

Vibrio cholerae, the causative agent of the diarrheal disease cholera, benefits from a sessile biofilm lifestyle that enhances survival outside the host but also contributes to host colonization and infectivity. The bacterial second messenger c-di-GMP has been identified as a central regulator of biofilm formation, including in V. cholerae; however, our understanding of the pathways that contribute to this process is incomplete. Here, we define a conserved signaling system that controls the stability of large adhesion proteins at the cell surface of V. cholerae, which are important for cell attachment and biofilm formation. Insight into the regulatory circuit underlying biofilm formation may inform targeted strategies to interfere with a process that renders this bacterium remarkably adaptable to changing environments.
KEYWORDS: Vibrio cholerae, adhesins, biofilms, cell signaling, proteases
ABSTRACT
The dinucleotide second messenger c-di-GMP has emerged as a central regulator of reversible cell attachment during bacterial biofilm formation. A prominent cell adhesion mechanism first identified in pseudomonads combines two c-di-GMP-mediated processes: transcription of a large adhesin and its cell surface display via posttranslational proteolytic control. Here, we characterize an orthologous c-di-GMP effector system and show that it is operational in Vibrio cholerae, where it regulates two distinct classes of adhesins. Through structural analyses, we reveal a conserved autoinhibition mechanism of the c-di-GMP receptor that controls adhesin proteolysis and present a structure of a c-di-GMP-bound receptor module. We further establish functionality of the periplasmic protease controlled by the receptor against the two adhesins. Finally, transcription and functional assays identify physiological roles of both c-di-GMP-regulated adhesins in surface attachment and biofilm formation. Together, our studies highlight the conservation of a highly efficient signaling effector circuit for the control of cell surface adhesin expression and its versatility by revealing strain-specific variations.
INTRODUCTION
The biofilm growth mode is a lifestyle preferred by microorganisms (1). Biofilm-forming ability enhances environmental survival, transmission, and infectivity of microbes, including Vibrio cholerae, the causative agent of the severe diarrheal disease cholera (2, 3). Hallmarks of biofilms include interactions of cells with each other, with biotic or abiotic surfaces, and with their surrounding extracellular matrix. For such interactions, bacteria rely on the presence of adhesive, often proteinaceous, appendages on their surface, which include the growing family of repeats-in-toxin (RTX) adhesins that mediate surface attachment of microorganisms (4, 5). Production, cell-surface localization, retention, and regulatory mechanisms for this class of adhesins have been well characterized for a Pseudomonas protein, LapA, whose function is controlled by the bacterial second messenger c-di-GMP (Fig. 1A) (6, 7). LapA is transported to the cell surface by a specific type 1 secretion system (T1SS) where it remains anchored in the system’s outer membrane-associated TolC-like subunit LapE (8, 9). In many bacteria, the RTX adhesin gene is linked to a bacterial transglutaminase-like cysteine proteinase (BTLCP), called LapG in Pseudomonas fluorescens (10–13). LapG acts on the transport intermediate, the adhesin spanning the LapE outer membrane channel, by recognizing and cleaving a specific sequence motif between an N-terminal, highly stable retention domain and the membrane-spanning portion of the adhesin (4, 7, 9, 14). LapG-containing operons also harbor a gene that encodes a conserved c-di-GMP receptor, LapD. LapD is composed of a periplasmic Cache domain, flanked by single-transmembrane helices, followed in the cytoplasm by a juxtamembrane HAMP domain with a signaling (or S) helix that leads into a GGDEF and EAL domain module (11, 15–17). GGDEF domains usually function as enzymatic units with diguanylate cyclase activity that produce c-di-GMP from two molecules of GTP (Fig. 1B); EAL domains, on the other hand, act as phosphodiesterases that degrade c-di-GMP to linear di-GMP (18). However, in LapD, both domains are enzymatically inactive, yet the EAL domain retains its ability to bind c-di-GMP (17).
FIG 1.

Structure of the autoinhibited cytoplasmic module of V. cholerae LapD. (A) Model of posttranslational LapA regulation via LapDG and c-di-GMP in P. fluorescens. (B) Canonical model for the transcriptional regulation of adhesin proteins via c-di-GMP-responsive transcription factors based on P. fluorescens and P. aeruginosa. (C) Sequence conservation between P. fluorescens LapDG and the putative V. cholerae orthologs. (D) Crystal structure of the S helix-GGDEF-EAL domain module of V. cholerae LapD in the absence of c-di-GMP. The bottom panel shows the corresponding structure of P. fluorescens LapD for comparison. The insets show the residues comprising the S helix-EAL domain interface. (E) Superposition of the cytoplasmic modules of V. cholerae and P. fluorescens LapD in their apo state. Structures were superimposed using the EAL domain as reference. Two orthogonal views are shown.
Through detailed structure-function studies, a mechanism for the regulation of LapG via LapD has emerged (Fig. 1A): LapD adopts an autoinhibited conformation that is characterized by the GGDEF domain functioning as a lid atop the EAL domain, which occludes the c-di-GMP binding site (16). This conformation is stabilized by an intramolecular interaction between the S helix of the HAMP domain and the homodimerization interface observed in c-di-GMP-bound EAL domains, thus also preventing EAL domain dimerization (Fig. 1A, left). LapD’s periplasmic domain has a low affinity for the protease LapG in this state, and as a result, LapG can diffuse freely in the periplasm and proteolytically process LapA, releasing the adhesin from the cell surface (16, 19). When c-di-GMP is produced by specific diguanylate cyclases, the second messenger binds to and dimerizes LapD receptors, which increases LapD’s affinity for LapG (Fig. 1A, right) (20–22). LapG that is sequestered to the surface of the inner membrane via LapD is unable to reach and cleave LapA efficiently. Consequently, LapA remains associated with the TolC-like component of the T1SS, which supports cell adhesion (9). The events can be reversed by specific phosphodiesterases that degrade c-di-GMP (23), converting LapD back to an autoinhibited state with low affinity for LapG. Thus, this system controls the reversible adhesion of bacteria through a posttranslational mechanism using c-di-GMP. Closely related, lapD-lapG-containing operons and corresponding adhesins have been predicted in many proteobacteria (11, 16). Regulatory mechanisms similar in part to those described above have been determined experimentally for orthologous systems in Pseudomonas putida (13, 24), Shewanella oneidensis (25, 26), and Bordetella bronchiseptica (27). In Pseudomonas aeruginosa, the corresponding LapD/LapG pair controls surface anchorage of an unrelated type Vb secretion system comprising the outer membrane transporter CdrB and the adhesin CdrA (11, 28, 29).
In addition to posttranslational regulation, c-di-GMP levels also impact transcriptional control of biofilm matrix formation (6, 30–32) (Fig. 1B). For example, expression of the cdrAB operon in P. aeruginosa increases steeply when c-di-GMP is produced by the diguanylate cyclase WspR (28, 33). Likewise, LapA expression in P. putida is controlled by c-di-GMP (34, 35). In both cases, expression is regulated through the conserved AAA+ domain-containing transcriptional regulator FleQ that binds c-di-GMP directly, resulting in a structural change and altered DNA binding properties (36, 37).
Genes encoding LapD and LapG orthologs have been predicted to be present in V. cholerae (11, 16) (Fig. 1C). However, the operon containing these putative orthologs lacks genes for adhesins that could be regulated by this system. Based on our previous work on V. cholerae regulatory networks, we surmised that the flagellum-regulated hemagglutinin FrhA and VCA0849 (named c-di-GMP-regulated adhesin A or CraA from here onwards) would be the adhesins for the V. cholerae LapDG/c-di-GMP signaling module (7, 9, 16, 38). FrhA, which belongs to the RTX adhesin family, facilitates attachment of V. cholerae (O1 classical strain) to mammalian cells and chitin beads (38). Expression of frhA is regulated by a diguanylate cyclase, CdgD. Likewise, transcriptomic analysis of cells with altered levels of c-di-GMP revealed that expression of craA, which encodes a large protein predicted to be secreted by a T1SS, increases at a high level of c-di-GMP (30). Furthermore, craA expression is coregulated with biofilm matrix genes and depends on the presence of the biofilm regulator VpsR (39, 40). The role of craA’s gene product is unclear; however, its transcriptional regulation suggests a contribution to biofilm formation. Finally, CraA in V. cholerae contains a periplasmic consensus sequence that can be cleaved artificially by P. fluorescens LapG (9, 16), hinting at regulation akin to the LapADG system in pseudomonads, a hypothesis we set out to test in this study.
Here, we characterize the transcriptional and posttranslational regulation of two large adhesins in V. cholerae, FrhA and CraA, by the second messenger c-di-GMP. We present structural insight into the autoinhibition and activation of V. cholerae LapD by c-di-GMP. We also show that the BTLCP/LapG ortholog of this organism functions as a calcium-dependent protease that cleaves FrhA and CraA adhesins at consensus sites. In addition to these conserved features, distinct regulation of the two adhesins by c-di-GMP is apparent at transcriptional and functional levels that are also dependent on the genetic background. Together, our study uncovers an expansive regulatory network for controlling reversible cell adhesion in V. cholerae strains via c-di-GMP.
RESULTS
Structural basis for Vibrio cholerae LapD autoinhibition and activation by c-di-GMP.
We previously predicted LapD and LapG orthologs in V. cholerae, albeit with sequence identities of only 27% and 50%, respectively, compared to their P. fluorescens counterpart (Fig. 1C) (11, 16). The low sequence conservation of the LapD ortholog in particular renders predictions regarding the conservation of regulatory features described first for P. fluorescens LapD unreliable. To gain unbiased insight into the structure and regulation of V. cholerae LapD, we determined a crystal structure of its cytoplasmic domains comprising the S helix followed by GGDEF and EAL domains in the absence of c-di-GMP. Crystals diffracted X rays to a maximum resolution of 2.7 Å (see Table S1 in the supplemental material). Phases were obtained by using molecular replacement with the individual GGDEF and EAL domains of the corresponding P. fluorescens structure (PDB code 3pjx [16]) as the search models. The electron density maps resolved the S helix with the final refined model spanning residues 221 to 638 (Fig. 1D, top).
Data collection and refinement statistics. Download Table S1, DOCX file, 0.02 MB (16.1KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Despite the low sequence identity, the structures of V. cholerae and P. fluorescens LapD superimpose with an overall root mean square deviation (RMSD) of 1.7 Å, indicating structural conservation of their apo states (Fig. 1D and E). The major difference pertains to a rigid body rotation of the GGDEF domain of approximately 12° relative to the EAL domain (Fig. 1E). However, the autoinhibitory S helix-EAL domain interaction responsible for P. fluorescens LapD autoinhibition at low cellular c-di-GMP levels is well preserved in the structure of the V. cholerae ortholog. More specifically, residues F223, A227, and R234 of the S helix (corresponding to F222, A226, and R233 of P. fluorescens LapD) nestle against a partially hydrophobic interface on the EAL domain (Fig. 1D). As seen with P. fluorescens LapD, this contact places the GGDEF domain atop the putative c-di-GMP binding site on the EAL domain.
A high-resolution model for the second messenger-activated state of LapD has been elusive. To provide insight into the conformational changes the cytosolic module undergoes when c-di-GMP binds, we here report a structure of the S helix/GGDEF/EAL domain-containing module of V. cholerae LapD in the presence of c-di-GMP at 2.6 Å resolution (Fig. 2 and Table S1). The asymmetric unit of the crystals contained two molecules of LapD that align with an RMSD of 0.4 Å. A minor difference pertains to the angle at which the S helix projects from the GGDEF domain, which differs by 18° between the two molecules. The two protomers form a dimer via homotypic interactions of their S helices and EAL domains (interface areas: S helices, 343 Å2; EAL domains, 525 Å2 [41]) (Fig. 2A). The homotypic S helix interaction appears to be rather weak with only two major hydrophobic contacts (L233 and A237). It remains to be seen whether the helices can pack more tightly, in particular, when the regulatory homodimeric HAMP domain would be present. Based on secondary structure predictions and a recent crystal structure of a HAMP-S helix-containing regulatory module of a bacterial sensor histidine kinase (42), the S helix forms a direct helical extension with the second helix of the HAMP domain, which may establish more favorable helical packing interactions. Hydrophobic residues A227, F223, and A230 of LapD’s S helix are candidate residues for a more extended interaction surface. The GGDEF domains are not involved in intermolecular protein-protein interactions; however, there is a c-di-GMP molecule bound to each of the GGDEF domains that contributes to homodimerization (Fig. 2A and B). This c-di-GMP binding site on V. cholerae LapD’s GGDEF domain is located below the degenerate active site, distinct from nucleotide-binding sites observed in other GGDEF domain structures (see Fig. S1). Several of the residues forming this c-di-GMP binding site are conserved in LapD orthologs (Fig. S1A).
FIG 2.

Structure of the c-di-GMP-bound cytoplasmic module of V. cholerae LapD. (A) Comparison between the c-di-GMP-bound and unbound structures of V. cholerae LapD’s cytoplasmic module. The c-di-GMP-bound protein crystallized with two protein molecules per asymmetric unit (left) (molecule A: S helix, dark blue; GGDEF domain, light green; EAL domain, orange with red α6 helix; molecule B: shades of gray). GGDEF and EAL domains bind a single c-di-GMP molecule each. The insets show the S helix dimerization interface in the c-di-GMP-bound dimer (left inset) or the S helix-α6 helix (EAL domain) interface of the apo state (right inset). The apo state is shown for comparison in a similar view as the in the left panel with regard to the S helix-GGDEF domain fragment (right) (S helix, slate; GGDEF domain, dark green; EAL domain, tan with red α6 helix). LapD’s periplasmic domains are not shown. (B) Detailed view of the c-di-GMP binding site on the GGDEF domain of V. cholerae LapD (colored as in panel A, left). (C) Superposition the structures of apo- and c-di-GMP-bound V. cholerae LapD. The respective GGDEF domains were superimposed to highlight the conformational change of this regulatory module. The relative position of the S helix would be fixed by the preceding HAMP and transmembrane domains (not shown). Two views separated by a 220° rotation along the y axis are shown. The apo state and c-di-GMP-bound (molecule B) protomers are colored as in panel A.
Sequence conservation and comparison of LapD’s c-di-GMP binding site at the GGDEF domain. (A) Surface sequence conservation of the c-di-GMP binding site in LapD orthologs. A sequence alignment was generated using Clustal Omega (1). Conservation scores were mapped on the solvent-accessible surface of a c-di-GMP-bound LapD monomer. The second molecule in the crystal lattice is shown as a grey ribbon. The inset shows a different view of the binding site without the second LapD molecule shown. (B) Comparison of c-di-GMP binding poses on GGDEF domains. Crystal structures of nucleotide-bound GGDEF domains of LapD, XCC4471 (2), and PleD (3, 4) are shown in the same orientation. Residues involved in c-di-GMP binding to LapD (and their corresponding positions in other GGDEF domains) are shown as sticks. Download FIG S1, DOCX file, 0.8 MB (854.2KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A c-di-GMP molecule is also bound to the canonical binding site on the EAL domain. In previously reported c-di-GMP-bound structures, EAL domains homodimerize via a conserved interface involving helix α6 (16, 43–45). In contrast, c-di-GMP binding to V. cholerae LapD appears nonobligatory to such EAL dimerization, since the interface involving helix α6 is involved in neither intradimer contacts nor other packing contacts in the lattice of this crystal form (Fig. 2A). S helix dimerization in the c-di-GMP-bound state utilizes the same face of the helix that rests against the EAL domain in the autoinhibited state (Fig. 2A, insets) (16), suggesting that LapD activation involves a rotation of the cytoplasmic module when c-di-GMP binds to the receptor.
We previously used size exclusion chromatography coupled with multiangle light scattering (SEC-MALS) to determine the molar mass and hence oligomerization in solution of the regulatory cytoplasmic module of P. fluorescens LapD, comprised of S helix, GGDEF, and EAL domains (46). The protein is monomeric in the absence of nucleotide but dimerizes when c-di-GMP is included in the mobile phase of the chromatography (16). To assess the impact of c-di-GMP binding to the GGDEF domain and the homotypic S helix interactions on the structure of LapD, we determined the oligomeric states of the corresponding V. cholerae LapD fragment by the same method (Fig. 3). A comparable result was observed with wild-type V. cholerae LapD, where the protein module is monomeric in the absence of c-di-GMP but forms transient dimers when c-di-GMP is included in the buffer. Mutation of a conserved arginine residue that coordinates c-di-GMP at the GGDEF domain to alanine (R248A) rendered the protein monomeric even in the presence of c-di-GMP. In contrast, mutation of a major c-di-GMP-interacting residue at the canonical EAL domain binding site (R423A) did not affect the oligomerization characteristics of the protein.
FIG 3.

Molecular weight determination shows c-di-GMP-mediated dimerization of the cytoplasmic V. cholerae LapD module. Absolute molecular weights (red and green data points across elution peaks) are plotted on the right axes. Theoretical monomer and dimer molecular weights for the purified S helix-GGDEF-EAL domain-containing LapD fragment are indicated as horizontal dashed lines. Wild-type protein and variants with a point mutation at the c-di-GMP binding site of either the GGDEF (R248A) or EAL (R423A) domain were analyzed using SEC-MALS (90° light scattering, gray solid lines; refractive index signals, black dashed lines; plotted on the left axis).
Together, our structural analysis revealed that the autoinhibited conformation of LapD is a conserved feature in this class of transmembrane c-di-GMP receptors. Activation by c-di-GMP appears to proceed through a large conformational change during which the EAL domain dislodges from the S helix and moves as a rigid body under the GGDEF domain (Fig. 2C). The orientation of the GGDEF domain relative to the S helix appears invariable, stabilized by a conserved salt bridge (D240-R311 in V. cholerae LapD) (16). The observed S helix dimerization upon c-di-GMP binding would require a rotation of this module, likely impacting the conformation of the HAMP, transmembrane, and periplasmic domains as a consequence.
The V. cholerae BTLCP functions as a bona fide LapG ortholog.
Previous bioinformatic predictions identified two large proteins, VC1620/FrhA and VCA0849/CraA, that contain putative N-terminal periplasmic retention domains followed by sequence motifs resembling LapG substrate sites (9, 11, 16, 47) (Fig. 4A). FrhA and CraA differ in their primary sequences but also the types of repeat domains they display at the cell surface. CraA resembles repeats-in-toxin adhesins (RtxA) and contains at least 6 domains that have been classified as T1SS-143 repeat-containing domains (IPR019959) (48–50). FrhA, on the other hand, encompasses Calx-beta and cadherin repeat domains. Importantly, the purified N-terminal fragment of CraA is cleaved by LapG, a BTLCP from P. fluorescens (9). However, the activity of the BTLCP encoded by the V. cholerae genome has not been validated to date, constituting a gap in our understanding of adhesin processing in this organism.
FIG 4.

V. cholerae LapG is an active calcium-dependent protease. (A) Domain organization of CraA and FrhA. The N-terminal domain comprises the periplasmic retention module and predicted consensus sequence for cleavage by LapG. (B) V. cholerae LapG proteolyzes the N-terminal fragments of P. fluorescens LapA, FrhA, and CraA in a calcium-dependent manner. Processing of CraA (relative to LapA and FrhA) required 10-fold more LapG (0.5 versus 5 μM LapG; LapG is indicated by # in the gel showing CraA processing). Representative gels from three independent experiments are shown. (C) Characterization of LapG specificity with the pCleevR assay. A diagram is shown of the pCleevR constructs in which various adhesin sequences containing their respective LapG cleavage sites are flanked N terminally by a His6-tagged SUMO protein and C terminally by sfGFP-A226R. The SDS-PAGE, imaged by in-gel fluorescence to detect fragments containing the sfGFP module, shows LapG activity toward peptides from CraA and FrhA. Proteolysis was prevented in the presence of EGTA or when the consensus di-alanine motif, the site where LapG cleaves its substrates, was mutated to a di-arginine motif. Representative gels from three independent experiments are shown.
Here, we compared the activities of BTLCP (or LapG) orthologs from P. fluorescens and V. cholerae on the N-terminal fragments of the various adhesins (Fig. 4B). As a control and as shown previously (11, 19), the P. fluorescens LapA fragment was processed over time to two smaller subfragments upon incubation with substoichiometric amounts of the endogenous LapG in a calcium ion-dependent manner. No proteolysis was observed in the presence of the calcium-chelating agent EGTA. Incubation of LapA with V. cholerae LapG yielded comparable results with regard to cleavage products and calcium dependence. Notably, V. cholerae LapG also processed the N-terminal fragments of FrhA and CraA in a calcium-dependent manner. Under the chosen conditions, FrhA appeared to be a better substrate than CraA, since 10-fold more LapG (500 nM for FrhA compared to 5 μM for CraA) was required to match similar proteolysis kinetics.
To confirm the exact cleavage site in FrhA and CraA, we assayed V. cholerae LapG activity against peptides spanning the predicted consensus protease recognition and cleavage sites, flanked by SUMO and a superfolder green fluorescent protein (sfGFP) variant (11) (Fig. 4C). The SUMO moiety renders the peptide soluble, while sfGFP enables detection of tagged proteins in SDS-PAGE via in-gel fluorescence (51). Peptide selection was guided by our previous work, in which we showed that optimal proteolysis of LapA by LapG is achieved when the consensus sequence is extended N terminally by a cryptic, poorly conserved motif that is predicted to adopt a helical fold (11). Similar to the results obtained with the N-terminal fragments that also included the adhesin’s retention domain, we observed proteolysis of the FrhA and CraA peptides over time when calcium was available but no cleavage in the presence of EGTA. In contrast to the previous experiment using the N-terminal fragments, V. cholerae LapG cleaved FrhA and CraA peptides with similar efficiencies. LapG is predicted to cleave a di-alanine sequence, and mutation of the motif PAAG to PRRG or AAAG to ARRG in FrhA or CraA, respectively, renders the peptides insensitive to LapG.
In summary, we show that the BTLCP gene adjacent to V. cholerae lapD encodes an active, calcium-dependent LapG-like protease with specificity for both FrhA and CraA, despite variations in the consensus substrate sequences (Fig. 4C).
Expression of frhA and craA is regulated by changes in c-di-GMP levels.
Our previous studies suggest that expression of frhA is modulated by CdgD, a diguanylate cyclase (38), and that craA expression is under the control of an established c-di-GMP-dependent regulatory system (30, 39). Here, we built on the earlier reports and extend our investigation into the transcriptional regulation of the two distinct V. cholerae adhesins, particularly with respect to c-di-GMP signaling.
To provide better insights into c-di-GMP-mediated regulation of FrhA and CraA, we first measured expression of frhA and craA using transcriptional reporters in a V. cholerae strain in which c-di-GMP production is controlled through expression of a diguanylate cyclase CdgF (VCA0956) from an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible promoter (PlacIq-lacI-Ptac-cdgF) (52). We determined the relative levels of c-di-GMP in strains harboring Ptac-cdgF grown to exponential or stationary phase by using a fluorescence-based ratiometric c-di-GMP reporter (53). The relative fluorescence intensity (RFI) values obtained with this reporter are directly proportional to c-di-GMP levels (53, 54). Cells grown to exponential or stationary phase in the presence of 100 μM IPTG showed a 1.3- and 3.3-fold increase in c-di-GMP levels, respectively, compared to those in uninduced cells (Fig. 5A). These results showed that c-di-GMP levels are significantly higher in the presence of induction and that the difference in c-di-GMP levels between uninduced and induced cells is more dramatic in stationary-phase-grown cells.
FIG 5.
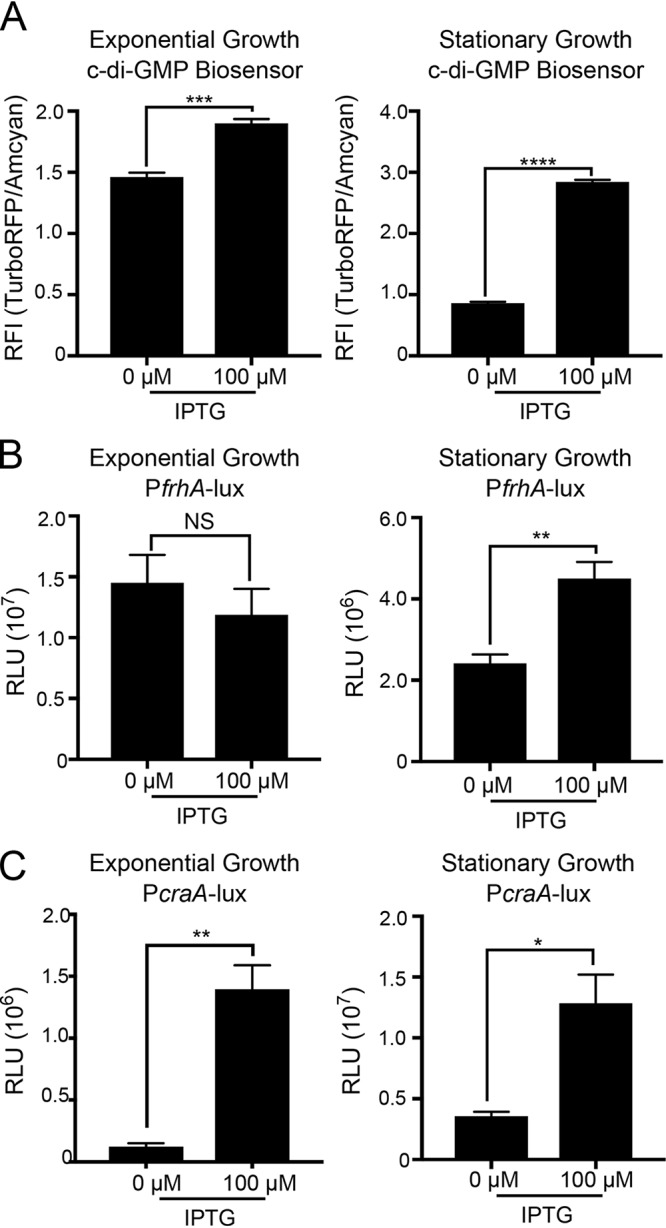
Changes in c-di-GMP levels affect expression of frhA and craA in the strain Ptac-cdgF. (A) Production of c-di-GMP in strain Ptac-cdgF was measured during exponential (left) and stationary (right) phases of growth in the absence or presence of 100 μM IPTG using a c-di-GMP reporter. Fluorescence intensity (FI) of AmCyan is used as a normalizer and FI of TurboRFP is an indicator of c-di-GMP level. Expressions of the transcriptional fusions PfrhA-lux (B) and PcraA-lux (C) were analyzed during exponential (left) and stationary (right) phases of growth in a genetic background where c-di-GMP levels are modulated using inducer IPTG (strain Ptac-cdgF). Cells were grown in the absence or presence of 100 μM IPTG. The graphs represent the means and standard deviations of the relative fluorescent intensity (RFI) (FI TurboRFP/AmCyan) or relative luminescence units (RLU) from at least three independent biological replicates. Means were compared using an unpaired t test with Welch’s correction. Mean differences with a P value of ≤0.05 were deemed significant. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001; NS, not significant.
We next analyzed frhA and craA expression using PfrhA-lux and PcraA-lux in cells grown to exponential or stationary phase in the absence or presence of inducer. While frhA expression was unaffected in exponentially grown cells, frhA expression showed a 1.9-fold increase in cells grown to stationary phase in the presence of inducer in comparison to that in uninduced cells (Fig. 5B). In contrast, c-di-GMP-dependent expression of craA was higher in cells grown either to exponential or stationary phase (an increase of 3.6- or 11.3-fold, respectively, compared to that in uninduced cells) (Fig. 5C). These results indicate that c-di-GMP levels positively regulate expression of frhA in stationary phase and of craA in both exponential and stationary phases.
c-di-GMP receptors regulate transcription of frhA and craA.
c-di-GMP regulates transcription via specific c-di-GMP receptors. In V. cholerae, the transcriptional regulators VpsR, VpsT, and FlrA have been shown to bind to c-di-GMP (32, 55, 56) and regulate gene expression. Therefore, we analyzed the expression of the transcriptional fusions PfrhA-lux and PcraA-lux in wild-type (WT) and ΔvpsR, ΔvpsT, and ΔflrA strains (Fig. 6). Expression of frhA was comparable in all strains when the cells were grown to exponential phase (Fig. 6A). In cells grown to stationary phase, frhA expression was unaltered in ΔvpsR and ΔvpsT strains. In contrast, we observed a 5.5-fold increase of expression in the ΔflrA strain (Fig. 6B). Introduction of a wild-type copy of flrA at the Tn7 site of the ΔflrA strain partially complemented this phenotype, showing only a 2.4-fold increase in frhA expression compared to that in the wild-type strain.
FIG 6.
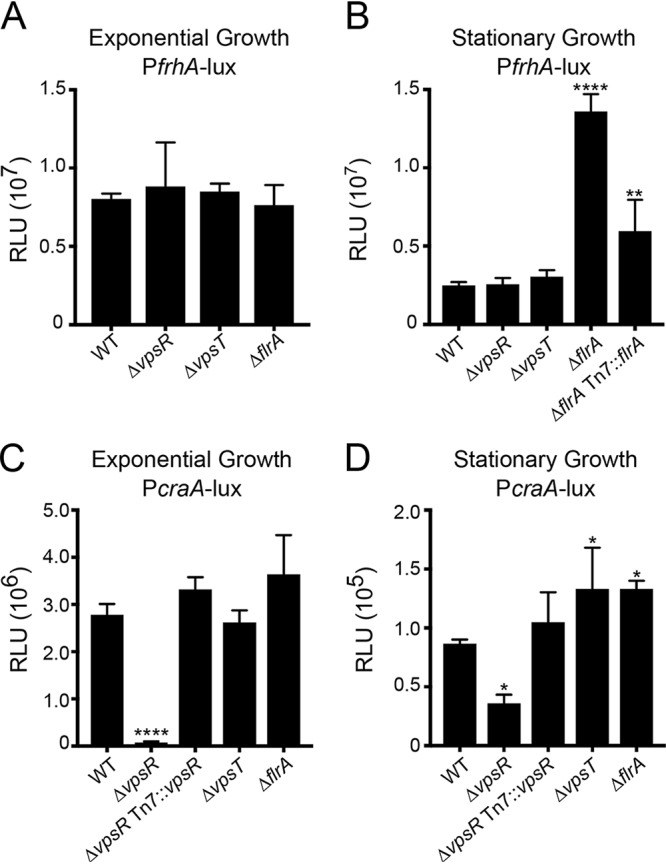
Different c-di-GMP receptors modulate expression of frhA and craA. Expressions of the transcriptional fusion PfrhA-lux (A and B) and PcraA-lux (C and D) were analyzed during exponential (left) and stationary (right) phases of growth in the indicated strains. The graphs represent the means and standard deviations of the relative luminescence units (RLU) from at least three independent biological replicates. Means were compared to WT using a one-way ANOVA and Dunnett’s multiple-comparison test. Mean differences with an adjusted P value of ≤0.05 were deemed significant. *, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001.
In exponentially grown cells, expression of craA decreased 39-fold in the ΔvpsR strain compared to that in the wild-type strain (Fig. 6C). This decrease in expression of craA was fully complemented when vpsR was expressed in trans. The expression of craA in ΔvpsT and ΔflrA strains was unaffected under these conditions. In cells grown to stationary phase, craA expression decreased 2.3-fold in the ΔvpsR strain compared to that in the wild-type strain, and this defect was complemented by the expression of vpsR in trans (Fig. 6D). We also found expression of craA was increased (1.5-fold) in both the ΔvpsT and ΔflrA strains. (Fig. 6D). Taken together, these results show that VpsR is the main activator of craA expression. The roles of VpsT and FlrA are less clear and appear to depend on the growth phase.
We did not observe an effect of the absence of either VpsR or VpsT on frhA expression at basal levels of c-di-GMP. We speculated that the activity of these regulators on frhA expression might require elevated c-di-GMP levels. Hence, we analyzed expression of these genes in a rugose (R) strain of V. cholerae that is documented to have higher cellular c-di-GMP levels (57) and in the corresponding RΔvpsR, RΔvpsT, RΔvpsRT, and RΔflrA strains (see Fig. S2). In cells grown to exponential phase, frhA expression did not change in RΔvpsR, RΔvpsRT, and RΔflrA strains compared to that in the rugose strain (Fig. S2A). However, we observed a 2-fold increase in frhA expression in the RΔvpsT strain (Fig. S2A). In cells grown to stationary phase, we did not observe any difference in frhA expression in RΔvpsR, RΔvpsT, and RΔvpsRT strains (Fig. S2B). In the RΔflrA strain during stationary phase, frhA expression was increased 1.8-fold. These results corroborate FlrA as a repressor of frhA expression during stationary-phase growth.
VpsR and VpsT play different roles in the regulation of frhA and craA in the rugose phase variant of strain A1552. Expressions of the transcriptional fusion PfrhA-lux (A and B) and PcraA-lux (C and D) in rugose genetic backgrounds during exponential (left) and stationary (right) phases of growth. The graphs represent the means and standard deviations of the relative luminescence units (RLU) from at least three independent biological replicates. Means were compared to rugose strain using a one-way ANOVA and Dunnett’s multiple-comparison test. Mean differences with an adjusted P value of ≤0.05 were deemed significant. **, P ≤ 0.01; ****, P ≤ 0.0001. Download FIG S2, DOCX file, 0.06 MB (58.2KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In cells grown to exponential phase, expression of craA decreased 163-fold in the RΔvpsR strain, 2-fold in the RΔvpsT strain, and 149-fold in the RΔvpsR ΔvpsT strain compared to that in the rugose strain (Fig. S2C). Similar changes to craA expression were observed in cells grown to late stationary phase, where expression of craA decreased 48-fold in the RΔvpsR strain, 3-fold in the RΔvpsT strain, and 48-fold in RΔvpsR ΔvpsT strain (Fig. S2D). These results corroborate the role of VpsR as the main regulator of craA and show that VpsT acts as a positive regulator of craA in the rugose strains with high c-di-GMP levels.
The LapDG/c-di-GMP signaling module regulates hemagglutination.
Since FrhA supports hemagglutination (38) and, as we show above, is a target of LapG in vitro (Fig. 4), we investigated next whether the V. cholerae LapDG system impacts hemagglutination of human red blood cells (Fig. 7). We observed no deficiencies in hemagglutination with the ΔlapG, ΔlapD, and ΔcraA mutants but also not with a ΔfrhA mutant (Fig. 7A). One reason for this apparent discrepancy to the previous report (38) regarding the involvement of frhA in this process pertains to the strain background. There are more than 200 serogroups of V. cholerae, and they differ in the expression of virulence factors, surface antigens, and other factors. The V. cholerae O1 serogroup is further divided into classical and El Tor biotypes. Involvement of FrhA in hemagglutination was established in V. cholerae O1 classical strain O395, while our experiments were carried out in V. cholerae O1 El Tor strain A1552. Notably, in V. cholerae El Tor strains, mannose-sensitive hemagglutinin (MSHA), a type 4 pilus, is involved in hemagglutination (58, 59). MSHA pili are assembled on the surfaces of V. cholerae cells of the O1 El Tor strain but not the O1 classical strain. A ΔmshA mutant in the El Tor strain A1552 background was included as a control, which showed significantly reduced hemagglutination (Fig. 7A), confirming that MSHA pili are the dominant adhesion structure driving the hemagglutination phenotype in this strain. We next examined the hemagglutination abilities of the ΔlapG, ΔlapD, ΔcraA, and ΔfrhA mutants generated in the V. cholerae O1 classical strain O395 (Fig. 7B). The absence of the LapG protease led to increased hemagglutination by 3.1-fold, while the absence of the LapD receptor led to a 1.6-fold decrease. In this genetic background, red blood cell hemagglutination was abolished in the ΔfrhA strain. Without LapD, LapG will cleave outer membrane-anchored adhesins constitutively, while a LapG deletion prevents release of the adhesins from the cell surface, explaining the inverse phenotypes of the two mutants observed in the hemagglutination assay. Taken together, these results show that the V. cholerae LapDG/c-di-GMP signaling module controls the presentation of FrhA on the cell surface and, in turn, red blood cell hemagglutination. However, this phenotype is dependent upon the genetic background, likely impacted by the expression of other adhesin-type proteins such as MSHA pili.
FIG 7.
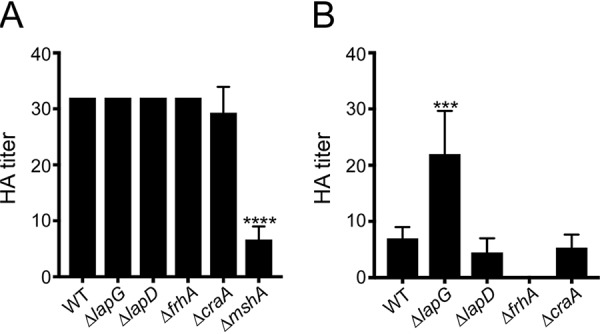
The LapDG system regulates hemagglutination. Hemagglutination assays were performed with V. cholerae O1 El Tor A1552 (A) or O1 classical O395 (B) strains. Either wild-type (WT) or indicated deletion mutants were used. The reciprocal of the lowest fold dilution (hemagglutinin [HA] titer) at which cells were able to agglutinate red blood cells was recorded. The graphs represent the means and standard deviations of HA titers from at least three biological replicates. One-way ANOVA and Dunnett’s multiple-comparison test were used to compare the mean of each mutant to WT. Mean differences with a P value of ≤0.05 were deemed significant. ***, P ≤ 0.001; ****, P ≤ 0.0001.
The LapDG/c-di-GMP signaling module regulates biofilm formation in V. cholerae.
c-di-GMP is a key signaling molecule controlling biofilm formation, and LapDG signaling modules are critical for biofilm formation in P. fluorescens, P. putida, and P. aeruginosa (11, 13, 17, 19, 29). Thus, we next asked whether this function is conserved in V. cholerae. In particular, we analyzed the contributions of LapD, LapG, FrhA, and CraA to V. cholerae biofilm formation when cells were grown under flow conditions. In the V. cholerae O1 classical strain O395, we observed markedly different biofilm formation dynamics for the strains lacking the LapDG signaling module and associated adhesins (Fig. 8A). Deletion of LapG and LapD had inverse effects, showing increased and decreased biofilm formation, respectively. FrhA appeared to contribute more prominently to biofilm formation of this strain than CraA. Quantitative analysis of biofilm formation was performed using COMSTAT to determine biomass, average/maximum thickness, and substrate coverage (see Table S2). For the ΔlapG strain at 24 h, we found that biomass, average thickness, and substratum coverage were 8.3-, 2.1-, and 2.5-fold greater, respectively, than in the wild-type strain. In contrast, in the ΔlapD strain, we observed 14.9-, 1.2-, and 14.9-fold lower biomass, thickness, and substratum coverage, respectively, than in the wild type. While both adhesins contribute to biofilm formation, trends that FrhA deletion has a more pronounced effect on biofilm formation than a CraA deletion are also reflected in this quantitative analysis (fold decreases in biomass, thickness, and coverage in the ΔfrhA strain: 42.9, 1.3, and 41.9, respectively; fold decreases in biomass, thickness, and coverage in the ΔcraA strain: 6.4, 1.2, and 5.0, respectively).
FIG 8.

The LapDG system regulates biofilm formation. Biofilm formation was analyzed using once-through flow cell system using wild-type (WT) and indicated mutants in V. cholerae O1 El Tor A1552 (A) or V. cholerae O1 classical O395 (B) strains. COMSTAT analysis of the biofilms is provided in Table S2 in the supplemental material.
COMSTAT analysis of biofilm properties. Download Table S2, DOCX file, 0.02 MB (16.3KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In the V. cholerae O1 El Tor strain A1552, biofilm formation defects were less pronounced than those in the V. cholerae O1 classical strain O395. In the A1552 strain, only the lack of LapG resulted in a significant change in biofilm properties, where the ΔlapG mutant formed biofilms with increased surface colonization and small but reproducible increases in biomass and thickness (Fig. 8B and Table S2). These results indicate that V. cholerae biofilm formation is modulated by the LapDG signaling module and its associated adhesions but that strain differences contribute to the relative importance of these adhesion systems for biofilm formation, mirroring the results from the hemagglutination assay.
Though biofilm formation on glass is a very fruitful environment for studying biomass accumulation and architectural development, V. cholerae forms biofilms on numerous other surfaces in natural contexts (3, 60). As the differences in biofilm formation for ΔcraA and ΔfrhA backgrounds relative to that for the wild type were only modest on glass for V. cholerae O1 El Tor A1552, we wondered if the contributions of the FrhA and CraA adhesins to biofilm formation would be more evident on chitin, a surface actively sought by V. cholerae in aquatic environments. To occupy and successfully digest chitin for consumption via exoenzyme secretion, V. cholerae must produce biofilms on particles of chitin polymer (61). To test for biofilm growth on chitin, we used a customized microfluidic assay in which pieces of chitin (sterilized shrimp shell) were immobilized, colonized with V. cholerae inoculum, and perfused with a defined seawater medium (61, 62). Attachment and subsequent biofilm growth on the chitin can then be quantified using confocal microscopy (Fig. 9). As shown previously (63, 64), ΔmshA and ΔpilA strains had dramatic 10-fold reductions in chitin attachment in the A1552 El Tor background. We found that on chitinous surfaces, the ΔfrhA and ΔcraA strains showed 4.2-fold and 2-fold reduced biomass, respectively, compared to that of the A1552 wild-type strain. A double deletion strain of both ΔfrhA and ΔcraA showed biomass accumulation (3.9-fold reduction) and a visual phenotype indistinguishable from those of the ΔfrhA single deletion mutant. These results suggest that both adhesins play a role in initial biofilm formation of V. cholerae on chitin in its natural marine environment.
FIG 9.

ΔcraA and ΔfrhA mutants in V. cholerae O1 El Tor A1552 show attachment defects on chitin. All images are top down 3D renderings with dimensions of 212 μm by 212 μm by 33 μm (length by width by depth); red indicates cellular biomass and cyan shows the chitin particles. Quantification of the biomass in of each strain was performed after 48 h of growth of 3 biological replicates. Wilcoxon signed-ranks tests were performed to compare each mutant to the wild type (WT). *, P < 0.05. All other mutants shown were found to not be significantly different from WT.
DISCUSSION
Biofilm formation is a multifactorial and highly regulated process. A central modulator of biofilm formation in a diverse group of microorganisms is c-di-GMP. In this study, we characterized a conserved c-di-GMP receptor, LapD, and its associated periplasmic protease, LapG, and established the pair’s role in controlling the stability of two c-di-GMP-regulated V. cholerae adhesins, FrhA and CraA. We also demonstrated that the V. cholerae LapG ortholog is an active calcium-dependent protease that processes both FrhA and CraA (Fig. 4), thus controlling cell adhesion and biofilm formation.
Based on structural work, we show that the Vibrio cholerae LapD ortholog adopts a similar autoinhibited conformation utilizing the same intramolecular interfaces as determined for the related P. fluorescens LapD, despite low sequence conservation (Fig. 1). We also determined for V. cholerae LapD the hitherto elusive structure of this cytoplasmic module bound to c-di-GMP, which identified a second c-di-GMP binding site on the GGDEF domain in addition to the canonical site located on the EAL domain (Fig. 2). While the noncanonical site appears to be crucial for V. cholerae LapD dimerization of the GGDEF-EAL domains (Fig. 3), its exact function remains to be determined. With regard to the canonical site, c-di-GMP binding does not appear to establish a conformation that supports EAL domain dimerization via the α6 helix-containing interface that has been observed in other LapD and EAL domain protein structures (16, 43–45). Instead, the backsides of the EAL domains contact each other, exposing the c-di-GMP binding site of the EAL domain to the cytosol. Whether this feature correlates with c-di-GMP binding to the GGDEF domain requires further investigation.
The c-di-GMP-bound structure of V. cholerae LapD is consistent with solution scattering-based modeling of full-length P. fluorescens LapD, which suggested a conformation with an exposed EAL domain that, in the fully activated state, can bridge two LapD dimers through EAL domain dimerization (see model in Fig. 1A) (21). But even in the context of P. fluorescens LapD, EAL domain dimerization via the canonical interface is puzzling: it clearly occurs in solution with the isolated EAL domain and the full-length receptor, but biofilm phenotypes of mutant receptors indicate an ancillary role in the regulation of LapD in vivo (16). It is quite possible the phenotypes are complex, since mutations that destabilize the EAL domain dimer are likely to also impact the main autoinhibitory interface between the S helix and the EAL domain (16, 21). A cytosol-facing EAL domain may also enable interactions with other proteins, for example, diguanylate cyclases that produce c-di-GMP (21, 65, 66). Such protein-protein interactions between enzymes and c-di-GMP-binding receptors have been shown to facilitate signaling specificity in complex signaling networks (67, 68).
A second level of c-di-GMP regulation in this system pertains to transcription of the adhesin proteins. We show that transcription of frhA and craA is governed by c-di-GMP-dependent transcriptional activators FlrA, VpsT, and VpsR. In this context, it is important to note that cellular c-di-GMP in V. cholerae is modulated by the growth state; exponentially grown cells have higher c-di-GMP levels than those in stationary phase (69). Transcription of craA is strongly induced by c-di-GMP regardless of the growth stage. Expression of craA depends on the presence of VpsR but is mostly independent of VpsT and FlrA.
In the V. cholerae classical strain O395, expression of frhA has been shown to be regulated as a class 4 gene from the flagellar regulatory hierarchy (38). The expression of frhA was downregulated in the absence of the flagellar regulators (FlrA, FlrBC, and FliA) as well as in the absence of another flagellum-regulated gene, cdgD, which encodes a diguanylate cyclase that negatively impacts motility (38, 54). It is, however, unclear how this c-di-GMP-synthesizing enzyme regulates frhA expression. In the El Tor strain A1552, we found that expression of frhA is upregulated by the absence of the master regulator of flagellar genes, FlrA, only during late stationary phase. It has yet to be determined if FlrA can directly regulate frhA or if it acts indirectly. Coincidently, expression of frhA was upregulated by increased c-di-GMP levels only during late stationary phase. Since FlrA is allosterically inhibited by c-di-GMP, it is possible that high c-di-GMP levels partially release frhA transcription from the negative control exerted by FlrA during late stationary phase.
The V. cholerae A1552 rugose genetic background is a phase variant locked in a mode of constitutively high c-di-GMP and biofilm matrix production. In the rugose genetic background, VpsR and VpsT are strongly activated by c-di-GMP and, in turn, activate the transcription of multiple regulatory targets (40). One of these targets is craA, which has a highly conserved VpsR binding site 222 bp upstream of its translational start site (TTTCACAATTGAGA) (70). frhA, on the other hand, appears to be negatively regulated by VpsT but not by VpsR in this genetic background. The extent by which VpsR and VpsT act independently is yet to be fully determined. It has been shown that when c-di-GMP levels are elevated, VpsT can antagonize the negative regulation of a few targets of H-NS (70, 71). It is possible that in the rugose background, VpsT releases the repression of a negative regulator of frhA that may be under the control of H-NS or a similar nucleoid-associated protein. The reported consensus sequence for VpsT binding is not present in the regulatory region of frhA. This observation does not rule out a direct involvement of VpsT, since its binding sites tend to be variable (70, 72). Together, these observations suggest that the cell has evolved mechanisms to limit frhA expression under conditions where craA and biofilm matrix gene expression are elevated.
Another layer of control arises from a recent chromatin immunoprecipitation sequencing (ChIP-seq) experiment that revealed cAMP receptor protein (CRP) binding to the regulatory region of frhA, craA, and vpsT—genes that were previously found to be regulated by CRP in a whole-genome expression analysis study designed to identify the CRP regulon (73, 74). It is possible that some of the variations in expression of these genes, and especially frhA, involve changes in cAMP and CRP abundance and/or CRP activity. In addition, frhA expression is also repressed by the transcriptional regulator TfoY (75). TfoY expression itself is controlled by at least two mechanisms, involving either a Vc2 c-di-GMP binding riboswitch or transcriptional control via the c-di-GMP binding protein VpsR, depending on the cellular c-di-GMP levels (76).
Regarding posttranslational regulation of the adhesins, we found that the N-terminal domains of CraA and FrhA were cleaved by V. cholerae LapG, which strongly suggests that release of these proteins from the cell surface functions in analogy to the system encoded by the lap operon in P. fluorescens (6, 7). The mechanisms by which FrhA and CraA are secreted and retained on the cell surface of V. cholerae are not well understood. CraA is related to P. fluorescens LapA. Secretion of LapA is linked to a specific T1SS that anchors the adhesin at the cell surface. In P. fluorescens, the genes encoding LapA, LapG, LapD, and the specialized T1SS (LapE, LapB, and LapC) are located in close proximity. The gene encoding FrhA (VC1620) in V. cholerae is in close proximity only to the gene VC1621, which encodes a LapE-like protein that in P. fluorescens forms the conduit for anchoring LapA at the cell surface (7, 9). Orthologues of lapG, lapD, lapB, and lapC are located in the second chromosome of V. cholerae (VCA1081, VCA1082-3, VCA1084, and VCA1080, respectively). The genomic context of craA (VCA0849) includes a diguanylate cyclase (VCA0848), which could be functionally linked to this c-di-GMP-regulated adhesin, and genes predicted to encode ABC-type transport proteins (VCA0854 and VCA0855) but lacks a gene encoding a LapE-like protein. Whether FrhA and CraA utilize the same T1SS for transport and outer membrane anchorage or adhesin-specific systems requires further characterization.
We found that the LapDG system as well as FrhA and CraA plays a role in biofilm formation in a strain- and surface-specific manner (Fig. 8 and 9; see also Table S2 in the supplemental material). Functionally, the lack of FrhA prevents the V. cholerae O1 classical strain from forming biofilms or hemagglutinating red blood cells. The lack of CraA in the O1 classical strain has a negative impact on biofilm formation but does not affect hemagglutination. The absence of the protease LapG results in a marked increase in biofilm formation and hemagglutination compared to that for the wild type, indicating the regulation of the two adhesins via this protease in vivo. In contrast, in the El Tor strains, the contributions of the LapDG system and FrhA and CraA adhesins to biofilm formation were more evident when biofilms were grown on chitin, the natural substratum and growth substrate for Vibrio spp. in aquatic environments.
Differences in the regulation of the same gene in the classical versus El Tor strains are not uncommon, and perhaps, in this particular case, these differences have been selected due to particular requirements for the frhA product for the survival and/or adaptation of these two strains. It is important to bear in mind that MSHA pili are not produced in the classical strain O395. As a result, FrhA and perhaps other adhesins are needed to substitute for MSHA function in this strain. Furthermore, Vibrio strains can differ with regard to the numbers of repeat units in their adhesins (e.g., in the case of FrhA), which also could contribute to differences in surface adhesion. The nature of the surface to which V. cholerae cells adhere may dictate what combination of adhesins need to be used in order to attach strongly enough to be able to colonize the surface. For instance, it is not known if c-di-GMP, FrhA, and/or CraA contribute to the enhanced attachment and growth on chitinous surfaces of the filamentous V. cholerae O139 strain CVD112 (62). Another open question pertains to the relevance of the distinct domain architecture of the various adhesins, which could add another layer of regulation, for example, through calcium binding to FrhA’s extracellular domains. In analogy to the adhesin P. aeruginosa CdrA interacting with Psl polysaccharide, which enhances stability and packing of the biofilm matrix (77), one may also speculate that V. cholerae adhesins engage in cooperative interactions with each other or Vibrio polysaccharide (Vps). In the future, it will be interesting to elucidate the relative or distinct contributions of Vibrio cholerae adhesins and their molecular regulation in the context of different strains of this biomedically relevant organism. Strain differences concerning adhesion factors may shape microbial fitness, function, and/or ecology, thus contributing to specific host-microbe interactions.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The strains and plasmids used in this study are listed in Table S3 in the supplemental material. Escherichia coli CC118λpir strains were used for DNA manipulation, and E. coli S17-1 (λ pir) and SM10 (λ pir) strains were used for conjugation with V. cholerae. V. cholerae and E. coli strains were grown aerobically at 30°C and 37°C, respectively, unless otherwise stated. Cultures were grown in lysogeny broth (LB) (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl [pH 7.5]). LB agar medium contains granulated agar (BD Difco, Franklin Lakes, NJ) at 1.5% (wt/vol). Antibiotics and inducers were used, when necessary, at the following concentrations: ampicillin (Ap), 100 μg/ml; kanamycin (Kan), 50 μg/ml; rifampin (Rif), 100 μg/ml; gentamicin (Gm), 15 μg/ml; chloramphenicol (Cm), 20 μg/ml for E. coli and 5 μg/ml for V. cholerae; and isopropyl β-d-1-thiogalactopyranoside (IPTG), 0.1 mM. For large-scale protein expression, E. coli BL21(DE3) strains (New England BioLabs, Ipswich, MA) were grown in Terrific broth (TB) (24 g/liter yeast extract, 20 g/liter tryptone, 17 mM KH2PO4, 72 mM K2HPO4, 8 ml/liter glycerol) (BD Difco, Franklin Lakes, NJ).
Strains and plasmids. Download Table S3, DOCX file, 0.03 MB (35.3KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Recombinant DNA techniques and plasmid and strain constructions.
DNA manipulations were carried out by standard molecular techniques according to the manufacturers’ instructions. Restriction and DNA modification enzymes were purchased from New England BioLabs (Ipswich, MA). PCRs were carried out using primers purchased from ELIM Biopharmaceuticals, Inc. (Hayward, CA) and the PfuUltra II Fusion HS DNA polymerase (Agilent Technologies, Santa Clara, CA). Sequences of the primers used in the present study are available upon request. Plasmid sequences were verified via DNA Sanger sequencing (Cornell Genomics Facility, Ithaca, NY). Site-directed point mutations were introduced using a QuikChange II kit (Agilent Technologies, Santa Clara, CA).
Plasmids were constructed using standard molecular cloning techniques or the Gibson assembly recombinant DNA technique (New England BioLabs, Ipswich, MA). In-frame gene deletions were generated through allelic exchange of a native open reading frame (ORF) with the truncated ORF, as described previously (78). Both fluorescent protein-tagged and chromosomal complementation strains were generated through a Tn7-based system that inserts in the genomic region between loci VC0487 and VC0488, as described previously (78). Triparental conjugation was performed, with donor E. coli S17-1 (λ pir) cells carrying either pGP704::Tn7-GFP (or desired promoter-gene in place of GFP), as well as a second helper E. coli S17-1 (λ pir) harboring pUX-BF13 carrying the Tn7 transposase gene. Transconjugants were selected for on thiosulfate-citrate-bile salts-sucrose (Difco) agar medium containing 15 μg/ml gentamicin at 30°C. Tn7 insertion strains were verified by PCR analysis. Transcriptional fusions were generated by cloning the upstream regulatory region (∼300 to 500 bp) of genes of interest into the promoterless pBBRlux plasmid.
For c-di-GMP induction assays, allelic exchange was used to introduce an IPTG-inducible promoter in place of the native promoter of the diguanylate cyclase VCA0956. The promoter swap was conducted in strain A1552 (this genetic variant is designated Ptac-cdgF). A plasmid-based dual-fluorescent c-di-GMP biosensor was used for c-di-GMP measurements. A Pbe promoter regulated constitutive expression of an AmCyan-Bc3-5-TurboRFP cassette, in which Bc3-5 is a triple-tandem c-di-GMP-sensing riboswitch adapted from that described in reference 53 that enables TurboRFP expression in the presence of c-di-GMP. AmCyan is used as a normalizer. A toxin-antitoxin system (hok-sok) was introduced to the plasmid to remove the need for antibiotics during the assays (54).
Protein expression and purification.
DNA fragments encoding the soluble portion of the V. cholerae LapD ortholog (residues 221 to 636 or 23 to 636 of VCA1082/1083) were amplified from V. cholerae chromosomal DNA by PCR and cloned into a pET28-based vector, which adds an N-terminal Ulp1-cleavable His6 SUMO tag. E. coli BL21(DE3) cells harboring the expression plasmid were grown shaking in in Terrific broth (TB) medium (12 g/liter tryptone, 24 g/liter yeast extract, 5 g/liter glycerol, 0.017 M KH2PO4, and 0.072 M K2HPO4) supplemented with 50 μg/ml kanamycin at 37°C. When cultures reached an optical density at 600 nm (OD600) of 0.8, the temperature was reduced to 18°C and protein expression was induced by addition of 0.5 mM IPTG. Proteins were expressed for 16 h, after which, cells were harvested by centrifugation, resuspended in nickel-nitrilotriacetic acid (Ni-NTA) buffer A (25 mM Tris-HCl [pH 8.5], 500 mM NaCl, and 20 mM imidazole), and flash frozen in liquid nitrogen. A DNA fragment encoding the V. cholerae BTLCP protein lacking the signal peptide (residues 54 to 220 of VCA1081) was amplified from V. cholerae genomic DNA, cloned into the pET28a derivative, and expressed as described above.
DNA fragments encoding segments of VCA0849 (CraA) and VC1620 (FrhA) containing the putative LapG cleavage sites (Fig. 4C) were amplified from V. cholerae genomic DNA and cloned into the pCleevR plasmid (11). Expression from the pCleevR construct creates fusion protein with an N-terminal His6 SUMO domain and a C-terminal superfolder green fluorescent protein variant (sfGFP-A226R) tag used for in-gel visualization of cleavage reactions (51). Fragments corresponding to the predicted N-terminal retention domain and the LapG cleavage sites of CraA and FrhA (residues 1 to 212 of CraA and 1 to 210 of FrhA) were amplified from genomic DNA and cloned into pET21 or the pET28 derivative, respectively. A corresponding expression plasmid for P. fluorescens LapA (LapA N terminus) was described previously (11, 19). Adhesin proteins fragments were expressed as described above.
For purification of proteins expressed in E. coli, frozen cell suspensions were thawed, and cells were lysed by sonication. After centrifugation, clarified lysates were incubated with Ni-NTA Superflow resin (Qiagen, Hilden, Germany) that was equilibrated with Ni-NTA buffer A. The resin was washed with 20 column volumes of buffer A, followed by elution of protein using 3 column volumes of Ni-NTA buffer B (Ni-NTA buffer A supplemented with 300 mM imidazole [pH 7.5]). Eluted proteins were buffer exchanged into Ulp-buffer (25 mM Tris-HCl [pH 7.5], 500 mM NaCl, 20 mM imidazole) using a fast-desalting column (GE Healthcare Life Science, Marlborough, MA). For BTLCPs and LapD orthologs, Ulp1 (a sumo protease) was added, followed by incubation at 4°C overnight. The reaction was subjected to a second Ni-NTA column step with the flowthrough containing the Ulp1-cleaved untagged target proteins. All proteins were concentrated using Amicon Ultra filters (Millipore, Burlington, MA) with a 10-kDa cutoff, and concentrated protein was subjected to size exclusion chromatography on a Superdex 200 column (GE Healthcare Life Science, Marlborough, MA) that was equilibrated in gel filtration buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl). Purified proteins were concentrated using 10-kDa-cutoff Amicon Ultra filters, flash frozen in liquid nitrogen, and stored at −80°C.
Crystallization, data collection, and structure solution.
Protein crystals of LapD220–636 were obtained by hanging-drop vapor diffusion, mixing equal volumes (1 μl) of protein (concentrations of 200 to 650 μM) and reservoir solution. For apo-LapD220–636, the reservoir solution consisted of 2.5 M sodium acetate trihydrate, pH 7.0. For c-di-GMP-bound LapD220–636, the reservoir solution consisted of 0.2 M sodium formate, 0.1 M bicine (pH 8.5), 20% (wt/vol) polyethylene glycol (PEG) MME 5000, and 0.5 to 1 mM c-di-GMP (Biolog, Bremen, Germany). Crystals were cryoprotected by soaking them in their respective reservoir solutions supplemented with 15% glycerol (apo) or 20% xylitol (c-di-GMP bound), followed by flash freezing and storage in liquid nitrogen. Data were collected at beamline A1 at the Cornell High Energy Synchrotron Source (CHESS; Cornell University, Ithaca, NY).
Data reduction was carried out with the software package HKL2000 (79) or iMosflm/Aimless (80, 81). Phases for the initial structure were obtained from molecular replacement using the software package Phenix (82) with the isolated GGDEF and EAL domains from the crystal structure of P. aeruginosa LapD (PDB ID 3pjw [16]) as the search models. Refinement in Phenix and COOT (83) yielded the final models. Data collection and refinement statistics are summarized in Table S1. Structural illustrations were made in PyMOL (Schrödinger, LLC, New York, NY). All crystallographic software was distributed by SBGrid (84).
Size exclusion chromatography coupled with multiangle light scattering.
Molecular masses of LapD protein samples were determined using size exclusion chromatography coupled with static multiangle light scattering (SEC-MALS) (46). Purified protein at concentrations between 1 and 15 mg/ml (20 to 300 μM) was subjected to gel filtration on a Bio Sep-SEC-s 3000 column (Phenomenex, Torrance, CA) that was equilibrated in MALS buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl). For samples with c-di-GMP in the mobile phase, 50 μM c-di-GMP was added to the MALS buffer. The SEC was coupled to a static 18-angle light-scattering detector (DAWN HELEOS-II) and a refractive index detector (Optilab T-rEX; Wyatt Technology, Goleta, CA). Data were collected at 25°C each second for 30 min at a flow rate of 1 ml/min. Data analysis was carried out using the program ASTRA V. The detectors were normalized using a sample of 5 mg/ml bovine serum albumin (BSA) (monomeric fraction; Sigma-Aldrich, St. Louis, MO).
LapG proteolysis of substrates.
To assess LapG cleavage of purified peptide substrates (expressed from the pCleevR plasmid as His6-SUMO-peptide-sfGFP fusion proteins), 5 μM BTLCP/LapG was preincubated for 10 min with either 20 mM EGTA or CaCl2 in reaction buffer containing 25 mM Tris HCl (pH 7.5) and 150 mM NaCl (11, 51). Reactions were initiated by addition of BTLCP (0.5 μM final concentration) into 20 μM pCleevR substrates in reaction buffer. Reactions were quenched at 2, 5, and 20 min by mixing an aliquot directly into SDS sample buffer (without sample boiling), and products were analyzed by SDS-PAGE. Gels were imaged by fluorescence (detecting folded sfGFP) using a ChemiDoc imager (Bio-Rad, Hercules, CA). Similar conditions were used for the protease assays with the N-terminal adhesin fragments as the substrates, except for VC0849, for which 5 μM (final concentration) LapG was used. N-terminal adhesin fragments and their proteolytic products were resolved by SDS-PAGE followed by Coomassie staining.
c-di-GMP biosensor assay.
The V. cholerae strain Ptac-cdgF harboring a c-di-GMP dual-fluorescent biosensor was grown aerobically overnight in LB supplemented with gentamicin. Overnight cultures were diluted 1:200 into fresh LB with or without 0.1 mM IPTG. Cells were grown aerobically at 30°C to exponential phase (OD600 of 0.3 to 0.4), and then fluorescence was measured using a PerkinElmer Victor3 multilabel counter (PerkinElmer, Waltham, MA). Another fluorescence measurement was taken in stationary phase. Fluorescence is reported as relative fluorescence intensity (RFI) (FI TurboRFP/FI Amcyan). Assays were repeated for a minimum of three independent biological replicates, with three technical replicates measured for all assays. Statistical significance was determined using an unpaired t test with Welch’s correction.
Luminescence assay.
V. cholerae strains harboring transcriptional reporters were grown aerobically overnight in LB supplemented with 5 μg/ml chloramphenicol. Overnight cultures were diluted 1:200 into fresh LB containing chloramphenicol. When the strain Ptac-cdgF was used, cells were grown in the presence or absence of 0.1 mM IPTG. Cells were grown aerobically at 30°C to exponential phase (OD600 of 0.3 to 0.4), and luminescence was measured using a PerkinElmer Victor3 multilabel counter (PerkinElmer, Waltham, MA). Another luminescence measurement was taken in stationary phase. Luminescence expression is reported as relative luminescence units (RLU; counts min−1 ml−1/OD600 unit). Assays were repeated for a minimum of three independent biological replicates, with three technical replicates measured for all assays. For Ptac-cdgF experiments, statistical significance was determined using an unpaired t test with Welch’s correction.
Hemagglutination assay.
Hemagglutination of V. cholerae strains was tested using human red blood cells (RBCs) (R407-0050; Rockland Immunochemicals, Limerick, PA). Ten percent human RBCs were centrifuged (2,000 × g, 5 min, 4°C) and washed with gentle resuspension in Krebs-Ringer Tris (KRT) buffer (7.5 g/liter NaCl, 0.383 g/liter KCl, 0.318 g/liter MgSO4·7H2O, and 0.305 g/liter CaCl2 in 10 mM Tris-HCl [pH 7.4]) until the supernatant was clear. RBCs were gently resuspended in KRT buffer to a final concentration of 2% (vol/vol). RBCs were kept on ice throughout. Overnight cultures of V. cholerae strains were diluted 1:200 in fresh LB and grown with aeration to an OD600 of 0.3 to 0.4. Five milliliters of each culture was centrifuged (4,000 × g, 10 min, 4°C), washed twice in KRT buffer with resuspension, and concentrated 10-fold in 500 μl KRT buffer. Round-bottom 96-well plates were used for the hemagglutination assay. Two technical replicates were performed per biological replicate. The top row of each plate was left blank of bacterial cells and was used as a negative control. Two hundred microliters of KRT buffer was added to A1, while 200 μl of the 10-fold concentrated V. cholerae cells were added to wells B1 to H1, with a different strain for each row. One hundred microliters of KRT buffer was added to all other wells in the plate. Two-fold serial dilutions were performed horizontally across the plate. For column 12, 100 μl was discarded, resulting in an equal volume per well across the entire 96-well plate. Seventy-five microliters of the 2% human RBCs was added rapidly to each well of the plate with minimal pipetting to prevent RBC lysis. The plates were incubated at 4°C overnight, and images were taken the next morning. The last dilution not showing clear RBC button formation at the bottom of the well was used as the hemagglutination titer for that strain. One-way analysis of variance (ANOVA) and Dunnett’s multiple-comparison test were used for determination of statistical significance.
Flow cell biofilm experiments and confocal laser scanning microscopy.
Overnight cultures of gfp-tagged V. cholerae strains were diluted 1:200 and used as flow cell inoculum. One hundred fifty microliters of diluted cells was injected into an Ibidi μ-Slide VI 0.4 (Ibidi 80606; Ibidi LLC, Verona, WI) and left for 1 h at room temperature with no flow to allow adherence to the substratum. A flow of 10% LB (with full strength NaCl) was initiated at a rate of 4.5 ml/h. Confocal laser scanning microscopy (CLSM) images of the biofilms were captured with an LSM 880 (Zeiss, Jena, Germany), using an excitation wavelength of 488 nm and an emission wavelength of 543 nm. Three-dimensional (3D) images of the biofilms were processed using Imaris software (Bitplane, Zurich, Switzerland). All image processing parameters in Imaris (opacity, min, max, etc.) were kept identical for each set of images for accurate comparison. Quantitative analysis was done using COMSTAT2.1 (http://www.comstat.dk) (85). For calculation of substrate coverage with z-stack images, the highest substratum coverage from the bottom 20 stacks was chosen, representing the surface of the slide.
Chitin microfluidic assay.
Microfluidic devices bonded to size 1.5 36-mm by 60-mm coverslips were created using standard soft lithography techniques (86). A columnar chamber design was used to trap the chitin flakes within the device. To place chitin in the device as well as establish flow, the same techniques were used as described in reference 62. Briefly, chitin was placed in the chamber under a high flow rate, and then a syringe pump was used to perfuse the device with defined seawater medium. Strains were grown overnight at 37°C in shaking culture and then normalized to an OD600 of 1 prior to inoculation. Chambers were then inoculated with culture and allowed to rest for 30 min. After this time, a flow regime of 0.2 μl/min was established, and biofilms were allowed to grow for 48 h prior to imaging. Biomass was quantified using custom MATLAB scripts (MathWorks, Natick, MA) as in references 61, 62, and 87.
Data availability.
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (https://www.rcsb.org/) under PDB identifier (ID) codes 6PWJ and 6PWK.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) via grants R01GM123609 (to H.S.) and R01AI114261 (to F.H.Y.). We acknowledge support from the Brown Foundation (to K.E.K.). C.D.N. is supported by the National Science Foundation (MCB 1817342), a Burke Award from Dartmouth College, a pilot award from the Cystic Fibrosis Foundation (STANTO15RO), and NIH grant P20-GM113132 to the Dartmouth BioMT COBRE. This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation under award DMR-1332208, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award GM-103485 from the National Institute of General Medical Sciences, NIH.
We thank Laura Kwuan, Sinem Beyhan, and Nick Fong for their involvement in the generation of a set of strains and plasmids used in this work.
Footnotes
This article is a direct contribution from Holger Sondermann, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Matthew Parsek, University of Washington, and Clay Fuqua, Indiana University Bloomington.
Citation Kitts G, Giglio KM, Zamorano-Sánchez D, Park JH, Townsley L, Cooley RB, Wucher BR, Klose KE, Nadell CD, Yildiz FH, Sondermann H. 2019. A conserved regulatory circuit controls large adhesins in Vibrio cholerae. mBio 10:e02822-19. https://doi.org/10.1128/mBio.02822-19.
REFERENCES
- 1.Flemming HC, Wuertz S. 2019. Bacteria and archaea on Earth and their abundance in biofilms. Nat Rev Microbiol 17:247–260. doi: 10.1038/s41579-019-0158-9. [DOI] [PubMed] [Google Scholar]
- 2.Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. 2016. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol 14:563–575. doi: 10.1038/nrmicro.2016.94. [DOI] [PubMed] [Google Scholar]
- 3.Teschler JK, Zamorano-Sanchez D, Utada AS, Warner CJ, Wong GC, Linington RG, Yildiz FH. 2015. Living in the matrix: assembly and control of Vibrio cholerae biofilms. Nat Rev Microbiol 13:255–268. doi: 10.1038/nrmicro3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo S, Vance TDR, Stevens CA, Voets I, Davies PL. 2019. RTX adhesins are key bacterial surface megaproteins in the formation of biofilms. Trends Microbiol 27:453–467. doi: 10.1016/j.tim.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Satchell KJ. 2011. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu Rev Microbiol 65:71–90. doi: 10.1146/annurev-micro-090110-102943. [DOI] [PubMed] [Google Scholar]
- 6.Krasteva PV, Sondermann H. 2017. Versatile modes of cellular regulation via cyclic dinucleotides. Nat Chem Biol 13:350–359. doi: 10.1038/nchembio.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith TJ, Sondermann H, O'Toole GA. 2018. Type 1 does the two-step: type 1 secretion substrates with a functional periplasmic intermediate. J Bacteriol 200:e00168-18. doi: 10.1128/JB.00168-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinsa SM, Espinosa-Urgel M, Ramos JL, O'Toole GA. 2003. Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol Microbiol 49:905–918. doi: 10.1046/j.1365-2958.2003.03615.x. [DOI] [PubMed] [Google Scholar]
- 9.Smith TJ, Font ME, Kelly CM, Sondermann H, O'Toole GA. 2018. An N-terminal retention module anchors the giant adhesin LapA of Pseudomonas fluorescens at the cell surface: a novel subfamily of type I secretion systems. J Bacteriol 200:e00734-17. doi: 10.1128/JB.00734-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyd CD, Chatterjee D, Sondermann H, O'Toole GA. 2012. LapG, required for modulating biofilm formation by Pseudomonas fluorescens Pf0-1, is a calcium-dependent protease. J Bacteriol 194:4406–4414. doi: 10.1128/JB.00642-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooley RB, Smith TJ, Leung W, Tierney V, Borlee BR, O'Toole GA, Sondermann H. 2016. Cyclic di-GMP-regulated periplasmic proteolysis of a Pseudomonas aeruginosa type Vb secretion system substrate. J Bacteriol 198:66–76. doi: 10.1128/JB.00369-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginalski K, Kinch L, Rychlewski L, Grishin NV. 2004. BTLCP proteins: a novel family of bacterial transglutaminase-like cysteine proteinases. Trends Biochem Sci 29:392–395. doi: 10.1016/j.tibs.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Gjermansen M, Nilsson M, Yang L, Tolker-Nielsen T. 2010. Characterization of starvation-induced dispersion in Pseudomonas putida biofilms: genetic elements and molecular mechanisms. Mol Microbiol 75:815–826. doi: 10.1111/j.1365-2958.2009.06793.x. [DOI] [PubMed] [Google Scholar]
- 14.Guo S, Stevens CA, Vance TDR, Olijve LLC, Graham LA, Campbell RL, Yazdi SR, Escobedo C, Bar-Dolev M, Yashunsky V, Braslavsky I, Langelaan DN, Smith SP, Allingham JS, Voets IK, Davies PL. 2017. Structure of a 1.5-MDa adhesin that binds its Antarctic bacterium to diatoms and ice. Sci Adv 3:e1701440. doi: 10.1126/sciadv.1701440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinsa SM, O'Toole GA. 2006. Biofilm formation by Pseudomonas fluorescens WCS365: a role for LapD. Microbiology 152:1375–1383. doi: 10.1099/mic.0.28696-0. [DOI] [PubMed] [Google Scholar]
- 16.Navarro MVAS, Newell PD, Krasteva PV, Chatterjee D, Madden DR, O'Toole GA, Sondermann H. 2011. Structural basis for c-di-GMP-mediated inside-out signaling controlling periplasmic proteolysis. PLoS Biol 9:e1000588. doi: 10.1371/journal.pbio.1000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newell PD, Monds RD, O'Toole GA. 2009. LapD is a bis-(3’,5’)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc Natl Acad Sci U S A 106:3461–3466. doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conner JG, Zamorano-Sanchez D, Park JH, Sondermann H, Yildiz FH. 2017. The ins and outs of cyclic di-GMP signaling in Vibrio cholerae. Curr Opin Microbiol 36:20–29. doi: 10.1016/j.mib.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newell PD, Boyd CD, Sondermann H, O'Toole GA. 2011. A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol 9:e1000587. doi: 10.1371/journal.pbio.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatterjee D, Cooley RB, Boyd CD, Mehl RA, O'Toole GA, Sondermann H. 2014. Mechanistic insight into the conserved allosteric regulation of periplasmic proteolysis by the signaling molecule cyclic-di-GMP. Elife 3:e03650. doi: 10.7554/eLife.03650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooley RB, O’Donnell JP, Sondermann H. 2016. Coincidence detection and bi-directional transmembrane signaling control a bacterial second messenger receptor. Elife 5:e21848. doi: 10.7554/eLife.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newell PD, Yoshioka S, Hvorecny KL, Monds RD, O'Toole GA. 2011. Systematic analysis of diguanylate cyclases that promote biofilm formation by Pseudomonas fluorescens Pf0-1. J Bacteriol 193:4685–4698. doi: 10.1128/JB.05483-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monds RD, Newell PD, Gross RH, O'Toole GA. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol Microbiol 63:656–679. doi: 10.1111/j.1365-2958.2006.05539.x. [DOI] [PubMed] [Google Scholar]
- 24.Gjermansen M, Ragas P, Sternberg C, Molin S, Tolker-Nielsen T. 2005. Characterization of starvation-induced dispersion in Pseudomonas putida biofilms. Environ Microbiol 7:894–906. doi: 10.1111/j.1462-2920.2005.00775.x. [DOI] [PubMed] [Google Scholar]
- 25.Theunissen S, De Smet L, Dansercoer A, Motte B, Coenye T, Van Beeumen JJ, Devreese B, Savvides SN, Vergauwen B. 2010. The 285 kDa Bap/RTX hybrid cell surface protein (SO4317) of Shewanella oneidensis MR-1 is a key mediator of biofilm formation. Res Microbiol 161:144–152. doi: 10.1016/j.resmic.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Zhou G, Yuan J, Gao H. 2015. Regulation of biofilm formation by BpfA, BpfD, and BpfG in Shewanella oneidensis. Front Microbiol 6:790. doi: 10.3389/fmicb.2015.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ambrosis N, Boyd CD, O Toole GA, Fernández J, Sisti F. 2016. Homologs of the LapD-LapG c-di-GMP effector system control biofilm formation by Bordetella bronchiseptica. PLoS One 11:e0158752. doi: 10.1371/journal.pone.0158752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. 2010. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol 75:827–842. doi: 10.1111/j.1365-2958.2009.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rybtke M, Berthelsen J, Yang L, Hoiby N, Givskov M, Tolker-Nielsen T. 2015. The LapG protein plays a role in Pseudomonas aeruginosa biofilm formation by controlling the presence of the CdrA adhesin on the cell surface. Microbiologyopen 4:917–930. doi: 10.1002/mbo3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beyhan S, Tischler AD, Camilli A, Yildiz FH. 2006. Transcriptome and phenotypic responses of Vibrio cholerae to increased cyclic di-GMP level. J Bacteriol 188:3600–3613. doi: 10.1128/JB.188.10.3600-3613.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickman JW, Harwood CS. 2008. Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol 69:376–389. doi: 10.1111/j.1365-2958.2008.06281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H. 2010. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science 327:866–868. doi: 10.1126/science.1181185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, Wozniak DJ, Harwood CS, Parsek MR. 2009. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martínez-Gil M, Ramos-González MI, Espinosa-Urgel M. 2014. Roles of cyclic di-GMP and the Gac system in transcriptional control of the genes coding for the Pseudomonas putida adhesins LapA and LapF. J Bacteriol 196:1484–1495. doi: 10.1128/JB.01287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao Y, Nie H, Liu H, Luo X, Chen W, Huang Q. 2016. C-di-GMP regulates the expression of lapA and bcs operons via FleQ in Pseudomonas putida KT2440. Environ Microbiol Rep 8:659–666. doi: 10.1111/1758-2229.12419. [DOI] [PubMed] [Google Scholar]
- 36.Baraquet C, Murakami K, Parsek MR, Harwood CS. 2012. The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res 40:7207–7218. doi: 10.1093/nar/gks384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuyama BY, Krasteva PV, Baraquet C, Harwood CS, Sondermann H, Navarro MV. 2016. Mechanistic insights into c-di-GMP-dependent control of the biofilm regulator FleQ from Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 113:E209–E218. doi: 10.1073/pnas.1523148113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Syed KA, Beyhan S, Correa N, Queen J, Liu J, Peng F, Satchell KJ, Yildiz F, Klose KE. 2009. The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J Bacteriol 191:6555–6570. doi: 10.1128/JB.00949-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beyhan S, Bilecen K, Salama SR, Casper-Lindley C, Yildiz FH. 2007. Regulation of rugosity and biofilm formation in Vibrio cholerae: comparison of VpsT and VpsR regulons and epistasis analysis of vpsT, vpsR, and hapR. J Bacteriol 189:388–402. doi: 10.1128/JB.00981-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yildiz FH, Liu XS, Heydorn A, Schoolnik GK. 2004. Molecular analysis of rugosity in a Vibrio cholerae O1 El Tor phase variant. Mol Microbiol 53:497–515. doi: 10.1111/j.1365-2958.2004.04154.x. [DOI] [PubMed] [Google Scholar]
- 41.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 42.Gushchin I, Melnikov I, Polovinkin V, Ishchenko A, Yuzhakova A, Buslaev P, Bourenkov G, Grudinin S, Round E, Balandin T, Borshchevskiy V, Willbold D, Leonard G, Buldt G, Popov A, Gordeliy V. 2017. Mechanism of transmembrane signaling by sensor histidine kinases. Science 356:eaah6345. doi: 10.1126/science.aah6345. [DOI] [PubMed] [Google Scholar]
- 43.Barends TR, Hartmann E, Griese JJ, Beitlich T, Kirienko NV, Ryjenkov DA, Reinstein J, Shoeman RL, Gomelsky M, Schlichting I. 2009. Structure and mechanism of a bacterial light-regulated cyclic nucleotide phosphodiesterase. Nature 459:1015–1018. doi: 10.1038/nature07966. [DOI] [PubMed] [Google Scholar]
- 44.Minasov G, Padavattan S, Shuvalova L, Brunzelle JS, Miller DJ, Basle A, Massa C, Collart FR, Schirmer T, Anderson WF. 2009. Crystal structures of YkuI and its complex with second messenger cyclic di-GMP suggest catalytic mechanism of phosphodiester bond cleavage by EAL domains. J Biol Chem 284:13174–13184. doi: 10.1074/jbc.M808221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tchigvintsev A, Xu X, Singer A, Chang C, Brown G, Proudfoot M, Cui H, Flick R, Anderson WF, Joachimiak A, Galperin MY, Savchenko A, Yakunin AF. 2010. Structural insight into the mechanism of c-di-GMP hydrolysis by EAL domain phosphodiesterases. J Mol Biol 402:524–538. doi: 10.1016/j.jmb.2010.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De N, Navarro MV, Wang Q, Krasteva PV, Sondermann H. 2010. Biophysical assays for protein interactions in the Wsp sensory system and biofilm formation. Methods Enzymol 471:161–184. doi: 10.1016/S0076-6879(10)71010-7. [DOI] [PubMed] [Google Scholar]
- 47.Gu J, Wang Y, Lilburn T. 2009. A comparative genomics, network-based approach to understanding virulence in Vibrio cholerae. J Bacteriol 191:6262–6272. doi: 10.1128/JB.00475-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Geer LY, Bryant SH. 2017. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45:D200–D203. doi: 10.1093/nar/gkw1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI, Gough J, Haft DR, Huang H, Letunic I, Lopez R, Luciani A, Madeira F, Marchler-Bauer A, Mi H, Natale DA, Necci M, Nuka G, Orengo C, Pandurangan AP, Paysan-Lafosse T, Pesseat S, Potter SC, Qureshi MA, Rawlings ND, Redaschi N, Richardson LJ, Rivoire C, Salazar GA, Sangrador-Vegas A, Sigrist CJA, Sillitoe I, Sutton GG, Thanki N, Thomas PD, Tosatto SCE, Yong SY, Finn RD. 2019. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47:D351–D360. doi: 10.1093/nar/gky1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cooley RB, Sondermann H. 2017. Probing protein-protein interactions with genetically encoded photoactivatable cross-linkers. Methods Mol Biol 1657:331–345. doi: 10.1007/978-1-4939-7240-1_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones CJ, Utada A, Davis KR, Thongsomboon W, Zamorano Sanchez D, Banakar V, Cegelski L, Wong GC, Yildiz FH. 2015. C-di-GMP regulates motile to sessile transition by modulating MshA pili biogenesis and near-surface motility behavior in Vibrio cholerae. PLoS Pathog 11:e1005068. doi: 10.1371/journal.ppat.1005068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou H, Zheng C, Su J, Chen B, Fu Y, Xie Y, Tang Q, Chou SH, He J. 2016. Characterization of a natural triple-tandem c-di-GMP riboswitch and application of the riboswitch-based dual-fluorescence reporter. Sci Rep 6:20871. doi: 10.1038/srep20871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zamorano-Sanchez D, Xian W, Lee CK, Salinas M, Thongsomboon W, Cegelski L, Wong GCL, Yildiz FH. 2019. Functional specialization in Vibrio cholerae diguanylate cyclases: distinct modes of motility suppression and c-di-GMP production. mBio 10:e00670-19. doi: 10.1128/mBio.00670-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsieh ML, Hinton DM, Waters CM. 2018. VpsR and cyclic di-GMP together drive transcription initiation to activate biofilm formation in Vibrio cholerae. Nucleic Acids Res 46:8876–8887. doi: 10.1093/nar/gky606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srivastava D, Hsieh ML, Khataokar A, Neiditch MB, Waters CM. 2013. Cyclic di-GMP inhibits Vibrio cholerae motility by repressing induction of transcription and inducing extracellular polysaccharide production. Mol Microbiol 90:1262–1276. doi: 10.1111/mmi.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beyhan S, Yildiz FH. 2007. Smooth to rugose phase variation in Vibrio cholerae can be mediated by a single nucleotide change that targets c-di-GMP signalling pathway. Mol Microbiol 63:995–1007. doi: 10.1111/j.1365-2958.2006.05568.x. [DOI] [PubMed] [Google Scholar]
- 58.Hanne LF, Finkelstein RA. 1982. Characterization and distribution of the hemagglutinins produced by Vibrio cholerae. Infect Immun 36:209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jonson G, Lebens M, Holmgren J. 1994. Cloning and sequencing of Vibrio cholerae mannose-sensitive haemagglutinin pilin gene: localization of mshA within a cluster of type 4 pilin genes. Mol Microbiol 13:109–118. doi: 10.1111/j.1365-2958.1994.tb00406.x. [DOI] [PubMed] [Google Scholar]
- 60.Meibom KL, Li XB, Nielsen AT, Wu CY, Roseman S, Schoolnik GK. 2004. The Vibrio cholerae chitin utilization program. Proc Natl Acad Sci U S A 101:2524–2529. doi: 10.1073/pnas.0308707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drescher K, Nadell CD, Stone HA, Wingreen NS, Bassler BL. 2014. Solutions to the public goods dilemma in bacterial biofilms. Curr Biol 24:50–55. doi: 10.1016/j.cub.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wucher BR, Bartlett TM, Hoyos M, Papenfort K, Persat A, Nadell CD. 2019. Vibrio cholerae filamentation promotes chitin surface attachment at the expense of competition in biofilms. Proc Natl Acad Sci U S A 116:14216–14221. doi: 10.1073/pnas.1819016116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adams DW, Stutzmann S, Stoudmann C, Blokesch M. 2019. DNA-uptake pili of Vibrio cholerae are required for chitin colonization and capable of kin recognition via sequence-specific self-interaction. Nat Microbiol 4:1545–1557. doi: 10.1038/s41564-019-0479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiavelli DA, Marsh JW, Taylor RK. 2001. The mannose-sensitive hemagglutinin of Vibrio cholerae promotes adherence to zooplankton. Appl Environ Microbiol 67:3220–3225. doi: 10.1128/AEM.67.7.3220-3225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dahlstrom KM, Giglio KM, Sondermann H, O'Toole GA. 2016. The inhibitory site of a diguanylate cyclase is a necessary element for interaction and signaling with an effector protein. J Bacteriol 198:1595–1603. doi: 10.1128/JB.00090-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giacalone D, Smith TJ, Collins AJ, Sondermann H, Koziol LJ, O’Toole GA. 2018. Ligand-mediated biofilm formation via enhanced physical interaction between a diguanylate cyclase and its receptor. mBio 9:e01254-18. doi: 10.1128/mBio.01254-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dahlstrom KM, Collins AJ, Doing G, Taroni JN, Gauvin TJ, Greene CS, Hogan DA, O'Toole GA. 2018. A multimodal strategy used by a large c-di-GMP network. J Bacteriol 200:e00703-17. doi: 10.1128/JB.00703-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarenko O, Klauck G, Wilke FM, Pfiffer V, Richter AM, Herbst S, Kaever V, Hengge R. 2017. More than enzymes that make or break cyclic di-GMP-local signaling in the interactome of GGDEF/EAL domain proteins of Escherichia coli. mBio 8:e01639-17. doi: 10.1128/mBio.01639-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Massie JP, Reynolds EL, Koestler BJ, Cong JP, Agostoni M, Waters CM. 2012. Quantification of high-specificity cyclic diguanylate signaling. Proc Natl Acad Sci U S A 109:12746–12751. doi: 10.1073/pnas.1115663109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zamorano-Sanchez D, Fong JC, Kilic S, Erill I, Yildiz FH. 2015. Identification and characterization of VpsR and VpsT binding sites in Vibrio cholerae. J Bacteriol 197:1221–1235. doi: 10.1128/JB.02439-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ayala JC, Wang H, Silva AJ, Benitez JA. 2015. Repression by H-NS of genes required for the biosynthesis of the Vibrio cholerae biofilm matrix is modulated by the second messenger cyclic diguanylic acid. Mol Microbiol 97:630–645. doi: 10.1111/mmi.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H, Ayala JC, Benitez JA, Silva AJ. 2014. The LuxR-type regulator VpsT negatively controls the transcription of rpoS, encoding the general stress response regulator, in Vibrio cholerae biofilms. J Bacteriol 196:1020–1030. doi: 10.1128/JB.00993-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fong JC, Yildiz FH. 2008. Interplay between cyclic AMP-cyclic AMP receptor protein and cyclic di-GMP signaling in Vibrio cholerae biofilm formation. J Bacteriol 190:6646–6659. doi: 10.1128/JB.00466-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manneh-Roussel J, Haycocks JRJ, Magan A, Perez-Soto N, Voelz K, Camilli A, Krachler AM, Grainger DC. 2018. cAMP receptor protein controls Vibrio cholerae gene expression in response to host colonization. mBio 9:e00966-18. doi: 10.1128/mBio.00966-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Metzger LC, Stutzmann S, Scrignari T, Van der Henst C, Matthey N, Blokesch M. 2016. Independent regulation of type VI secretion in Vibrio cholerae by TfoX and TfoY. Cell Rep 15:951–958. doi: 10.1016/j.celrep.2016.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pursley BR, Maiden MM, Hsieh ML, Fernandez NL, Severin GB, Waters CM. 2018. Cyclic di-GMP regulates TfoY in Vibrio cholerae to control motility by both transcriptional and posttranscriptional mechanisms. J Bacteriol 200:e00578-17. doi: 10.1128/JB.00578-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reichhardt C, Wong C, Passos da Silva D, Wozniak DJ, Parsek MR. 2018. CdrA interactions within the Pseudomonas aeruginosa biofilm matrix safeguard it from proteolysis and promote cellular packing. mBio 9:e01376-18. doi: 10.1128/mBio.01376-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fong JC, Karplus K, Schoolnik GK, Yildiz FH. 2006. Identification and characterization of RbmA, a novel protein required for the development of rugose colony morphology and biofilm structure in Vibrio cholerae. J Bacteriol 188:1049–1059. doi: 10.1128/JB.188.3.1049-1059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 80.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Evans PR, Murshudov GN. 2013. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 84.Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P. 2013. Collaboration gets the most out of software. Elife 2:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, Molin S. 2000. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146:2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- 86.Sia SK, Whitesides GM. 2003. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis 24:3563–3576. doi: 10.1002/elps.200305584. [DOI] [PubMed] [Google Scholar]
- 87.Nadell CD, Drescher K, Wingreen NS, Bassler BL. 2015. Extracellular matrix structure governs invasion resistance in bacterial biofilms. ISME J 9:1700–1709. doi: 10.1038/ismej.2014.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data collection and refinement statistics. Download Table S1, DOCX file, 0.02 MB (16.1KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Sequence conservation and comparison of LapD’s c-di-GMP binding site at the GGDEF domain. (A) Surface sequence conservation of the c-di-GMP binding site in LapD orthologs. A sequence alignment was generated using Clustal Omega (1). Conservation scores were mapped on the solvent-accessible surface of a c-di-GMP-bound LapD monomer. The second molecule in the crystal lattice is shown as a grey ribbon. The inset shows a different view of the binding site without the second LapD molecule shown. (B) Comparison of c-di-GMP binding poses on GGDEF domains. Crystal structures of nucleotide-bound GGDEF domains of LapD, XCC4471 (2), and PleD (3, 4) are shown in the same orientation. Residues involved in c-di-GMP binding to LapD (and their corresponding positions in other GGDEF domains) are shown as sticks. Download FIG S1, DOCX file, 0.8 MB (854.2KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
VpsR and VpsT play different roles in the regulation of frhA and craA in the rugose phase variant of strain A1552. Expressions of the transcriptional fusion PfrhA-lux (A and B) and PcraA-lux (C and D) in rugose genetic backgrounds during exponential (left) and stationary (right) phases of growth. The graphs represent the means and standard deviations of the relative luminescence units (RLU) from at least three independent biological replicates. Means were compared to rugose strain using a one-way ANOVA and Dunnett’s multiple-comparison test. Mean differences with an adjusted P value of ≤0.05 were deemed significant. **, P ≤ 0.01; ****, P ≤ 0.0001. Download FIG S2, DOCX file, 0.06 MB (58.2KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
COMSTAT analysis of biofilm properties. Download Table S2, DOCX file, 0.02 MB (16.3KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains and plasmids. Download Table S3, DOCX file, 0.03 MB (35.3KB, docx) .
Copyright © 2019 Kitts et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (https://www.rcsb.org/) under PDB identifier (ID) codes 6PWJ and 6PWK.