

Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and the leading monogenetic cause of autism. One symptom of FXS and autism is sensory hypersensitivity (also called sensory over-responsivity). Perhaps related to this, the audiogenic seizure (AGS) is arguably the most robust behavioral phenotype in the FXS mouse model—the Fmr1 knock-out (KO) mouse. Therefore, the AGS may be considered a mouse model of sensory hypersensitivity.
Keywords: auditory, Fmr1, fragile X, hypersensitivity, mouse, seizure
Abstract
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and the leading monogenetic cause of autism. One symptom of FXS and autism is sensory hypersensitivity (also called sensory over-responsivity). Perhaps related to this, the audiogenic seizure (AGS) is arguably the most robust behavioral phenotype in the FXS mouse model—the Fmr1 knock-out (KO) mouse. Therefore, the AGS may be considered a mouse model of sensory hypersensitivity. Hyperactive circuits are hypothesized to underlie dysfunction in a number of brain regions in patients with FXS and Fmr1 KO mice, and the AGS may be a result of this. But the specific cell types and brain regions underlying AGSs in the Fmr1 KO are unknown. We used conditional deletion or expression of Fmr1 in different cell populations to determine whether Fmr1 deletion in those cells was sufficient or necessary, respectively, for the AGS phenotype in males. Our data indicate that Fmr1 deletion in glutamatergic neurons that express vesicular glutamate transporter 2 (VGlut2) and are located in subcortical brain regions is sufficient and necessary to cause AGSs. Furthermore, the deletion of Fmr1 in glutamatergic neurons of the inferior colliculus is necessary for AGSs. When we demonstrate necessity, we show that Fmr1 expression in either the larger population of VGlut2-expressing glutamatergic neurons or the smaller population of inferior collicular glutamatergic neurons—in an otherwise Fmr1 KO mouse—eliminates AGSs. Therefore, targeting these neuronal populations in FXS and autism may be part of a therapeutic strategy to alleviate sensory hypersensitivity.
SIGNIFICANCE STATEMENT Sensory hypersensitivity in fragile X syndrome (FXS) and autism patients significantly interferes with quality of life. Audiogenic seizures (AGSs) are arguably the most robust behavioral phenotype in the FXS mouse model—the Fmr1 knockout—and may be considered a model of sensory hypersensitivity in FXS. We provide the clearest and most precise genetic evidence to date for the cell types and brain regions involved in causing AGSs in the Fmr1 knockout and, more broadly, for any mouse mutant. The expression of Fmr1 in these same cell types in an otherwise Fmr1 knockout eliminates AGSs indicating possible cellular targets for alleviating sensory hypersensitivity in FXS and other forms of autism.
Introduction
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and the leading monogenetic cause of autism (Bassell and Warren, 2008). It is caused by loss-of-function mutations in FMR1, which encodes an RNA binding protein, FMRP (fragile X mental retardation protein). Many of the impairments in FXS are reproduced in the FXS mouse model, the Fmr1 knock-out (KO) mouse (Bakker et al., 1994).
Sensory hypersensitivity (or sensory over-responsivity) and abnormal sensory processing occur in 70–90% of FXS and autistic patients, and these traits can significantly disrupt behavior (Musumeci et al., 1994; Miller et al., 1999; Rojas et al., 2001; Baranek et al., 2008; Ben-Sasson et al., 2009; Hagerman et al., 2009). FXS patients display increased physiological auditory responses as observed by the event-related potential (ERP) amplitude and reduced habituation of the ERP in response to repeated sounds (Castrén et al., 2003; Van der Molen et al., 2012; Ethridge et al., 2016). These ERP changes correlate with sensory hypersensitivity and communication deficits in FXS patients, suggesting that hyperexcitability of auditory pathways contributes to these symptoms (Ethridge et al., 2016). Fmr1 KO mice also have an enhanced auditory ERP in the form of reduced habituation and enhanced sound-evoked firing of auditory cortical neurons (Rotschafer and Razak, 2013). The mice also have audiogenic seizures (AGSs; Musumeci et al., 2000; Chen and Toth, 2001).
As a result of these and other observations, it is hypothesized that brain circuits are hyperexcitable in FXS (Contractor et al., 2015). In support of this idea, individuals with FXS have an increased incidence of epilepsy (Musumeci et al., 1999; Sabaratnam et al., 2001; Berry-Kravis et al., 2010). Indeed, in the Fmr1 KO mouse, circuit hyperexcitability and potential underlying mechanisms have been well demonstrated in neocortex and hippocampus (Chuang et al., 2005; Galvez and Greenough, 2005; Gonçalves et al., 2013; Cea-Del Rio and Huntsman, 2014; Zhang et al., 2014a; Contractor et al., 2015). However, establishing a link between a specific hyperexcitable circuit and altered behavior in the Fmr1 KO mouse has been elusive.
We consider the AGS in the Fmr1 KO mice to be a model of sensory stimulus hypersensitivity in FXS. AGSs occur in other autism mouse models, such as with Syngap1 and Ube3a deletion (Jiang et al., 1998; Clement et al., 2012). The AGS is arguably the most robust behavioral phenotype in the Fmr1 KO mouse and has been reproduced in 49 original research articles since 2000 (Table 1) and in multiple strain backgrounds. But interpretations from all these studies are limited by a lack of knowledge of the circuits or cell types in which Fmr1 functions to cause the AGS.
Table 1.
Citations of original research articles reproducing the AGS phenotype
From studies of seizure-prone rat and mouse strains, AGSs likely originate from hyperexcitable circuits in the brainstem (Faingold, 2002, 2004; Ribak, 2017). In the Fmr1 KO, studies using c-fos expression to mark active neurons during the AGS implicate cells in the midbrain and pons (Chen and Toth, 2001), but this experimental approach cannot determine whether FMRP deletion in these active neurons causes the AGS or whether they are simply indirectly activated.
By crossing mice with conditional deletion or expression of Fmr1 with cell type and/or brain region-specific Cre lines, we determined the locus in which Fmr1 deletion causes AGSs. Fmr1 is expressed in many cell types throughout the brain, as follows: neurons, astrocytes, oligodendrocytes, and endothelial cells (Zhang et al., 2014b). Our results indicate that Fmr1 deletion in subcortical glutamatergic neurons that express vesicular glutamate transporter 2 (VGlut2) underlies AGSs. Fmr1 deletion in glutamatergic neurons in the inferior colliculus is necessary for the phenotype, which represents the most precise genetic localization to date for causing AGSs in mice. This latter finding pertaining to the inferior colliculus implicates a potentially hyperexcitable, localized circuit underlying a behavioral phenotype in the Fmr1 KO mouse. Finally, selective Fmr1 expression in glutamatergic neurons in an otherwise Fmr1 KO mouse eliminates AGSs, suggesting that targeting these neurons may be part of a strategy to alleviate sensory hypersensitivity in FXS and in autism.
Materials and Methods
Mice.
For conditional Fmr1 deletion, experimental mice were produced by crossing a sire expressing Cre recombinase (Cre) in a specific neuronal population to an Fmr1loxP/+ dam (Mientjes et al., 2006). We refer to the loxP allele as “conditional off,” or cOFF, and the genotype as Fmr1cOFF/+. For conditional expression of, or “turning on,” the Fmr1 gene, we crossed a Cre sire with an Fmr1loxP-Neo/+ dam (Guo et al., 2011). We refer to the loxP-Neo allele as “conditional on,” or cON, and the genotype as Fmr1cON/+. Without any Cre expression, the male Fmr1cOFF/y and Fmr1cON/y mice used in all experiments are functionally equivalent to wild-type (WT) and Fmr1 KO mice, respectively. The Fmr1cON/y line has some residual FMRP expression at either 1.5% or 10% nominal levels (Guo et al., 2011) (Dr. David Nelson, personal communication). The Fmr1cOFF/+ and Fmr1cON/+ mice were provided by Dr. David Nelson (Baylor College of Medicine, Houston, Texas) as part of the FRAXA Resource Foundation program (https://www.fraxa.org/toward-a-cure/resources/).
We used the following Cre-expressing mice: (1) Emx1Cre/+ (Emx1-Cre KIΔNeo) provided by Drs. Takuji Iwasato (National Institute of Genetics, Mishima, Japan) and Shigeyoshi Itohara (Riken BSI, Wako, Japan) (Iwasato et al., 2000); (2) NexCre/+ provided by Dr. Klaus-Armin Nave (Max Planck Institute, Gottingen) (Goebbels et al., 2006); (3) Vglut2Cre/+ from The Jackson Laboratory (Slc17a6-IRES-Cre; Vong et al., 2011); (4) Vglut1Cre/+ from The Jackson Laboratory (Slc17a7-IRES2-Cre; Harris et al., 2014); (5) Hoxb1Cre/+ (Hoxb1-IRES-Cre) provided by Drs. Russell Ray and Benjamin Arenkiel (Baylor College of Medicine, Houston, TX) and Dr. Mario Capecchi (University of Utah, Salt Lake City, UT; Arenkiel et al., 2003); (6) Egr2Cre/+ from The Jackson Laboratory (Krox20-Cre; Voiculescu et al., 2000); and (7) Ntsr1Cre/+ (GN209, BAC transgenic) from Gensat (for expression pattern, see http://www.gensat.org/creGeneView.jsp?founder_id=44880&gene_id=511&backcrossed=false; Gong et al., 2007) and purchased from the Mouse Mutant Resource and Research Center (Davis, CA). All Cre mice are “knock-in” except for Ntsr1Cre/+. Expression patterns for Vglut2Cre/+ and Vglut1Cre/+ lines are illustrated in the Allen Brain Atlas (http://connectivity.brain-map.org/transgenic). For the examination of Cre expression in the above lines, we used the following two reporter mice created by the Allen Brain Institute and obtained from The Jackson Laboratory: (1) Rosa26tdTomato/+, commonly known as the Ai9 tdTomato reporter (Madisen et al., 2010); and (2) Rosa26EYFP/+, commonly known as the Ai3 EYFP (enhanced yellow fluorescent protein) reporter (Madisen et al., 2010). We provide a table summarizing all Cre lines used, their pattern of expression, and whether Fmr1 deletion in the targeted cells was sufficient or necessary for AGS (Table 2).
Table 2.
Summary table of mouse Cre lines, their targeted cells, and results
| Mouse Cre line | Main target structures/cells | Deletion sufficient for AGS (× cOFF) | Deletion necessary for AGS (× cON) |
|---|---|---|---|
| Emx1Cre/+ | Cortical glutamatergic neurons and glia | No | No |
| vGlut2Cre/+ | Most brain glutamatergic neurons, SGNs | Yes | Yes |
| vGlut1Cre/+ | Glutamatergic neurons: in cortex and some brainstem, SGNs | No | No |
| Hoxb1Cre/+ | Starting at rhombomere 4 and caudal into spinal cord, auditory nuclei in medulla and pons | No | No |
| Egr2Cre/+ | Rhombomeres 3- and 5-derived cells, auditory nuclei in medulla and pons | No | No |
| Ntsr1Cre/+ | Inferior colliculus glutamatergic neurons, pyriform cortex | No | Yes |
Results presented in terms of whether Fmr1 deletion in Cre-targeted neurons was sufficient (from cOFF) or necessary (from cON) for AGS. SGN, Spiral ganglion neuron.
The following strains have been maintained on a C57BL/6J background for at least eight generations, as follows: Fmr1cOFF/+, Fmr1cON/+, Emx1Cre/+, NexCre/+, Hoxb1Cre/+, Ntsr1Cre/+, and Rosa26tdTomato/+. Vglut2Cre/+, Vglut1Cre/+, and Egr2Cre/+ mice were maintained for two to three generations after initial purchase as a C57BL/6J strain. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Audiogenic seizures.
AGSs were induced in 3-week-old male mice [postnatal day 18 (P18) to P24], as described previously (Ronesi et al., 2012; Guo et al., 2016). Briefly, mice were placed in a plastic chamber (30 × 19 × 12 cm) containing a door alarm (GE 50246 personal security alarm) and covered with a plastic lid. A 110–120 dB siren sound was presented to mice for 3 min. Sound intensity was calibrated approximately every 20 experiments with a sound level meter (bandwidth, 300–8000 Hz; model 407730, Extech). Mice were scored for behavioral phenotype based on experimenter observation, as follows: 0 = no response; 1 = wild running; 2 = tonic-clonic seizures; 3 = status epilepticus/death.
For a subset of Fmr1cON/y experiments (those crossed to vGlut1Cre/+, Hoxb1Cre/+, Egr2Cre/+, and Ntsr1Cre/+), we measured the time after sound initiation at which wild running began for all mice with a nonzero AGS score. We pooled all these times over all of the experiments. These times were 32.4 ± 2.6 and 32.1 ± 3.8 s for cON and Cre:cON genotypes, respectively (N = 63 and N = 48, respectively; see genotypic description below). These times and the average AGS scores in these same mice (2.2 ± 0.1 and 2.4 ± 0.1) were not statistically different.
Experimental design and statistical analysis.
Each individual dataset for AGS was defined by the particular Cre mouse line used, and each set used “littermate” comparisons. For example, in conditional deletion experiments using Cre-expressing and Fmr1cOFF/y mice, male mice in a single litter all underwent the AGS protocol and could be any one of the following four genotypes based on their expression of two alleles: (1) WT with no mutant allele, (2) Cre only, (3) cOFF only, and (4) Cre:cOFF. The first three are considered “WT controls.” It was uncommon for a single litter to have three or all four genotypes, but we still refer to the experimental design as littermate comparisons. Conditional expression experiments using Fmr1cON/y mice underwent the identical design. In these cON experiments, WT and Cre mice were considered to be WT controls, but the cON mice were considered to be Fmr1 KO controls and were referred to as the “cON-KO control.” Mutant alleles were always expressed as 1 copy, which was sufficient since Fmr1 is an X-linked gene (Verkerk et al., 1991; O'Donnell and Warren, 2002). AGS experiments were done blind to genotype. Average seizure scores for controls in the different Cre lines varied, but the use of this littermate comparison design decreased any interpretation problems caused by this variability.
AGS scores are ordinal data. Therefore, we used the nonparametric Kruskal–Wallis (K-W) ANOVA followed by the Dunn's multiple-comparisons test to compare AGS scores among the four genotypic groups in each experiment. For another analysis, we calculated the AGS fraction for each genotypic group, which was the fraction of mice with behavioral symptoms of AGS (or in other words, the fraction with a nonzero AGS score). The same Kruskal-Wallis ANOVA followed by the Dunn's multiple-comparisons test was used. In the figures, statistical comparisons are shown only between the gene manipulation group (Cre:cOFF or Cre:cON) and the other control groups. All statistical analyses were performed with GraphPad Prism version 8.0. Data are plotted as the mean ± SE. The sample number (N) is the mouse number, unless stated otherwise.
Organ of Corti dissection for tdTomato reporter expression.
Cochlear tissue containing the organ of Corti and the apical-most spiral ganglion was obtained from two mice expressing Vglut2-Cre and tdTomato reporter alleles and two mice expressing Vglut1-Cre and tdTomato reporter alleles. The procedure was based on methods used in a previous study (King et al., 2014). Mice were killed at age P17 under anesthesia, their tympanic bullas opened, the inner ear localized, and the oval and round windows dislodged. The perilymphatic space was perfused with room temperature 4% formalin before the otic capsule (inner ear) was dissected away and placed in formalin at 4°C overnight. The following day, the ears were rinsed in 0.1 m sodium PBS, at pH 7.4 before microdissection. The partially calcified bone was picked away from the apical half of the cochlea, and the apical portion of the organ of Corti and corresponding modiolar tissue were harvested. The basal portion of the bony otic capsule was then removed exposing the remaining organ of Corti, which was then dissected away from the basal portion of the modiolus. The harvested tissue was mounted in glycerin and coverslipped before observation under a fluorescent microscope.
Immunohistochemistry.
Mice (age, P21) were given intraperitoneal injections of 0.08 ml of ketamine mixed with xylazine (4 mg/ml xylazine in 30 mg/ml ketamine) and perfused intracardially with PBS followed by 4% PFA in 0.01 m PBS. Brains were postfixed in 4% PFA/PBS overnight at 4°C and were cryoprotected in a 30% sucrose/PBS solution until equilibrated. Tissue was sectioned at 25 μm using a CM1950 Leica cryostat. Coronal sections of midbrain were taken and stored in PBS for 24–48 h. Sections were mounted on Fisher Scientific ProbeOn Plus Microscope Slides (Thermo Fisher Scientific), then rinsed and placed in blocking solution (0.01% Triton X-100, 5% goat serum, 1% bovine serum albumin) at room temperature for 2 h. The mounted sections were rinsed and incubated with the following primary antibodies in the blocking solution: mouse anti-NeuN (1:500; catalog #MAB377, Millipore); rabbit anti-GABA (1:1000; catalog #A2052, Sigma-Aldrich); or chicken anti-GFP (1:500; GFP-1020, Aves Labs; Hays et al., 2011). For FMRP labeling, we followed a similar protocol to that previously published (Hodges et al., 2017; Siegel et al., 2017). Mounted sections were rinsed 3 × 5 min in PBS and then transferred to a hot sodium citrate bath for antigen retrieval at 85–95°C and pH 6.0, and incubated for 30 min. The mounted sections were rinsed and placed in blocking solution (0.01% Triton X-100, 5% goat serum, 1% bovine serum albumin) at room temperature for 2 h, then rinsed and incubated with mouse supernatant anti-FMRP (1:1; catalog #2F5–1, Developmental Studies Hybridoma Bank) at 4°C overnight. After rinsing, mounted sections were incubated with Invitrogen Alexa Fluor 488- and 555-conjugated secondary antibodies (1:500; Thermo Fisher Scientific) for 2 h at room temperature, rinsed several times, and coverslipped with Aqua-Poly/Mount (Polysciences).
Optical imaging and image analysis.
Confocal images were acquired on a Zeiss LSM880 confocal microscope with a 5×/numerical aperture (NA) 0.16 objective, a 20×/NA 0.8 objective, or a 63×/NA 1.4 oil-immersion objective. Confocal images were used to analyze EYFP-positive cells colabeled with various cell-specific markers by automated counting in ImageJ.
Results
Fmr1 deletion in cortical excitatory neurons and glial cells are neither sufficient nor necessary for recapitulating the AGS phenotype
Sensory cortices in both Fmr1 KO mice and FXS patients have been reported to have enhanced sensory responses (Rotschafer and Razak, 2013; Zhang et al., 2014a), and sensory neocortical circuits are hyperexcitable, as measured in acute brain slices (Hays et al., 2011; Goswami et al., 2019). Therefore, it is possible that Fmr1 deletion in cortical structures results in enhanced responses in the cortex, which in turn, cause AGSs. Therefore, we first determined whether Fmr1 expression in cortical structures plays a role in the AGS phenotype. We used the Emx1Cre/+ mouse, which expresses Cre mainly in excitatory neurons and glia in cortical structures, as follows: neocortex, piriform cortex, basolateral complex of the amygdala, and hippocampus (Gorski et al., 2002; Iwasato et al., 2004). There is very minor expression in the thalamus, cerebellum, and brainstem. Cre expression and subsequent recombination begins at embryonic day 10 (E10; Iwasato et al., 2004). In Emx1Cre/+:Fmr1cOFF/y male mice, immunohistochemistry for FMRP indicated Fmr1 deletion in cortical structures, but subcortical regions, such as the midbrain, still had normal FMRP expression (Fig. 1A,B; N = 2 mice). We also observed no change in FMRP expression in striatum and thalamus with high-power images (images not shown). GABAergic neurons in the cortex still expressed FMRP (Fig. 1C). Therefore, recombination occurred as expected.
Figure 1.
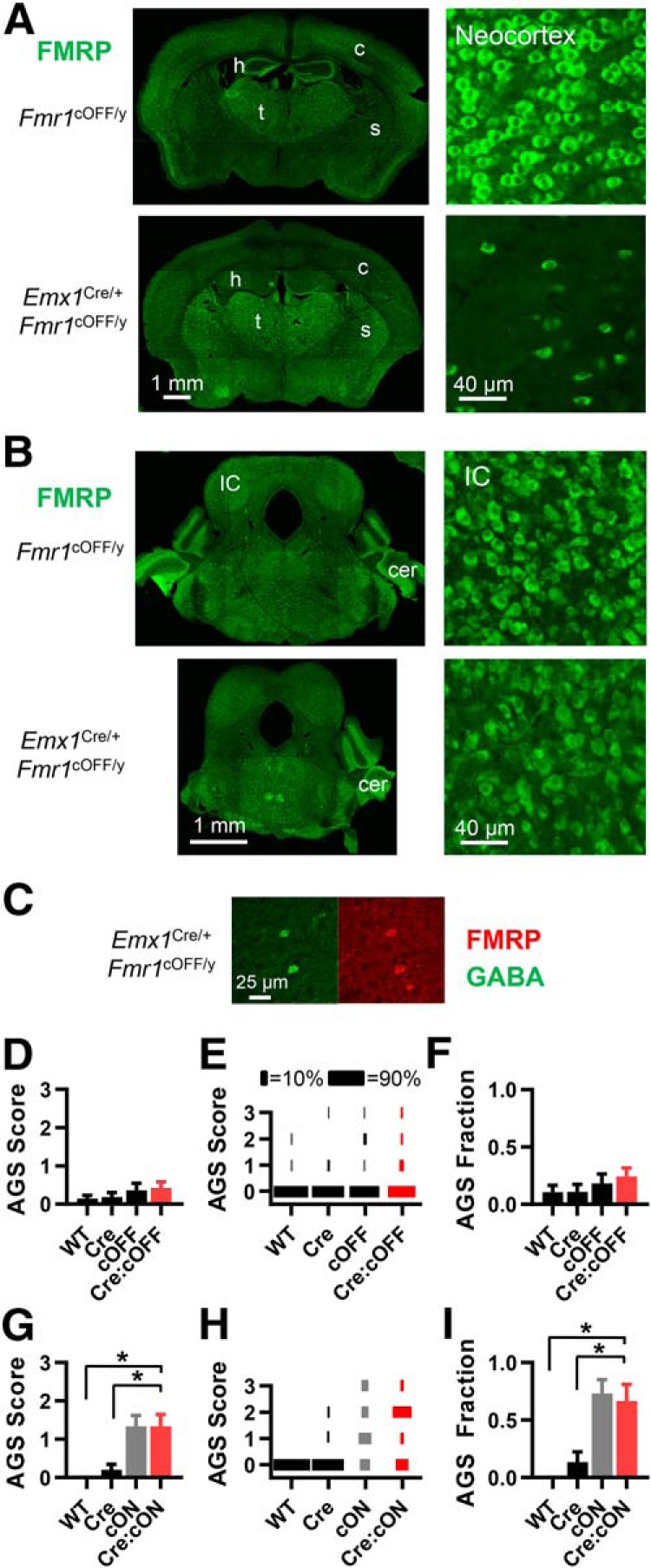
Fmr1 deletion in Emx1-expressing cells, primarily cortical excitatory neurons and cortical glia, is neither sufficient nor necessary for recapitulating the AGS phenotype. A, FMRP immunohistochemistry in forebrain in brain sections obtained from Fmr1cOFF/y and Emx1Cre/+:Fmr1cOFF/y mice. FMRP expression is dramatically reduced in neocortex (c) and hippocampus (h), but not striatum (s) or thalamus (t). B, FMRP expression is unchanged in the midbrain as seen in the inferior colliculus (IC) and cerebellum (cer). Scale bars at bottom apply to the whole column. C, High-power images of cortex showing immunohistochemistry for GABA (green) and FMRP (red) illustrating the selective deletion of Fmr1 in neocortical cells that are not GABAergic. D–F, AGS data for mice derived from Emx1Cre/+ and Fmr1cOFF/y cross-breeding. Deletion in cortex (red bar) does not result in a change in AGS measurements compared with WT controls (black bars). Therefore, deletion was not sufficient for the AGS phenotype. D, AGS scores for the four different possible genotypic combinations. E, Distribution of AGS scores by percentage. F, AGS fraction (the fraction of all mice that had nonzero AGS scores). G–I, Same analysis applied for mice derived from Emx1Cre/+ and Fmr1cON/y cross-breeding. AGS measurements resulting from Fmr1 expression in cortex are no different from cON-KO controls (gray) and increased compared with WT controls (black). Therefore, deletion was not necessary for the AGS phenotype. For all figures, black indicates WT controls, gray indicates the cON-KO control, and red indicates the gene expression manipulation group. N values for AGS data were as follows: cOFF = 28, 27, 22, 33 mice; and cON = 9, 15, 15, and 12 mice. *p < 0.05, K-W ANOVA followed by Dunn's test.
To test whether cortical Fmr1 deletion is sufficient to induce AGSs, we crossed Emx1Cre/+ sires with Fmr1cOFF/+ dams to produce male littermates of all four possible allelic combinations, which were tested for AGSs. We found that deletion in cortical structures did not induce AGSs. When compared with WT control genotypes (WT, Cre, and cOFF), deletion did not increase the average AGS score or AGS fraction—the latter being the fraction of mice with nonzero AGS scores (Fig. 1D–F). We repeated this experiment with another “cortical” Cre line with a very similar spatial and temporal expression profile—the NexCre/+ mouse—except that cortical expression is restricted to glutamatergic neurons (Goebbels et al., 2006; Kazdoba et al., 2012; Itoh et al., 2016). This yielded the same result. For WT, Cre, cOFF, and Cre:cOFF genotypic groups, AGS scores were 0.29 ± 0.29, 0.09 ± 0.09, 0.33 ± 0.33, and 0.5 ± 0.25, respectively (N = 7, 11, 9, and 11, respectively). In summary, these data indicate that Fmr1 deletion in cortical excitatory neurons and glia is not sufficient to recapitulate the AGS phenotype in the Fmr1 KO.
Next, we determined whether cortical Fmr1 deletion was necessary for AGSs. We crossed Emx1Cre/+ sires with Fmr1cON/+ dams to produce male littermates of all four possible allelic combinations. We considered the cON genotypic group as an Fmr1 KO control—referred to as “cON-KO control.” We tested for AGSs and found that deletion in cortical excitatory neurons and glia was not necessary for the AGS phenotype. Mice with selective cortical expression of FMRP had average AGS scores and AGS fractions that were no different from those of the cON-KO control and were significantly larger than those of the two WT controls (WT, Cre; Fig. 1G–I).
These experiments indicate one of two most likely possibilities, as follows: (1) subcortical Fmr1 deletion underlies AGSs; or (2) Fmr1 deletion in GABAergic neurons underlies AGSs.
Fmr1 deletion in VGlut2-expressing glutamatergic neurons is both sufficient and necessary for recapitulating the AGS phenotype
Next, we tested whether glutamatergic neurons could underlie AGSs. If this is the case, this would eliminate the possibility of a cortical GABAergic neuron role indicated by the AGS results in Emx1Cre/+:Fmr1cOFF/y mice and support a role for subcortical structures.
We used the vGlut2Cre/+ mouse to delete Fmr1 in a large proportion of glutamatergic excitatory neurons throughout the brain—forebrain, brainstem, and spinal cord. VGlut2 expression begins before birth (Boulland et al., 2004), and, consistent with this, fluorescent reporter expression induced by the Vglut2-Cre allele is very strong at P4 (Allen Brain Atlas; see Materials and Methods). The vast majority of glutamatergic neurons in the mature brain express VGlut1 (Slc17a7), VGlut2 (Slc17a6), or both (Fremeau et al., 2001; Takamori, 2006). For a large number of VGlut1-expressing neurons, VGlut2 is expressed early in development, but only VGlut1 is expressed later in development (Fremeau et al., 2004). Therefore, the vGlut2Cre/+ mouse will cause Fmr1 deletion in a large fraction of mature VGlut1-expressing neurons (including those in cortical structures), and this is indicated by fluorescent reporter expression (Allen Brain Atlas; see Materials and Methods). We confirmed this widespread Cre expression in vGlut2Cre/+:Rosa26ctdTomato/+ mice based on tdTomato fluorescence, which included cortical structures, thalamus, superior colliculus (SC), and inferior colliculus (Fig. 2A). We also saw tdTomato fluorescence in the spiral ganglion (the ganglia for the auditory nerve; Fig. 2A), which is probably due to early developmental expression of VGlut2 since mature ganglion neurons express only VGlut1 (Zhou et al., 2007; Petitpré et al., 2018). We saw no tdTomato fluorescence in auditory hair cells, which is consistent with previous studies (Seal et al., 2008; Yu and Goodrich, 2014). As indicated previously, Cre expression in these mice occurs in glutamatergic neurons, and not in GABAergic or glycinergic neurons (Vong et al., 2011; Xu et al., 2015; Wozny et al., 2018; Allen Brain Atlas; see Materials and Methods). Moreover, it is unlikely that Cre expression occurs in glia since oligodendrocytes and microglia express VGlut1 (Zhang et al., 2014b) and astrocytes express little or none of either VGlut1 or VGlut2 (Li et al., 2013; Zhang et al., 2014b).
Figure 2.
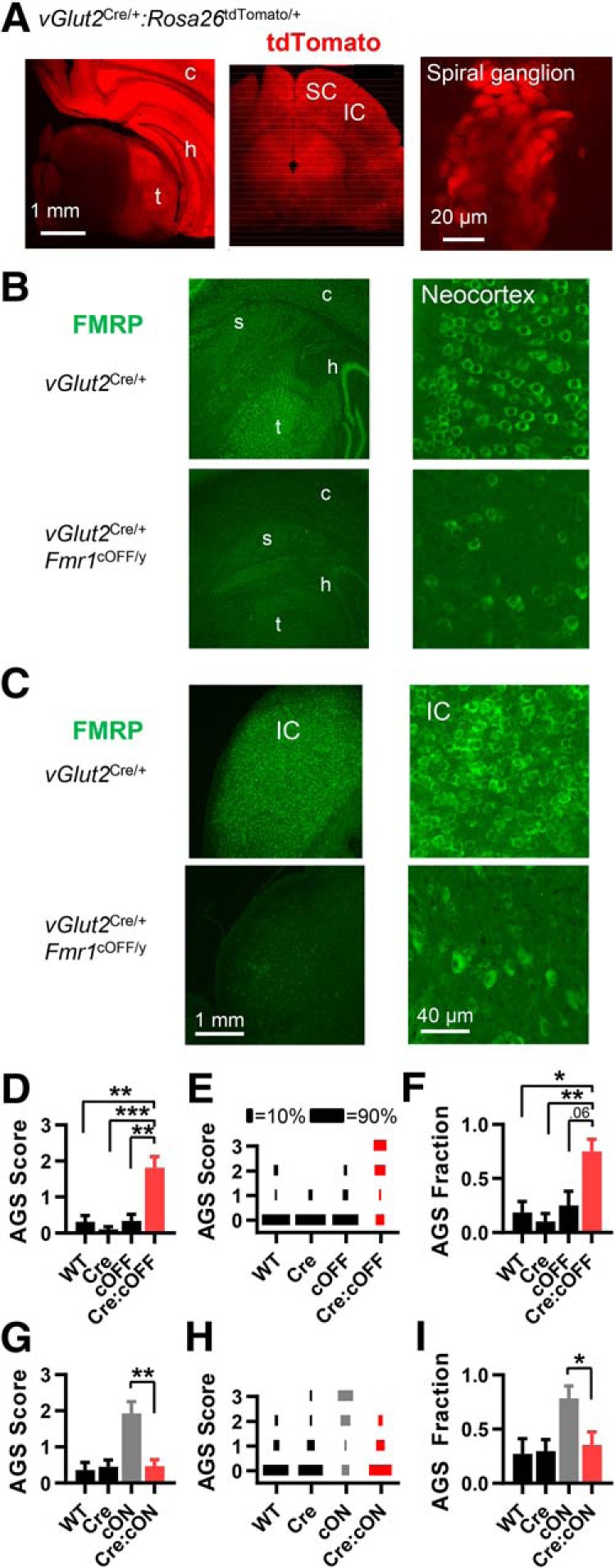
Fmr1 deletion in VGlut2-expressing glutamatergic neurons is sufficient and necessary for recapitulating the AGS phenotype. A, tdTomato fluorescence in live coronal slices obtained from vGlut2Cre/+:Rosa26tdTomato/+ mice. Left, middle, Fluorescence indicates abundant Cre expression in cortical, thalamic, and midbrain regions. Right, With the organ of Corti preparation, fluorescence indicates Cre expression in spiral ganglion neurons. Other abbreviations are stated in Figure 1. B, C, FMRP immunohistochemistry in forebrain (B) and in the inferior colliculus (C) obtained from vGlut2Cre/+ and vGlut2Cre/+:Fmr1cOFF/y mice. Consistent with the tdTomato reporter expression in A, FMRP expression is dramatically reduced in both of these regions. D–F, AGS data for mice derived from vGlut2Cre/+ and Fmr1cOFF/y cross-breeding. Deletion in vGlut2-expressing neurons (in red) results in increased AGS measurements compared with WT controls (black). Therefore, deletion was sufficient for the AGS phenotype. G–I, AGS data for mice derived from vGlut2Cre/+ and Fmr1cON/y cross-breeding. AGS measurements resulting from Fmr1 expression in VGlut2-expressing neurons are no different from WT controls (black) and are reduced compared with the cON-KO control (gray). Therefore, deletion was necessary for the AGS phenotype. N values for AGS data were as follows: cOFF = 16, 19, 12, and 16; cON = 11, 20, 14, and 17. *p < 0.05, **p < 0.01, ***p < 0.001. K-W ANOVA followed by Dunn's test.
We performed FMRP immunohistochemistry on brain slices obtained from vGlut2Cre/+:Fmr1cOFF/y mice and confirmed that FMRP was indeed lost in many brain structures containing glutamatergic neurons, including the neocortex, hippocampus, thalamus, and inferior colliculus (Fig. 2B,C; N = 2 mice). Regions containing primarily GABAergic neurons, such as the striatum, did not differ in FMRP expression. Staining for the neuronal marker NeuN in the inferior colliculus indicated that neuron numbers were not altered with loss of FMRP when compared with controls (data not shown).
Fmr1 deletion in glutamatergic neurons induced the full AGS phenotype, indicating that deletion in these cells was sufficient (Fig. 2D–F). Selective expression of Fmr1 in these neurons normalized the AGS phenotype, indicating that deletion in these neurons is necessary (Fig. 2G–I). These data indicate that Fmr1 deletion in a large subpopulation of excitatory neurons in the brain accounts for AGSs in the Fmr1 KO mouse.
These data rule out the possibility from the Emx1Cre/+ experiments that deletion in GABAergic neurons plays a role. Instead, the other reasonable alternative from the Emx1Cre/+ experiments remains—that subcortical Fmr1 deletion induces AGSs. And, more specifically, the Emx1Cre/+ and vGlut2Cre/+ experiments provide strong evidence that deletion in subcortical VGlut2-expressing glutamatergic neurons underlies AGSs in Fmr1 KO mice.
Fmr1 deletion in VGlut1-expressing glutamatergic neurons is neither sufficient nor necessary for recapitulating the AGS phenotype
As explained above, Fmr1 deletion likely occurs in both VGlut1- and VGlut2-expressing neurons when using the vGlut2Cre/+ mouse. To determine whether the VGlut1- or VGlut2-expressing population plays the most significant role in AGSs, we manipulated Fmr1 expression with the vGlut1Cre/+ mouse. VGlut1 begins to be expressed right after birth (Boulland et al., 2004), and, consistent with this, fluorescent reporter expression in vGlut1Cre/+ mice is clearly observed at P4 (Allen Brain Atlas; see Materials and Methods). In line with a previous study (Fremeau et al., 2001), Cre expression is limited to glutamatergic neurons mainly in cortical structures, but also in a few subcortical structures (Allen Brain Atlas; see Materials and Methods). We also observed that Cre is expressed in the spiral ganglia (Fig. 3A) but not in auditory hair cells, which is consistent with the known expression of VGlut1 (Zhou et al., 2007; Seal et al., 2008; Petitpré et al., 2018). These expression data indicate that Cre expression in vGlut1Cre/+ mice is most likely limited to VGlut1-expressing glutamatergic neurons.
Figure 3.
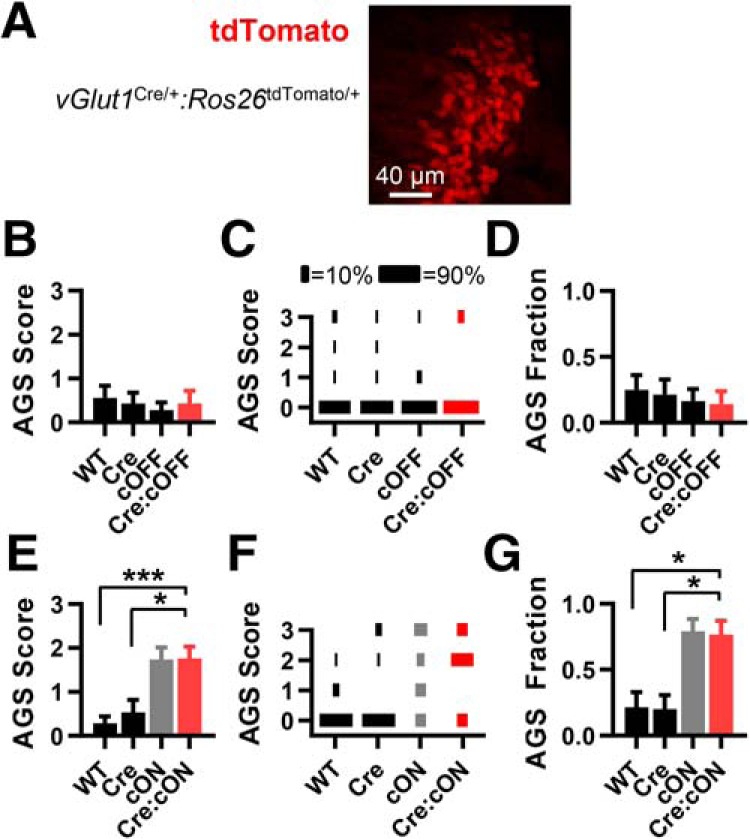
Fmr1 deletion in VGlut1-expressing glutamatergic neurons is neither sufficient nor necessary for recapitulating the AGS phenotype. A, tdTomato fluorescence in spiral ganglion neurons in the cochlea from vGlut1Cre/+:Rosa26 tdTomato/+ mice indicating Cre expression in these cells. B–D, AGS data for mice derived from vGlut1Cre/+ and Fmr1cOFF/y cross-breeding. Deletion in cortex does not result in a change in AGS measurements compared with WT controls. E–G, Data from vGlut1Cre/+ and Fmr1cON/y cross-breeding. AGS measurements resulting from Cre-dependent Fmr1 expression are no different from the cON-KO control and increased compared with WT controls. N values for AGS data were as follows: cOFF = 16, 14, 18, and 14; cON = 14, 15, 19, and 17. *p < 0.05, ***p < 0.001. K-W ANOVA followed by Dunn's test.
We found that Fmr1 deletion limited to VGlut1-expressing neurons was not sufficient to induce AGSs (Fig. 3B–D). And deletion was not necessary in these neurons (Fig. 3E–G). By a process of elimination, these data indicate that Fmr1 deletion in neuron types that express VGlut2, but never express VGlut1, underlies AGSs in the Fmr1 KO.
Based on our vGlut2Cre/+ experiments, it was possible that deletion in spiral ganglion neurons could play a role in AGSs since Cre was expressed in those neurons (Fig. 2A). But in these vGlut1Cre/+ experiments, Cre is also expressed in spiral ganglion neurons, and, therefore, it is unlikely that Fmr1 expression in spiral ganglion neurons plays a role in AGSs.
Fmr1 deletion in subpopulations of neurons in the pons and medulla is neither sufficient nor necessary for recapitulating the AGS phenotype
Since experiments indicate that Fmr1 deletion in subcortical excitatory neuron populations underlies AGSs, we examined the role of Fmr1 expression in different, but overlapping, populations of cells in the caudal pons and medulla.
First, we used the Hoxb1Cre/+ mouse line, which has selective Cre expression starting at rhombomere 4 (r4) and extending into caudal rhombomeres starting at approximately E9. These cells are progenitors to a large subpopulation of neurons in the mature caudal pons down through the spinal cord (Maricich et al., 2009). Related to the auditory system, this line induces recombination in a large proportion of neurons located in the superior olive, nucleus of the trapezoid body, ventral lateral lemniscus, and cochlear nuclei (Arenkiel et al., 2003; Maricich et al., 2009; Di Bonito et al., 2013; Di Bonito and Studer, 2017). Using Hoxb1Cre/+:Rosa26tdTomato/+ mice, we confirmed Cre expression extending from the caudal pons into the spinal cord (Fig. 4A). We found that Fmr1 deletion in these neurons was neither sufficient nor necessary for the AGS phenotype (Fig. 4B–G).
Figure 4.
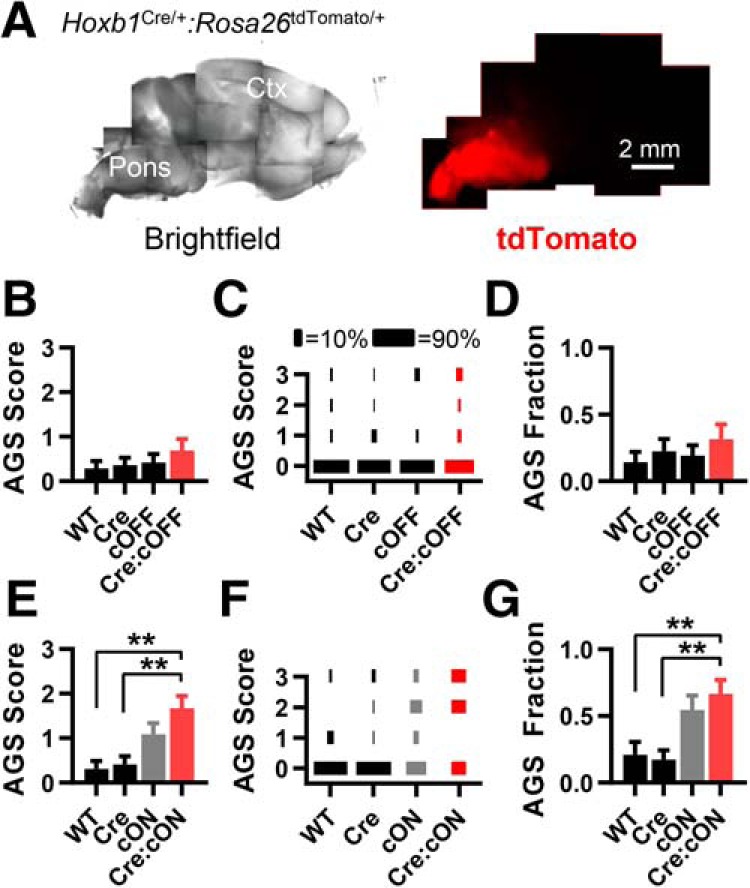
Fmr1 deletion in Hoxb1-expressing cells in the brainstem and spinal cord is neither sufficient nor necessary for recapitulating the AGS phenotype. A, A live sagittal section obtained from Hoxb1Cre/+:Rosa26 tdTomato/+ mice. tdTomato fluorescence indicates Cre expression in caudal Pons and posterior into the spinal cord. B–D, AGS data for mice derived from Hoxb1Cre/+ and Fmr1cOFF/y cross-breeding. Deletion in cortex does not result in a change in AGS measurements compared with WT controls. E–G, Data derived from Hoxb1Cre/+ and Fmr1cON/y cross-breeding. AGS measurements resulting from Cre-dependent Fmr1 expression are no different from the cON-KO control and are increased compared with WT controls. N values for AGS data were as follows: cOFF = 21, 22, 26, and 19; cON = 19, 29, 22, and 21. **p < 0.01. K-W ANOVA followed by Dunn's test.
Next, we used the Egr2Cre/+ mouse line, which has selective Cre expression in r3- and r5-derived cells starting at approximately E9 and populating similar, but not completely overlapping, mature brainstem structures as the Hoxb1Cre/+ mice. This includes the superior olive and cochlear nuclei, but not the spinal cord (Voiculescu et al., 2000; Maricich et al., 2009). Using Egr2Cre/+:Rosa26tdTomato/+ mice, we confirmed Cre expression in limited portions of the caudal pons and medulla, but, unexpectedly, some expression occurred in the superficial layers of motor and somatosensory cortex (data not shown). We found that Fmr1 deletion in these neurons was neither sufficient nor necessary for AGSs (Fig. 5A–F).
Figure 5.
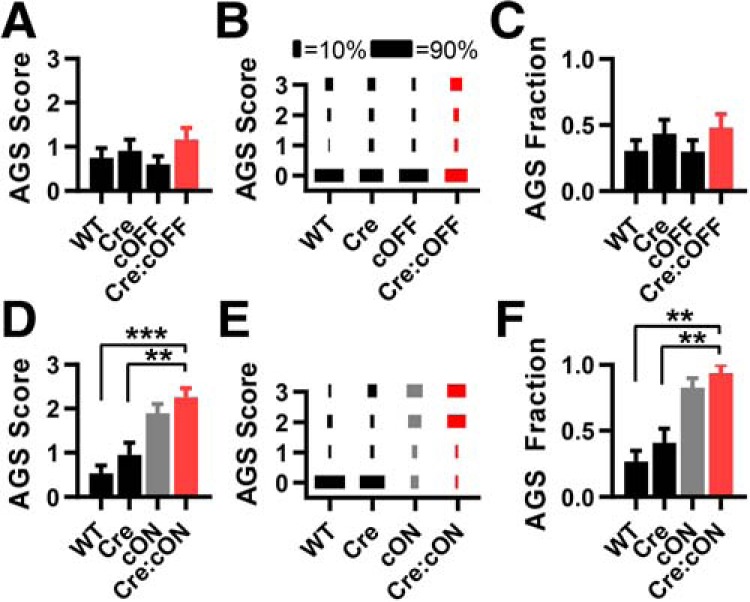
Fmr1 deletion in Egr2-expressing cells in the brainstem and neocortex is neither sufficient nor necessary for recapitulating the AGS phenotype. A–C, AGS data for mice derived from Egr2Cre/+ and Fmr1cOFF/y cross-breeding. Deletion in cortex does not result in a change in AGS measurements compared with WT controls. D–F, Data derived from Egr2Cre/+ and Fmr1cON/y cross-breeding. AGS measurements resulting from expression resulting from Cre-dependent Fmr1 expression are no different from the cON-KO control and increased compared with WT controls. N values for AGS data were as follows: cOFF = 33, 23, 30, and 25; cON = 30, 22, 29, and 16. **p < 0.01, ***p < 0.001. K-W ANOVA followed by Dunn's test.
Fmr1 deletion in the inferior colliculus is necessary for recapitulating the AGS phenotype
The inferior colliculus is hypothesized to contain the circuits that initiate AGSs in genetically epilepsy-prone rats (Faingold, 2002, 2017; Ribak, 2017). With this in mind, we tested the hypothesis that Fmr1 deletion in the inferior colliculus is important for inducing AGSs.
We used the Ntsr1Cre/+ mouse line, which expresses Cre most strongly in the inferior colliculus, but it also has strong expression in the pyriform cortex and moderate expression in the superficial layer of the SC. We consider the pyriform cortex to be irrelevant since it is not considered important for AGS, and since it expressed Cre in the Emx1Cre/+ and vGlut1Cre/+ experiments, where no link to AGSs was observed. While the neurons in the superficial layer of the SC are close to the deep layers of the SC (DLSCs), which is an AGS-relevant structure in rats, these Cre-expressing neurons in the superficial layer are known not to interact with the DLSCs. Instead, they project to the thalamus (Gale and Murphy, 2014).
There is also weak, sparse expression in other cortical regions, the DLSCs, and the periaqueductal gray (Gale and Murphy, 2014; see Materials and Methods). We confirmed this expression using Ntsr1Cre/+:Rosa26tdTomato/+ mice (Fig. 6A). Based on the rat literature, the DLSC and periaqueductal gray are AGS-relevant structures (Faingold, 2002, 2017; Ribak, 2017). Therefore, our attributions of FMRP function in the inferior colliculus may be confounded. However, the much stronger Cre expression in the inferior colliculus makes it much more likely that FMRP expression in this structure contributes the most to AGS.
Figure 6.
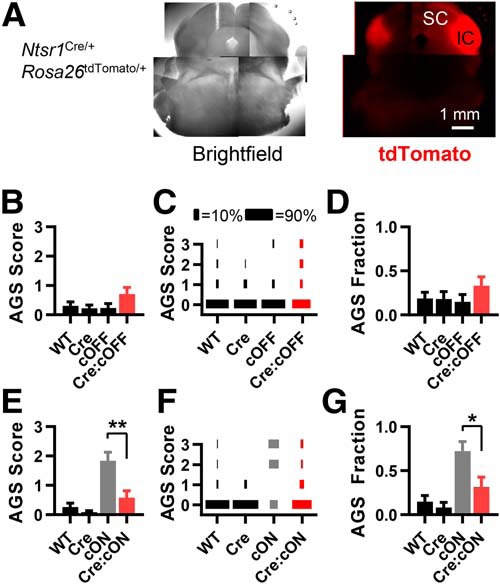
Fmr1 deletion primarily in the inferior colliculus is necessary for recapitulating the AGS phenotype. A, A live coronal section obtained from Ntsr1Cre/+:Rosa26tdTomato/+ mice. tdTomato fluorescence indicates strong Cre expression that is primarily in the inferior colliculus. B–D, AGS data for mice derived from Ntsr1Cre/+ and Fmr1cOFF/y cross-breeding. Deletion in cortex does not result in a change in AGS measurements compared with WT controls. E–G, Data derived from Ntsr1Cre/+ and Fmr1cON/y cross-breeding. AGS measurements resulting from Fmr1 expression in the inferior colliculus are no different from WT controls and are reduced compared with the cON-KO control. Therefore, deletion in inferior colliculus neurons is necessary for the AGS phenotype. N values for AGS data were as follows: cOFF = 32, 22, 20, and 24; cON = 27, 24, 18, and 19. *p < 0.05, **p < 0.01. K-W ANOVA followed by Dunn's test.
While we found that Fmr1 deletion in the inferior colliculi was not sufficient to induce AGSs (Fig. 6B–D), deletion was necessary (Fig. 6E–G). We performed FMRP immunohistochemistry and confirmed that FMRP was indeed selectively lost in the inferior colliculus of Ntsr1Cre/+:Fmr1cOFF/y mice and selectively expressed in Ntsr1Cre/+:Fmr1cON/y mice (Fig. 7A,B).
Figure 7.
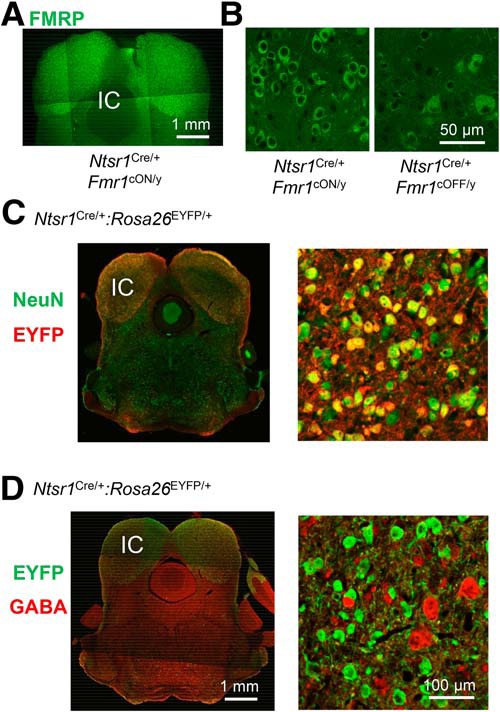
Fmr1 deletion in glutamatergic neurons in the inferior colliculus is the most likely cell type necessary for recapitulating the AGS phenotype. A, FMRP immunohistochemistry in a midbrain section obtained from an Ntsr1Cre/+:Fmr1cON/y mouse indicating selective expression in the inferior colliculus. B, High-power images of FMRP immunohistochemistry in the inferior colliculus of Ntsr1Cre/+:Fmr1cON/y and Ntsr1Cre/+:Fmr1cOFF/y mice. C, A coronal section from an Ntsr1Cre/+:Rosa26EYFP/+ mouse immunostained for NeuN (green) and the EYFP reporter (red). The high-power image of the inferior colliculus (right) shows a large overlap of NeuN and EYFP staining, indicating that the most Cre-expressing cells are neurons. D, A coronal section from an Ntsr1Cre/+:Rosa26EYFP/+ mouse immunostained for the EYFP reporter (green) and GABA (red). The high-power image of the inferior colliculus (right) shows essentially no overlap of EYFP and GABA staining, indicating that the most Cre-expressing cells are not GABAergic. Therefore, Cre-expressing neurons are most likely glutamatergic. Scale bars in D apply to C.
The cell types expressing Cre in the inferior colliculus of the Ntsr1Cre/+ mice have not been precisely determined. To determine the cell types expressing Cre, we performed immunohistochemistry in sections obtained from Ntsr1Cre/+:Rosa26EYFP/+ mice. First, we costained for the neuronal marker NeuN and the Cre-dependent reporter YFP (Fig. 7C). There was almost a 1:1 overlap of these markers. Of all YFP-positive cells, 84% were NeuN positive (544 of 644, two sections, three loci). And YFP-positive cells constituted 80% of all NeuN-positive cells (535 of 666). Second, we costained for GABA and YFP, and found that YFP-positive cells were never GABAergic (Fig. 7D; 0 of 467 cells; two sections, three loci). These data indicate that Cre expression is occurring, as follows: (1) mainly in neurons, (2) in a majority of the neurons, and (3) not in GABAergic neurons. In these same sections, the number of GABAergic neurons was much smaller compared with YFP-positive neurons (76 and 467 cells, respectively)—consistent with the known ratio of GABAergic to glutamatergic neurons if we assume that YFP-positive neurons are glutamatergic (Ito and Oliver, 2012). Therefore, our data indicate that Fmr1 deletion in excitatory neurons of the inferior colliculus is necessary for AGSs in the Fmr1 KO.
Discussion
We have used a conditional deletion and expression strategy to determine the specific cell types and brain regions where Fmr1 deletion is sufficient and necessary, respectively, to induce AGSs in the Fmr1 KO mouse. We provide evidence that Fmr1 deletion in VGlut2-expressing neurons in subcortical brain regions is fully sufficient to cause AGSs. Deletion of Fmr1 in glutamatergic neurons in the inferior colliculus, while not sufficient to cause AGSs, is necessary for the phenotype. We did this precise localization by conducting 13 Fmr1 conditional-expression experiments (7 using Fmr1cOFF/y, 6 using Fmr1cON/y) in which all four possible genotypic controls were examined. This is unprecedented for studying behavior in the Fmr1 KO and only possible due to the robustness of the AGS phenotype.
Our results are consistent with previous findings that Fmr1 deletion in glutamatergic neurons results in changes in synaptic and circuit function that may lead to hyperexcitability in the Fmr1 KO brain (Hays et al., 2011; Deng et al., 2013; Patel et al., 2013, 2014). Therefore, accumulating evidence implicates glutamatergic neurons in inducing circuit and behavioral dysfunction related to hyperexcitability in the Fmr1 KO mouse.
The ability of FMRP expression in the inferior colliculus to affect the AGS phenotype is particularly striking considering the limited number of neurons involved and the restricted spatial locus of these neurons. Of 109 million cells in the mouse brain (Williams, 2000), glutamatergic neurons in the two inferior colliculi number ∼0.5 million (Kulesza et al., 2002; Ito and Oliver, 2012). Because Fmr1 deletion in these neurons was not sufficient to induce AGS, our data suggest that deletion in inferior collicular glutamatergic neurons together with other subcortical glutamatergic neuron types is necessary for AGSs. To date, this is the smallest and most spatially restricted cell population for conditional gene expression found for regulating the AGS. The next closest was the finding that that deletion of Ube3a in all brain GABAergic neurons was sufficient for inducing AGSs in the Angelman syndrome mouse model (Judson et al., 2016). These findings relating to the inferior colliculus represent the first potential link between a localized, hyperexcitable circuit and a behavioral phenotype in the Fmr1 KO mouse.
Our finding that Fmr1 deletion in VGlut2-expressing neurons in subcortical brain structures underlies the AGS is consistent with what is known about genetically epilepsy-prone rats (GEPRs) and about VGlut2 expression. First, the brainstem is thought to be the main, if not the exclusive, site for mediating the AGS in GEPRs, and the inferior colliculus is thought to be the initiation site (Faingold, 2002, 2004; Ribak, 2017). Second, our finding that Fmr1 deletion in VGlut2-expressing glutamatergic neurons that never express VGlut1 underlies the AGS is consistent with our finding that deletion in subcortical regions is required. This subtype of glutamatergic neuron is primarily located in subcortical structures with few or none in cortical structures, and they are the exclusive type of glutamatergic neuron in the inferior colliculus (Fremeau et al., 2001; Ito et al., 2011; Allen Brain Atlas; see Materials and Methods).
Our strategy combining both conditional “on” and “off” Fmr1 mice with many Cre lines practically eliminates roles for Fmr1 in other cell types in AGSs. Because the vGlut2Cre/+ mouse experiments demonstrated that Fmr1 deletion was both sufficient and necessary for the AGS phenotype, this indicates that deletion in all other cell types plays a small role, if any. These other irrelevant types include non-neuronal cells and the following other neuron types: GABAergic, glycinergic, serotonergic, and cholinergic.
The known expression of VGlut2 supports the elimination of these other cell types based on interpretations of the vGlut2Cre/+ experiments. For example, it is unlikely that Cre is expressed in glial cells in the vGlut2Cre/+ mouse since oligodendrocytes and microglia express VGlut1 (Zhang et al., 2014b) and astrocytes express little or none of either VGlut1 or VGlut2 (Li et al., 2013; Zhang et al., 2014b). Therefore, it is unlikely that Fmr1 deletion in glial cells was involved in causing the AGS. Serotonergic and cholinergic neurons have been reported to release glutamate, but VGlut3 is the transporter in these neurons (Higley et al., 2011; Wang et al., 2019). There is a small subpopulation of dopaminergic neurons in the ventral tegmental area that express VGlut2, so we cannot rule out the possibility that they play a role in AGSs (Papathanou et al., 2018). Finally, Fmr1 deletion in the auditory system periphery does not play a role since auditory hair cells did not express Cre in our vGlut2Cre/+ experiments and since AGS induction was unaffected in the vGlut1Cre/+ experiments, even though spiral ganglion cells expressed Cre.
Based on previous studies of the developmental time course of VGlut1 and VGlut2, Fmr1 deletion in vGlut2Cre/+ mice may occur before that occurring in vGlut1Cre/+ mice (Boulland et al., 2004; Fremeau et al., 2004). Because of this, we cannot completely rule out the possibility that the effectiveness of the vGlut2Cre/+ mice in completely mimicking the AGS phenotype is due to early Fmr1 deletion in VGlut1-expressing neurons (remember that vGlut2Cre/+ mice induce recombination in both VGlut1- and VGlut2-expressing cells). In this scenario, Fmr1 is deleted early enough in VGlut1-expressing neurons in vGlut2Cre/+ mice to induce AGS, but not early enough in vGlut1Cre/+ mice. But we think this scenario is unlikely. We have used two other Cre lines in this study with Cre expression patterns that are very similar to those in vGlut1Cre/+, but with early expression onset similar to that of vGlut2Cre/+—Emx1Cre/+ and NexCre/+—and AGS results were similar to that of vGlut1Cre/+.
While our data indicate that Fmr1 deletion in VGlut2-positive neurons is important for AGSs, they do not indicate that hyperexcitability of glutamatergic neurons directly cause AGS. For example, cell-autonomous deletion of Fmr1 in neocortical excitatory neurons results in weak excitatory synapses onto parvalbumin-positive inhibitory neurons, which results in reduced feedback inhibition in cortical circuits (Patel et al., 2013). A similar “presynaptic” function of FMRP in glutamatergic neurons in the inferior colliculus may result in reduced inhibition and hyperexcitability. Therefore, further investigation is needed to identify the specific cell types and circuit mechanisms in the inferior colliculus. These future investigations will be expedited by our finding of established Cre-mouse lines targeting the relevant neurons in the AGS.
We assert that AGSs are a model of sensory hypersensitivity found in many forms of autism and in FXS. Because AGSs are likely primarily mediated by brainstem circuits, this model implies that sensory hypersensitivity is mediated by brainstem circuits and not cortical circuits. For studies observing enhanced responses in sensory cortical areas, it is unknown whether the enhancement is due to cortical or subcortical Fmr1 deletion (Zhang et al., 2014a; Lovelace et al., 2016; Wen et al., 2018), and some studies do not observe enhanced cortical sensory responses (Goel et al., 2018; Antoine et al., 2019). This leaves open the possibility that enhanced physiological responses mediating sensory hypersensitivity are occurring subcortically in FXS.
Because evidence obtained by us and others indicates that AGSs in the Fmr1 KO involve alterations in brainstem circuits, we would expect the auditory brainstem response (ABR) to be enhanced in the mouse and in FXS patients. Interestingly, recent studies report no changes in the ABR in FXS patients (Roberts et al., 2005; but see Arinami et al., 1988) and mixed results in the Fmr1 KO mouse (Rotschafer et al., 2015; El-Hassar et al., 2019). How might this relate to the AGS and sensory hypersensitivity? First, the ABR, being measured from the scalp, may not be sensitive enough to detect enhanced responses occurring in higher stages of auditory brainstem processing such as in the inferior colliculus. Second, sensory hypersensitivity might only occur under certain circumstances, and not under the conditions under which ABRs are measured. For example, in mice, the ABR is in response to short duration stimuli and are recorded under anesthesia. The AGS experiments use prolonged stimuli (on the order of minutes) in alert mice, and in our experiments, AGS induction required an average of 30 s with respect to stimulus onset (see Materials and Methods). These points suggest that time-dependent processes or particular brain states like those achieved during an AGS in mouse may be needed to observe the physiological and behavioral manifestations of sensory hypersensitivity in FXS patients.
Our Fmr1cON/y experiments, while described in the context of “deletion necessity,” also show which cell populations normalize the AGS phenotype when FMRP is expressed in an otherwise Fmr1 KO mouse (Figs. 2I, 6G). These data indicate that FMRP expression restricted to VGlut2-expressing neurons and glutamatergic inferior colliculus neurons are sufficient to remedy the AGS phenotype. Interestingly, while our data indicate that the inferior collicular neurons are only a subset of all the neurons that underlie the AGS phenotype in the Fmr1 KO mouse (i.e., Fmr1 deletion in them is necessary but not sufficient), we show that targeting this subset is enough to rescue the AGS phenotype. Therefore, we demonstrate that not all the relevant neurons need to be manipulated to normalize a behavior. In summary, if the AGS is an effective model for sensory hypersensitivity, our data indicate that targeting brainstem structures in autism and FXS patients may be an effective strategy for the treatment of sensory hypersensitivity.
Footnotes
This research was supported by National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant 1U54-HD-082008–01 (to J.R.G. and K.M.H.). This facility is supported by the Neuroscience Department at the University of Texas Southwestern Medical Center and the Peter O'Donnell Jr. Brain Institute. We thank Patricia Hahn, Bianca Walker, and Manasa Sarma for technical assistance. We also thank the Neuroscience Microscopy Facility for the use of confocal microscopes.
The authors declare no competing financial interests.
References
- Antoine MW, Langberg T, Schnepel P, Feldman DE (2019) Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101:648–661.e4. 10.1016/j.neuron.2018.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenkiel BR, Gaufo GO, Capecchi MR (2003) Hoxb1 neural crest preferentially form glia of the PNS. Dev Dyn 227:379–386. 10.1002/dvdy.10323 [DOI] [PubMed] [Google Scholar]
- Arinami T, Sato M, Nakajima S, Kondo I (1988) Auditory brain-stem responses in the fragile X syndrome. Am J Hum Genet 43:46–51. [PMC free article] [PubMed] [Google Scholar]
- Bakker DB, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen AT, Oostra BA, Reyniers E, De Boule K, D'Hooge R, Cras P, van Velzen D, Nagels G, Martin J-J, De Deyn PP, Darby JK, Willems PJ (1994) Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78:23–33. 10.1016/0092-8674(94)90569-X [DOI] [PubMed] [Google Scholar]
- Baranek GT, Roberts JE, David FJ, Sideris J, Mirrett PL, Hatton DD, Bailey DB Jr (2008) Developmental trajectories and correlates of sensory processing in young boys with fragile X syndrome. Phys Occup Ther Pediatr 28:79–98. 10.1300/J006v28n01_06 [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST (2008) Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60:201–214. 10.1016/j.neuron.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sasson A, Hen L, Fluss R, Cermak SA, Engel-Yeger B, Gal E (2009) A meta-analysis of sensory modulation symptoms in individuals with autism spectrum disorders. J Autism Dev Disord 39:1–11. 10.1007/s10803-008-0593-3 [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, Bailey DB (2010) Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil 115:461–472. 10.1352/1944-7558-115.6.461 [DOI] [PubMed] [Google Scholar]
- Boulland JL, Qureshi T, Seal RP, Rafiki A, Gundersen V, Bergersen LH, Fremeau RT Jr, Edwards RH, Storm-Mathisen J, Chaudhry FA (2004) Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J Comp Neurol 480:264–280. 10.1002/cne.20354 [DOI] [PubMed] [Google Scholar]
- Busquets-Garcia A, Gomis-González M, Guegan T, Agustín-Pavón C, Pastor A, Mato S, Pérez-Samartin A, Matute C, de la Torre R, Dierssen M, Maldonado R, Ozaita A (2013) Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19:603–607. 10.1038/nm.3127 [DOI] [PubMed] [Google Scholar]
- Castrén M, Pääkkönen A, Tarkka IM, Ryynänen M, Partanen J (2003) Augmentation of auditory N1 in children with fragile X syndrome. Brain Topogr 15:165–171. 10.1023/A:1022606200636 [DOI] [PubMed] [Google Scholar]
- Cea-Del Rio CA, Huntsman MM (2014) The contribution of inhibitory interneurons to circuit dysfunction in fragile X syndrome. Front Cell Neurosci 8:245. 10.3389/fncel.2014.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee M, Kurup PK, Lundbye CJ, Hugger Toft AK, Kwon J, Benedict J, Kamceva M, Banke TG, Lombroso PJ (2018) STEP inhibition reverses behavioral, electrophysiologic, and synaptic abnormalities in Fmr1 KO mice. Neuropharmacology 128:43–53. 10.1016/j.neuropharm.2017.09.026 [DOI] [PubMed] [Google Scholar]
- Chen L, Toth M (2001) Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103:1043–1050. 10.1016/S0306-4522(01)00036-7 [DOI] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK (2005) Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci 25:8048–8055. 10.1523/JNEUROSCI.1777-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, Xu X, Rumbaugh G (2012) Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151:709–723. 10.1016/j.cell.2012.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, Portera-Cailliau C (2015) Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87:699–715. 10.1016/j.neuron.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curia G, Gualtieri F, Bartolomeo R, Vezzali R, Biagini G (2013) Resilience to audiogenic seizures is associated with p-ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front Cell Neurosci 7:46. 10.3389/fncel.2013.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansie LE, Phommahaxay K, Okusanya AG, Uwadia J, Huang M, Rotschafer SE, Razak KA, Ethell DW, Ethell IM (2013) Long-lasting effects of minocycline on behavior in young but not adult fragile X mice. Neuroscience 246:186–198. 10.1016/j.neuroscience.2013.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, Klyachko VA (2013) FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77:696–711. 10.1016/j.neuron.2012.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bonito M, Studer M (2017) Cellular and molecular underpinnings of neuronal assembly in the central auditory system during mouse development. Front Neural Circuits 11:18. 10.3389/fncir.2017.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bonito M, Narita Y, Avallone B, Sequino L, Mancuso M, Andolfi G, Franzè AM, Puelles L, Rijli FM, Studer M (2013) Assembly of the auditory circuitry by a Hox genetic network in the mouse brainstem. PLoS Genet 9:e1003249. 10.1371/journal.pgen.1003249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Sethna F, Wang H (2014) Behavioral analysis of male and female Fmr1 knockout mice on C57BL/6 background. Behav Brain Res 271:72–78. 10.1016/j.bbr.2014.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY, Tonegawa S (2013) Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci U S A 110:5671–5676. 10.1073/pnas.1219383110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF (2007) Correction of fragile X syndrome in mice. Neuron 56:955–962. 10.1016/j.neuron.2007.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hassar L, Song L, Tan WJT, Large CH, Alvaro G, Santos-Sacchi J, Kaczmarek LK (2019) Modulators of Kv3 potassium channels rescue the auditory function of fragile X mice. J Neurosci 39:4797–4813. 10.1523/JNEUROSCI.0839-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethridge LE, White SP, Mosconi MW, Wang J, Byerly MJ, Sweeney JA (2016) Reduced habituation of auditory evoked potentials indicate cortical hyper-excitability in fragile X syndrome. Transl Psychiatry 6:e787. 10.1038/tp.2016.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold CL. (2002) Role of GABA abnormalities in the inferior colliculus pathophysiology—audiogenic seizures. Hear Res 168:223–237. 10.1016/S0378-5955(02)00373-8 [DOI] [PubMed] [Google Scholar]
- Faingold CL. (2004) Emergent properties of CNS neuronal networks as targets for pharmacology: application to anticonvulsant drug action. Prog Neurobiol 72:55–85. 10.1016/j.pneurobio.2003.11.003 [DOI] [PubMed] [Google Scholar]
- Faingold CL. (2017) Anticonvulsant drugs are neuronal network-modifying agents (NMAs). In: Reference module in neuroscience and biobehavioral psychology, Vol 2017 (Stein F, ed), pp 1–13. Amsterdam: Elsevier; 10.1016/B978-0-12-809324-5.00009-2 [DOI] [Google Scholar]
- Fremeau RT Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH (2001) The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31:247–260. 10.1016/S0896-6273(01)00344-0 [DOI] [PubMed] [Google Scholar]
- Fremeau RT Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH (2004) Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304:1815–1819. 10.1126/science.1097468 [DOI] [PubMed] [Google Scholar]
- Gale SD, Murphy GJ (2014) Distinct representation and distribution of visual information by specific cell types in mouse superficial superior colliculus. J Neurosci 34:13458–13471. 10.1523/JNEUROSCI.2768-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez R, Greenough WT (2005) Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A 135:155–160. 10.1002/ajmg.a.30709 [DOI] [PubMed] [Google Scholar]
- Gantois I, Khoutorsky A, Popic J, Aguilar-Valles A, Freemantle E, Cao R, Sharma V, Pooters T, Nagpal A, Skalecka A, Truong VT, Wiebe S, Groves IA, Jafarnejad SM, Chapat C, McCullagh EA, Gamache K, Nader K, Lacaille JC, Gkogkas CG, Sonenberg N (2017) Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat Med 23:674–677. 10.1038/nm.4335 [DOI] [PubMed] [Google Scholar]
- Gholizadeh S, Arsenault J, Xuan IC, Pacey LK, Hampson DR (2014) Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology 39:3100–3111. 10.1038/npp.2014.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA (2006) Genetic targeting of principal neurons in neocortex and hippocampus of NEX-cre mice. Genesis 44:611–621. 10.1002/dvg.20256 [DOI] [PubMed] [Google Scholar]
- Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, Lombroso PJ (2012) Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav 11:586–600. 10.1111/j.1601-183X.2012.00781.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Cantu DA, Guilfoyle J, Chaudhari GR, Newadkar A, Todisco B, de Alba D, Kourdougli N, Schmitt LM, Pedapati E, Erickson CA, Portera-Cailliau C (2018) Impaired perceptual learning in a mouse model of fragile X syndrome is mediated by parvalbumin neuron dysfunction and is reversible. Nat Neurosci 21:1404–1411. 10.1038/s41593-018-0231-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves JT, Anstey JE, Golshani P, Portera-Cailliau C (2013) Circuit level defects in the developing neocortex of fragile X mice. Nat Neurosci 16:903–909. 10.1038/nn.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR (2007) Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci 27:9817–9823. 10.1523/JNEUROSCI.2707-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22:6309–6314. 10.1523/JNEUROSCI.22-15-06309.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S, Cavalier S, Sridhar V, Huber KM, Gibson JR (2019) Local cortical circuit correlates of altered EEG in the mouse model of fragile X syndrome. Neurobiol Dis 124:563–572. 10.1016/j.nbd.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Chang CW, Kelly SM, Bhattacharya A, McBride SM, Danielson SW, Jiang MQ, Chan CB, Ye K, Gibson JR, Klann E, Jongens TA, Moberg KH, Huber KM, Bassell GJ (2015a) Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep 11:727–736. 10.1016/j.celrep.2015.03.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Raj N, Molinaro G, Allen AG, Whyte AJ, Gibson JR, Huber KM, Gourley SL, Bassell GJ (2015b) Selective role of the catalytic PI3K subunit p110beta in impaired higher order cognition in fragile X syndrome. Cell Rep 11:681–688. 10.1016/j.celrep.2015.03.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Banerjee A, Tiwari D, Longo F, White AR, Allen AG, Schroeder-Carter LM, Krzeski JC, Elsayed NA, Puckett R, Klann E, Rivero RA, Gourley SL, Bassell GJ (2019) Isoform-selective phosphoinositide 3-kinase inhibition ameliorates a broad range of fragile X syndrome-associated deficits in a mouse model. Neuropsychopharmacology 44:324–333. 10.1038/s41386-018-0150-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL, Eisch AJ, Oostra BA, Nelson DL, Jin P, Zhao X (2011) Ablation of fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med 17:559–565. 10.1038/nm.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Molinaro G, Collins KA, Hays SA, Paylor R, Worley PF, Szumlinski KK, Huber KM (2016) Selective disruption of metabotropic glutamate receptor 5-homer interactions mimics phenotypes of fragile X syndrome in mice. J Neurosci 36:2131–2147. 10.1523/JNEUROSCI.2921-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, Picker J, Gane L, Tranfaglia M (2009) Advances in the treatment of fragile X syndrome. Pediatrics 123:378–390. 10.1542/peds.2008-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, Bohn P, Mortrud M, Ouellette B, Kidney J, Smith KA, Dang C, Sunkin S, Bernard A, Oh SW, Madisen L, Zeng H (2014) Anatomical characterization of cre driver mice for neural circuit mapping and manipulation. Front Neural Circuits 8:76. 10.3389/fncir.2014.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SA, Huber KM, Gibson JR (2011) Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci 31:14223–14234. 10.1523/JNEUROSCI.3157-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR, Brynczka C, Rush R, Thomas A, Paylor R, Warren ST, Vanderklish PW, Kind PC, Carpenter RL, Bear MF, Healy AM (2012) Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABA(B) receptors with arbaclofen. Sci Transl Med 4:152ra128. 10.1126/scitranslmed.3004218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heulens I, D'Hulst C, Van Dam D, De Deyn PP, Kooy RF (2012) Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res 229:244–249. 10.1016/j.bbr.2012.01.031 [DOI] [PubMed] [Google Scholar]
- Higley MJ, Gittis AH, Oldenburg IA, Balthasar N, Seal RP, Edwards RH, Lowell BB, Kreitzer AC, Sabatini BL (2011) Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PLoS One 6:e19155. 10.1371/journal.pone.0019155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges JL, Yu X, Gilmore A, Bennett H, Tjia M, Perna JF, Chen CC, Li X, Lu J, Zuo Y (2017) Astrocytic contributions to synaptic and learning abnormalities in a mouse model of fragile X syndrome. Biol Psychiatry 82:139–149. 10.1016/j.biopsych.2016.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Oliver DL (2012) The basic circuit of the IC: tectothalamic neurons with different patterns of synaptic organization send different messages to the thalamus. Front Neural Circuits 6:48. 10.3389/fncir.2012.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bishop DC, Oliver DL (2011) Expression of glutamate and inhibitory amino acid vesicular transporters in the rodent auditory brainstem. J Comp Neurol 519:316–340. 10.1002/cne.22521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Higuchi M, Oishi K, Kishi Y, Okazaki T, Sakai H, Miyata T, Nakajima K, Gotoh Y (2016) PDK1-akt pathway regulates radial neuronal migration and microtubules in the developing mouse neocortex. Proc Natl Acad Sci U S A 113:E2955–E2964. 10.1073/pnas.1516321113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knöpfel T, Erzurumlu RS, Itohara S (2000) Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406:726–731. 10.1038/35021059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Nomura R, Ando R, Ikeda T, Tanaka M, Itohara S (2004) Dorsal telencephalon-specific expression of cre recombinase in PAC transgenic mice. Genesis 38:130–138. 10.1002/gene.20009 [DOI] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL (1998) Mutation of the angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21:799–811. 10.1016/S0896-6273(00)80596-6 [DOI] [PubMed] [Google Scholar]
- Judson MC, Wallace ML, Sidorov MS, Burette AC, Gu B, van Woerden GM, King IF, Han JE, Zylka MJ, Elgersma Y, Weinberg RJ, Philpot BD (2016) GABAergic neuron-specific loss of Ube3a causes angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron 90:56–69. 10.1016/j.neuron.2016.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazdoba TM, Sunnen CN, Crowell B, Lee GH, Anderson AE, D'Arcangelo G (2012) Development and characterization of NEX- pten, a novel forebrain excitatory neuron-specific knockout mouse. Dev Neurosci 34:198–209. 10.1159/000337229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KA, Gordon-Salant S, Pawlowski KS, Taylor AM, Griffith AJ, Houser A, Kurima K, Wassif CA, Wright CG, Porter FD, Repa JJ, Brewer CC (2014) Hearing loss is an early consequence of Npc1 gene deletion in the mouse model of Niemann-Pick disease, type C. J Assoc Res Otolaryngol 15:529–541. 10.1007/s10162-014-0459-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulesza RJ, Viñuela A, Saldaña E, Berrebi AS (2002) Unbiased stereological estimates of neuron number in subcortical auditory nuclei of the rat. Hear Res 168:12–24. 10.1016/S0378-5955(02)00374-X [DOI] [PubMed] [Google Scholar]
- Li D, Hérault K, Silm K, Evrard A, Wojcik S, Oheim M, Herzog E, Ropert N (2013) Lack of evidence for vesicular glutamate transporter expression in mouse astrocytes. J Neurosci 33:4434–4455. 10.1523/JNEUROSCI.3667-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovelace JW, Wen TH, Reinhard S, Hsu MS, Sidhu H, Ethell IM, Binder DK, Razak KA (2016) Matrix metalloproteinase-9 deletion rescues auditory evoked potential habituation deficit in a mouse model of fragile X syndrome. Neurobiol Dis 89:126–135. 10.1016/j.nbd.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricich SM, Xia A, Mathes EL, Wang VY, Oghalai JS, Fritzsch B, Zoghbi HY (2009) Atoh1-lineal neurons are required for hearing and for the survival of neurons in the spiral ganglion and brainstem accessory auditory nuclei. J Neurosci 29:11123–11133. 10.1523/JNEUROSCI.2232-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74:49–56. 10.1016/j.neuron.2012.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, Zu T, Hoogeveen-Westerveld M, Severijnen L, Rifé M, Willemsen R, Nelson DL, Oostra BA (2006) The generation of a conditional Fmr1 knock out mouse model to study fmrp function in vivo. Neurobiol Dis 21:549–555. 10.1016/j.nbd.2005.08.019 [DOI] [PubMed] [Google Scholar]
- Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, Tassone F, Neitzel K, Stackhouse T, Hagerman RJ (1999) Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet 83:268–279. [DOI] [PubMed] [Google Scholar]
- Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS, Bauchwitz RP (2009) Elevated glycogen synthase kinase-3 activity in fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56:463–472. 10.1016/j.neuropharm.2008.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscas M, Louros SR, Osterweil EK (2019) Lovastatin, not simvastatin, corrects core phenotypes in the fragile X mouse model. eNeuro 6:ENEURO.0097–19.2019. 10.1523/ENEURO.0097-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci SA, Elia M, Ferri R, Scuderi C, Del Gracco S (1994) Evoked spikes and giant somatosensory evoked potentials in a patient with fragile-X syndrome. Ital J Neurol Sci 15:365–368. 10.1007/BF02339934 [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA, De Sarro GB, Elia M (1999) Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40:1092–1099. 10.1111/j.1528-1157.1999.tb00824.x [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, Oostra BA (2000) Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41:19–23. 10.1111/j.1528-1157.2000.tb01499.x [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Calabrese G, Bonaccorso CM, D'Antoni S, Brouwer JR, Bakker CE, Elia M, Ferri R, Nelson DL, Oostra BA, Catania MV (2007) Audiogenic seizure susceptibility is reduced in fragile X knockout mice after introduction of FMR1 transgenes. Exp Neurol 203:233–240. 10.1016/j.expneurol.2006.08.007 [DOI] [PubMed] [Google Scholar]
- O'Donnell WT, Warren ST (2002) A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci 25:315–338. 10.1146/annurev.neuro.25.112701.142909 [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF (2010) Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 30:15616–15627. 10.1523/JNEUROSCI.3888-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF (2013) Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77:243–250. 10.1016/j.neuron.2012.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey LK, Heximer SP, Hampson DR (2009) Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol Pharmacol 76:18–24. 10.1124/mol.109.056127 [DOI] [PubMed] [Google Scholar]
- Pacey LK, Tharmalingam S, Hampson DR (2011) Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J Pharmacol Exp Ther 338:897–905. 10.1124/jpet.111.183327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papathanou M, Creed M, Dorst MC, Bimpisidis Z, Dumas S, Pettersson H, Bellone C, Silberberg G, Lüscher C, Wallén-Mackenzie Å (2018) Targeting VGLUT2 in mature dopamine neurons decreases mesoaccumbal glutamatergic transmission and identifies a role for glutamate corelease in synaptic plasticity by increasing baseline AMPA/NMDA ratio. Front Neural Circuits 12:64. 10.3389/fncir.2018.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Hays SA, Bureau I, Huber KM, Gibson JR (2013) A target cell-specific role for presynaptic Fmr1 in regulating glutamate release onto neocortical fast-spiking inhibitory neurons. J Neurosci 33:2593–2604. 10.1523/JNEUROSCI.2447-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Loerwald KW, Huber KM, Gibson JR (2014) Postsynaptic FMRP promotes the pruning of cell-to-cell connections among pyramidal neurons in the L5A neocortical network. J Neurosci 34:3413–3418. 10.1523/JNEUROSCI.2921-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitpré C, Wu H, Sharma A, Tokarska A, Fontanet P, Wang Y, Helmbacher F, Yackle K, Silberberg G, Hadjab S, Lallemend F (2018) Neuronal heterogeneity and stereotyped connectivity in the auditory afferent system. Nat Commun 9:3691. 10.1038/s41467-018-06033-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Kang J, Smith CB (2005) A null mutation for Fmr1 in female mice: effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience 135:999–1009. 10.1016/j.neuroscience.2005.06.081 [DOI] [PubMed] [Google Scholar]
- Ribak CE. (2017) An abnormal GABAergic system in the inferior colliculus provides a basis for audiogenic seizures in genetically epilepsy-prone rats. Epilepsy Behav 71:160–164. 10.1016/j.yebeh.2015.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts J, Hennon EA, Anderson K, Roush J, Gravel J, Skinner M, Misenheimer J, Reitz P (2005) Auditory brainstem responses in young males with fragile X syndrome. J Speech Lang Hear Res 48:494–500. 10.1044/1092-4388(2005/034) [DOI] [PubMed] [Google Scholar]
- Rojas DC, Benkers TL, Rogers SJ, Teale PD, Reite ML, Hagerman RJ (2001) Auditory evoked magnetic fields in adults with fragile X syndrome. Neuroreport 12:2573–2576. 10.1097/00001756-200108080-00056 [DOI] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, Huber KM (2012) Disrupted homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci 15:431–440, S1. 10.1038/nn.3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotschafer S, Razak K (2013) Altered auditory processing in a mouse model of fragile X syndrome. Brain Res 1506:12–24. 10.1016/j.brainres.2013.02.038 [DOI] [PubMed] [Google Scholar]
- Rotschafer SE, Marshak S, Cramer KS (2015) Deletion of Fmr1 alters function and synaptic inputs in the auditory brainstem. PLoS One 10:e0117266. 10.1371/journal.pone.0117266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabaratnam M, Vroegop PG, Gangadharan SK (2001) Epilepsy and EEG findings in 18 males with fragile X syndrome. Seizure 10:60–63. 10.1053/seiz.2000.0492 [DOI] [PubMed] [Google Scholar]
- Saré RM, Song A, Loutaev I, Cook A, Maita I, Lemons A, Sheeler C, Smith CB (2018) Negative effects of chronic rapamycin treatment on behavior in a mouse model of fragile X syndrome. Front Mol Neurosci 10:452. 10.3389/fnmol.2017.00452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka K, Pyronneau A, Chao M, Bennett MV, Zukin RS (2016) Elevated ERK/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in fragile X mice. Proc Natl Acad Sci U S A 113:E6290–E6297. 10.1073/pnas.1610812113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer TL, Davenport MH, Grainger LM, Robinson CK, Earnheart AT, Stegman MS, Lang AL, Ashworth AA, Molinaro G, Huber KM, Erickson CA (2017) Acamprosate in a mouse model of fragile X syndrome: modulation of spontaneous cortical activity, ERK1/2 activation, locomotor behavior, and anxiety. J Neurodev Disord 9:6. 10.1186/s11689-017-9184-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal RP, Akil O, Yi E, Weber CM, Grant L, Yoo J, Clause A, Kandler K, Noebels JL, Glowatzki E, Lustig LR, Edwards RH (2008) Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron 57:263–275. 10.1016/j.neuron.2007.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna F, Feng W, Ding Q, Robison AJ, Feng Y, Wang H (2017) Enhanced expression of ADCY1 underlies aberrant neuronal signalling and behaviour in a syndromic autism model. Nat Commun 8:14359. 10.1038/ncomms14359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JJ, Chitwood RA, Ding JM, Payne C, Taylor W, Gray R, Zemelman BV, Johnston D (2017) Prefrontal cortex dysfunction in fragile X mice depends on the continued absence of fragile X mental retardation protein in the adult brain. J Neurosci 37:7305–7317. 10.1523/JNEUROSCI.0571-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ, Bear MF (2017) beta-Arrestin2 couples metabotropic glutamate receptor 5 to neuronal protein synthesis and is a potential target to treat fragile X. Cell Rep 18:2807–2814. 10.1016/j.celrep.2017.02.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S. (2006) VGLUTs: “exciting” times for glutamatergic research? Neurosci Res 55:343–351. 10.1016/j.neures.2006.04.016 [DOI] [PubMed] [Google Scholar]
- Thomas AM, Bui N, Graham D, Perkins JR, Yuva-Paylor LA, Paylor R (2011) Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav Brain Res 223:310–321. 10.1016/j.bbr.2011.04.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, Bui N, Perkins JR, Yuva-Paylor LA, Paylor R (2012) Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology (Berl) 219:47–58. 10.1007/s00213-011-2375-4 [DOI] [PubMed] [Google Scholar]
- Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJA, Hardingham GE, Kind PC, Osterweil EK (2017) Cell-type-specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron 95:550–563.e5. 10.1016/j.neuron.2017.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anilkumar S, Ivshina M, Hurt JA, Nagaoka K, Nalavadi VC, Lorenz LJ, Bassell GJ, Akbarian S, Chattarji S, Klann E, Richter JD (2013) Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med 19:1473–1477. 10.1038/nm.3353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Molen MJ, Van der Molen MW, Ridderinkhof KR, Hamel BC, Curfs LM, Ramakers GJ (2012) Auditory and visual cortical activity during selective attention in fragile X syndrome: a cascade of processing deficiencies. Clin Neurophysiol 123:720–729. 10.1016/j.clinph.2011.08.023 [DOI] [PubMed] [Google Scholar]
- Veeraragavan S, Bui N, Perkins JR, Yuva-Paylor LA, Carpenter RL, Paylor R (2011a) Modulation of behavioral phenotypes by a muscarinic M1 antagonist in a mouse model of fragile X syndrome. Psychopharmacology (Berl) 217:143–151. 10.1007/s00213-011-2276-6 [DOI] [PubMed] [Google Scholar]
- Veeraragavan S, Bui N, Perkins JR, Yuva-Paylor LA, Paylor R (2011b) The modulation of fragile X behaviors by the muscarinic M4 antagonist, tropicamide. Behav Neurosci 125:783–790. 10.1037/a0025202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraragavan S, Graham D, Bui N, Yuva-Paylor LA, Wess J, Paylor R (2012) Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS). Behav Brain Res 228:1–8. 10.1016/j.bbr.2011.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914. 10.1016/0092-8674(91)90397-H [DOI] [PubMed] [Google Scholar]
- Voiculescu O, Charnay P, Schneider-Maunoury S (2000) Expression pattern of a krox-20/Cre knock-in allele in the developing hindbrain, bones, and peripheral nervous system. Genesis 26:123–126. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154. 10.1016/j.neuron.2011.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Zhang S, Qi J, Wang H, Cachope R, Mejias-Aponte CA, Gomez JA, Mateo-Semidey GE, Beaudoin GMJ, Paladini CA, Cheer JF, Morales M (2019) Dorsal raphe dual serotonin-glutamate neurons drive reward by establishing excitatory synapses on VTA mesoaccumbens dopamine neurons. Cell Rep 26:1128–1142.e7. 10.1016/j.celrep.2019.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, Castellani RJ, Casadesus G, Smith MA, Zhu X (2012) Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile X-syndrome. J Neurochem 121:672–679. 10.1111/j.1471-4159.2012.07722.x [DOI] [PubMed] [Google Scholar]
- Wen TH, Afroz S, Reinhard SM, Palacios AR, Tapia K, Binder DK, Razak KA, Ethell IM (2018) Genetic reduction of matrix metalloproteinase-9 promotes formation of perineuronal nets around parvalbumin-expressing interneurons and normalizes auditory cortex responses in developing Fmr1 knock-out mice. Cereb Cortex 28:3951–3964. 10.1093/cercor/bhx258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Westmark PR, Malter JS (2009) MPEP reduces seizure severity in fmr-1 KO mice over expressing human abeta. Int J Clin Exp Pathol 3:56–68. [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Westmark PR, O'Riordan KJ, Ray BC, Hervey CM, Salamat MS, Abozeid SH, Stein KM, Stodola LA, Tranfaglia M, Burger C, Berry-Kravis EM, Malter JS (2011) Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS One 6:e26549. 10.1371/journal.pone.0026549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark PR, Dekundy A, Gravius A, Danysz W, Westmark CJ (2018) Rescue of Fmr1(KO) phenotypes with mGluR5 inhibitors: MRZ-8456 versus AFQ-056. Neurobiol Dis 119:190–198. 10.1016/j.nbd.2018.08.008 [DOI] [PubMed] [Google Scholar]
- Williams RW. (2000) Mapping genes that modulate mouse brain development: a quantitative genetic approach. Results Probl Cell Differ 30:21–49. 10.1007/978-3-540-48002-0_2 [DOI] [PubMed] [Google Scholar]
- Wozny C, Beed P, Nitzan N, Pössnecker Y, Rost BR, Schmitz D (2018) VGLUT2 functions as a differential marker for hippocampal output neurons. Front Cell Neurosci 12:337. 10.3389/fncel.2018.00337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Chung S, Zhang S, Zhong P, Ma C, Chang WC, Weissbourd B, Sakai N, Luo L, Nishino S, Dan Y (2015) Basal forebrain circuit for sleep-wake control. Nat Neurosci 18:1641–1647. 10.1038/nn.4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP (2004) A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav 3:337–359. 10.1111/j.1601-183X.2004.00087.x [DOI] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP (2005) Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49:1053–1066. 10.1016/j.neuropharm.2005.06.004 [DOI] [PubMed] [Google Scholar]
- Yu WM, Goodrich LV (2014) Morphological and physiological development of auditory synapses. Hear Res 311:3–16. 10.1016/j.heares.2014.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang JB, Nosyreva ED, Spencer CM, Volk LJ, Musunuru K, Zhong R, Stone EF, Yuva-Paylor LA, Huber KM, Paylor R, Darnell JC, Darnell RB (2009) A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet 5:e1000758. 10.1371/journal.pgen.1000758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bonnan A, Bony G, Ferezou I, Pietropaolo S, Ginger M, Sans N, Rossier J, Oostra B, LeMasson G, Frick A (2014a) Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1(-/y) mice. Nat Neurosci 17:1701–1709. 10.1038/nn.3864 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014b) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wang J, Song S, Li F, Yuan F (2015) Reduction of alpha1GABAA receptor mediated by tyrosine kinase C (PKC) phosphorylation in a mouse model of fragile X syndrome. Int J Clin Exp Med 8:13219–13226. [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Chuang SC, Bianchi R, Zhao W, Paul G, Thakkar P, Liu D, Fenton AA, Wong RK, Tiedge H (2010) Regulatory BC1 RNA and the fragile X mental retardation protein: convergent functionality in brain. PLoS One 5:e15509. 10.1371/journal.pone.0015509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Nannapaneni N, Shore S (2007) Vessicular glutamate transporters 1 and 2 are differentially associated with auditory nerve and spinal trigeminal inputs to the cochlear nucleus. J Comp Neurol 500:777–787. 10.1002/cne.21208 [DOI] [PubMed] [Google Scholar]