of Pseudomonas aeruginosa. J Am Chem Soc 135:10318–10321. doi: 10.1021/ja405464b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rivera I, Molina R, Lee M, Mobashery S, Hermoso JA. 2016. Orthologous and paralogous AmpD peptidoglycan amidases from Gram-negative bacteria. Microb Drug Resist 22:470–476. doi: 10.1089/mdr.2016.0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Juan C, Moyá B, Pérez JL, Oliver A. 2006. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level β-lactam resistance involves three AmpD homologues. Antimicrob Agents Chemother 50:1780–1787. doi: 10.1128/AAC.50.5.1780-1787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moya B, Dötsch A, Juan C, Blázquez J, Zamorano L, Haussler S, Oliver A. 2009. β-Lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog 5:e1000353. doi: 10.1371/journal.ppat.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desmarais SM, De Pedro MA, Cava F, Huang KC. 2013. Peptidoglycan at its peaks: how chromatographic analyses can reveal bacterial cell wall structure and assembly. Mol Microbiol 89:1–13. doi: 10.1111/mmi.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ropy A, Cabot G, Sánchez-Diener I, Aguilera C, Moya B, Ayala JA, Oliver A. 2015. Role of Pseudomonas aeruginosa low-molecular-mass penicillin-binding proteins in AmpC expression, β-lactam resistance, and peptidoglycan structure. Antimicrob Agents Chemother 59:3925–3934. doi: 10.1128/AAC.05150-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ursinus A, Steinhaus H, Holtje JV. 1992. Purification of a nocardicin A-sensitive LD-carboxypeptidase from Escherichia coli by affinity chromatography. J Bacteriol 174:441–446. doi: 10.1128/jb.174.2.441-446.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Templin MF, Ursinus A, Höltje JV. 1999. A defect in cell wall recycling triggers autolysis during the stationary growth phase of Escherichia coli. EMBO J 18:4108–4117. doi: 10.1093/emboj/18.15.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korza HJ, Bochtler M. 2005. Pseudomonos aeruginosa LD-carboxypeptidase, a serine peptidase with a Ser-His-Glu Triad and a nucleophilic elbow. J Biol Chem 280:40802–40812. doi: 10.1074/jbc.M506328200. [DOI] [PubMed] [Google Scholar]
- 63.Korat B, Mottl H, Keck W. 1991. Penicillin‐binding protein 4 of Escherichia coli: molecular cloning of the dacB gene, controlled overexpression, and alterations in murein composition. Mol Microbiol 5:675–684. doi: 10.1111/j.1365-2958.1991.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 64.Meberg BM, Paulson AL, Priyadarshini R, Young KD. 2004. Endopeptidase penicillin-binding proteins 4 and 7 play auxiliary roles in determining uniform morphology of Escherichia coli. J Bacteriol 186:8326–8336. doi: 10.1128/JB.186.24.8326-8336.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghosh AS, Chowdhury C, Nelson DE. 2008. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol 16:309–317. doi: 10.1016/j.tim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 66.Aguilera Rossi CG, Gómez-Puertas P, Ayala Serrano JA. 2016. In vivo functional and molecular characterization of the penicillin-binding protein 4 (DacB) of Pseudomonas aeruginosa. BMC Microbiol 16:234. doi: 10.1186/s12866-016-0853-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiedemann B, Pfeifle D, Wiegand I, Janas E. 1998. Beta-lactamase induction and cell wall recycling in gram-negative bacteria. Drug Resist Updat 1:223–226. doi: 10.1016/s1368-7646(98)80002-2. [DOI] [PubMed] [Google Scholar]
- 68.Francis VI, Stevenson EC, Porter SL. 2017. Two-component systems required for virulence in Pseudomonas aeruginosa. FEMS Microbiol Lett 364:fnx104. doi: 10.1093/femsle/fnx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zamorano L, Moyà B, Juan C, Mulet X, Blázquez J, Oliver A. 2014. The Pseudomonas aeruginosa CreBC two-component system plays a major role in the response to β-lactams, fitness, biofilm growth, and global regulation. Antimicrob Agents Chemother 58:5084–5095. doi: 10.1128/AAC.02556-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Avison MB, Horton RE, Walsh TR, Bennett PM. 2001. Escherichia coli CreBC is a global regulator of gene expression that responds to growth in minimal media. J Biol Chem 276:26955–26961. doi: 10.1074/jbc.M011186200. [DOI] [PubMed] [Google Scholar]
- 71.Huang HH, Lin YT, Chen WC, Huang YW, Chen SJ, Yang TC. 2015. Expression and functions of CreD, an inner membrane protein in Stenotrophomonas maltophilia. PLoS One 10:e0145009. doi: 10.1371/journal.pone.0145009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Höltje JV, Kopp U, Ursinus A, Wiedemann B. 1994. The negative regulator of beta-lactamase induction AmpD is a N-acetyl-anhydromuramyl-L-alanine amidase. FEMS Microbiol Lett 122:159–164. doi: 10.1111/j.1574-6968.1994.tb07159.x. [DOI] [PubMed] [Google Scholar]
- 73.Moya B, Juan C, Albertí S, Pérez JL, Oliver A. 2008. Benefit of having multiple ampD genes for acquiring β-lactam resistance without losing fitness and virulence in Pseudomonas aeruginosa. Antimicrob Agents Chemother 52:3694–3700. doi: 10.1128/AAC.00172-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pérez-Gallego M, Torrens G, Castillo-Vera J, Moya B, Zamorano L, Cabot G, Hultenby K, Albertí S, Mellroth P, Henriques-Normark B, Normark S, Oliver A, Juan C. 2016. Impact of AmpC derepression on fitness and virulence: the mechanism or the pathway? mBio 7:e01783-16. doi: 10.1128/mBio.01783-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Torrens G, Pérez-Gallego M, Moya B, Munar-Bestard M, Zamorano L, Cabot G, Blázquez J, Ayala JA, Oliver A, Juan C. 2017. Targeting the permeability barrier and peptidoglycan recycling pathways to disarm Pseudomonas aeruginosa against the innate immune system. PLoS One 12:e0181932. doi: 10.1371/journal.pone.0181932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Korfmann G, Sanders CC. 1989. ampG is essential for high-level expression of AmpC beta-lactamase in Enterobacter cloacae. Antimicrob Agents Chemother 33:1946–1951. doi: 10.1128/aac.33.11.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Normark S. 1995. Beta-lactamase induction in gram-negative bacteria is intimately linked to peptidoglycan recycling. Microb Drug Resist 1:111–114. doi: 10.1089/mdr.1995.1.111. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Y, Bao Q, Gagnon LA, Huletsky A, Oliver A, Jin S, Langaee T. 2010. ampG gene of Pseudomonas aeruginosa and its role in β-lactamase expression. Antimicrob Agents Chemother 54:4772–4779. doi: 10.1128/AAC.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang Q, Wu C, Lin C, Li P, Zhang K, Xu L, Liu Y, Lu J, Cheng C, Bao Q, Hu Y, Lu S, Li J. 2018. The structure of ampG gene in Pseudomonas aeruginosa and its effect on drug resistance. Can J Infect Dis Med Microbiol 2018:7170416. doi: 10.1155/2018/7170416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mallik D, Pal S, Ghosh AS. 2018. Involvement of AmpG in mediating a dynamic relationship between serine beta-lactamase induction and biofilm-forming ability of Escherichia coli. FEMS Microbiol Lett 365:fny065. doi: 10.1093/femsle/fny065. [DOI] [PubMed] [Google Scholar]
- 81.Herlihey FA, Moynihan PJ, Clarke AJ. 2014. The essential protein for bacterial flagella formation FlgJ functions as a β-N-acetylglucosaminidase. J Biol Chem 289:31029–31042. doi: 10.1074/jbc.M114.603944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Folkesson A, Eriksson S, Andersson M, Park JT, Normark S. 2005. Components of the peptidoglycan-recycling pathway modulate invasion and intracellular survival of Salmonella enterica serovar Typhimurium. Cell Microbiol 7:147–155. doi: 10.1111/j.1462-5822.2004.00443.x. [DOI] [PubMed] [Google Scholar]
- 83.Garcia DL, Dillard JP. 2008. Mutations in ampG or ampD affect peptidoglycan fragment release from Neisseria gonorrhoeae. J Bacteriol 190:3799–3807. doi: 10.1128/JB.01194-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adin DM, Engle JT, Goldman WE, McFall-Ngai MJ, Stabb EV. 2009. Mutations in ampG and lytic transglycosylase genes affect the net release of peptidoglycan monomers from Vibrio fischeri. J Bacteriol 191:2012–2022. doi: 10.1128/JB.01547-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Juan C, Maciá MD, Gutiérrez O, Vidal C, Pérez JL, Oliver A. 2005. Molecular mechanisms of β-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother 49:4733–4738. doi: 10.1128/AAC.49.11.4733-4738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quénée L, Lamotte D, Polack B. 2005. Combined sacB-based negative selection and cre-lox antibiotic marker recycling for efficient gene deletion in Pseudomonas aeruginosa. Biotechniques 38:63–67. doi: 10.2144/05381ST01. [DOI] [PubMed] [Google Scholar]