

The combination of hypoxia and hypercapnia during sleep produces arousal, which helps restore breathing and normalizes blood gases. Hypercapnia and hypoxia produce arousal in mammals by activating central (pH-sensitive) and peripheral (primarily O2-sensitive) chemoreceptors. The relevant chemoreceptors and the neuronal circuits responsible for arousal are largely unknown.
Keywords: arousal systems, chemoreception, hypercapnia, hypoxia, respiration, sleep apnea
Abstract
The combination of hypoxia and hypercapnia during sleep produces arousal, which helps restore breathing and normalizes blood gases. Hypercapnia and hypoxia produce arousal in mammals by activating central (pH-sensitive) and peripheral (primarily O2-sensitive) chemoreceptors. The relevant chemoreceptors and the neuronal circuits responsible for arousal are largely unknown. Here we examined the contribution of two lower brainstem nuclei that could be implicated in CO2 and hypoxia-induced arousal: the retrotrapezoid nucleus (RTN), a CO2-responsive nucleus, which mediates the central respiratory chemoreflex; and the C1 neurons, which are hypoxia activated and produce arousal and blood pressure increases when directly stimulated. Additionally, we assessed the contribution of the carotid bodies (CBs), the main peripheral chemoreceptors in mammals, to hypoxia and CO2-induced arousal. In unanesthetized male rats, we tested whether ablation of the RTN, CBs, or C1 neurons affects arousal from sleep and respiratory responses to hypercapnia or hypoxia. The sleep–wake pattern was monitored by EEG and neck EMG recordings and breathing by whole-body plethysmography. The latency to arousal in response to hypoxia or hypercapnia was determined along with changes in ventilation coincident with the arousal. RTN lesions impaired CO2-induced arousal but had no effect on hypoxia-induced arousal. CB ablation impaired arousal to hypoxia and, to a lesser extent, hypercapnia. C1 neuron ablation had no effect on arousal. Thus, the RTN contributes to CO2-induced arousal, whereas the CBs contribute to both hypoxia and CO2-induced arousal. Asphyxia-induced arousal likely requires the combined activation of RTN, CBs and other central chemoreceptors.
SIGNIFICANCE STATEMENT Hypercapnia and hypoxia during sleep elicit arousal, which facilitates airway clearing in the case of obstruction and reinstates normal breathing in the case of hypoventilation or apnea. Arousal can also be detrimental to health by interrupting sleep. We sought to clarify how CO2 and hypoxia cause arousal. We show that the retrotrapezoid nucleus, a brainstem nucleus that mediates the effect of brain acidification on breathing, also contributes to arousal elicited by CO2 but not hypoxia. We also show that the carotid bodies contribute predominantly to hypoxia-induced arousal. Lesions of the retrotrapezoid nucleus or carotid bodies attenuate, but do not eliminate, arousal to CO2 or hypoxia; therefore, we conclude that these structures are not the sole trigger of CO2 or hypoxia-induced arousal.
Introduction
Hypercapnia and hypoxia during sleep elicit arousal, which facilitates airway clearing in the case of obstruction and reinstates normal breathing in the case of hypoventilation or apnea (Berry and Gleeson, 1997; Younes, 2008; Eckert and Younes, 2014). Impaired arousal to hypercapnia and hypoxia is characteristic of congenital central hypoventilation syndrome (Weese-Mayer et al., 2010) and contributes to sudden unexpected death from epilepsy and sudden infant death syndrome (Buchanan, 2019). In obstructive sleep apnea, excessively frequent arousal contributes to poor sleep quality and daytime somnolence (Eckert and Younes, 2014; Malhotra and Jordan, 2016).
Hypoxia and CO2 cause sleep disruption by activating central and peripheral chemoreceptors that ultimately leads to the activation of neurons in the brainstem and hypothalamus that regulate subcortical structures responsible for forebrain arousal (Guyenet and Abbott, 2013; Kaur et al., 2017; Buchanan, 2019). The relevant chemoreceptors and the pathways linking these chemoreceptors and arousal are largely conjectural. Studies in humans have shown that the point of arousal induced by CO2, hypoxia, resistive loading, and sleep disordered breathing is closely linked with ventilatory effort (Gleeson et al., 1990; Kimoff et al., 1994; Li et al., 2019). Neural ventilatory drive may cause arousal via unidentified components of the brainstem respiratory network or through increased sensory afferent feedback from the lungs, airways, or chest receptors, resulting from increased ventilatory effort (Berry and Gleeson, 1997). However, the brain contains many types of acid-responsive neurons (Nattie and Li, 2012; Guyenet and Abbott, 2013), some of which could facilitate arousal without contributing to the increase in breathing. For example, mouse midbrain serotonergic neurons seem to preferentially regulate CO2-induced arousal, whereas serotonergic cells in the medullary raphe regulate the ventilatory response to CO2 (Smith et al., 2018).
The retrotrapezoid nucleus (RTN) is CO2-responsive, innervates brain regions that are implicated in the control of breathing (Guyenet and Bayliss, 2015), and mediates the hypercapnic ventilatory reflex (Marina et al., 2010; Basting et al., 2015; Souza et al., 2018). These neurons could therefore contribute to CO2-induced arousal by virtue of their ability to increase ventilatory drive or via their axonal projections to structures other than the respiratory pattern generator. Interestingly, in congenital central hypoventilation syndrome, a condition characterized by major deficits of the ventilatory and arousal response to hypercapnia, RTN development is suspected to fail (Weese-Mayer et al., 2010; Ramanantsoa et al., 2011). Here we examine whether ablating the RTN in rats impairs CO2-induced arousal. Since RTN lesion disrupts the hypercapnic ventilatory response (Souza et al., 2018), the persistence of CO2 arousal after RTN lesion would suggest that chemoreceptors other than the RTN contribute to CO2-induced arousal.
We also examined the contribution of the carotid bodies (CBs) to CO2 and hypoxia-induced arousal from sleep. The CBs are synergistically activated by hypoxia and CO2 and mediate the acute ventilatory and cardiovascular responses to hypoxia (Prabhakar and Semenza, 2015; Smith et al., 2015; López-Barneo et al., 2016). The CBs contribute to hypoxia-induced arousal in dogs (Bowes et al., 1981) and lambs (Fewell et al., 1989), but similar evidence does not exist for rats. Furthermore, whether the CBs contribute to CO2-induced arousal remains unknown.
The C1 catecholaminergic neurons of the rostral ventrolateral medulla are another population of cells that could contribute to hypoxia-induced arousal. C1 neurons of rats are robustly activated by CB stimulation and brainstem hypoxia (Guyenet et al., 2013; Angelova et al., 2015), and stimulation of these cells produces arousal and activates breathing (Burke et al., 2014). Accordingly, we tested whether C1 neuron ablation impairs CO2 and hypoxia-induced arousal. In sum, this study is designed to examine the contribution of the CBs and a selection of O2 or CO2-responsive lower brainstem neurons to arousal from sleep in response to hypercapnia or hypoxia.
Materials and Methods
Animals.
All experiments were conducted in accordance with the National Institutes of Health's Guide for the care and use of laboratory animals and approved by the University of Virginia Animal Care and Use Committee (#31870617). We used 33 male adult Sprague Dawley rats (Taconic Biosciences) and 13 male adult Th-cre rats with a targeted insertion of IRES-Cre immediately after the translational stop in the open reading frame of Th (Sprague Dawley background; SD-Th-cretm1sage; TGRA8400), procured from Horizon Discovery and maintained as Cre homozygous as recommended by the company. Rats weighed 275–300 g at the time of surgery and injections and 400–550 g at the time of experiments. Animals were housed at 23°C–24°C under a standard artificial 12 h light-dark cycle with water and food provided ad libitum.
Animal preparation.
All surgical procedures were conducted under aseptic conditions, and body temperature was maintained at 37°C with a servo-controlled heating pad. The depth of anesthesia was judged by the lack of reflex in response to a firm tail pinch. For brain injections and EEG/EMG electrode implantation, rats were anesthetized with a mixture of ketamine (75 mg/kg), xylazine (5 mg/kg), and acepromazine (1 mg/kg, administered i.m.). Additional anesthetic was administered if required (25% of the original dose, i.m.). Brain injections were performed on a stereotaxic frame (David Kopf Instruments), with the bite bar set at 3.5 mm below the interaural line for a flat skull. In all rats, incisions were closed in two layers using absorbable sutures for internal closures, and steel clips and Vetbond adhesive for skin. Antibiotic (ampicillin, 125 mg/kg, i.p.) and analgesic (ketoprofen, 3–5 mg/kg, s.c.) were administered every 24 h for 3 d following surgery.
Lesions with substance P-conjugated saporin.
To ablate the RTN, we injected ([Sar9, Met (O2)11]-substance P-saporin (SSP-SAP; Advanced Targeting Systems) bilaterally. This toxin destroys neurons that express substance P receptors (NK1R) (Nattie and Li, 2002; Takakura et al., 2008). Bilateral microinjections were performed using glass pipettes (25 μm external diameter) via a dorsal craniotomy as described previously (Souza et al., 2018). The RTN is located under the facial motor nucleus (on average, −10.5 to −11.6 mm caudal to bregma, 1.8–2.0 mm lateral, 8.3–8.8 mm ventral from dorsal brain surface) (Paxinos and Watson, 1998). Because stereotaxic coordinates are not reliable to accurately target this nucleus, field potentials elicited in the facial motor nucleus by electrical stimulation of the mandibular branch of the facial nerve were monitored via the injection pipette, and injections were placed 200 μm ventral to the location of these field potentials (120 nl or 2.4 ng of SSP-SAP in 3 sites separated by 200 μm along the anterior–posterior axis, bilaterally) (Brown and Guyenet, 1985; Souza et al., 2018). Control rats received injections of PBS (same volume, number, and location). EEG/EMG electrodes were implanted during the same surgery. Rats recovered for 4 weeks before experimentation.
Lesions of C1 catecholaminergic neurons.
SD-Th-cretm1sage (Th-cre) rats were used in these experiments. C1 neurons, a subtype of catecholaminergic neurons located in the rostral ventrolateral medulla (RVLM), were ablated by microinjecting a Cre-dependent vector (AAV5-FLEX-taCasP3-TEVp, 4.6 × 1012 virus molecules/ml, UNC GTC Vector Core, Chapel Hill, NC) (Yang et al., 2013). This vector expresses the apoptotic enzyme caspase-3 (taCasp3) and its activation protease, the tobacco etch virus protease (TEVp) in Cre-expressing cells (Yang et al., 2013; Morgan et al., 2014). As a control, Th-cre rats received AAV5-hSyn-DIO-hM3D(Gq)-mCherry (8.6 × 1012 molecules/ml, Addgene, 44361-AAV5). This vector was used solely to control for possible nonselective effects of viral transfection in this region of the brain. The effects of activating hM3Dq with clozapine-N-oxide and the distribution of cells expressing mCherry were not examined. Injections of virus were targeted to the RVLM by mapping the facial nucleus as described above. The most rostral injections of vector (120 nl) were placed under the caudal edge of the facial motor nucleus. Two additional injections/side (120 nl each) were placed 400 and 800 μm, respectively, caudal to the first one along the anterior–posterior axis at the same depth and mediolateral coordinate. EEG/EMG electrodes were implanted during the same surgery and rats recovered for 4 weeks before experimentation.
Surgical removal of the CBs.
CB ablation (CBx) and surgery without CBx (CBx-Sham) were conducted under isoflurane anesthesia (2.5% in O2) in aseptic conditions as previously described (Basting et al., 2016). Briefly, a midline incision in the ventral surface of the neck was performed to access the carotid bifurcation. CBs were identified, isolated, and physically removed on both sides. The incisions were then closed in two layers (muscle and skin) with absorbable sutures. Rats received postoperative ampicillin (125 mg/kg, i.p.) and ketoprofen (3–5 mg/kg, s.c.) and were monitored daily. CBx-Sham involved exposing the carotid bifurcation bilaterally without damaging the CBs or the surrounding nerves. CBx or CBx-Sham groups recovered for >1 week and were then reanesthetized using ketamine/xylazine/acepromazine to implant EEG/EMG electrodes. Experiments were conducted after another 2 weeks of recovery from EEG/EMG implantation (i.e., at least 3 weeks and up to 5 weeks from CBx or CBx-Sham surgery).
Implantation of electrodes for EEG and neck EMG recordings.
In all rats, bipolar EEG recordings were performed through stainless-steel jeweler screws (Plastics One) implanted over the left frontal and right parietal cortex, with a ground screw implanted in the occipital bone as previously described (Abbott et al., 2013). Additional screws were implanted for structural support of the head stage. Teflon-coated braided stainless-steel wire connected screws for EEG to Amphenol pins in a plastic head stage. Two additional wires were connected to the head stage, stripped at the tips, and implanted in the superficial muscles of the neck for EMG recordings. The head stage was fixed to the skull using Fast-Cure dental acrylic.
Physiological recordings.
All the experiments were performed between 0900 and 1600 h, at an ambient room temperature of 27°C–28°C. Rats were habituated to the experimental conditions for 2 d before recordings. On the day of the recordings, rats were briefly anesthetized with isoflurane (2.0% in room air for < 5 min) to connect the head stage to an amplifier and then placed unrestrained in a whole-body plethysmography chamber (5 L, EMKA Technologies). Recordings began after 1 h of habituation. Respiratory flow was recorded with a differential pressure transducer. The signal was filtered and amplified (0.1–100 Hz, ×500), and digitized at 1 kHz. Respiratory frequency (fR, in breath/min) and tidal volume (VT) were derived from the flow signal in Spike 2 (version 8, CED). VT (area of the curve of the inspiratory flow) was calibrated by injecting 5 mL of dry air into the chamber with a syringe and was expressed as mL/100 g body weight. Minute-ventilation (VE), the product of fR × VT, was expressed in ml/min/100 g. EEG and EMG signals were bandpass filtered (EEG: 0.5–100 Hz, EMG: 100–3000 Hz), amplified (×1000), and digitized at 1 kHz.
EEG, EMG, and breathing pattern were used to determine sleep–wake state. NREM sleep was identified by the presence of slow-wave EEG activity (delta, 1–4 Hz), low EMG activity, and a regular breathing pattern. REM sleep was characterized by stable theta rhythm (6–8 Hz), EMG atonia, and high breathing variability. Quiet wake was identified by the absence of slow-wave EEG activity, generalized EEG desynchronization, increased and variable EMG activity, increased breathing variability, and the presence of behavioral breathing patterns (sniffing).
Arousal protocols.
Rats were placed in an unrestrained whole-body plethysmography chamber with a continuous air flow of 2.5 L/min. Recording sessions lasted 6 h. The chamber was continuously perfused with room air or, as required by the protocols, with various mixtures of O2, N2, and CO2 (total flow: 2.5 L/min) (Fig. 1A). The gas mixtures were produced by mass flow controllers computer-operated by FlowVision software (Alicat Scientific). To test the arousal response to hypercapnia, 15% CO2 was administered for 60 s into the chamber. The concentration of CO2 used to produce arousal was chosen to produce a rapid elevation in FICO2 that resulted in arousal in every trial in control rats. We measured FICO2 at the level of the rats' head in the plethysmography chamber during 15% CO2 administration using a high-resolution capnograph (Columbus Instruments) and found that FICO2 reached 10% ∼20 s after the hypercapnic gas mixture was switched on (Fig. 1A,B). Based on extrapolation, the level of CO2 inside the chamber reaches an FICO2 plateau of 0.15 for <10 s before rapidly returning to prestimulus levels (Fig. 1B).
Figure 1.

Model of CO2-induced arousal. A, Experimental setup: a distribution manifold on the floor of the plethysmography chamber was used to evenly distribute gases. Air was exhausted through the top of the chamber. Note the location of CO2 sampling point reported in B. B, Mean (±SD) chamber FICO2 during 6 trials measured at the level of the rat (black). Theoretical CO2 curve at sampling point based on nonlinear regression of CO2 readout between 5 and 25 s after stimulus onset (one-phase association, R2 = 0.999; used to extrapolate FICO2 beyond the limit of the sensor used for measurement at 12%). C, Recordings made in a representative intact rat (Retrotrapezoid Nucleus (RTN)-Control) exposed to recurrent hypercapnia. Top to bottom, Streaming FFT of EEG, raw EEG, neck EMG, and respiratory flow. CO2 (15%) was administered to the chamber for 1 min every 10 min. D, Excerpt from C showing an arousal event (bottom, *) manifested in EEG desynchronization, increased EMG activity, and a sigh. The respiratory flow trace represents a biphasic artifact caused by switching of gas valves indicated by gray area overflow trace at the start and end of the CO2 trial.
To test the arousal response to hypoxia, 100% N2 was delivered into the chamber for 60 s, resulting in a nadir FIO2 between 0.08 and 0.10 measured with an electrochemical sensor (KE-50, Figaro). To assess the probability of spontaneous arousal, and control for noise and pressure changes associated with valve switching, trials were conducted in which valve switching occurred, but the gas mixture was unchanged (air-to-air) (FIO2 = 0.21, FICO2 = 0). Stimuli were applied at 10 min intervals over a 6 h recording session on separate days with at least 3 d between recording sessions.
Testing ventilatory reflexes.
The ventilatory response to CO2 and hypoxia were tested in three ways. First, the change in total ventilation (ΔVE) that occurred during CO2 and hypoxia trials used to test arousal was measured at the peak of the response, that is, in the final 10 s of the 60 s trials (for specific parameters of trials, see above). For clarity, this measure is referred to as the peak ΔVE during trials testing arousal. These values were assessed for all groups. Second, the ΔVE at the point of arousal, calculated as the difference between baseline VE before the stimulus and the 5 breaths immediately preceding arousal, was determined for RTN-Lesion and RTN-Control for CO2 trials, and CBx and CBx sham for hypoxia trials. Third, steady-state ventilation during hyperoxia (FICO2 = 0.00, FIO2 = 0.65 for 10–15 min) and during hyperoxic hypercapnia (FICO2 = 0.06, FIO2 = 0.65 for 10–15 min) was examined in the RTN-Lesion rats and RTN-Controls (Souza et al., 2018). Hyperoxia was used in these tests to reduce the activity of the CBs and determine deficits in the central component of the hypercapnic ventilatory reflex (Souza et al., 2018). Steady-state ventilation during normoxia (FICO2 = 0.00, FIO2 = 0.21 for 10–15 min) and during poikilocapnic hypoxia (FICO2 = 0.00, FIO2 = 0.12 for 10–15 min) was examined in CBx and CBX-Sham rats.
Blood gases.
We determined the effects of hypoxia and hypercapnia on blood gases as used in our arousal protocols. These parameters were measured at the 60 s time point when FICO2 would have been the highest or FiO2 lowest in the plethysmography chamber and therefore represent the maximum perturbation, not the blood gases at the point of arousal, which would have been usually much lower. As blood collection required an invasive procedure (artery cannulation), measurements were taken only after completion of the arousal protocol as follows: rats were anesthetized with isoflurane (2.5% via nose cone) in room air and a polyethylene catheter (P-10 connected to P-50, Clay Adams) was implanted in the descending aorta via the right femoral artery, and then exteriorized through the nape of the neck. The analgesic ketoprofen (3–5 mg/kg, s.c.) and the antibiotic ampicillin (125 mg/kg, i.p.) were administered postoperatively, and rats recovered overnight. The following day, the rats were placed in the plethysmography chamber, and arterial blood gases, pH, and bicarbonate were measured using a hand-held iStat configured with CG8+ cartridges (Abbott Point of Care). Three blood samples per rat (0.1–0.2 ml per sample) were drawn over the course of 3 h for normoxia (FIO2 = 0.21), and during peak VE in CO2 and hypoxia trials testing arousal (i.e., 60 s after the beginning of each trial).
Data analysis.
Breathing (fR, VT, and VE) was determined via plethysmography calibrated to 5 ml of air injected into the chamber using a syringe. Sleep–wake behavior was scored as wake, NREM, and REM sleep according to standard criteria based on EEG, EMG, and breathing pattern collected in Spike 2. An arousal event was defined as a continuous period of ≥10 s of EEG desynchronization, with or without EMG activation. Arousal latency was defined as the interval between the beginning of the stimulus (hypoxia or hypercapnia) and the point at which the EEG desynchronized (Fig. 1C,D). We only considered stimulus trials that were preceded by ≥ 20 s of stable NREM sleep. Due to the limited number of trials occurring during REM sleep, we did not examine arousals from this sleep state. At least 7 trials (actual values given in figure legends) were first averaged within a rat to produce individual sleep probability curves for each condition, representing the probability that a rat was asleep at a given time point during a trial. Sleep probability curves were then grouped to generate a grand average for statistical comparison.
To determine the percentage of time spent in each state (NREM, REM, and wake), NREM sleep bout duration, and delta power, were manually scored using a publicly available script in the Spike 2 software (http://ced.co.uk/downloads/spike2scripts). Recordings were scored in 5 s segments and collated as 20 s epochs. Epochs for NREM, REM, and wake were collated over a 6 h period during days used to test the probability of spontaneous arousal. Average delta power was determined by performing a FFT of EEG (FFT size1024, Hanning window, bin resolution 0.9965 Hz), then summing the power (μV2) of the bins within the delta band (0.00–3.88 Hz), and normalizing this value to total EEG power between 0.00 and 32.1 Hz.
Histology.
After the experiments, animals were deeply anesthetized and then perfused transcardially with 4% PFA (Burke et al., 2014; Souza et al., 2018). Brains were removed and postfixed in the same fixative for 12–16 h at 4°C. Brains were sectioned (30 μm; transverse plane) the next morning on a vibratome, and brain slices were stored in cryoprotectant at −20°C. RTN neurons were identified by the presence of Neuromedin B transcripts (NMB). According to Shi et al. (2017), the presence of NMB mRNA is a more selective marker of RTN-chemosensitive neurons than Phox2b immunoreactivity, which is also present in nearby catecholaminergic and cholinergic neurons (Stornetta et al., 2006). To identify NMB mRNA, we used multiplex ISH with RNAscope (Advanced Cell Diagnostics) according to the manufacturer's directions. Sections were briefly washed in sterile PBS, mounted on charged slides, and dried overnight. After two rinses in RNase-free water, sections were incubated with Protease IV (Advanced Cell Diagnostics) for 30 min at 40°C. Sections were rinsed twice in sterile water and incubated in RNAscope catalog oligonucleotide probes for Nmb transcripts for 2 h at 40°C. After incubation with probe, tissue was treated according to the manufacturer's protocol.
C1 neurons were defined by the expression of TH and phenylethanolamine-N-methyl transferase (PNMT) within a region extending from the split of the lateral reticular nucleus to ∼300 μm rostral to the caudal pole of the facial motor nucleus. To identify C1 neurons, we used either Th mRNA detected with RNAScope as described above or immunohistochemistry with antibodies against TH (sheep anti-TH, MilliporeSigma, 1:1000 dilution, catalog number AB1542, RRID:AB_90755) or PNMT (rabbit anti-PNMT, gift from Dr. M. Bohn, Northwestern University, Evanston, IL; dilution 1:5000, RRID:AB_2315181).
Immunohistochemistry was performed on free-floating sections. All immunohistochemistry procedures were conducted at room temperature unless otherwise noted. For fluorescence immunohistochemistry, sections were rinsed, then incubated in a Tris-buffered solution of 10% horse serum, 0.1% Triton-X, and 150 mm sodium chloride, and then rinsed and incubated with primary antibody at 4°C overnight. Sections were then rinsed and incubated with secondary antibody for 60 min and rinsed again before mounting on slides. Slides were covered with ProLong Gold with DAPI anti-fade mounting medium (P36931, Thermo Fisher Scientific). Cells were mapped and counted with a computer assisted stage controlled with Neurolucida software (version 11, MBF Bioscience) using an AxioImager M2 (Carl Zeiss) after methods previously described (Stornetta et al., 2006). Digital photomicrographs using the same microscope were made with a Hamamatsu C11440 Orca-Flash 4.0LT digital camera (MBF Bioscience). Filter settings for Atto 550, Alexa-488, or Atto 647 fluorophores were as follows: Alexa-488, excitation of 500 nm, emission of 535 nm; Atto 550, excitation of 545, emission of 605 nm; Atto 647, excitation of 640 nm, emission of 690 nm. Only cell profiles that included a nucleus in the plane of section were counted and/or mapped. Between 9 and 12 sections were counted per rat.
Experimental design and statistical analysis.
Statistical comparisons were made in Prism software (version 7.03, GraphPad Software). Following either D'Agostino-Pearson or Shapiro–Wilk tests for normality, differences within and between groups were determined using unpaired Student's t test or repeated-measures two-way ANOVA with the appropriate post-test. For the analysis of the sleep probability curves, we used two-way ANOVA for repeated measures followed by Bonferroni's multiple-comparisons test; and for the other comparisons, we used two-tailed unpaired Student's t test. Pearson's correlation and least-squares linear regression were used to assess the correlation between the latency to arousal and the ΔVE at the point of arousal. Plots were generated using 7 trials per animal for RTN-Lesion and RTN-Control for CO2 trials, and CBx and CBx-Sham for hypoxia trials. This analysis does not include individual trials where arousal did not occur within 60 s.
All F and p values for the interaction effects and individual treatment effects are reported either in the text or in corresponding figure legends. Degrees of freedom for each F value are designated by subscripts as per convention. Data are reported as mean ± SD, unless stated otherwise; and differences were considered significant when p < 0.05.
Results
RTN lesion impairs arousal to hypercapnia but not hypoxia
Targeted injection of the toxin SSP-SAP below the facial motor nucleus reduced the number of RTN neurons identified by the presence of Nmb mRNA by 93 ± 5% relative to saline-injected (RTN-Control) rats (Fig. 2A,B,D). To determine the extent of the collateral damage caused by SSP-SAP beyond the destruction of RTN neurons, we examined the loss of NK1R-immunoreactive neurons in regions adjacent to the RTN. Injections of SSP-SAP in the RTN reduced the number of Th mRNA-positive neurons located next to the RTN, in both the rostral C1 adrenergic cell group and caudal A5 noradrenergic cell group (Fig. 2A,C,D), consistent with reports that a proportion of catecholamine cells express NK1R (Makeham et al., 2001). There was also a significant loss of NK1R neurons in the most rostral aspect of the Bötzinger complex (BötC), the region of the ventrolateral medulla adjacent and immediately caudal to the RTN (<500 μm; Fig. 2E, bottom) (Del Negro et al., 2018). However, the NK1R-immunoreactive neurons located 600–1000 μm caudal to the facial motor nucleus (i.e., within the Pre-Bötzinger Complex (pre-BötC)) were intact (Fig. 2E, top). The NKR1-immunoreactive neurons of the pre-BötC are required for breathing rhythm generation (Gray et al., 2001; Del Negro et al., 2018). Thus, targeted injections of SSP-SAP eliminated on average >93% of RTN neurons and caused damage to NK1R-immunoreactive neurons in regions immediately adjacent to the RTN (C1, A5, and BötC), but not in the more caudal pre-BötC.
Figure 2.

Histological analysis of RTN lesions. A, NMB mRNA-expressing RTN neurons (magenta) and Th mRNA-expressing C1 neurons (green) in a transverse section (bregma level −11.6 mm; all bregma references after Paxinos and Watson, 1998) from a saline (left, RTN-Control) and SSP-Saporin (SSP-SAP, right, RTN-Lesion) treated rat. White dashed line indicates the ventral surface of the medulla oblongata. There is absence of NMB+ neurons in the RTN and preservation of C1 catecholaminergic neurons. B, Bilateral counts of NMB+ neurons in the RTN by bregma level. C, Bilateral counts of Th+ neurons by bregma level. D, Total bilateral counts of NMB and TH neurons throughout the RTN in saline and SSP-SAP treated rats (unpaired two-tailed Student's t test, for NMB: t(14) = 7.1, p < 0.0001 (****); for TH: t(14) = 3, p = 0.0093 (**). E, Neurokinin 1 receptor (NK1R, magenta) and TH (green) expression in transverse sections of the Bötzinger Complex (BötC) (top, bregma level −11.8 mm) and Pre-Bötzinger Complex (Pre-BötC) (bottom; bregma level −12.8 mm) in a saline rat (left) and SSP-SAP (right) treated rat. Amb, Nucleus ambiguus. F, Bilateral counts of NK1R-immunoreactive neuronal cell bodies in a single transverse section containing the BötC and pre-BötC. Two-way ANOVA, effect of region (pre-BötC vs BötC), F(1,6) = 114.7, p < 0.0001; effect of lesion, F(1,6) = 23.1, p = 0.003; interaction between lesion and regions, F(1,6) = 43.6, p = 0.0006. Bonferroni-corrected post hoc t test: effect of lesion in pre-BötC, t(12) = 1.2, p = 0.48; effect of lesion in BötC, t(12) = 7.3, p < 0.0001 (****).
RTN lesions had no effect on sleep probability during control (air-to-air) trials (Fig. 3A). In RTN-Control rats, hypercapnia in 21% FiO2 caused arousal in all trials within 40 s of the stimulus onset, whereas, at this time point, only 62 ± 11% of trials resulted in arousal in RTN-Lesion rats (Fig. 3B). Indeed, RTN-Lesion rats had a higher probability to remain asleep at almost every time point during CO2 trials (Fig. 3B). In contrast, the arousal response to hypoxia was unaffected by RTN lesions (Fig. 3C). The peak ΔVE during CO2 trials testing arousal (maximum breathing stimulation elicited by hypercapnia at the 60 s time point of the trials) was greatly reduced in RTN-Lesion rats, whereas the peak ΔVE during hypoxia trials testing arousal was unchanged (Fig. 3D). At rest in a normoxic environment, RTN-Lesion rats had an elevated PaCO2 and a reduced PaO2 relative to controls (Table 1); however, the peak change in PaCO2 during CO2 trials testing arousal was similar in RTN-Lesion and RTN-Control rats (Table 2). The steady-state ventilatory response to hyperoxic hypercapnia (6% FICO2, 65% O2), an approximation of the breathing stimulation caused by brain acidification (based on the presumption of reduced peripheral chemoreceptor activation by CO2), was reduced on average by 66% in RTN-Lesion rats (Fig. 3E), as previously described (Souza et al., 2018). In summary, RTN lesions impaired CO2-induced arousal and markedly reduced the hypercapnic ventilatory response. In contrast, RTN lesions had no effect on hypoxia-induced arousal or the hypoxic ventilatory response (Table 3).
Figure 3.

RTN lesions impairs CO2-induced arousal. A, Sleep probability curves in RTN-Lesion (N = 9) and RTN-Control (N = 10) rats during air-to-air trials (Control: 14 ± 3 trials/rat; RTN-Lesion: 15 ± 3 trials/rat). Repeated-measures two-way ANOVA; interaction between RTN-Lesion and time, F(13,221) = 1.55, p = 0.10; time effect, F(13,221) = 165.3, p < 0.0001; lesion effect, F(1.17) = 1.0, p = 0.32. These curves indicate the rats' propensity to wake up spontaneously during a 2 min period and are represented in light gray without error bars (B, C). B, Sleep probability curves during CO2 trials in RTN-Lesion (N = 9) and RTN-Control (N = 9) rats (Control: 11 ± 2 trials/rat; RTN-Lesion 16 ± 5 trials/rat). Repeated-measures two-way ANOVA; interaction between RTN-Lesion and time, F(13,208) = 15.6, p < 0.0001; time effect, F(13,208) = 401.3, p < 0.0001; lesion effect F(1,16) = 59.1, p < 0.0001. C, Sleep probability curves in RTN-Lesion (N = 7) and RTN-Control (N = 7) rats during hypoxia trials (Control: 10 ± 2 trials/rat; RTN-Lesion: 14 ± 2 trials/rat). Repeated-measures two-way ANOVA; interaction between RTN-Lesion and time, F(13,156) = 0.5, p = 0.92; time effect, F(13,156) = 184.3, p < 0.0001; lesion effect, F(1.12) = 1.1, p = 0.31. D, Peak ΔVE during trials testing arousal in RTN-Lesion and control rats (N = 9 per group for CO2, N = 7 per group for hypoxia; unpaired Student's t test for CO2 trials: t(16) = 9.2). ****p < 0.0001. For hypoxia trials: t(12) = 0.5, p = 0.60. E, Steady-state ventilation during sustained (10–15 min) hyperoxia and hyperoxic hypercapnia in RTN-Lesion (N = 8) and RTN-Control rats (N = 6). Absolute breathing frequency (fR, during hyperoxia, unpaired t(12) = 1.9, p = 0.09; during hypercapnia, unpaired t(12) = 5.9, p < 0.0001); absolute tidal volume (VT, hyperoxia unpaired t(12) = 6.9, p < 0.0001, during hypercapnia, unpaired t(12) = 6.9, p < 0.0001), absolute minute-volume (VE, during hyperoxia, unpaired t(12) = 11.0, p < 0.0001, during hypercapnia, unpaired t(12) = 8.4, p < 0.0001). **p < 0.01; ****p < 0.0001; RTN-Lesion versus RTN-Control by Bonferroni's multiple comparisons (A–C) or unpaired t test (D, E).
Table 1.
Resting blood gases in experimental groups in normoxiaa
| n | pH | PaCO2 | PaO2 | HCO3− | |
|---|---|---|---|---|---|
| RTN-Control | 6 | 7.48 ± 0.02 | 42.4 ± 2 | 84.5 ± 7 | 31.9 ± 2 |
| RTN-Lesion | 7 | 7.45 ± 0.03 | 54.4 ± 8* | 74.4 ± 8* | 37.9 ± 4* |
| CBx-Sham | 5 | 7.47 ± 0.03 | 43.4 ± 3 | 84.9 ± 6 | 31.3 ± 2 |
| CBx | 5 | 7.48 ± 0.01 | 45.0 ± 5 | 80.0 ± 7 | 33.5 ± 4 |
| C1-Control | 4 | 7.47 ± 0.01 | 42.1 ± 2 | 82.3 ± 6 | 30.4 ± 2 |
| C1-Lesion | 5 | 7.46 ± 0.02 | 43.0 ± 1 | 84.0 ± 6 | 30.1 ± 1 |
aData are mean ± SD.
*p < 0.05. RTN-Lesion versus RTN-Control, unpaired t test for pH, t(11) = 2.1, p = 0.06; PaCO2, t(11) = 3.7, p = 0.0035; PaO2, t(11) = 2.4, p = 0.034; HCO3−, t(11) = 3.8, p = 0.0032. CBx-Sham versus CBx, unpaired t test for pH, t(9) = 0.9, p = 0.41; PaCO2, t(9) = 0.7, p = 0.53; PaO2, t(9) = 1.1, p = 0.30; HCO3−, t(9) = 1.2, p = 0.25. C1-Lesion versus C1-Control, unpaired t test for pH, t(7) = 0.7, p = 0.49; PaCO2, t(7) = 0.8, p = 0.47; PaO2, t(7) = 0.4, p = 0.68; HCO3−, t(7) = 1, p = 0.92.
Table 2.
Peak changes in PaCO2 and PaO2 during CO2 and hypoxia trials testing arousala
| n | Hypercapnia |
Hypoxia |
||||
|---|---|---|---|---|---|---|
| Δ PaCO2 | Δ PaO2 | Δ PaCO2 | Δ PaO2 | |||
| RTN-Control | 5 | 20.9 ± 4 | 38.6 ± 5 | −14.4 ± 3 | −43.6 ± 8 | |
| RTN-Lesion | 6–7 | 18.1 ± 5 | 30.6 ± 9 | −16.5 ± 3 | −33.3 ± 3* | |
| CBx-Sham | 5 | 11.6 ± 5 | 29.8 ± 14 | −13.6 ± 2 | −43.4 ± 6 | |
| CBx | 5 | 16.8 ± 7 | 40.6 ± 6 | −6.0 ± 3* | −47.0 ± 7 | |
| C1-Control | 3 | 9.5 ± 3 | 27.6 ± 3 | −10 ± 1 | −36.6 ± 5 | |
| C1-Lesion | 5 | 9.1 ± 4 | 29.8 ± 6 | −13 ± 4 | −45.0 ± 8 | |
aData are mean ± SD.
*p < 0.0001 for comparisons within the same experimental groups. RTN-Lesion versus RTN-Control during CO2 trials, unpaired t test for PaCO2, t(10) = 0.9, p = 0.37; PaO2, t(10) = 1.8, p = 0.10. RTN-Lesion versus RTN-Control during hypoxia trials, unpaired t test for PaCO2, t(9) = 1.1, p = 0.31; for PaO2, t(9) = 2.8, p = 0.02. CBx-Sham versus CBx during CO2 trials, unpaired t test for PaCO2, t(8) = 1.3, p = 0.24; PaO2, t(8) = 1.5, p = 0.19. CBx-Sham versus CBx during hypoxia trials, unpaired t test for PaCO2, t(8) = 5.7, p = 0.0005; PaO2, t(8) = 0.9, p = 0.40. C1-Control versus C1-Lesion during CO2 trials, unpaired t test for PaCO2, t(6) = 0.2, p = 0.88; PaO2, t(6) = 0.06, p = 0.58. C1-Control versus C1-Lesion during CO2 trials, unpaired t test for PaCO2, t(6) = 1.5, p = 0.20; PaO2, t(6) = 1.6, p = 0.16.
Table 3.
Respiratory parameters at rest (room air) and exactly 1 min after introducing a hypercapnic (FICO2 = 15%; O2 = 21%) or hypoxic gas mixture (FIO2 = 10%; no CO2 added) into the plethysmography chambera
| n | RTN-Control | RTN-Lesion | CBx-Sham | CBx | C1-Control | C1-Lesion |
|---|---|---|---|---|---|---|
| Resting | ||||||
| fR (bpm) | 68 ± 6 | 97 ± 13* | 69 ± 7 | 61 ± 10 | 86 ± 16 | 81 ± 4 |
| VT (ml/100 g) | 0.45 ± 0.1 | 0.22 ± 0.1* | 0.49 ± 0.1 | 0.41 ± 0.1 | 0.34 ± 0.1 | 0.34 ± 0.1 |
| VE (ml/kg/min) | 30.6 ± 4 | 21.1 ± 6* | 34.1 ± 6 | 24.9 ± 5 | 28.3 ± 4 | 28.1 ± 4 |
| Hypercapnia | ||||||
| fR (bpm) | 130 ± 6 | 106 ± 12* | 128 ± 10 | 118 ± 14 | 123 ± 8 | 120 ± 8 |
| VT (ml/100 g) | 0.75 ± 0.1 | 0.32 ± 0.1* | 0.75 ± 0.1 | 0.68 ± 0.1 | 0.63 ± 0.1 | 0.65 ± 0.2 |
| VE (ml/kg/min) | 98.5 ± 15 | 34.7 ± 11* | 96.7 ± 10 | 80.8 ± 15* | 78.6 ± 18 | 77.8 ± 14 |
| Hypoxia | ||||||
| fR (bpm) | 108 ± 6 | 111 ± 17 | 103 ± 13 | 79 ± 11* | 120 ± 10 | 118 ± 11 |
| VT (ml/100 g) | 0.54 ± 0.1 | 0.51 ± 0.2 | 0.63 ± 0.1 | 0.42 ± 0.1* | 0.52 ± 0.1 | 0.62 ± 0.2 |
| VE (ml/kg/min) | 58.8 ± 10 | 55.7 ± 16 | 64.4 ± 13 | 33.0 ± 7* | 62.3 ± 10 | 75.6 ± 19 |
aData are mean ± SD.
*p < 0.0001 for Bonferroni's multiple comparisons between groups in the same condition: resting, hypercapnia, or hypoxia. Only the two-way ANOVA significant differences were described below. RTN-Control versus RTN-Lesion, two-way repeated-measures ANOVA for fR; interaction between RTN-Lesion and hypercapnia, F(1,16) = 95.5, p < 0.0001; lesion effect, F(1,16) = 0.24, p = 0.6248; hypercapnia effect, F(1,16) = 180.4, p < 0.0001; two-way repeated-measures ANOVA for VT; interaction between RTN-Lesion and hypercapnia, F(1,8) = 37,1, p = 0.0003; lesion effect, F(1,8) = 164.5, p < 0.0001; hypercapnia effect, F(1,8) = 62.2, p < 0.0001; two-way repeated-measures ANOVA for VE; interaction between RTN-Lesion and hypercapnia, F(1,8) = 131.7, p < 0.0001; lesion effect, F(1,8) = 258.6, p < 0.0001; hypercapnia effect, F(1,8) = 139.4, p < 0.0001; CBx-Sham versus CBx, two-way repeated-measures ANOVA for fR; interaction between CBx and hypoxia, F(1,12) = 13.1, p = 0.0035; CBx effect, F(1,12) = 10.8, p = 0.0065; hypoxia effect, F(1,12) = 118.1, p < 0.0001; two-way repeated-measures ANOVA for VT; interaction between CBx and hypoxia, F(1,12) = 35.9, p < 0.0001; CBx effect, F(1,12) = 17.6, p = 0.0012; hypoxia effect, F(1,12) = 52.3, p < 0.0001; two-way repeated-measures ANOVA for VE; interaction between CBx and hypoxia, F(1,12) = 34.6, p < 0.0001; CBx effect, F(1,12) = 23.4, p = 0.0004; hypoxia effect, F(1,12) = 103.1, p < 0.0001.
CB ablation impairs arousal to both hypoxia and hypercapnia
CB excision (CBx) had no effect on sleep probability during control (air to air) trials (Fig. 4A). The probability of arousal in response to CO2 on a normoxic background was slightly but significantly impaired in CBx rats compared with CBx-Sham (Fig. 4B). There was also a pronounced reduction in the probability of arousal in response to hypoxia in CBx rats (Fig. 4C). Indeed, the probability of arousal during the first 50 s of hypoxia trials was not different from the probability of spontaneous arousal; however, the sleep probability curve during hypoxia trials diverged significantly from the air-to-air trials between 60 and 120 s (Fig. 4B). This suggests that hypoxia may also cause arousal by a mechanism independent of the CBs. At rest, CBx rats had similar blood gases and ventilation as CBx-Sham rats (Table 1). The peak changes in PaCO2 and PaO2 observed during CO2 and hypoxia trials testing arousal were similar between CBx and CBx-Sham-operated rats (Table 2). Consistent with the absence of CBs, peak ΔVE after 1 min of hypoxia (as in trials testing arousal) was reduced by 73% in CBx rats relative to CBx-Sham rats, whereas peak ΔVE during CO2 trials was unchanged (Fig. 4D). By contrast, the ΔVE-induced by longer periods of hypoxia (12% O2 for 10–15 min) was not significantly reduced by CBx (CBx vs CBx-Sham; 11 ± 4 vs 16 ± 7 ml/min/100 g; unpaired two-tailed t test, t(11) = 1.7, p = 0.12) (Fig. 4E).
Figure 4.

Carotid body excision impairs hypoxia and CO2-induced arousal. A, Sleep probability curves during air-to-air trials in CBx (N = 7) and CBx-Sham rats (N = 7). CBx-Sham: 14 ± 4 trials/rat; CBx:11 ± 2 trials/rat. Repeated-measures two-way ANOVA; interaction between CBx and time, F(13,156) = 0.6, p = 0.88; time effect, F(13,156) = 165.9, p < 0.0001; CBx effect F(1,12) = 0.8, p = 0.39. B, C, Light gray represents air-to-air controls, without error bars. B, Sleep probability curves during CO2 trials in CBx (N = 6) and CBx-Sham (N = 6) Sprague Dawley rats (CBx-Sham: 12 ± 1 trials/rat; CBx: 12 ± 3 trials/rat). Repeated-measures two-way ANOVA; interaction between CBx and time, F(13,130) = 2.3, p = 0.008; time effect, F(13,130) = 349.0, p < 0.0001; lesion effect, F(1,10) = 4.4, p = 0.06. C, Sleep probability curves during hypoxia trials in CBx (N = 7) and CBx-Sham rats (N = 7). CBx-Sham: 15 ± 4 trials/rat; CBx: 16 ± 4 trials/rat. Repeated-measures two-way ANOVA; interaction between CBx and time, F(13,156) = 8.6, p < 0.0001; time effect, F(13,156) = 338.8, p < 0.0001; CBx effect, F(1,12) = 12.6, p = 0.004. Repeated-measures two-way ANOVA comparing air-air versus air-hypoxia in CBx rats; interaction between gas challenge and time, F(13,156) = 9.9, p < 0.0001; gas challenge effect, F(1,12) = 12.3, p = 0.044; time effect, F(13,156) = 196.6, p < 0.0001. D, Peak ΔVE during trials testing arousal in CBx and CBx-Sham rats (CO2, N = 6 per group, unpaired t(10) = 1.0, p = 0.34; hypoxia, N = 7 per group, unpaired t(12) = 5.9, p < 0.0001). E, Steady-state ventilation during normoxia and sustained (10–15 min) hypoxia in CBx (n = 7) and CBx-Sham (n = 6) rats. Absolute breathing frequency (fR, CBx-Sham vs CBx for normoxia, unpaired t(11) = 0.7, p = 0.50; for hypoxia, unpaired t(11) = 1.0, p = 0.36), absolute tidal volume (VT, CBx-Sham vs CBx for normoxia, unpaired t(11) = 1.3, p = 0.22; for hypoxia, unpaired t(11) = 1.3, p = 0.24), absolute minute-volume (VE, CBx-Sham vs CBx for normoxia, unpaired t(11) = 0.7, p = 0.49; for hypoxia, unpaired t(11) = 2.2, p = 0.047). *p < 0.05; **p < 0.01; ****p < 0.0001; CBx versus CBx-Sham: Bonferroni's multiple comparisons (A–C) or unpaired t test (D, E). #p < 0.05; ##p < 0.01; ###p < 0.001; air versus hypoxia trials in CBx rats (B, Bonferroni's multiple comparisons).
In summary, CBx impaired CO2 and hypoxia-induced arousal, and reduced the rapid breathing stimulation elicited by hypoxia during trials testing arousal. Interestingly, the ventilatory response to sustained hypoxia was only modestly reduced by CBx (Fig. 4E), consistent with the possibility that sensors other than the CBs contribute to the ventilatory effects produced by sustained (>10 min) hypoxia (Gourine and Funk, 2017).
C1 lesions do not affect arousal from sleep in response to hypercapnia or hypoxia
For these experiments, we used a strain of Sprague Dawley rats that expresses Cre-recombinase under the control of the Th promoter. C1 neurons (identified as PNMT-immunoreactive neurons located in the rostral ventrolateral medulla from 180 μm rostral to the caudal pole of the facial nucleus to 1260 μm caudal to the caudal pole of facial motor nucleus) were ablated bilaterally by local injections of AAV5-Flex-taCasp3-TEVp (Yang et al., 2013). This procedure resulted in 86 ± 12% reduction of the C1 cells bilaterally (C1-Lesion vs C1-Intact rats: 47 ± 43 vs 342 ± 47 PNMT-immunoreactive neurons; unpaired t test, t(11) = 12, p < 0.0001; Fig. 5A,B). C1-Lesion had no effect on sleep probability during control (air to air) trials (Fig. 5C). Contrary to expectations, CO2- and hypoxia-induced arousal were both unaffected by C1 ablation (Fig. 5D,E). C1 lesion had no effect either on the peak ventilatory stimulation elicited by either hypercapnia or hypoxia (ΔVE at 60 s) during trials testing arousal (Fig. 5F). Finally, C1 lesion had no effect on arterial blood gases at rest (Table 1).
Figure 5.

C1 neuron ablation has no effect on arousal or ventilatory stimulation during hypercapnia or hypoxia. A, Transverse sections showing phenylethanolamine-N-methyl transferase-immunoreactive neurons in the rostral ventrolateral medulla (RVLM) (C1 region, top, bregma level −12.0 mm) and dorsomedial medulla (C3 region, bottom, bregma level −11.4 mm) in a TH-Cre rat injected with AAV2-DIO-hM3D(Gq)-mCherry (C1-Control, left) or AAV5-Flex-taCasp3-TEVp (C1-Lesion, right). B, Counts of PNMT-immunoreactive cells in the ventrolateral medulla by bregma level (N = 6 for C1-Control, N = 7 for C1-Lesion). C, Sleep probability curves during air-to-air trials (C1-Control: 11 ± 3 trials/rat; C1-Lesion: 11 ± 2 trials/rat). Repeated-measures two-way ANOVA; interaction between C1-Lesion and time, F(13,143) = 0.4, p = 0.98; time effect, F(13,143) = 106.9, p < 0.0001; lesion effect, F(1,11) = 0.01, p = 0.91. Light gray represents air-to-air trials. D, E, Error bars. D, Sleep probability curves during CO2 trials (C1-Control: 11 ± 2 trials/rat; C1-Lesion: 13 ± 5 trials/rat). Repeated-measures two-way ANOVA; interaction between lesion and time, F(13,143) = 0.12, p = 0.99; time effect, F(13,143) = 361.9, p < 0.0001; lesion effect, F(1,11) = 0.02, p = 0.88. E, Sleep probability curves during hypoxia trials (C1-Control: 12 ± 4 trials/rat; C1-Lesion: 13 ± 4 trials/rat). Repeated-measures two-way ANOVA; interaction between C1-Lesion and time, F(13,143) = 1.4, p = 0.17; time effect, F(13,143) = 268.1, p < 0.0001; lesion effect, F(1,11) = 1.3, p = 0.27. F, Peak ΔVE during trials testing arousal for C1-Lesion (N = 7) versus C1-Control (N = 6) rats (CO2, unpaired t(11) = 0.22, p = 0.83; hypoxia, unpaired t(11) = 1.2, p = 0.28).
Ablation of the RTN, CBs, and C1 neurons does not influence basal sleep–wake pattern
Based on a 6 h period of recording, ablation of the RTN, CBs, or C1 neurons had no effect on the proportion of NREM, REM, and waking (Fig. 6A,B), normalized delta power during NREM sleep (Fig. 6C), or the duration of NREM sleep episodes (Fig. 6D).
Figure 6.

Sleep–wake behavior is unaffected by ablation of the RTN, CBs, or C1 neurons. A, Hypnograms from RTN-Control, RTN-Lesion, CBx-Sham, CBx, C1-Control, and C1-Lesion rats. Recording time starting at 0900–1000 h and ending at 1500–1600 h. B, Percentage of time spent in NREM sleep, waking, and REM sleep in a 6 h period of testing during the light phase in all groups of rats. Repeated-measures two-way ANOVA; interaction between RTN-Lesion and sleep stage, F(2,34) = 1.1, p = 0.36; sleep stage effect, F(2,34) = 308.3, p < 0.0001; RTN-Lesion effect, F(1,17) = 4.9, p = 0.99. Repeated-measures two-way ANOVA; interaction between CBx and sleep stage, F(2,24) = 0.02, p = 0.98, sleep stage effect, F(1,14) = 125.6, p < 0.0001; CBx effect, F(1,12) = 0.18, p = 0.68. Interaction between C1-lesion and sleep stage, F(2,22) = 0.2, p = 0.86, sleep stage effect, F(1,14) = 130.4, p < 0.0001, C1-lesion effect, F(1,11) = 0.2, p = 0.68. C, Delta power during NREM sleep in all groups of rats (RTN-Control vs RTN-Lesion, t(10) = 0.2, p = 0.81; CBx-Sham vs CBx, t(10) = 0.002, p = 1.0; C1-Control vs C1-Lesion, t(10) = 1.0, p = 0.36). D, Average duration of NREM bouts in all groups of rats (unpaired t tests, RTN-Control vs RTN-Lesion, t(14) = 0.07, p = 0.94; CBx-Sham vs CBx, t(10) = 0.49, p = 0.63; C1-Control vs C1-Lesion, t(10) = 0.71, p = 0.49).
Relationship between arousal latency and ventilatory stimulation in rats with lesions of RTN or CBs
We measured the ΔVE at the point of arousal (average of the 5 breaths preceding EEG desynchronization) for rats subjected to hypoxia or hypercapnia. In intact rats (RTN-Controls and CBx-Sham rats), the ΔVE at the point of arousal was similar during CO2 and hypoxia trials (Fig. 7A). By contrast, ΔVE at the point of arousal during CO2 and hypoxia trials was greatly reduced in RTN-Lesion rats (Fig. 7A).
Figure 7.
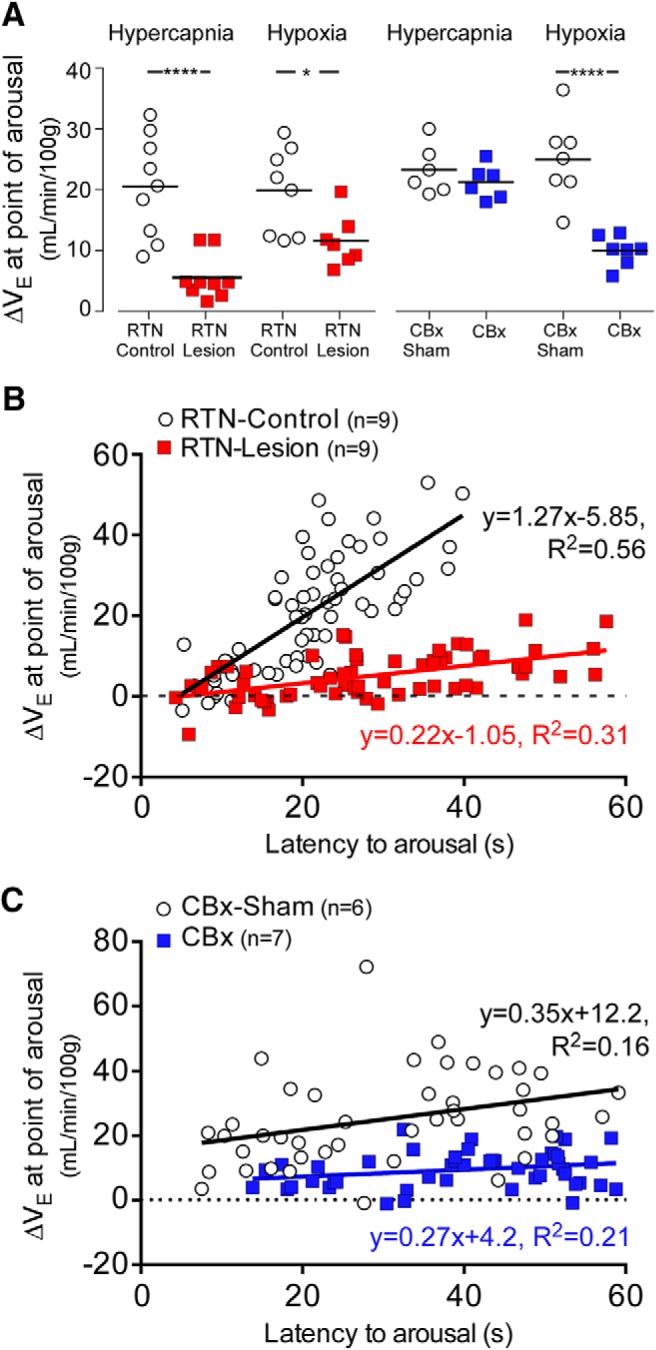
Ventilatory stimulation at the point of arousal in rats exposed to hypercapnia or hypoxia: effects of RTN lesion or CBx. A, Average ΔVE at the point of arousal for RTN-Lesion, RTN-Control, CBx, and CBx-sham rats during CO2 and hypoxia trials used to test arousal. *p < 0.05; ****p < 0.001; unpaired t test. B, Plots of ΔVE at the point of arousal versus latency to arousal during CO2 trials in RTN-Lesion and RTN-Control rats: RTN-Control vs RTN-Lesion; N = 9, 63 data points for both; Pearson r = 0.75, p < 0.0001 versus Pearson r = 0.56, p < 0.0001; linear fit slope (95% CI), 1.27 (0.99–1.56) versus 0.21 (0.13–0.30), F(1,122) = 65.0, p < 0.0001. C, plots of ΔVE at the point of arousal versus latency to arousal during hypoxia trials in CBx and CBx-Sham rats: CBx versus CBx-Sham; N = 7, 49 data points for both; Pearson r = 0.40, p = 0.0045 versus Pearson r = 0.46, p = 0.0009; linear fit slope (95% CI), 0.36 (0.12–0.59) versus 0.27 (0.12–0.42), F(1,94) = 0.55, p = 0.55, Y intercept (95% CI), 12.17 (4.2–19.73) versus 4.22 (−0.34–8.78), F(1,95) = 25.5, p < 0.0001.
We examined in more detail how RTN lesions affected the relationship between latency to arousal and ΔVE at the point of arousal during CO2 trials and how CB ablation changed this relationship during hypoxia trials. During CO2 trials (Fig. 7B), ΔVE at the point of arousal and latency to arousal were strongly correlated in both RTN-Control rats and RTN-Lesion rats, but the slope of the line of best fit was significantly reduced in RTN-Lesion compared with RTN-Control rats (Fig. 7B). During hypoxia trials (Fig. 7C), ΔVE at the point of arousal and latency to arousal were also significantly correlated in both CBx-Sham and CBx rats. The slope of the line of best fit was not statistically different between CBx-Sham and CBx rats, but there was a significant decrease in the Y intercept (Fig. 7C). This analysis showed that ΔVE at the point of arousal is highly variable from trial to trial within and between animals, suggesting that factors other than breathing intensity play a preeminent role in defining the arousal latency to hypercapnia or hypoxia. Second, the results suggest that the contribution of ΔVE to arousal is considerably reduced after lesion of the RTN.
Discussion
This study demonstrates that the RTN and the CBs contribute to CO2-induced arousal, whereas hypoxia-induced arousal requires the CBs but not the RTN. Contrary to expectations, ablation of RVLM C1 neurons had no effect on arousal to either CO2 or hypoxia. These results are compatible with the possibility that RTN neurons and CBs promote arousal partly by increasing ventilatory drive since RTN lesion and CBx greatly reduced the ventilatory stimulation to CO2 and hypoxia, respectively. However, in the absence of RTN, CO2-induced arousal still occurred despite the quasi-absence of ventilatory stimulation and hypoxia-induced arousal persisted in CBx rats. We conclude that neither respiratory network activation nor increased mechanosensation is absolutely required for CO2-induced arousal, and suggest that nonrespiratory chemoreceptors and mechanisms other than ventilatory stimulation contribute to hypercapnia or hypoxia-induced arousal.
Contribution of RTN to CO2-induced arousal
Central chemoreceptors are crucial for CO2-induced arousal (Berry and Gleeson, 1997). Theoretically, central chemoreceptors could elicit arousal via four nonmutually exclusive mechanisms. First (Mechanism 1), a subset of CO2-responsive neurons with minimal influence on the respiratory network (nonrespiratory chemoreceptors) could directly interact with arousal-promoting structures. Second, CO2-induced arousal could be triggered by activation of respiratory chemoreceptors, in which case arousal could result from one of three mechanisms: direct connections between these chemoreceptors and arousal centers (Mechanism 2), indirect connections between these chemoreceptors and arousal centers via the respiratory pattern generator (Mechanism 3), or even more indirect connections via sensory feedback from lung, chest, and airway afferents (Mechanism 4) (Gleeson et al., 1990).
RTN neurons are central respiratory chemoreceptors; they derive a large portion of their CO2 sensitivity from local effects of acidification and their activation produces robust increases in breathing (Abbott et al., 2009; Gourine et al., 2010; Guyenet et al., 2016). Since RTN lesions attenuated CO2-induced arousal, we presume that the activation of these neurons by hypercapnia contributes to arousal in intact animals. An alternative explanation is that the increased sleep probability of RTN-Lesion rats during hypercapnia trials results from a general reduction in arousability. RTN lesions produce chronic mild hypercapnia, which could conceivably raise the arousal threshold (Fraigne et al., 2008). This interpretation is very unlikely, however, given that RTN lesions had no effect in control (air-to-air) trials, no effect on hypoxia-induced arousal, and no effect on sleep architecture.
The present study does not identify by which mechanism (2, 3, or 4) RTN activation causes arousal. Based on their axonal projections to the nucleus of the solitary tract, medullary reticular formation, and lateral parabrachial region (Abbott et al., 2009; Kaur et al., 2017), RTN neurons could promote arousal by activating neurons that have no direct role in breathing regulation (Mechanism 2). However, RTN neurons stimulate breathing powerfully (Abbott et al., 2009) and could trigger arousal by increasing ventilatory effort (Mechanisms 3 and 4). Consistent with this interpretation, in intact rats, ventilatory stimulation at the point of arousal during hypoxia and CO2 trials was similar. This observation bears a resemblance to classic human studies, which show that arousal occurs at the same level of ventilatory effort regardless of the stimulus responsible for increasing ventilation (Gleeson et al., 1990; Kimoff et al., 1994). However, such correlation does not imply causality. Indeed, arousal was still observed in 62% of hypercapnia trials in RTN-Lesion rats despite a near absence of breathing stimulation at the point of arousal. Experiments conducted in artificially ventilated tetraplegic humans in whom hypercapnia elicits arousal, but no ventilatory change have indicated that hypercapnia can induce arousal in the absence of altered respiratory mechanoreception (Ayas et al., 2000). Based on our observation in RTN-Lesion rats, neither mechanosensory feedback nor central respiratory pattern activation (Mechanism 3) is absolutely required for CO2-induced arousal.
The persistence of arousals to hypercapnia despite the virtually complete elimination of RTN neurons and quasi-absence of respiratory stimulation at the point of arousal could have several explanations. First, these arousal events could be triggered by the activation of nonrespiratory central chemoreceptors (Mechanism 1). For example, acidification of the dorsal raphe in mice produces arousal with only a modest increase in breathing (Smith et al., 2018). Second, the permanent loss of RTN neurons could increase the pH sensitivity of other central chemoreceptors or cause a reorganization of the circuitry controlling breathing and/or arousal. Third, the contribution of the CBs to hypercapnia-induced arousal could be enhanced in RTN-Lesion rats because these animals hypoventilate and experience mild hypoxemia and hypercapnia at rest (Souza et al., 2018).
From a clinical perspective, the present results suggest that the reduced CO2-induced arousal and loss of hypercapnic ventilatory reflex observed in congenital central hypoventilation syndrome patients could both be explained, at least in part, by the postulated congenital absence of RTN (Weese-Mayer et al., 2010; Ramanantsoa et al., 2011).
In summary, CO2-induced arousal is partially mediated by RTN neurons, and this effect is possibly, but not necessarily, the consequence of an increase in breathing. In the absence of the RTN, CO2-induced arousal seems driven primarily by mechanisms that are breathing-independent.
Experimental limitations
SSP-SAP damaged other types of NK1R-expressing neurons in addition to RTN. The partial lesion of C1 neurons does not explain the deficit in CO2-induced arousal because specific and far more extensive C1 neuron ablation had no impact on this response. There was a significant loss of NK1R+ neurons in the BötC. The BötC contains neurons that inhibit breathing (Wang et al., 2002). Whether these neurons are NK1R+, hence vulnerable to SSP-SAP, is unknown. The hypoxic ventilatory reflex was not detectably altered in RTN-Lesion rats; therefore, we assume that the loss of NK1R+ neurons in the BötC was inconsequential for breathing regulation. Whether these neurons contribute selectively to CO2-induced arousal cannot be excluded, however.
Mechanism of hypoxia-induced arousal
CBx reduced considerably the probability of hypoxia-induced arousal, consistent with previous studies (Bowes et al., 1981; Fewell et al., 1989). The peak ΔVE during hypoxia trials was reduced by 73% in CBx rats consistent with the possibility that the CBs could promote arousal through ventilatory stimulation.
RTN lesions had no effect on hypoxia-induced arousal. This result seems at odds with the ability of CB stimulation to vigorously activate RTN neurons under anesthesia (Takakura et al., 2006). The likely explanation is that, in unanesthetized rats, the dominant effect of hypoxia on RTN neurons is inhibition due to hyperventilation and resulting hypocapnia (Basting et al., 2015).
Interestingly, CBx reduced, but did not eliminate, hypoxia-induced arousal or the ventilatory response to steady-state hypoxia. The residual arousal and ventilatory effects of hypoxia may be attributed to peripheral oxygen sensors other than the CBs (Piskuric and Nurse, 2012) or to O2 sensors located in the medulla oblongata (Angelova et al., 2015; Gourine and Funk, 2017). In sum, hypoxia-induced arousal is largely mediated by the CBs in intact rats, but activation of central O2 sensors, possibly astrocytes, or peripheral sensors other than the CBs may also produce arousal.
Comprehensive ablation of RVLM C1 neurons had no effect on arousal elicited by hypoxia or hypercapnia, suggesting that these cells are not required for these responses. This outcome was contrary to our expectations because C1 neurons of rats are vigorously activated by CB stimulation and brainstem hypoxia (Guyenet et al., 2013; Marina et al., 2015), and stimulation of these neurons activates the locus ceruleus, produces arousal, and activates breathing (Guyenet et al., 2013; Holloway et al., 2013; Burke et al., 2014; Marina et al., 2015). One interpretation is that the CBs are far more sensitive to hypoxia than the C1 cells and cause powerful arousal via a pathway that does not include the C1 neurons. Once arousal has occurred, the arousal potential of the C1 cells becomes irrelevant.
Finally, in rats with intact RTN, CBx slightly reduced CO2-induced arousal without detectably affecting the ventilatory response to CO2 (Figs. 4D, 7A), suggesting that the contribution of the CBs to CO2-induced arousal could be independent of their effect on ventilation (Zera et al., 2019).
In conclusion, we confirm that CBx impairs hypoxia-induced arousal. We add that CBx also reduces hypercapnia-induced arousal, and demonstrate that CBx does not eliminate hypoxia-induced arousal entirely. Additional hypoxia sensors located either centrally or peripherally are also capable of triggering arousal, at least after CBx.
We provide the first experimental evidence that RTN neurons contribute to CO2-induced arousal. The persistence of arousals to CO2 after RTN lesion also strongly supports the notion that CO2-responsive neurons without effects on breathing (“nonrespiratory” central chemoreceptors) contribute to CO2-induced arousal (Smith et al., 2018). The pathways responsible for the arousal-promoting effects of RTN neurons remain conjectural. Arousal could be mediated by direct connections between RTN neurons and arousal-promoting structures, such as the lateral parabrachial complex, or indirectly via the respiratory pattern generator and mechanosensory feedback (Bochorishvili et al., 2012). The lateral parabrachial complex is a plausible convergence point for inputs from RTN, nucleus tractus solitarius neurons activated by the CBs and nonrespiratory chemoreceptors (Song et al., 2011; Kaur et al., 2017; Smith et al., 2018).
Footnotes
This work was supported by National Institutes of Health Grants HL28785 and HL074011, and the Congenital Central Hypoventilation Syndrome Foundation to P.G.G.
The authors declare no competing financial interests.
References
- Abbott SB, Stornetta RL, Fortuna MG, Depuy SD, West GH, Harris TE, Guyenet PG (2009) Photostimulation of retrotrapezoid nucleus phox2b-expressing neurons in vivo produces long-lasting activation of breathing in rats. J Neurosci 29:5806–5819. 10.1523/JNEUROSCI.1106-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott SB, Coates MB, Stornetta RL, Guyenet PG (2013) Optogenetic stimulation of C1 and retrotrapezoid nucleus neurons causes sleep state-dependent cardiorespiratory stimulation and arousal in rats. Hypertension 61:835–841. 10.1161/HYPERTENSIONAHA.111.00860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova PR, Kasymov V, Christie I, Sheikhbahaei S, Turovsky E, Marina N, Korsak A, Zwicker J, Teschemacher AG, Ackland GL, Funk GD, Kasparov S, Abramov AY, Gourine AV (2015) Functional oxygen sensitivity of astrocytes. J Neurosci 35:10460–10473. 10.1523/JNEUROSCI.0045-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayas NT, Brown R, Shea SA (2000) Hypercapnia can induce arousal from sleep in the absence of altered respiratory mechanoreception. Am J Respir Crit Care Med 162:1004–1008. [DOI] [PubMed] [Google Scholar]
- Basting TM, Burke PG, Kanbar R, Viar KE, Stornetta DS, Stornetta RL, Guyenet PG (2015) Hypoxia silences retrotrapezoid nucleus respiratory chemoreceptors via alkalosis. J Neurosci 35:527–543. 10.1523/JNEUROSCI.2923-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basting TM, Abe C, Viar KE, Stornetta RL, Guyenet PG (2016) Is plasticity within the retrotrapezoid nucleus responsible for the recovery of the PCO2 set-point after carotid body denervation in rats? J Physiol 594:3371–3390. 10.1113/JP272046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry RB, Gleeson K (1997) Respiratory arousal from sleep: mechanisms and significance. Sleep 20:654–675. 10.1093/sleep/20.8.654 [DOI] [PubMed] [Google Scholar]
- Bochorishvili G, Stornetta RL, Coates MB, Guyenet PG (2012) pre-Botzinger complex receives glutamatergic innervation from galaninergic and other retrotrapezoid nucleus neurons. J Comp Neurol 520:1047–1061. 10.1002/cne.22769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes G, Townsend ER, Kozar LF, Bromley SM, Phillipson EA (1981) Effect of carotid body denervation on arousal response to hypoxia in sleeping dogs. J Appl Physiol Respir Environ Exerc Physiol 51:40–45. 10.1152/jappl.1981.51.1.40 [DOI] [PubMed] [Google Scholar]
- Brown DL, Guyenet PG (1985) Electrophysiological study of cardiovascular neurons in the rostral ventrolateral medulla in rats. Circ Res 56:359–369. 10.1161/01.RES.56.3.359 [DOI] [PubMed] [Google Scholar]
- Buchanan GF. (2019) Impaired CO2-induced arousal in SIDS and SUDEP. Trends Neurosci 42:242–250. 10.1016/j.tins.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke PG, Abbott SB, Coates MB, Viar KE, Stornetta RL, Guyenet PG (2014) Optogenetic stimulation of adrenergic C1 neurons causes sleep state-dependent cardiorespiratory stimulation and arousal with sighs in rats. Am J Respir Crit Care Med 190:1301–1310. 10.1164/rccm.201407-1262OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Negro CA, Funk GD, Feldman JL (2018) Breathing matters. Nat Rev Neurosci 19:351–367. 10.1038/s41583-018-0003-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert DJ, Younes MK (2014) Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol 116:302–313. 10.1152/japplphysiol.00649.2013 [DOI] [PubMed] [Google Scholar]
- Fewell JE, Kondo CS, Dascalu V, Filyk SC (1989) Influence of carotid denervation on the arousal and cardiopulmonary response to rapidly developing hypoxemia in lambs. Pediatr Res 25:473–477. 10.1203/00006450-198905000-00009 [DOI] [PubMed] [Google Scholar]
- Fraigne JJ, Dunin-Barkowski WL, Orem JM (2008) Effect of hypercapnia on sleep and breathing in unanesthetized cats. Sleep 31:1025–1033. [PMC free article] [PubMed] [Google Scholar]
- Gleeson K, Zwillich CW, White DP (1990) The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis 142:295–300. 10.1164/ajrccm/142.2.295 [DOI] [PubMed] [Google Scholar]
- Gourine AV, Funk GD (2017) On the existence of a central respiratory oxygen sensor. J Appl Physiol 123:1344–1349. 10.1152/japplphysiol.00194.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S (2010) Astrocytes control breathing through pH-dependent release of ATP. Science 329:571–575. 10.1126/science.1190721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PA, Janczewski WA, Mellen N, McCrimmon DR, Feldman JL (2001) Normal breathing requires preBotzinger complex neurokinin-1 receptor-expressing neurons. Nat Neurosci 4:927–930. 10.1038/nn0901-927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Abbott SB (2013) Chemoreception and asphyxia-induced arousal. Respir Physiol Neurobiol 188:333–343. 10.1016/j.resp.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Bayliss DA (2015) Neural control of breathing and CO2 homeostasis. Neuron 87:946–961. 10.1016/j.neuron.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Bochorishvili G, Depuy SD, Burke PG, Abbott SB (2013) C1 neurons: the body's EMTs. Am J Physiol Regul Integr Comp Physiol 305:R187–R204. 10.1152/ajpregu.00054.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Bayliss DA, Stornetta RL, Ludwig MG, Kumar NN, Shi Y, Burke PG, Kanbar R, Basting TM, Holloway BB, Wenker IC (2016) Proton detection and breathing regulation by the retrotrapezoid nucleus. J Physiol 594:1529–1551. 10.1113/JP271480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway BB, Stornetta RL, Bochorishvili G, Erisir A, Viar KE, Guyenet PG (2013) Monosynaptic glutamatergic activation of locus coeruleus and other lower brainstem noradrenergic neurons by the C1 cells in mice. J Neurosci 33:18792–18805. 10.1523/JNEUROSCI.2916-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Wang JL, Ferrari L, Thankachan S, Kroeger D, Venner A, Lazarus M, Wellman A, Arrigoni E, Fuller PM, Saper CB (2017) A genetically defined circuit for arousal from sleep during hypercapnia. Neuron 96:1153–1167.e5. 10.1016/j.neuron.2017.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimoff RJ, Cheong TH, Olha AE, Charbonneau M, Levy RD, Cosio MG, Gottfried SB (1994) Mechanisms of apnea termination in obstructive sleep apnea: role of chemoreceptor and mechanoreceptor stimuli. Am J Respir Crit Care Med 149:707–714. 10.1164/ajrccm.149.3.8118640 [DOI] [PubMed] [Google Scholar]
- Li Y, Orr J, Jen R, Sands SA, DeYoung P, Smales E, Edwards B, Owens RL, Malhotra A (2019) Is there a threshold that triggers cortical arousals in obstructive sleep apnea? Sleep 42:zsz047. 10.1093/sleep/zsz047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J, Ortega-Sáenz P, González-Rodríguez P, Fernández-Agüera MC, Macías D, Pardal R, Gao L (2016) Oxygen-sensing by arterial chemoreceptors: mechanisms and medical translation. Mol Aspects Med 47:90–108. 10.1016/j.mam.2015.12.002 [DOI] [PubMed] [Google Scholar]
- Makeham JM, Goodchild AK, Pilowsky PM (2001) NK1 receptor and the ventral medulla of the rat: bulbospinal and catecholaminergic neurons. Neuroreport 12:3663–3667. 10.1097/00001756-200112040-00012 [DOI] [PubMed] [Google Scholar]
- Malhotra A, Jordan A (2016) The importance of arousal in obstructive sleep apnea: updates from the American Thoracic Society 2016. J Thorac Dis 8:S542–S544. 10.21037/jtd.2016.06.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Abdala AP, Trapp S, Li A, Nattie EE, Hewinson J, Smith JC, Paton JF, Gourine AV (2010) Essential role of Phox2b-expressing ventrolateral brainstem neurons in the chemosensory control of inspiration and expiration. J Neurosci 30:12466–12473. 10.1523/JNEUROSCI.3141-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Ang R, Machhada A, Kasymov V, Karagiannis A, Hosford PS, Mosienko V, Teschemacher AG, Vihko P, Paton JF, Kasparov S, Gourine AV (2015) Brainstem hypoxia contributes to the development of hypertension in the spontaneously hypertensive rat. Hypertension 65:775–783. 10.1161/HYPERTENSIONAHA.114.04683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BJ, Adrian R, Bates ML, Dopp JM, Dempsey JA (2014) Quantifying hypoxia-induced chemoreceptor sensitivity in the awake rodent. J Appl Physiol 117:816–824. 10.1152/japplphysiol.00484.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E, Li A (2012) Central chemoreceptors: locations and functions. Compr Physiol 2:221–254. 10.1002/cphy.c100083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie EE, Li A (2002) Substance P-saporin lesion of neurons with NK1 receptors in one chemoreceptor site in rats decreases ventilation and chemosensitivity. J Physiol 544:603–616. 10.1113/jphysiol.2002.020032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates, Ed 4 San Diego: Academic. [DOI] [PubMed] [Google Scholar]
- Piskuric NA, Nurse CA (2012) Effects of chemostimuli on [Ca2+]i responses of rat aortic body type I cells and endogenous local neurons: comparison with carotid body cells. J Physiol 590:2121–2135. 10.1113/jphysiol.2012.229468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Semenza GL (2015) Oxygen sensing and homeostasis. Physiology (Bethesda) 30:340–348. 10.1152/physiol.00022.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanantsoa N, Hirsch MR, Thoby-Brisson M, Dubreuil V, Bouvier J, Ruffault PL, Matrot B, Fortin G, Brunet JF, Gallego J, Goridis C (2011) Breathing without CO(2) chemosensitivity in conditional Phox2b mutants. J Neurosci 31:12880–12888. 10.1523/JNEUROSCI.1721-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Stornetta RL, Stornetta DS, Onengut-Gumuscu S, Farber EA, Turner SD, Guyenet PG, Bayliss DA (2017) Neuromedin B expression defines the mouse retrotrapezoid nucleus. J Neurosci 37:11744–11757. 10.1523/JNEUROSCI.2055-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Blain GM, Henderson KS, Dempsey JA (2015) Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO2: role of carotid body CO2. J Physiol 593:4225–4243. 10.1113/JP270114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HR, Leibold NK, Rappoport DA, Ginapp CM, Purnell BS, Bode NM, Alberico SL, Kim YC, Audero E, Gross CT, Buchanan GF (2018) Dorsal raphe serotonin neurons mediate CO2-induced arousal from sleep. J Neurosci 38:1915–1925. 10.1523/JNEUROSCI.2182-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Xu H, Wang H, Macdonald SM, Poon CS (2011) Hypoxia-excited neurons in NTS send axonal projections to Kolliker-Fuse/parabrachial complex in dorsolateral pons. Neuroscience 175:145–153. 10.1016/j.neuroscience.2010.11.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza GM, Kanbar R, Stornetta DS, Abbott SB, Stornetta RL, Guyenet PG (2018) Breathing regulation and blood gas homeostasis after near complete lesions of the retrotrapezoid nucleus in adult rats. J Physiol 596:2521–2545. 10.1113/JP275866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA, Guyenet PG (2006) Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci 26:10305–10314. 10.1523/JNEUROSCI.2917-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG (2006) Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol 572:503–523. 10.1113/jphysiol.2005.103788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Stornetta RL, West GH, Gwilt JM, Guyenet PG (2008) Selective lesion of retrotrapezoid Phox2b-expressing neurons raises the apnoeic threshold in rats. J Physiol 586:2975–2991. 10.1113/jphysiol.2008.153163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Germanson TP, Guyenet PG (2002) Depressor and tachypneic responses to chemical stimulation of the ventral respiratory group are reduced by ablation of neurokinin-1 receptor-expressing neurons. J Neurosci 22:3755–3764. 10.1523/JNEUROSCI.22-09-03755.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H (2010) An official ATS clinical policy statement: congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med 181:626–644. 10.1164/rccm.200807-1069ST [DOI] [PubMed] [Google Scholar]
- Yang CF, Chiang MC, Gray DC, Prabhakaran M, Alvarado M, Juntti SA, Unger EK, Wells JA, Shah NM (2013) Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell 153:896–909. 10.1016/j.cell.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M. (2008) Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J Appl Physiol 105:1389–1405. 10.1152/japplphysiol.90408.2008 [DOI] [PubMed] [Google Scholar]
- Zera T, Moraes DJ, da Silva MP, Fisher JP, Paton JF (2019) The logic of carotid body connectivity to the brain. Physiology (Bethesda) 34:264–282. 10.1152/physiol.00057.2018 [DOI] [PubMed] [Google Scholar]