

An isoform of peroxisome proliferator-activated receptors (PPARs), PPARγ, is the receptor for the thiazolidinedione class of anti-diabetic medications including pioglitazone. Neuroanatomical data indicate PPARγ localization in brain areas involved in drug addiction. Preclinical and clinical data have shown that pioglitazone reduces alcohol and opioid self-administration, relapse to drug seeking, and plays a role in emotional responses.
Keywords: addiction, nicotine, pioglitazone, PPARγ, withdrawal
Abstract
An isoform of peroxisome proliferator-activated receptors (PPARs), PPARγ, is the receptor for the thiazolidinedione class of anti-diabetic medications including pioglitazone. Neuroanatomical data indicate PPARγ localization in brain areas involved in drug addiction. Preclinical and clinical data have shown that pioglitazone reduces alcohol and opioid self-administration, relapse to drug seeking, and plays a role in emotional responses. Here, we investigated the behavioral effect of PPARγ manipulation on nicotine withdrawal in male Wistar rats and in male mice with neuron-specific PPARγ deletion (PPARγ(−/−)) and their littermate wild-type (PPARγ(+/+)) controls. Real-time quantitative RT-PCR and RNAscope in situ hybridization assays were used for assessing the levels of expression and cell-type localization of PPARγ during nicotine withdrawal. Brain site-specific microinjections of the PPARγ agonist pioglitazone were performed to explore the role of this system on nicotine withdrawal at a neurocircuitry level. Results showed that activation of PPARγ by pioglitazone abolished the expression of somatic and affective nicotine withdrawal signs in rats and in (PPARγ(+/+)) mice. This effect was blocked by the PPARγ antagonist GW9662. During early withdrawal and protracted abstinence, the expression of PPARγ increased in GABAergic and glutamatergic cells of the amygdala and hippocampus, respectively. Hippocampal microinjections of pioglitazone reduced the expression of the physical signs of withdrawal, whereas excessive anxiety associated with protracted abstinence was prevented by pioglitazone microinjection into the amygdala. Our results demonstrate the implication of the neuronal PPARγ in nicotine withdrawal and indicates that activation of PPARγ may offer an interesting strategy for smoking cessation.
SIGNIFICANCE STATEMENT Smoking cessation leads the occurrence of physical and affective withdrawal symptoms representing a major burden to quit tobacco use. Here, we show that activation of PPARγ prevents the expression of both somatic and affective signs of nicotine withdrawal. At molecular levels results show that PPARγ expression increases in GABAergic cells in the hippocampus and in GABA- and glutamate-positive cells in the basolateral amygdala. Hippocampal microinjections of pioglitazone reduce the insurgence of the physical withdrawal signs, whereas anxiety linked to protracted abstinence is attenuated by pioglitazone injected into the amygdala. Our results demonstrate the implication of neuronal PPARγ in nicotine withdrawal and suggest that PPARγ agonism may represent a promising treatment to aid smoking cessation.
Introduction
Tobacco use is one of the leading causes of preventable disease and death worldwide, killing >7 million people each year (World Health Organization, 2017). Nicotine, the major psychoactive compound in tobacco, exerts its addictive properties by affecting neuronal circuits that control reward, motivation, and habit processes (De Biasi and Dani, 2011).
Smoking cessation leads to the insurgence of the physical and affective withdrawal symptoms, the major burden to quit tobacco use (Grunberg, 2007). Anxiety, depression, and impaired memory represent the major negative affective disturbances in nicotine withdrawal (Hughes et al., 1986; Caan et al., 1996; Jorenby et al., 1996; Kenny et al., 2001). Insurgence of the physical and affective withdrawal symptoms can be also be observed in rats and mice upon cessation of nicotine when administered via osmotic minipumps or chronic injections (Shiffman and Jarvik, 1976; Rossetti et al., 1999; Watkins et al., 2000; Malin et al., 2006).
Recently peroxisome proliferator-activated receptors (PPARs) have been proposed as new therapeutic targets in drug addiction including nicotine dependence (Le Foll et al., 2013). PPARs are a family of nuclear receptor proteins that regulate gene expression as ligand-activated transcription factors (Michalik et al., 2006). Although PPARs were first identified in peripheral tissue, there is now evidence of their abundant distribution in the brain (Sarruf et al., 2009; Schnegg and Robbins, 2011). Among the three isoforms (α, δ, and γ), PPARγ has the highest level of expression in the CNS. Initially, PPARγ expression in the brain was suggested mainly in astrocytes, and glial cells (Sarruf et al., 2009). Recent studies, however, suggest that PPARγ expression in the brain occurs almost exclusively in neurons and astrocytes (Warden et al., 2016). PPARγ agonists have been shown to modulate genes linked to synaptic transmission and neuronal function in regions such as the amygdala (AMY) and hippocampus (HIPP; Searcy et al., 2012; Ferguson et al., 2014). However, despite the evidence of a role of PPARγ in drug addiction, the neuronal contribution underlying its effects has not been yet explored. We previously showed that pioglitazone, a selective PPARγ agonist, was highly effective in reducing behavioral responses to stress and preventing stress-induced relapse to alcohol and opioid seeking in rats (Stopponi et al., 2011; Domi et al., 2016; de Guglielmo et al., 2017). Pioglitazone was also shown to reduce heroin craving and anxiety in humans (Jones et al., 2018), and most important, a recent clinical study reported that pioglitazone attenuates measures of nicotine craving (Jones et al., 2017).
Here, we explored the role of PPARγ in nicotine withdrawal to better determine the potential of this receptor system as a treatment target for nicotine abuse. To this end, we assessed the efficacy of pioglitazone on the expression of the physical and affective withdrawal symptoms in a rat model of nicotine dependence induced by application of nicotine patches (Cippitelli et al., 2011). To address the functional contribution of neuronal PPARγ in nicotine addiction we used mice with a neuronal deletion of PPARγ (PPARγ(−/−)) in a model of nicotine dependence using chronic subcutaneous injections of nicotine. The specificity of the effects of pioglitazone was confirmed by using the selective PPARγ antagonist GW9662. Based on our previous findings on the role of amygdaloid PPARγ on anxiety, and the anatomical distribution of PPARγ in limbic structures involved in nicotine addiction we investigated the regional, temporal, and cell-specific changes in PPARγ expression occurring in AMY and HIPP during nicotine withdrawal in PPARγ(+/+) mice.
Materials and Methods
Subjects.
Male Wistar rats (275–325 g at the beginning of the experiments) were single housed and kept on a reverse 12 h light/dark cycle (lights on 20:00–08:00 h). Male mice with neuronal-specific PPARγ deletion (PPARγ(−/−)) and their littermate wild-type (PPARγ(+/+); 20–25 g, ∼8 weeks of age at the beginning of the experiments), were housed in groups of four to five and kept in a normal light/dark cycle (lights on 08:00–20:00 h). Experiments were conducted in separate groups of animals and during the dark phase of the light/dark cycle except for the physical spontaneous nicotine withdrawal assessment. Upon arrival, animals were weighed and handled daily. To obtain the conditional inactivation of PPARγ in neuronal cells transgenic mice expressing the Cre recombinase under the control of rat Nestin (Nes) promoter were bred to homozygous PPARγ loxP/loxP mice. The resulting heterozygous F1 offspring (PPARγ+/loxP) were either positive or negative for Nes–Cre. From mating of PPARγ+/loxP with PPARγ+/loxPNes–Cre mice, F2 generation of the desired genotypes [PPARγ loxP/loxPNes–Cre (PPARγNestinCre) and PPARγ loxP/loxP] were obtained, which were then intercrossed to obtain F3 generation. PPARγ loxP/loxP mice were used as control littermates (PPARγ(+/+)) for conditional PPARγ mice (PPARγ(−/−)). The mice used for this study, were bred at the School of Pharmacy of the University of Camerino and originally provided by Dr. K. Niswender (Vanderbilt University, Nashville, NT) were on a C57BL/6J background (Jones et al., 2002). All animals were housed at constant temperature (20–22°C) and humidity (45–55°). Ad libitum food and water were provided for the entire duration of the experiments.
All procedures followed the EU Directive for Care and Use of Laboratory Animals and were approved by the Ethical Committee of the University of Camerino.
Drugs.
Nicotine patches (NIQUITIN CQ Step 1,21 mg/d, Glaxo) were used for the induction of nicotine dependence in rats (Cippitelli et al., 2011). (-)-Nicotine hydrogen tartrate salt (Sigma-Aldrich) was dissolved in sterile saline and administered by subcutaneous injections in mice (10 ml/kg). Pioglitazone (Actos, Takeda Pharmaceuticals) was suspended in distilled water and administered orally (1 ml/kg or 10 ml/kg) in rats and mice, respectively. GW9662 (Tocris Bioscience) was dissolved in 5% DMSO, 5% Tween 20, and 90% distilled water and administered intraperitoneally (1 ml/kg or 10 ml/kg), in rats and mice, respectively. For brain site-specific microinjections, pioglitazone was purchased from Molcan and dissolved in 15% DMSO, 10% Cremophor and the final volume was adjusted adding sterile saline. Pioglitazone (5 μg/0.6 μl) was administered in a volume of 0.3 μl per site. Drugs were prepared immediately before administration.
Nicotine dependence and spontaneous nicotine withdrawal.
Rats was thoroughly shaved on the back, depilated with a depilatory lotion, and cleansed with water as previously described (Cippitelli et al., 2011). Patches were divided into four equal parts so that 5.2 mg/rat/d of nicotine was administered by patch applied to the shaved region. Pieces of flexible fabric Band-Aid and waterproof tape were used to wrap the nicotine patch and to improve its adherence to the rat's back. Control rats were shaved and depilated, but only the Band-Aid and waterproof tape were placed on their backs. Comparable doses were previously shown to produce sufficiently high blood nicotine and cotinine levels to elicit the occurrence of reliable nicotine abstinence symptoms (Cippitelli et al., 2011). This application procedure was repeated for 7 consecutive days and on Day 8, transdermal patches were removed to induce spontaneous nicotine withdrawal. To assess the physical withdrawal signs, 16 h after removal of the transdermal nicotine patches, rats were placed into transparent cylinders and the frequency of nicotine abstinence signs was counted for 10 min by an observer blind to treatment condition. The most frequently observed categories included teeth-chattering/chews, writhes/gasps, wet dog shakes/tremors, and yawns. For statistical analysis withdrawal signs were compounded and the total score was evaluated as previously described (Cippitelli et al., 2011). Previous research, reported that a similar dose of nicotine (5.0 mg/kg) induced blood nicotine levels averaged 88.5 ± 21.5 ng/ml and blood cotinine levels averaged 647.6 ± 123.2 ng/ml after administration with the same procedure (Slawecki and Ehlers, 2002).
In mice nicotine dependence was induced by four daily subcutaneous injections of 2 mg/kg for 8 consecutive days. On Day 9 and 20 h after the last nicotine injection, mice were placed into transparent cylinders (diameter 28.5 cm) and then observed for 30 min for the occurrence of the following signs: paw tremors, chewing, genital licks, scratches, teeth chattering, head shakes, abdominal constrictions, and jumps. All signs were compounded and the total score was evaluated (Kenny and Markou, 2001).
Elevated plus maze.
The elevated plus maze (EPM) test was performed 6 d following the removal of nicotine patches in rats and 6 d after the last nicotine injections in mice. This time point corresponds to protracted abstinence during which affective withdrawal signs (i.e., anxiety) are exhibited. Each trial lasted 5 min and animals were recorded using an EthoVision video tracking system (Noldus Information Technology). Percentage OAT = (time in open arm/time in “open arm” + time in “closed arm”) × 100 was considered an index of anxiety, whereas total number of entries was considered a locomotion index.
Real-time quantitative RT-PCR.
Tissue samples were collected from PPARγ(+/+) mice 20 h and 6 d after last nicotine injection corresponding to the early and late abstinence time points. Brains were rapidly removed and dissected to harvest HIPP and AMY. Tissues were immediately frozen in dry ice and stored at −80°C until RNA extraction. Total mRNA was isolated using TRIZOL reagent (Life Technologies) and its integrity was checked by gel electrophoresis. The amounts of RNA were determined by measuring optical densities and only RNA samples with an OD260/OD280 ratio 1.8–2.0 were used. Total RNA was reverse transcribed using random hexamers and MMLV Reverse Transcriptase (Life Technologies) in a final volume of 20 μl, according to the manufacturer's instructions. Quantitative real-time PCR was performed using TaqMan Gene Expression Master Mix on an Applied Biosystems Step One System as previously reported (Caputi et al., 2015, 2016). Relative expression of PPARγ gene transcripts (Mm 01184322_m1 FAM, Applied Biosystems) was calculated by Delta–Delta Ct (DDCt) method and converted to relative expression ratio (2-DDCt) for statistical analysis (Livak and Schmittgen, 2001). Data were normalized to the endogenous reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression (Mm 99999915_g1 VIC, Applied Biosystems). Data are expressed as mean ± SEM of six samples/group (each sample run in triplicate). After PCR, a dissociation curve (melting curve) was constructed in the range of 60–95°C to evaluate the specificity of the amplification products (Lyon, 2001). Results are represented as fold-change in mRNA levels.
RNAscope in situ hybridization assay.
We performed RNA in situ hybridization in HIPP and AMY for PPARγ (accession number NM_011146.3, target nucleotide region 170–1490), Slc17a7 (VGlut1, accession number NM_182993.2, target nucleotide region 464–1415), and Gad1 (GAD67, accession number NM_008077.4, target nucleotide region 62–3113) mRNAs as described previously (Rubio et al., 2015). Brains were removed and quickly frozen in isopentane on dry ice and stored at −80°C. Brains slices (20 μm) were collected approximately at the bregma level −1.7 mm (Paxinos and Franklin, 2003) were mounted directly onto Superfrost Plus microscope slides (Fisher Scientific) and stored at −80°C until the in situ hybridization was performed. In situ hybridization was performed according to the RNAscope Fluorescent Multiplex Kit User Manual (Advanced Cell Diagnostics). Briefly, the slides were transferred to slide racks and fixed by immersion in 10% neutral buffered formalin for 15 min at 4°C, and then dehydrated in a series of 50, 70, and 100% ethanol at room temperature. A hydrophobic pen was used to create a barrier around each brain section. Sections were incubated for 30 min at room temperature with the protease pretreatment solution from the RNAscope Fluorescent Multiplex Kit (Advanced Cell Diagnostics) and then washed in PBS and incubated with the target probes for 2 h at 40°C using the HybEZ Hybridization System (Advanced Cell Diagnostics). Sections were then incubated with a series of four amplifier probes each for Steps 1 and 3, 30 min each for Steps 2 and 4) at 40°C, fluorescently labeled in Step 4 with AlexaFluor 488 (green), Atto 550 (red), and Atto 647 (far red) to visualize probes in each of the three channels in different colors, washing with the wash buffer provided with the kit between each step. Finally, sections were briefly incubated with DAPI to visualize nuclei in blue. Slides were coverslipped, air dried, and stored at 4°C.
Tissue sections were examined with a confocal microscope (Zeiss LSM 700) at 40× magnification to determine colocalization and to create images for quantification.
Tile scan images to visualize the entire brain section were taken with a fluorescent microscope (Leica, DMi8) at 10× magnification. For quantification of each PPARγ, GAD67, and VGlut1-positive cells (n = 5/per group, 2 sections) we counted the total pixels of the fluorescent signal (fluorescent “dots”), which represent a single molecule of mRNA above the sensitivity threshold of the assay, using ImageJ software (Rubio et al., 2015). RNAscope in situ hybridization analysis revealed measurable transcript of PPARγ expression in GABAergic (GAD67-positive) and glutamatergic (VGlut1-positive) cells of the HIPP, basolateral amygdala (BLA), and central amygdala (CeA) in PPARγ(+/+) mice (n = 5/group).
Intracranial surgery and histological analysis.
PPARγ(+/+) mice were anesthetized by intramuscular injection of 100–150 μl of a tiletamine chlorohydrate (58.17 mg/10 ml) and zolazepam chlorohydrate (57.5 mg/10 ml) and placed into a stereotaxic frame. The skull was exposed and stainless steel guide cannulae (diameter: 0.35 mm; length: 7 mm) were bilaterally implanted to reach the AMY or the HIPP (Paxinos and Franklin, 2003). Mice were bilaterally implanted to reach AMY or HIPP using the following coordinates: (1) AMY: 1.4 mm caudal from the bregma, ±3.0 mm mediolateral and −3.9 mm ventral from the dura; and (2) dorsal HIPP: 1.7 posterior to bregma, ± 1.5 mm mediolateral, and 1.3 mm ventral to skull surface (Paxinos and Franklin, 2003). The guide cannulae were fixed to the skull with dental cement and two anchoring screws.
The rate of injection was precisely controlled by an infusion pump (SPLab02, Baoding Shenchen Precision Pump Co.LTD). The stainless-steel injector protruding beyond the cannula tip 1.00 mm and 0.5 mm for the AMY and the HIPP, respectively, was allowed to remain in the brain 2 min/site before being retracted. Pioglitazone was administered via a 10 μl Hamilton syringe at a rate of 0.25 μl/min. At completion of the experiments, to verify the cannula placement, mice were lightly anesthetized with isoflurane and 0.3 μl/site Malachite green solution was injected into the area. After the animals were killed, the ink diffusion into the region was histologically evaluated.
The injection sites were confirmed for both AMY and HIPP by comparison with plates taken from a mouse brain atlas (Paxinos and Franklin, 2003). Histological analysis confirmed correct bilateral injections into the AMY in 39 mice and in the HIPP in 43 mice. Only these mice were used for statistical analysis (see Fig. 5). Behavioral tests were initiated following full recovery (5–6 d after surgery).
Figure 5.

PPARγ transcription levels in CeA. Representative images (40× magnification) in CeA for PPARγ (red) + GAD67 (white) and DAPI (blue) in control, early, and late nicotine withdrawal. A–C, Merge of PPARγ (red) + DAPI (blue) counts. (D–F) represents PPARγ+ GAD67 + cells and (G–I) represents the merge of PPARγ +, GAD67+, and DAPI cells. J, PPARγ counts in VGLUT1+ cells, (K) PPARγ counts in GAD67+ cells, and (L) coronal brain section illustrating CeA region sampled for RNAscope ISH analysis. Data are expressed as mean ± SEM values.
Statistical analysis.
Behavioral experiments were analyzed by mean of one- or two-way ANOVA. Differences were considered significant if p < 0.05. Post hoc comparisons were performed by Newman–Keuls or Dunnett tests, when appropriate. Data were first examined for significant violations to the assumption of homogeneity of variances using Bartlett's test. When deviation from homogeneity of variances was detected (RNAscope ISH assay), nonparametric Kruskall–Wallis analysis was performed followed by multiple comparisons between the independent groups.
Results
Systemic pioglitazone reduced the expression of the physical and affective nicotine withdrawal signs in rats
Physical withdrawal signs: rats (n = 27) received nicotine patches and were treated with pioglitazone (0, 15, or 30 mg/kg) 12 and 1 h before assessing physical nicotine withdrawal. Nicotine withdrawal increased the insurgence of the physical signs that were reduced by pioglitazone treatment.
One-way ANOVA showed a significant overall treatment effect (F(3,23) = 77.7; p = 0); η2 = 0.77=). Post hoc analysis showed a significant expression of physical nicotine withdrawal signs in rats subjected to nicotine and treated with vehicle versus control (p < 0.001). Withdrawal score was significantly reduced following administration of pioglitazone 15 mg/kg (p < 0.01) and 30 mg/kg (p < 0.001) versus vehicle (Fig. 1A).
Figure 1.
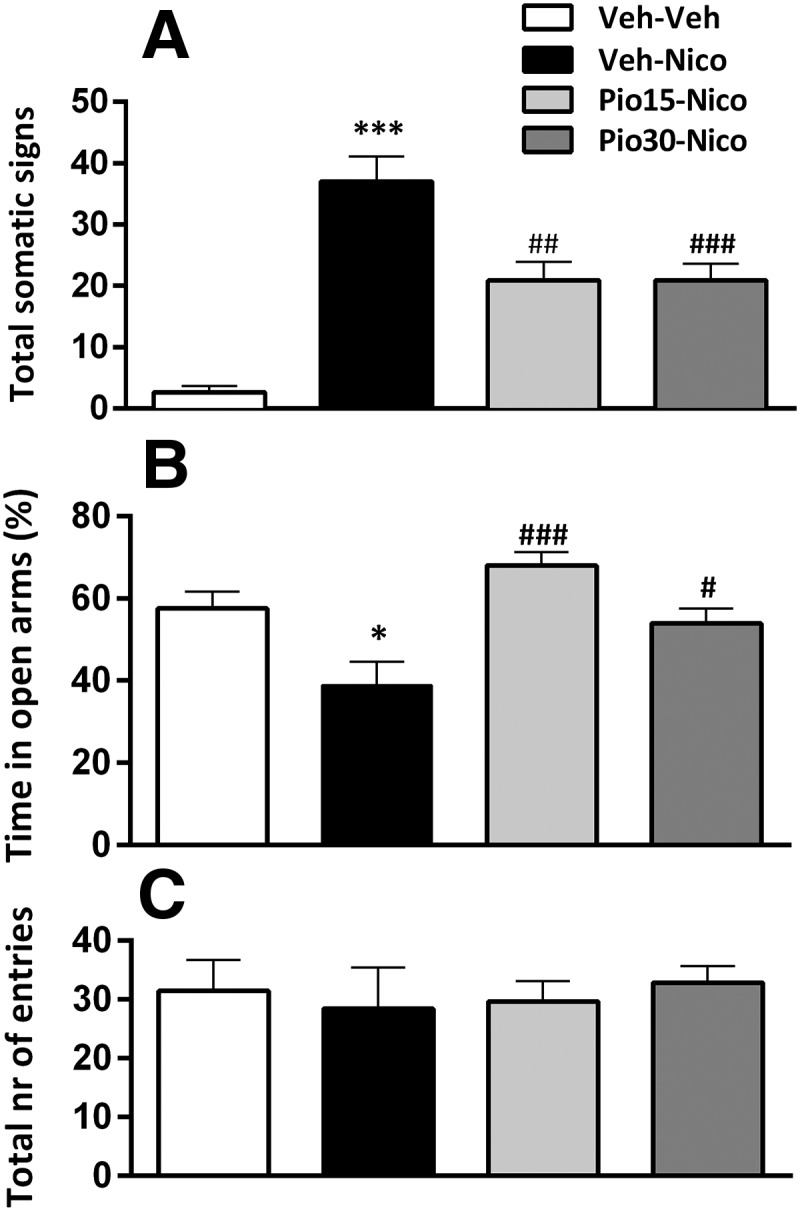
Pioglitazone (Pio) on nicotine (Nico) withdrawal-induced physical symptoms and anxiety-like behavior. A, Nicotine cessation elicited a robust score of the physical withdrawal signs (***p < 0.001) reduced by Pio (15 and 30 mg/kg; ##p < 0.01, ###p < 0.001). B, Percentage time in open arms in the EPM. Nicotine induced an anxiogenic effect (*p < 0.05) that was blocked by Pio 15 and 30 mg/kg (###p < 0.001, #p < 0.05). C, Total number of entries. Data are expressed as mean ± SEM values. Veh, Vehicle.
Affective withdrawal signs
EPM test was performed 6 d after the removal of nicotine patches and pioglitazone (0, 15, or 30 mg/kg) was given twice 12 and 1 h before the test.
Nicotine withdrawal decreased the percentage time in the open arms and this anxiogenic-like behavior was reversed by pioglitazone.
Overall ANOVA of the percentage time spent in open arms showed a significant effect of treatment (F(3,23) = 7.3; p = 0.001); η2 = 0.48. Post hoc comparisons showed a significant increase of anxiety-like behavior in nicotine-treated rats versus control (p < 0.05). Pioglitazone significantly attenuated anxiety-like behavior (15 mg/kg: p < 0.001; 30 mg/kg: p < 0.05; Fig. 1B).
No significant main effect of treatment was observed in the total number of entries in the EPM (F(3,23) = 0.1; p = 0.93; Fig. 1C).
Systemic pioglitazone reduced the expression of the physical and affective nicotine withdrawal signs in PPARγ(+/+) but not in PPARγ(−/−) mice
Physical withdrawal signs
PPARγ(+/+) and PPARγ(−/−) mice were divided into four groups (n = 6–8/group). Nicotine withdrawal induced a substantial increase of the physical withdrawal signs in PPARγ(+/+) and PPARγ(−/−) mice and pioglitazone reduced the expression only in the PPARγ(+/+) genotype. Factorial ANOVA showed a significant effect of genotype (F(1,54) = 25.4; p = 0.001); η2 = 0.48, treatment (F(3,54) = 5.9; p = 0.001); η2 = 0.24 and interaction genotype × treatment (F(3,54) = 2.77; p = 0.04); η2 = 0.13. Post hoc analysis showed a significant expression of physical withdrawal signs in PPARγ(+/+) (p < 0.05) and PPARγ(−/−) (p < 0.01) in nicotine-treated mice versus control. Pioglitazone 15 and 30 mg/kg significantly reduced physical withdrawal signs in PPARγ(+/+) mice (p < 0.05; Fig. 2A).
Figure 2.
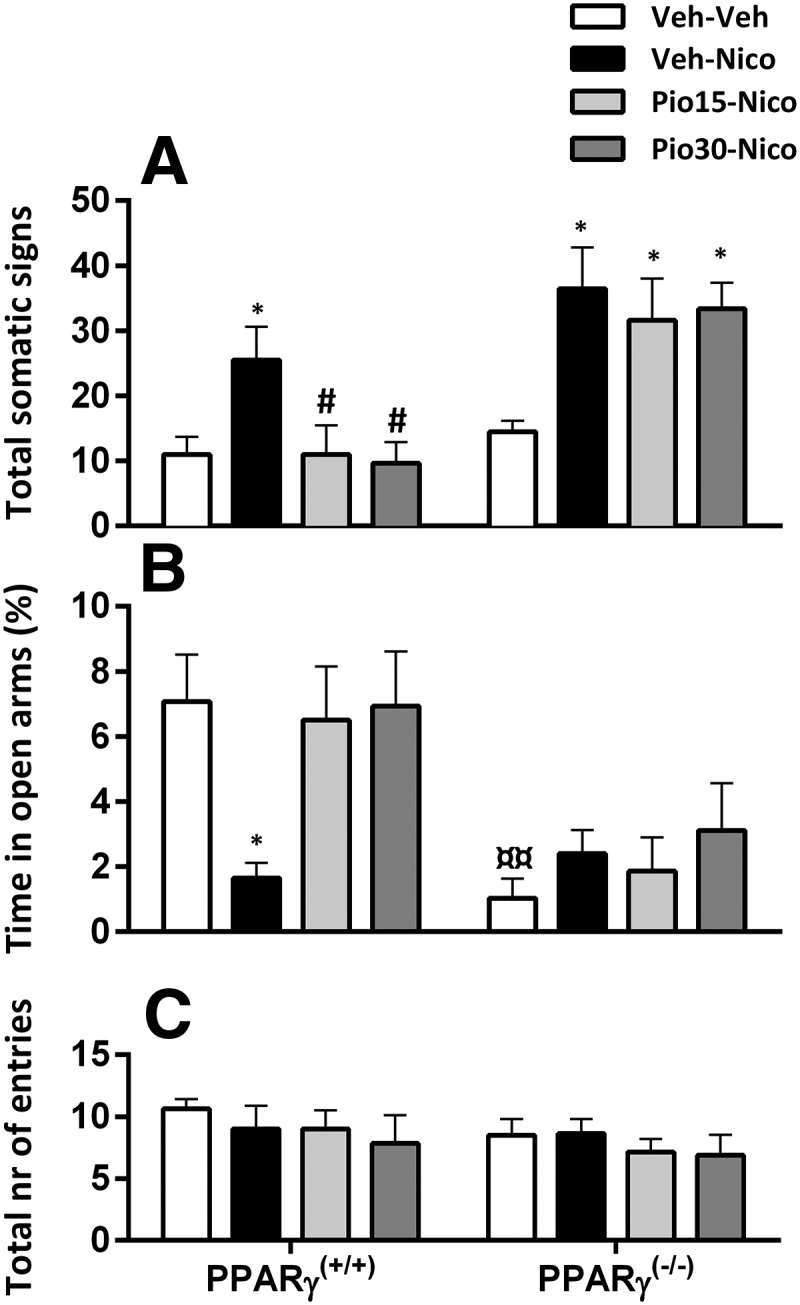
Effects of pioglitazone (Pio) on nicotine (Nico) withdrawal-induced physical signs and anxiety-like behavior in PPARγ(+/+) and PPARγ(−/−) mice. A, Nicotine induced significant expression of physical withdrawal symptoms (*p < 0.05) prevented by Pio in PPARγ(+/+) (#p < 0.05). B, Percentage time in open arms. Nicotine induced an anxiogenic effect in PPARγ(+/+) mice (*p < 0.05), prevented by Pio 15 and 30 mg/kg (#p < 0.05). PPARγ(−/−) mice exhibited a higher anxiety baseline compared with PPARγ(+/+) mice (¤¤p < 0.01). C, Total number of entries. Data are expressed as mean ± SEM values. Veh, Vehicle.
Affective withdrawal signs
PPARγ(−/−) spent less time in the open arms of the EPM compared with PPARγ(+/+) mice. Nicotine withdrawal decreased the percentage time in the open arms in PPARγ(+/+) mice and this effect was reversed by pioglitazone.
Factorial ANOVA showed a significant genotype effect (F(1,54) = 16.5; p = 0); η2 = 0.23, no effect of treatment (F(3,54) = 2.26; p = 0.09) but a significant interaction genotype × treatment (F(3,54) = 3.15; p = 0.03); η2 = 0.15. Post hoc analysis showed a higher basal anxiety-like behavior in PPARγ(−/−) compared with PPARγ(+/+) (p < 0.01). Nicotine withdrawal induced a significant decrease of percentage of time in the open arms in PPARγ(+/+) (p < 0.05; Fig. 2A). Pioglitazone, 15 and 30 mg/kg significantly reversed nicotine withdrawal-induced anxiety (p < 0.05) in PPARγ(+/+) mice. In PPARγ(−/−) mice anxiety-like response was significantly higher in all groups compared with PPARγ(+/+) mice (p < 0.05) and was not affected by nicotine or pioglitazone. (Fig. 2B). The total number of entries did not differ between genotypes and was not affected by the treatment. Factorial ANOVA did not show a significant effect of genotype (F(1,54) = 1.24; p = 0.26), treatment (F(3,54) = 0.62; p = 0.6), or interaction genotype × treatment in the total number of entries (F(1,54) = 0.2; p = 0.89; Fig. 2C).
Systemic GW9662 blocked the effect of pioglitazone on the physical and affective nicotine withdrawal signs in wild-type PPARγ(+/+) mice
A new cohort of PPARγ(+/+) mice (n = 51), divided into five groups (n = 8–12/group) was tested to verify the specificity of pioglitazone on PPARγ receptors by pretreatment with the selective PPARγ antagonist, GW9662. Nicotine withdrawal induced an increase of the physical signs that were reduced by pioglitazone. The effect of pioglitazone was prevented by GW9662.
Physical withdrawal signs
ANOVA showed a significant main effect of pioglitazone treatment (F(1,46) = 3.6; p = 0.012); η2 = 0.23. Post hoc analysis confirmed a significant expression of the physical withdrawal signs in nicotine-treated mice versus control (p < 0.05). Pioglitazone 30 mg/kg significantly reduced the expression of the physical withdrawal score (p < 0.05). GW9662 significantly blocked the effect of pioglitazone (p < 0.05; Fig. 3A).
Figure 3.
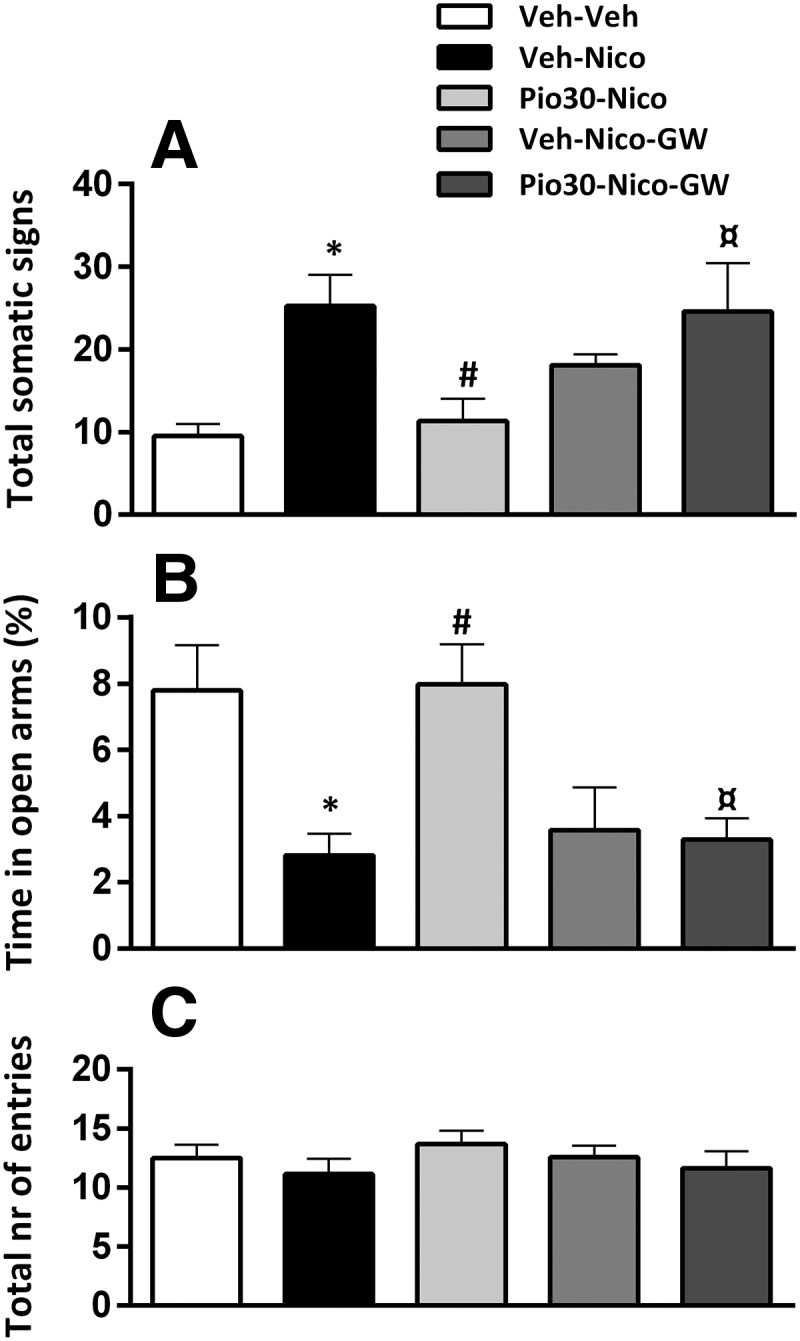
Effects of pioglitazone (Pio), GW9662 and the combination on the expression of the physical withdrawal signs and anxiety-like behavior in PPARγ(+/+) mice. A, Nicotine (Nico) induced a significant physical withdrawal score (*p < 0.05) prevented by Pio (30 mg/kg; #p < 0.05). The effect was blocked by GW9662 (¤p < 0.05). B, Nicotine reduced percentage time in open arms (*p < 0.05), effect reversed by Pio (30 mg/kg; #p < 0.05) and blocked by GW9662 (¤p < 0.05). C, Total number of entries. Data are expressed as Mean ± SEM values. Veh, Vehicle.
Affective withdrawal signs
Nicotine withdrawal decreased the percentage time in the open arms and this effect was reversed by pioglitazone. GW9662 prevented the effect of pioglitazone.
One-way ANOVA showed a significant main effect of treatment (F(1,46) = 6.7; p = 0); η2 = 0.37 on anxiety-like behavior. Post hoc analysis showed a significant reduction in percentage time in open arms in nicotine exposed mice vs control (p < 0.01). Pioglitazone 30 mg/kg significantly reversed the anxiogenic condition (p < 0.01). GW9662 and nicotine-treated mice showed a decrease in percentage spent in open arms compared with vehicle-treated mice (p < 0.05). The effect of pioglitazone was completely blocked by GW9662 (p < 0.01; Fig. 3B). Total number of entries did not differ between groups. ANOVA did not show a significant effect of treatment in total number of entries (F(1,46) = 0.7; p = 0.55; Fig. 3C).
PPARγ gene expression is increased in the early and late stage of nicotine withdrawal
PPARγ mRΝΑ levels were analyzed in HIPP and AMY in PPARγ(+/+) mice (n = 17). PPARγ expression increased during the early and late nicotine withdrawal in both dorsal HIPP and AMY. ANOVA showed a significant main difference of PPARγ expression in both dorsal HIPP (F(2,15) = 66.56; p = 0); η2 = 0.89 and AMY (F(2,14) = 21.95; p = 0); η2 = 0.23. Compared with the control group, Dunnett's post hoc tests showed a significant increase of PPARγ mRNA levels at both 20 h and 6 d into nicotine withdrawal (p < 0.001) in the HIPP (Fig. 4A). In AMY, PPARγ mRNA levels were also significantly increased at 20 h (p < 0.05) and 6 d (p < 0.001; Fig. 4B).
Figure 4.

PPARγ transcription levels in the (A, left) HIPP (B, left) AMY at 20 h and 6 d into nicotine withdrawal PPARγ(+/+) mice. Nicotine withdrawal increased PPARγ mRNA levels after 20 h and 6 d in HIPP (***p < 0.001) and AMY (*p < 0.05, **p < 0.01). Representative images (40× magnification) in the HIPP (top; a–c) and in the AMY (bottom; g–i) for PPARγ (red) + VGLUT1 (green) and GAD67 (white) in control, early, and late nicotine withdrawal. m, n, Coronal brain section of HIPP and BLA region from RNAscope ISH analysis. d, j, Merge of PPARγ (red) + GAD67 + VGLUT1 (green) and DAPI (blue) PPARγ counts in HIPP and AMY. e, f, PPARγ counts in VGlut1+ and GAD67 + cells in HIPP. Early nicotine withdrawal increased PPARγ+ cells in GAD67 + cells (*p < 0.05). k, In the BLA early nicotine withdrawal increased PPARγ+ cells in VGLUT1+ cells (#p < 0.05). l, PPARγ expression in GAD67 + cells increased in the late withdrawal phase compared with control and early withdrawal (#p < 0.05). Data are expressed as mean ± SEM values.
Nicotine withdrawal induced PPARγ expression changes in GABAergic and glutamatergic cells of HIPP and AMY
In dorsal HIPP nicotine withdrawal increased PPARγ expression in GAD67+ but not in VGlut1+ cells. Nonparametric analysis showed an increase of PPARγ in GAD67+ cells (χ2 = 10.17, df = 2, p = 0.006) and no significant difference in VGlut1+ cells (χ2= 0.5, df = 2, p = 0.7). Multiple pairwise comparison between the independent groups showed a significant increase of PPARγ+/GAD67+ in early withdrawal compared with control (p < 0.05; Fig. 4f). In the late nicotine withdrawal period PPARγ+/ GAD67+ cells did not differ compared with control or to the early time point.
Bartlett's test showed a significant violation assumption of homogeneity of variances in PPARγ expression in VGlut1+ and GAD67+ cells in BLA in the early withdrawal phase (Bartlett's χ2 = 10.43, df = 2, p = 0.005; χ2 = 10.92, df = 2, p = 0.004, respectively). The nonparametric Kruskal–Wallis test showed a significant difference between groups (χ2 = 6.9, df = 2, p = 0.03; χ2 = 8.6, df = 2, p = 0.01). Multiple comparisons between groups showed a significant increase in PPARγ+/VGlut1+ cells compared with control (p < 0.05; Fig. 4k). PPARγ expression increased significantly in GAD67+ cells of the BLA in the late withdrawal group compared with control and early withdrawal group (p < 0.05; Fig. 4l). Expression of PPARγ in VGlut1+ cells in the CeA was too low for statistical quantification, whereas the expression of PPARγ was well quantifiable in GAD67+ cells (Bartlett′s χ2 = 10.43, df = 2, p = 0.005) There was no significant difference between experimental groups in PPARγ+/GAD67+ cells of the CeA in both phases of nicotine withdrawal (χ2 = 2.142, df = 2, p = 0.34; see Fig. 5).
HIPP infusion of pioglitazone attenuated the expression of the physical but not affective nicotine withdrawal signs in PPARγ(+/+) mice
Physical withdrawal signs
PPARγ(+/+) mice (n = 45) were subjected to chronic nicotine or saline treatment followed by pioglitazone (0 or 0.3 μl). Four mice lost the cannula implant and were excluded from the statistical analysis.
Two-way ANOVA showed a significant effect of nicotine exposure (F(1,37) = 20.23; p = 0); η2 = 0.34, pioglitazone (F(1,37) = 7.34; p = 0.016); η2 = 0.15, and a significant interaction nicotine × pioglitazone (F(1,37) = 5.48; p = 0.02); η2 = 0.13 in the physical withdrawal score. Post hoc comparisons showed a significant increase of the physical withdrawal signs in nicotine mice compared with vehicles (p < 0.001) reduced by pioglitazone (p < 0.01; Fig. 6A).
Figure 6.

Effect of pioglitazone (Pio) in HIPP and AMY (left, right, respectively) on physical and affective nicotine withdrawal signs in PPARγ(+/+) mice. A, B, Nicotine (Nico) withdrawal induced physical withdrawal signs (***p < 0.001, **p < 0.01) prevented by Pio in HIPP (##p < 0.01). C, D, Nicotine withdrawal decreased percentage open arm time (**p < 0.01, *p < 0.05) versus control. D, Pio in the AMY reversed the anxiogenic behavior (#p < 0.05). E, F, Total number of arm entries. Data are expressed as mean ± SEM values. Histological reconstructions show correct (filled circles) injections into the AMY and dorsal HIPP, taken from Paxinos and Franklin (2003). Veh, Vehicle.
Affective withdrawal signs
Factorial ANOVA showed a significant effect of nicotine on the percentage time spent in the open arms (F(1,37) = 19.06; p = 0); η2 = 0.3, no effect of pioglitazone (F(1,37) = 0.24; p = 0.5) or interaction nicotine × pioglitazone (F(1,37) = 1.60; p = 0.2). Nicotine-treated mice spent less time in the open arms compared with control (p < 0.01; Fig. 6C). No difference was observed in the total number of arm entries by nicotine (F(1,37) = 1.36; p = 0.25), pioglitazone (F(1,37) = 0.1; p = 0.7), or their interaction (F(1,37) = 0.01; p = 1.2; Fig. 6E).
Infusion of pioglitazone into the AMY attenuated the expression of the affective but not physical nicotine withdrawal signs in PPARγ(+/+) mice
Physical withdrawal signs
PPARγ(+/+) mice (n = 46) were subjected to chronic nicotine or saline followed by administration of pioglitazone (0 or 0.3 μl). Mice that lost the cannula implant before assessing the affective withdrawal signs (n = 2) and mice with wrong cannula placement (n = 5) were excluded from the statistical analysis.
Two-way ANOVA showed a significant effect of nicotine (F(1,35) = 13.53; p = 0); η2 = 0.25; but not a significant pioglitazone treatment (F(1,35) = 2.62; p = 0.15) or nicotine × pioglitazone interaction (F(1,35) = 1.46; p = 0.19; Fig. 6B).
Affective withdrawal signs
Two-way ANOVA on percentage time spent in the open arms showed a significant effect of nicotine (F(1,35) = 4.9; p = 0.035); η2 = 0.13, pioglitazone (F(1,35) = 4.34; p < 0.04); η2 = 0.11 and interaction nicotine × pioglitazone (F(1,35) = 4.21; p = 0.42); η2 = 0.1. Post hoc analysis showed a significant reduction of the percentage of time spent in open arms in nicotine mice versus control (p < 0.05). Pioglitazone increased percentage time spent in the open arms compared the nicotine-vehicle group (p < 0.05; Fig. 6D). ANOVA did not show a significant main effect of nicotine (F(1,35) = 1.52; p = 0.24), pioglitazone (F(1,35) = 0.05; p = 0.86), or nicotine × pioglitazone interaction (F(1,35) = 0.19; p = 0.67) in total number of entries (Fig. 6F).
Discussion
Smoking cessation leads to aversive physical and affective withdrawal symptoms that contribute to the maintenance of tobacco use, and promote relapse (Slawecki et al., 2003; Cippitelli et al., 2011; Piper et al., 2011). In the nicotine dependence models used in our study, discontinuation from chronic nicotine exposure elicited the expression of marked physical and affective withdrawal signs (Robinson et al., 1994). PPARγ activation by pioglitazone attenuated the expression of the withdrawal symptoms through neuronal mechanisms involving PPARγ in HIPP and AMY.
Specifically, we found that pioglitazone reduced both the negative physical signs expressed in acute withdrawal and the anxiogenic response associated to the protracted withdrawal stage in both rats and mice. Previous data have shown that PPARγ activation does not affect locomotor activity in rodents, indicating the specificity of its anxiolytic action (Morgenweck et al., 2010; Sadaghiani et al., 2011).
Here to characterize the involvement of PPARγ in the modulation of nicotine withdrawal, we compared the effect of chronic nicotine exposure and pioglitazone treatment in mice with a restricted deletion of neuronal PPARγ (PPARγ(−/−)) with its wild-type (PPARγ(+/+)) counterpart. Spontaneous insurgence of nicotine withdrawal induced a marked increase in the physical withdrawal signs in both genotypes. Nicotine withdrawal also produced an anxiogenic-like behavior in PPARγ(+/+) mice, whereas in PPARγ(−/−) mice, anxiety level was already extremely high under the basal condition, limiting the possibility to observe the effect of nicotine withdrawal on this parameter. The hyper-anxious phenotype of PPARγ(−/−) mice was described in an earlier report in which we demonstrated that genetic deletion of the neuronal PPARγ reduces resilience to environmental changes, dampening the innate ability to adapt to stressful stimuli (Domi et al., 2016). In subsequent experiments we found that pioglitazone ameliorated both physical and affective withdrawal signs in PPARγ(+/+) but not in the PPARγ(−/−) mice. Moreover, the effect of pioglitazone in PPARγ(+/+) mice was completely blocked by prior administration of the selective PPARγ antagonist GW9662. Altogether, these findings provide robust evidence of the specificity of neuronal PPARγ activation as a mechanism to ameliorate symptoms associated with nicotine withdrawal and demonstrate that the effect of pioglitazone is mediated by this nuclear receptor.
Neuroanatomical evidence of PPARγ expression in AMY, HIPP, and other brain regions involved in the regulation of emotion and motivation have been previously described (Moreno et al., 2004; Inestrosa et al., 2005; Gofflot et al., 2007; de Guglielmo et al., 2015). Cortical and amygdalar PPARγ activation has been linked to the attenuation of the negative effects induced by acute and chronic stress exposure (García-Bueno et al., 2005; Domi et al., 2016) and specific hippocampal PPARγ activation appears to play a role in attenuating the cognitive deficits caused by alcohol intoxication (Cippitelli et al., 2017).
We decided to ascertain the impact of nicotine withdrawal on PPARγ expression and distribution in both HIPP and AMY during early (20 h) and protracted (6 d) nicotine withdrawal in PPARγ(+/+) mice. Nicotine withdrawal was associated with a significant increase of PPARγ mRNA levels in both regions in the examined time points.
It is well known that expression and transcriptional activity of PPARγ is affected by the cross talk with several kinases, phosphatases (i.e., ERK- and p38-MAPK, PKA, PKC GSK3; Rochette-Egly, 2003) and cAMP response element binding protein (CREB; Liu et al., 2016). All these intracellular signaling elements are also known for their role in mediating nicotine effects (Brunzell et al., 2003; Burns and Vanden Heuvel, 2007; Michalak and Biala, 2017; Wang et al., 2017). Notably, Fisher et al. (2017) highlighted distinct roles of CREB within the HIPP in mediating nicotine withdrawal phenotypes in animals chronically treated with nicotine and undergoing 24 h into the withdrawal phase. Hence, we can speculate that the increase in PPARγ gene expression observed into early or late withdrawal in the AMY and HIPP may represent an adaptive response counteracting the physical and emotional state associated with nicotine withdrawal, similarly to increased PPARγ levels in stress conditions previously described (García-Bueno et al., 2008).
Nicotine exerts its pharmacological effects through activation of the nicotinic acetylcholine receptors which in turn activate different neurotransmitter systems, including glutamate and GABA, in various brain regions that can regulate not only the reinforcing properties of nicotine but also modulate the negative state associated with drug withdrawal (Picciotto and Corrigall, 2002; Hadjiconstantinou and Neff, 2011). Notably, PPARγ activation in the AMY induces significant changes in the expression of several genes linked to the GABAergic and glutamatergic transmission, emphasizing the hypothetical role of PPARs in the modulation of these two neurotransmitters in this region (Ferguson et al., 2014). Nicotine withdrawal was associated with enhanced PPARγ mRNA expression in AMY and HIPP; therefore, we decided to investigate whether these changes occurred in GABAergic or in glutamatergic neurons. Results showed a site-specific and time-dependent increase in the expression of PPARγ in both GABAergic and glutamatergic cells of AMY and HIPP. Specifically, in the early phase of nicotine withdrawal PPARγ mRNA was increased in glutamatergic neurons of the BLA and in GABAergic neurons of the HIPP. During protracted abstinence PPARγ transcript was increased in GABAergic neurons only of the BLA.
Marked biochemical and intracellular signaling alteration have been described in AMY and HIPP neurotransmission during nicotine withdrawal (Pandey et al., 2001; Tzavara et al., 2002; Fisher et al., 2017). In particular, electrophysiological experiments demonstrated that unpleasant withdrawal symptoms are linked to profound alteration in glutamate and GABA neurotransmission in HIPP (Yamazaki et al., 2006).
In this framework, it has been shown that nicotine exposure causes the downregulation of mGluR2/3 resulting in the impairment of the negative feedback control on glutamatergic terminals in several cortical and limbic brain sites (Liechti et al., 2007). Accordingly, the metabotropic glutamate receptor agonist, LY354740, decreases glutamate levels and reduces physical nicotine withdrawal symptoms in the rat (Helton et al., 1997; Cartmell and Schoepp, 2000). Considering the coexpression of PPARγ with VGLUT-positive cells, which mostly colocalize with mGlu2/3 (Di Prisco et al., 2016), one possibility is that the increase of PPARγ in the AMY might counteract the expression of nicotine withdrawal syndrome through the modulation of glutamatergic transmission.
The role of GABA in nicotine withdrawal is not entirely clear (D'Souza and Markou, 2013). However, recent evidence suggested that GABAB1 subunit of the GABAB receptor is involved in the regulation of behavioral alterations induced by nicotine withdrawal. In particular, the severity of physical withdrawal signs and anxiety was markedly reduced in mice with GABAB1 deletion compared with wild-type littermates (Varani et al., 2015). Hence, the possibility that the activation of PPARγ may counteract the expression of nicotine withdrawal symptoms through the modulation of GABA transmission in HIPP or AMY also exists and requires further investigation.
To further understand the role of PPARγ receptors in the modulation of physical and affective nicotine withdrawal signs, we performed microinjection studies to selectively deliver pioglitazone in the dorsal HIPP and the AMY.
Results showed that intra-amygdalar injections of pioglitazone abolished the anxiogenic response linked to protracted nicotine abstinence but did not significantly modify the expression of the physical withdrawal signs. On the other hand, pioglitazone microinjected into the HIPP attenuated the expression of the physical but not the affective withdrawal signs. Overall, these results suggest a brain region-dependent role of PPARγ in the modulation of physical and affective aspects of nicotine withdrawal.
Based on expression data, at the mechanistic level it is tempting to speculate that in the HIPP expression of the physical withdrawal may involve PPARγ-mediated adaptations in the GABAergic transmission. Whereas, in AMY PPARγ appears to interact with glutamatergic transmission to control the expression of physical signs of nicotine withdrawal and with GABA to regulate affective responses linked to protracted abstinence. Because of the close apposition and the small size of the basolateral and central portion of the AMY, it was not possible to target specifically these two subregions with pioglitazone microinjections. However, nicotine withdrawal increased PPARγ expression only in the BLA (in both GABAergic and glutamatergic neurons) but did not alter the expression of PPARγ in the CeA neurons, suggesting that the effect of pioglitazone may probably involve the BLA.
The negative condition associated with nicotine withdrawal is one of the major factors that maintain tobacco use and that promotes resumption of smoking in patients attempting to quit smoking (D'Souza and Markou, 2011). Anxiolytic and antidepressant drugs have been proposed to ameliorate nicotine withdrawal symptoms and are used to aid smoking cessation (Hughes et al., 2000, 2014). Preclinical research has shown that activation of PPARγ may result in marked anxiolytic and antidepressant effects, in part because of the receptor's ability to modulate AMY neurotransmission and in part through inhibition of microglia function (Sadaghiani et al., 2011; Domi et al., 2016; Zhao et al., 2016; Guo et al., 2017). These data have been confirmed in clinical trials that demonstrated the efficacy of pioglitazone as mood stabilizer and in the treatment of unremitted depression (Kemp et al., 2012; Sepanjnia et al., 2012; Zeinoddini et al., 2015; Colle et al., 2017). Preclinical experiments demonstrated that pioglitazone attenuated alcohol, opioid, cocaine consumption, and reinstatement of drug seeking in rodents (Stopponi et al., 2013; de Guglielmo et al., 2017; Miller et al., 2018). Of note pilot clinical experiments confirmed that pioglitazone attenuates craving intensity and improves brain white matter integrity in cocaine-use disorder patients (Schmitz et al., 2017), and importantly, in a recent clinical trial pioglitazone reduced nicotine craving in heavy smokers (Jones et al., 2017). In contrast with our data on nicotine, in a recent small randomized clinical trial, pioglitazone was unable to prevent the expression of opioid withdrawal symptoms pointing to the possibility that the effects on withdrawal are substance dependent (Schroeder et al., 2018).
Together, the results of our study shed new light on neurobiological mechanisms responsible for the effect of PPARγ agonists on drug abuse and support the possibility of using these compounds as adjunct treatments for smoking cessation. In this regard, it is important to highlight that clinical studies revealed the efficacy of PPARγ agonists in treatment of chronic obstructive pulmonary disease, a pathological condition that is largely due to smoking (Lakshmi et al., 2017).
Footnotes
This work was supported by a fellowship (to E.D.) from the Italian Society of Pharmacology, and by NIH Grants RO1 AA017447 and AA014351, and Grant PRIN 2017 (2017SXEXT5) to R.C. We thank Dr. Kevin Niswender at Vanderbild University (Nashville, TN) for providing the PPARγ engineered mouse lines, Serena Stopponi for animal care supervision, and Alfredo Fiorelli for technical support.
G.D. is the Chairman and CEO of Omeros, G.G. is Chief Scientific Officer of Omeros, and R.C. is the inventor on several patent applications relating to the therapeutic use of PPARγ agonists in addiction. Omeros, through agreements with the University of Camerino and with R.C., exclusively controls the intellectual property rights directed to R.C.'s inventions related to the use of PPARγ receptor agonists for the treatment of addiction and addictive behaviors. Under these agreements, R.C. may be entitled to receive payments and royalties from Omeros. The remaining authors declare no competing financial interests.
References
- Brunzell DH, Russell DS, Picciotto MR (2003) In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57Bl/6J mice. J Neurochem 84:1431–1441. 10.1046/j.1471-4159.2003.01640.x [DOI] [PubMed] [Google Scholar]
- Burns KA, Vanden Heuvel JP (2007) Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 1771:952–960. 10.1016/j.bbalip.2007.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caan B, Coates A, Schaefer C, Finkler L, Sternfeld B, Corbett K (1996) Women gain weight 1 year after smoking cessation while dietary intake temporarily increases. J Am Diet Assoc 96:1150–1155. 10.1016/S0002-8223(96)00296-9 [DOI] [PubMed] [Google Scholar]
- Caputi FF, Carretta D, Lattanzio F, Palmisano M, Candeletti S, Romualdi P (2015) Proteasome subunit and opioid receptor gene expression down-regulation induced by paraquat and maneb in human neuroblastoma SH-SY5Y cells. Environ Toxicol Pharmacol 40:895–900. 10.1016/j.etap.2015.09.019 [DOI] [PubMed] [Google Scholar]
- Caputi FF, Palmisano M, Carboni L, Candeletti S, Romualdi P (2016) Opioid gene expression changes and post-translational histone modifications at promoter regions in the rat nucleus accumbens after acute and repeated 3,4-methylenedioxy-methamphetamine (MDMA) exposure. Pharmacol Res 114:209–218. 10.1016/j.phrs.2016.10.023 [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75:889–907. 10.1046/j.1471-4159.2000.0750889.x [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Astarita G, Duranti A, Caprioli G, Ubaldi M, Stopponi S, Kallupi M, Sagratini G, Rodrìguez de Fonseca F, Piomelli D, Ciccocioppo R (2011) Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PLoS One 6:e28142. 10.1371/journal.pone.0028142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cippitelli A, Domi E, Ubaldi M, Douglas JC, Li HW, Demopulos G, Gaitanaris G, Roberto M, Drew PD, Kane CJM, Ciccocioppo R (2017) Protection against alcohol-induced neuronal and cognitive damage by the PPARγ receptor agonist pioglitazone. Brain Behav Immun 64:320–329. 10.1016/j.bbi.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colle R, de Larminat D, Rotenberg S, Hozer F, Hardy P, Verstuyft C, Fève B, Corruble E (2017) Pioglitazone could induce remission in major depression: a meta-analysis. Neuropsychiatr Dis Treat 13:9–16. 10.2147/NDT.S121149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biasi M, Dani JA (2011) Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci 34:105–130. 10.1146/annurev-neuro-061010-113734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guglielmo G, Melis M, De Luca MA, Kallupi M, Li HW, Niswender K, Giordano A, Senzacqua M, Somaini L, Cippitelli A, Gaitanaris G, Demopulos G, Damadzic R, Tapocik J, Heilig M, Ciccocioppo R (2015) PPARγ activation attenuates opioid consumption and modulates mesolimbic dopamine transmission. Neuropsychopharmacology 40:927–937. 10.1038/npp.2014.268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guglielmo G, Kallupi M, Scuppa G, Demopulos G, Gaitanaris G, Ciccocioppo R (2017) Pioglitazone attenuates the opioid withdrawal and vulnerability to relapse to heroin seeking in rodents. Psychopharmacology 234:223–234. 10.1007/s00213-016-4452-1 [DOI] [PubMed] [Google Scholar]
- Di Prisco S, Merega E, Bonfiglio T, Olivero G, Cervetto C, Grilli M, Usai C, Marchi M, Pittaluga A (2016) Presynaptic, release-regulating mGlu2-preferring and mGlu3-preferring autoreceptors in CNS: pharmacological profiles and functional roles in demyelinating disease. Br J Pharmacol 173:1465–1477. 10.1111/bph.13442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domi E, Uhrig S, Soverchia L, Spanagel R, Hansson AC, Barbier E, Heilig M, Ciccocioppo R, Ubaldi M (2016) Genetic deletion of neuronal PPARγ enhances the emotional response to acute stress and exacerbates anxiety: an effect reversed by rescue of amygdala PPARγ function. J Neurosci 36:12611–12623. 10.1523/JNEUROSCI.4127-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza MS, Markou A (2011) Neuronal mechanisms underlying development of nicotine dependence: implications for novel smoking-cessation treatments. Addict Sci Clin Pract 6:4–16. [PMC free article] [PubMed] [Google Scholar]
- D'Souza MS, Markou A (2013) The “stop” and “go” of nicotine dependence: role of GABA and glutamate. Cold Spring Harb Perspect Med 3:a012146. 10.1101/cshperspect.a012146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson LB, Most D, Blednov YA, Harris RA (2014) PPAR agonists regulate brain gene expression: relationship to their effects on ethanol consumption. Neuropharmacology 86:397–407. 10.1016/j.neuropharm.2014.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher ML, LeMalefant RM, Zhou L, Huang G, Turner JR (2017) Distinct roles of CREB within the ventral and dorsal hippocampus in mediating nicotine withdrawal phenotypes. Neuropsychopharmacology 42:1599–1609. 10.1038/npp.2016.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Bueno B, Madrigal JL, Lizasoain I, Moro MA, Lorenzo P, Leza JC (2005) Peroxisome proliferator-activated receptor gamma activation decreases neuroinflammation in brain after stress in rats. Biol Psychiatry 57:885–894. 10.1016/j.biopsych.2005.01.007 [DOI] [PubMed] [Google Scholar]
- García-Bueno B, Madrigal JL, Pérez-Nievas BG, Leza JC (2008) Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology 149:1969–1978. 10.1210/en.2007-0482 [DOI] [PubMed] [Google Scholar]
- Gofflot F, Chartoire N, Vasseur L, Heikkinen S, Dembele D, Le Merrer J, Auwerx J (2007) Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell 131:405–418. 10.1016/j.cell.2007.09.012 [DOI] [PubMed] [Google Scholar]
- Grunberg NE. (2007) A neurobiological basis for nicotine withdrawal. Proc Natl Acad Sci U S A 104:17901–17902. 10.1073/pnas.0708964104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Li C, Lei Y, Xu S, Zhao D, Lu XY (2017) Role of the adipose PPARγ-adiponectin axis in susceptibility to stress and depression/anxiety-related behaviors. Mol Psychiatry 22:1056–1068. 10.1038/mp.2016.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjiconstantinou M, Neff NH (2011) Nicotine and endogenous opioids: neurochemical and pharmacological evidence. Neuropharmacology 60:1209–1220. 10.1016/j.neuropharm.2010.11.010 [DOI] [PubMed] [Google Scholar]
- Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ (1997) LY354740: a metabotropic glutamate receptor agonist which ameliorates symptoms of nicotine withdrawal in rats. Neuropharmacology 36:1511–1516. 10.1016/S0028-3908(97)00170-6 [DOI] [PubMed] [Google Scholar]
- Hughes JR, Hatsukami DK, Skoog KP (1986) Physical dependence on nicotine in gum: a placebo substitution trial. JAMA 255:3277–3279. 10.1001/jama.1986.03370230083036 [DOI] [PubMed] [Google Scholar]
- Hughes JR, Stead LF, Lancaster T (2000) Anxiolytics and antidepressants for smoking cessation. Cochrane Database Syst Rev 2:CD000031. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Stead LF, Hartmann-Boyce J, Cahill K, Lancaster T (2014) Antidepressants for smoking cessation. Cochrane Database Syst Rev 1:CD000031. 10.1002/14651858.CD000031.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Godoy JA, Quintanilla RA, Koenig CS, Bronfman M (2005) Peroxisome proliferator-activated receptor gamma is expressed in hippocampal neurons and its activation prevents beta-amyloid neurodegeneration: role of Wnt signaling. Exp Cell Res 304:91–104. 10.1016/j.yexcr.2004.09.032 [DOI] [PubMed] [Google Scholar]
- Jones JD, Comer SD, Metz VE, Manubay JM, Mogali S, Ciccocioppo R, Martinez S, Mumtaz M, Bisaga A (2017) Pioglitazone, a PPARγ agonist, reduces nicotine craving in humans, with marginal effects on abuse potential. Pharmacol Biochem Behav 163:90–100. 10.1016/j.pbb.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JD, Bisaga A, Metz VE, Manubay JM, Mogali S, Ciccocioppo R, Madera G, Doernberg M, Comer SD (2018) The PPARγ agonist pioglitazone fails to alter the abuse potential of heroin, but does reduce heroin craving and anxiety. J Psychoactive Drugs 50:390–401. 10.1080/02791072.2018.1508789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JR, Shelton KD, Guan Y, Breyer MD, Magnuson MA (2002) Generation and functional confirmation of a conditional null PPARγ allele in mice. Genesis 32:134–137. 10.1002/gene.10042 [DOI] [PubMed] [Google Scholar]
- Jorenby DE, Hatsukami DK, Smith SS, Fiore MC, Allen S, Jensen J, Baker TB (1996) Characterization of tobacco withdrawal symptoms: transdermal nicotine reduces hunger and weight gain. Psychopharmacology 128:130–138. 10.1007/s002130050118 [DOI] [PubMed] [Google Scholar]
- Kemp DE, Ismail-Beigi F, Ganocy SJ, Conroy C, Gao K, Obral S, Fein E, Findling RL, Calabrese JR (2012) Use of insulin sensitizers for the treatment of major depressive disorder: a pilot study of pioglitazone for major depression accompanied by abdominal obesity. J Affect Disord 136:1164–1173. 10.1016/j.jad.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Markou A (2001) Neurobiology of the nicotine withdrawal syndrome. Pharmacol Biochem Behav 70:531–549. 10.1016/S0091-3057(01)00651-7 [DOI] [PubMed] [Google Scholar]
- Kenny PJ, File SE, Rattray M (2001) Nicotine regulates 5-HT1A receptor gene expression in the cerebral cortex and dorsal hippocampus. Eur J Neurosci 13:1267–1271. 10.1046/j.0953-816x.2001.01501.x [DOI] [PubMed] [Google Scholar]
- Lakshmi SP, Reddy AT, Reddy RC (2017) Emerging pharmaceutical therapies for COPD. Int J Chron Obstruct Pulmon Dis 12:2141–2156. 10.2147/COPD.S121416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Di Ciano P, Panlilio LV, Goldberg SR, Ciccocioppo R (2013) Peroxisome proliferator-activated receptor (PPAR) agonists as promising new medications for drug addiction: preclinical evidence. Curr Drug Targets 14:768–776. 10.2174/1389450111314070006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liechti ME, Lhuillier L, Kaupmann K, Markou A (2007) Metabotropic glutamate 2/3 receptors in the ventral tegmental area and the nucleus accumbens shell are involved in behaviors relating to nicotine dependence. J Neurosci 27:9077–9085. 10.1523/JNEUROSCI.1766-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, Tsai YS, Lin SC, Liao NS, Jan MS, Liang CT, Hsu SW, Chen WC, Sung JM, Maeda N, Tsai PJ (2016) Quantitative PPARγ expression affects the balance between tolerance and immunity. Sci Rep 6:26646. 10.1038/srep26646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lyon E. (2001) Mutation detection using fluorescent hybridization probes and melting curve analysis. Expert Rev Mol Diagn 1:92–101. 10.1586/14737159.1.1.92 [DOI] [PubMed] [Google Scholar]
- Malin DH, Lake JR, Smith TD, Khambati HN, Meyers-Paal RL, Montellano AL, Jennings RE, Erwin DS, Presley SE, Perales BA (2006) Bupropion attenuates nicotine abstinence syndrome in the rat. Psychopharmacology 184:494–503. 10.1007/s00213-005-0135-z [DOI] [PubMed] [Google Scholar]
- Michalak A, Biala G (2017) Calcium homeostasis and protein kinase/phosphatase balance participate in nicotine-induced memory improvement in passive avoidance task in mice. Behav Brain Res 317:27–36. 10.1016/j.bbr.2016.09.023 [DOI] [PubMed] [Google Scholar]
- Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O'Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W (2006) International union of pharmacology: LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 58:726–741. 10.1124/pr.58.4.5 [DOI] [PubMed] [Google Scholar]
- Miller WR, Fox RG, Stutz SJ, Lane SD, Denner L, Cunningham KA, Dineley KT (2018) PPARγ agonism attenuates cocaine cue reactivity. Addict Biol 23:55–68. 10.1111/adb.12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Farioli-Vecchioli S, Cerù MP (2004) Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 123:131–145. 10.1016/j.neuroscience.2003.08.064 [DOI] [PubMed] [Google Scholar]
- Morgenweck J, Abdel-Aleem OS, McNamara KC, Donahue RR, Badr MZ, Taylor BK (2010) Activation of peroxisome proliferator-activated receptor gamma in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology 58:337–345. 10.1016/j.neuropharm.2009.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Xu T, Mittal N (2001) Effects of protracted nicotine exposure and withdrawal on the expression and phosphorylation of the CREB gene transcription factor in rat brain. J Neurochem 77:943–952. 10.1046/j.1471-4159.2001.00309.x [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2003) The mouse brain in stereotaxic coordinates, Ed 2 San Diego: Academic. [Google Scholar]
- Picciotto MR, Corrigall WA (2002) Neuronal systems underlying behaviors related to nicotine addiction: neural circuits and molecular genetics. J Neurosci 22:3338–3341. 10.1523/JNEUROSCI.22-09-03338.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper ME, Cook JW, Schlam TR, Jorenby DE, Baker TB (2011) Anxiety diagnoses in smokers seeking cessation treatment: relations with tobacco dependence, withdrawal, outcome and response to treatment. Addiction 106:418–427. 10.1111/j.1360-0443.2010.03173.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SF, Pauly JR, Marks MJ, Collins AC (1994) An analysis of response to nicotine infusion using an automated radiotelemetry system. Psychopharmacology 115:115–120. 10.1007/BF02244760 [DOI] [PubMed] [Google Scholar]
- Rochette-Egly C. (2003) Nuclear receptors: integration of multiple signalling pathways through phosphorylation. Cell Signal 15:355–366. 10.1016/S0898-6568(02)00115-8 [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Isola D, De Vry J, Fadda F (1999) Effects of nimodipine on extracellular dopamine levels in the rat nucleus accumbens in ethanol withdrawal. Neuropharmacology 38:1361–1369. 10.1016/S0028-3908(99)00039-8 [DOI] [PubMed] [Google Scholar]
- Rubio FJ, Liu QR, Li X, Cruz FC, Leão RM, Warren BL, Kambhampati S, Babin KR, McPherson KB, Cimbro R, Bossert JM, Shaham Y, Hope BT (2015) Context-induced reinstatement of methamphetamine seeking is associated with unique molecular alterations in Fos-expressing dorsolateral striatum neurons. J Neurosci 35:5625–5639. 10.1523/JNEUROSCI.4997-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaghiani MS, Javadi-Paydar M, Gharedaghi MH, Fard YY, Dehpour AR (2011) Antidepressant-like effect of pioglitazone in the forced swimming test in mice: the role of PPAR-gamma receptor and nitric oxide pathway. Behav Brain Res 224:336–343. 10.1016/j.bbr.2011.06.011 [DOI] [PubMed] [Google Scholar]
- Sarruf DA, Yu F, Nguyen HT, Williams DL, Printz RL, Niswender KD, Schwartz MW (2009) Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology 150:707–712. 10.1210/en.2008-0899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JM, Green CE, Hasan KM, Vincent J, Suchting R, Weaver MF, Moeller FG, Narayana PA, Cunningham KA, Dineley KT, Lane SD (2017) PPAR-gamma agonist pioglitazone modifies craving intensity and brain white matter integrity in patients with primary cocaine use disorder: a double-blind randomized controlled pilot trial. Addiction 112:1861–1868. 10.1111/add.13868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnegg CI, Robbins ME (2011) Neuroprotective mechanisms of PPARδ: modulation of oxidative stress and inflammatory processes. PPAR Res 2011:373560. 10.1155/2011/373560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JR, Phillips KA, Epstein DH, Jobes ML, Furnari MA, Kennedy AP, Heilig M, Preston KL (2018) Assessment of pioglitazone and proinflammatory cytokines during buprenorphine taper in patients with opioid use disorder. Psychopharmacology 235:2957–2966. 10.1007/s00213-018-4986-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searcy JL, Phelps JT, Pancani T, Kadish I, Popovic J, Anderson KL, Beckett TL, Murphy MP, Chen KC, Blalock EM, Landfield PW, Porter NM, Thibault O (2012) Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer's disease. J Alzheimers Dis 30:943–961. 10.3233/JAD-2012-111661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepanjnia K, Modabbernia A, Ashrafi M, Modabbernia MJ, Akhondzadeh S (2012) Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: randomized double-blind placebo-controlled trial. Neuropsychopharmacology 37:2093–2100. 10.1038/npp.2012.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiffman SM, Jarvik ME (1976) Smoking withdrawal symptoms in two weeks of abstinence. Psychopharmacology 50:35–39. 10.1007/BF00634151 [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Ehlers CL (2002) Lasting effects of adolescent nicotine exposure on the electroencephalogram, event related potentials, and locomotor activity in the rat. Brain Res Dev Brain Res 138:15–25. 10.1016/S0165-3806(02)00455-8 [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Gilder A, Roth J, Ehlers CL (2003) Increased anxiety-like behavior in adult rats exposed to nicotine as adolescents. Pharmacol Biochem Behav 75:355–361. 10.1016/S0091-3057(03)00093-5 [DOI] [PubMed] [Google Scholar]
- Stopponi S, Somaini L, Cippitelli A, Cannella N, Braconi S, Kallupi M, Ruggeri B, Heilig M, Demopulos G, Gaitanaris G, Massi M, Ciccocioppo R (2011) Activation of nuclear PPARgamma receptors by the antidiabetic agent pioglitazone suppresses alcohol drinking and relapse to alcohol seeking. Biol Psychiatry 69:642–649. 10.1016/j.biopsych.2010.12.010 [DOI] [PubMed] [Google Scholar]
- Stopponi S, de Guglielmo G, Somaini L, Cippitelli A, Cannella N, Kallupi M, Ubaldi M, Heilig M, Demopulos G, Gaitanaris G, Ciccocioppo R (2013) Activation of PPARγ by pioglitazone potentiates the effects of naltrexone on alcohol drinking and relapse in msP rats. Alcohol Clin Exp Res 37:1351–1360. 10.1111/acer.12091 [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Monory K, Hanoune J, Nomikos GG (2002) Nicotine withdrawal syndrome: behavioural distress and selective up-regulation of the cyclic AMP pathway in the amygdala. Eur J Neurosci 16:149–153. 10.1046/j.1460-9568.2002.02061.x [DOI] [PubMed] [Google Scholar]
- Varani AP, Pedrón VT, Machado LM, Antonelli MC, Bettler B, Balerio GN (2015) Lack of GABAB receptors modifies behavioural and biochemical alterations induced by precipitated nicotine withdrawal. Neuropharmacology 90:90–101. 10.1016/j.neuropharm.2014.11.013 [DOI] [PubMed] [Google Scholar]
- Wang J, He X, Guo F, Cheng X, Wang Y, Wang X, Feng Z, Vreugdenhil M, Lu C (2017) Multiple kinases involved in the nicotinic modulation of gamma oscillations in the rat hippocampal CA3 area. Front Cell Neurosci 11:57. 10.3389/fncel.2017.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden A, Truitt J, Merriman M, Ponomareva O, Jameson K, Ferguson LB, Mayfield RD, Harris RA (2016) Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep 6:27618. 10.1038/srep27618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SS, Stinus L, Koob GF, Markou A (2000) Reward and somatic changes during precipitated nicotine withdrawal in rats: centrally and peripherally mediated effects. J Pharmacol Exp Ther 292:1053–1064. [PubMed] [Google Scholar]
- World Health Organization (2017) Report on the global tobacco epidemic: monitoring tobacco use and prevention policies. Geneva: World Health Organization. [Google Scholar]
- Yamazaki Y, Fujii S, Jia Y, Sumikawa K (2006) Nicotine withdrawal suppresses nicotinic modulation of long-term potentiation induction in the hippocampal CA1 region. Eur J Neurosci 24:2903–2916. 10.1111/j.1460-9568.2006.05160.x [DOI] [PubMed] [Google Scholar]
- Zeinoddini A, Sorayani M, Hassanzadeh E, Arbabi M, Farokhnia M, Salimi S, Ghaleiha A, Akhondzadeh S (2015) Pioglitazone adjunctive therapy for depressive episode of bipolar disorder: a randomized, double-blind, placebo-controlled trial. Depress Anxiety 32:167–173. 10.1002/da.22340 [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wu X, Yan S, Xie X, Fan Y, Zhang J, Peng C, You Z (2016) The antidepressant-like effects of pioglitazone in a chronic mild stress mouse model are associated with PPARγ-mediated alteration of microglial activation phenotypes. J Neuroinflammation 13:259. 10.1186/s12974-016-0728-y [DOI] [PMC free article] [PubMed] [Google Scholar]